

The Chromenopyridine Scaffold: A Privileged Platform in Drug Design

Abstract
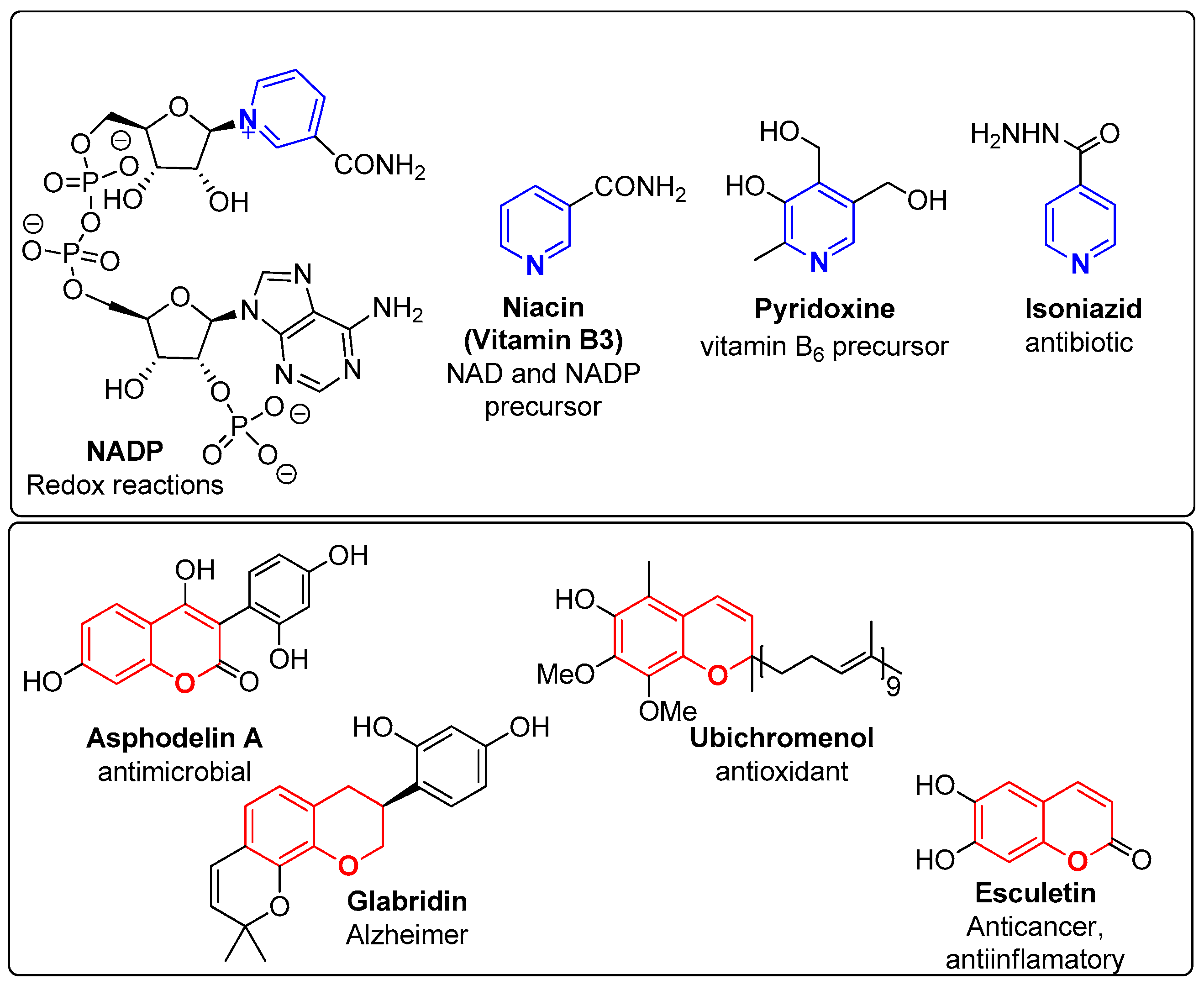
:1. Introduction
2. Synthesis and Biological Activity of Chromenopyridines
2.1. Salicylaldehydes
2.2. Chromones
2.3. Chromanones
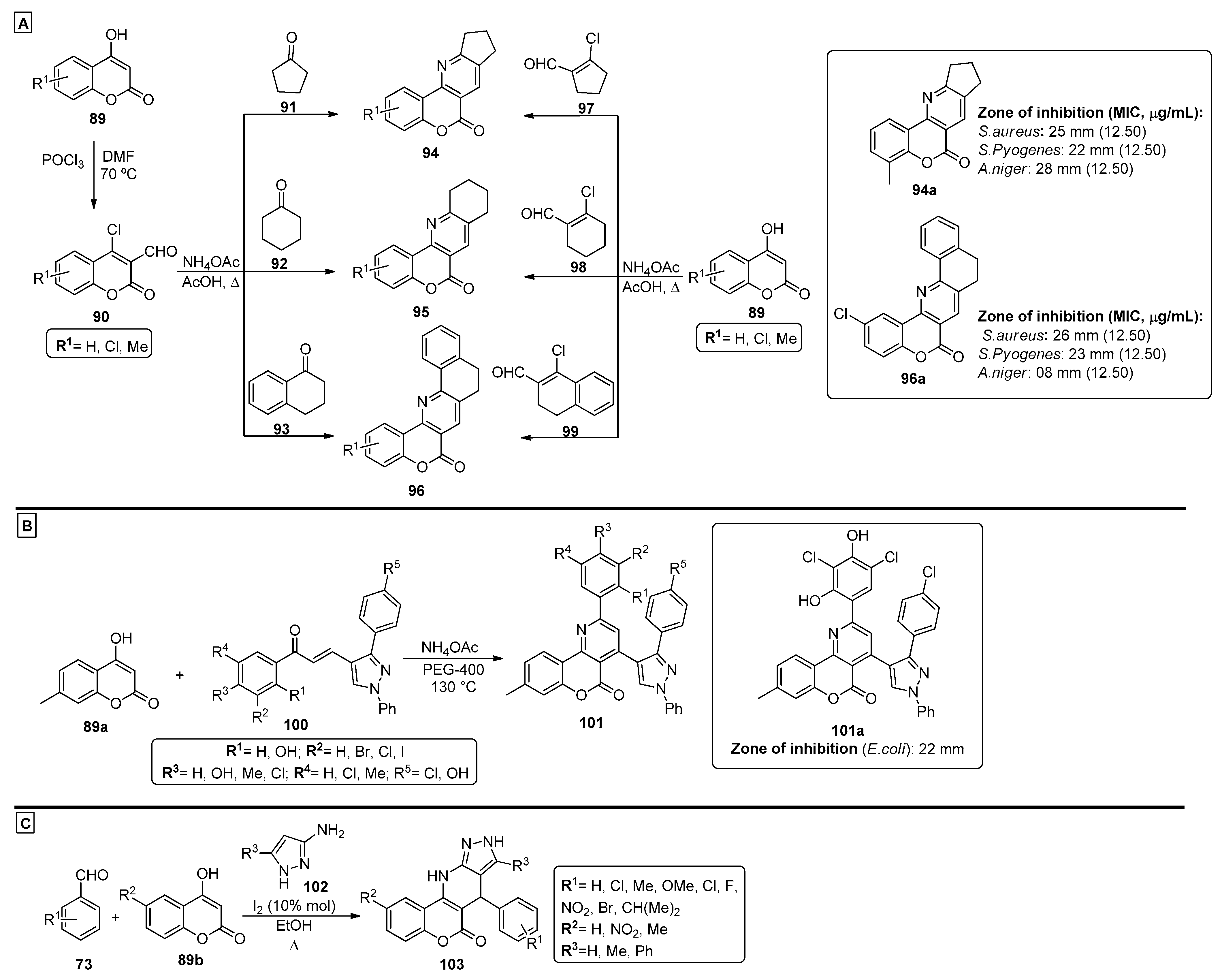
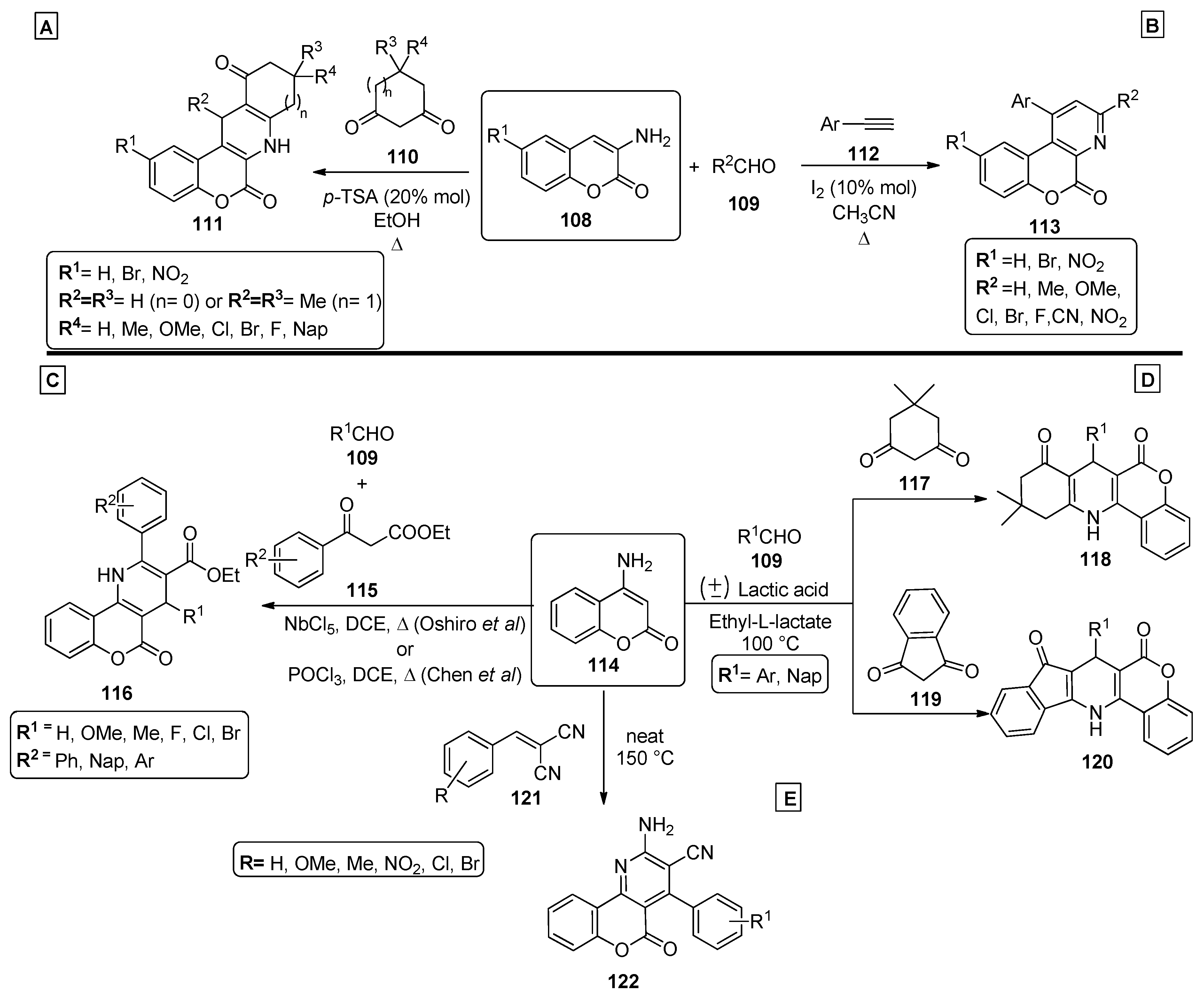
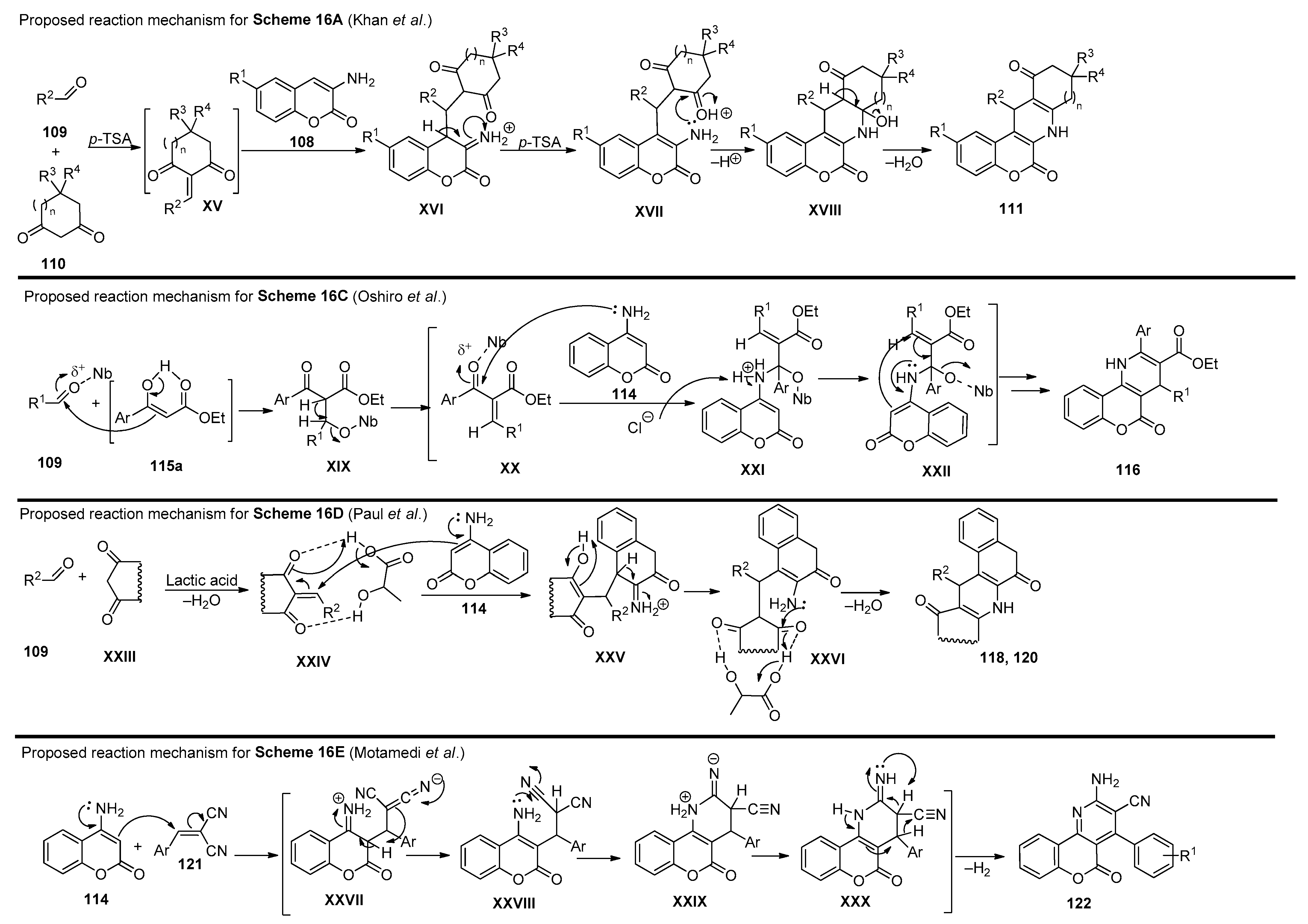
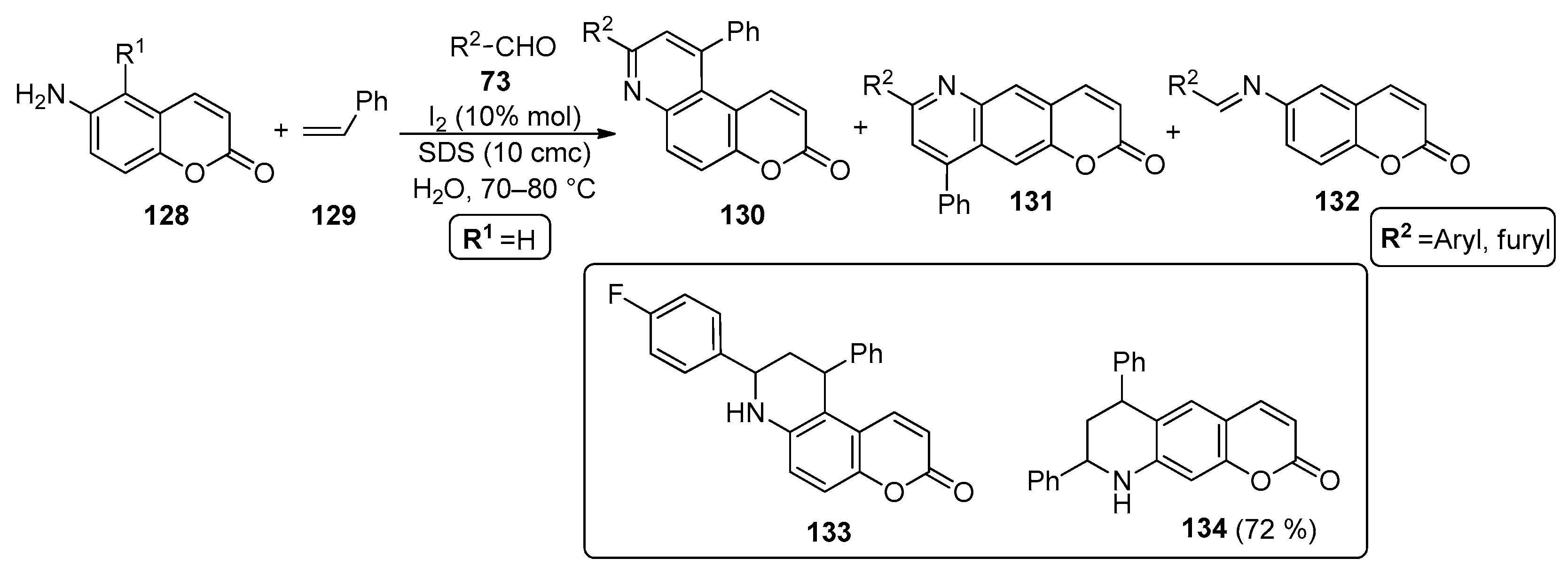
2.4. Coumarins
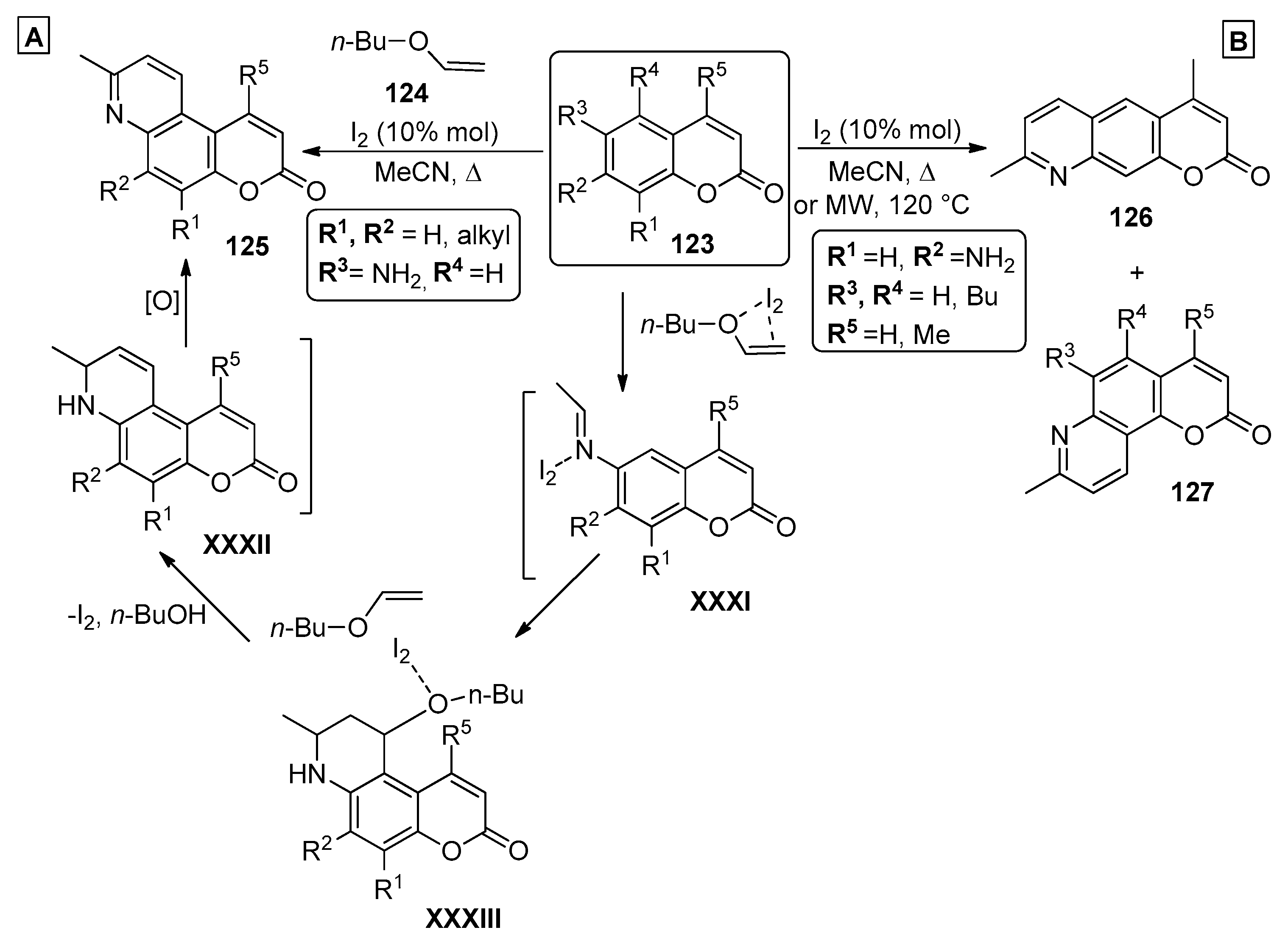
2.5. Miscellaneous
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and Drug Discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Budagumpi, S.; Pai, R.K.; Balakrishna, R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Park, S.B. Privileged Structures: Efficient Chemical “Navigators” toward Unexplored Biologically Relevant Chemical Spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, S.; Pingaew, R.; Worachartcheewan, A.; Sinthupoom, N.; Prachayasittikul, V.; Ruchirawat, S.; Prachayasittikul, V. Roles of Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Anticancer Agents. Mini-Rev. Med. Chem. 2017, 17, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagoniststs. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, C.; Sitkoff, D.; Cheadle, N.L.; Xu, S.; Muckelbauer, J.K.; Adam, L.P.; Wexler, R.R.; Quan, M.L. Identification of 5H-chromeno [3,4-c]pyridine and 6H-isochromeno[3,4-c]pyridine derivatives as potent and selective dual ROCK inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127474. [Google Scholar] [CrossRef] [PubMed]
- Marson, C.M. Targeting the histamine H4 receptor. Chem. Rev. 2011, 111, 7121–7156. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Tabrizi, M.A.; Gessi, S.; Borea, P.A. Adenosine receptor antagonists: Translating medicinal chemistry and pharmacology into clinical utility. Chem. Rev. 2008, 108, 238–263. [Google Scholar] [CrossRef]
- Martinez-Gualda, B.; Pu, S.Y.; Froeyen, M.; Herdewijn, P.; Einav, S.; De Jonghe, S. Structure-activity relationship study of the pyridine moiety of isothiazolo[4,3-b]pyridines as antiviral agents Targeting cyclin G-associated kinase. Bioorg. Med. Chem. 2020, 28, 115188. [Google Scholar] [CrossRef]
- Barreiro, E.J. Chapter 1: Privileged Scaffolds in Medicinal Chemistry: An Introduction. In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; The Royal Society of Chemistry: London, UK, 2016; pp. 1–15. ISBN 978-1-78262-030-3. [Google Scholar]
- Evdokimov, N.M.; Kireev, A.S.; Yakovenko, A.A.; Antipin, M.Y.; Magedov, I.V.; Kornienko, A. One-Step Synthesis of Heterocyclic Privileged Medicinal Scaffolds by a Multicomponent Reaction of Malononitrile with Aldehydes and Thiols. J. Org. Chem. 2007, 72, 3443–3453. [Google Scholar] [CrossRef] [PubMed]
- Henry, G.D. De Novo Synthesis of Substituted Pyridines. Tetrahedron 2004, 60, 6043–6061. [Google Scholar] [CrossRef]
- Edwards, J.P.; Kindrachuk, D.E.; Venable, J.D. Benzo-Imidazolyl Pyridines as Modulators of the Histamine H4 Receptor. Hong. Kong Patent HK1124767A1, 24 July 2009. [Google Scholar]
- Radwan, M.A.A.; Alshubramy, M.A.; Abdel-Motaal, M.; Hemdan, B.A.; El-Kady, D.S. Synthesis, molecular docking and antimicrobial activity of new fused pyrimidine and pyridine derivatives. Bioorg. Chem. 2020, 96, 103516. [Google Scholar] [CrossRef]
- Jian, X.E.; Yang, F.; Jiang, C.S.; You, W.W.; Zhao, P.L. Synthesis and biological evaluation of novel pyrazolo[3,4-b]pyridines as cis-restricted combretastatin A-4 analogues. Bioorg. Med. Chem. Lett. 2020, 30, 127025. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Dias, T.A.; Brito, A.; Proença, F. Biological importance of structurally diversified chromenes. Eur. J. Med. Chem. 2016, 123, 487–507. [Google Scholar] [CrossRef] [PubMed]
- Pontes, O.; Costa, M.; Santos, F.; Sampaio-Marques, B.; Dias, T.; Ludovico, P.; Proença, F.; Baltazar, F. Exploitation of new chalcones and 4H-chromenes as agents for cancer treatment. Eur. J. Med. Chem. 2018, 157, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Azm, F.S.M.; El-Shahawi, M.M.; Elgubbi, A.S.; Madkour, H.M.F. Design, synthesis, anti-proliferative activity, and molecular docking studies of novel benzo[f]chromene, chromeno[2,3-d]pyrimidines and chromenotriazolo[1,5-c]pyrimidines. Synth. Commun. 2020, 50, 669–683. [Google Scholar] [CrossRef]
- Alblewi, F.F.; Okasha, R.M.; Hritani, Z.M.; Mohamed, H.M.; El-Nassag, M.A.A.; Halawa, A.H.; Mora, A.; Fouda, A.M.; Assiri, M.A.; Al-Dies, A.A.M.; et al. Antiproliferative effect, cell cycle arrest and Aaoptosis generation of novel synthesized anticancer heterocyclic derivatives based 4H-benzo[h]chromene. Bioorg. Chem. 2019, 87, 560–571. [Google Scholar] [CrossRef]
- Halawa, A.H.; Elaasser, M.M.; El Kerdawy, A.M.; Abd El-Hady, A.M.A.I.; Emam, H.A.; El-Agrody, A.M. Anticancer activities, molecular docking and structure–activity relationship of novel synthesized 4H-chromene, and 5H-chromeno[2,3-d]pyrimidine candidates. Med. Chem. Res. 2017, 26, 2624–2638. [Google Scholar] [CrossRef]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef]
- Haiba, M.E.; Al-Abdullah, E.S.; Ahmed, N.S.; Ghabbour, H.A.; Awad, H.M. Efficient and easy synthesis of new Benzo[h]chromene and Benzo[h]quinoline derivatives as a new class of cytotoxic agents. J. Mol. Struct. 2019, 1195, 702–711. [Google Scholar] [CrossRef]
- Luque-Agudo, V.; Albarrán-Velo, J.; Light, M.E.; Padrón, J.M.; Román, E.; Serrano, J.A.; Gil, M.V. Synthesis and antiproliferative activity of new 2-glyco-3-nitro-2H-chromenes. Bioorg. Chem. 2019, 87, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Okasha, R.M.; Alsehli, M.; Ihmaid, S.; Althagfan, S.S.; El-Gaby, M.S.A.; Ahmed, H.E.A.; Afifi, T.H. First example of Azo-Sulfa conjugated chromene moieties: Synthesis, characterization, antimicrobial assessment, docking simulation as potent class I histone deacetylase inhibitors and antitumor agents. Bioorg. Chem. 2019, 92, 103262. [Google Scholar] [CrossRef] [PubMed]
- Sabry, N.M.; Mohamed, H.M.; Khattab, E.S.A.E.H.; Motlaq, S.S.; El-Agrody, A.M. Synthesis of 4H-Chromene, Coumarin, 12H-Chromeno[2,3-d]Pyrimidine Derivatives and Some of Their Antimicrobial and Cytotoxicity Activities. Eur. J. Med. Chem. 2011, 46, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.; Zachariah, S.M. Pharmacological Activities of Chromene Derivatives: An Overview. Asian J. Pharm. Clin. Res. 2013, 6, 11–15. [Google Scholar]
- Li, M.; Zhao, X.; Yang, W.; Zhong, F.; Yuan, L.; Ren, Q. Asymmetric synthesis and biological evaluation of 3-nitro-2H-chromenes as potential antibacterial agents. Tetrahedron Lett. 2018, 59, 3511–3515. [Google Scholar] [CrossRef]
- Thanh, N.D.; Hai, D.S.; Ngoc Bich, V.T.; Thu Hien, P.T.; Ky Duyen, N.T.; Mai, N.T.; Dung, T.T.; Toan, V.N.; Kim Van, H.T.; Dang, L.H.; et al. Efficient click chemistry towards novel 1H-1,2,3-triazole-tethered 4H-chromene−D-glucose conjugates: Design, synthesis and evaluation of in vitro antibacterial, MRSA and antifungal activities. Eur. J. Med. Chem. 2019, 167, 454–471. [Google Scholar] [CrossRef] [PubMed]
- Subbareddy, C.V.; Subashini, R.; Sumathi, S. Synthesis of substituted 2H-chromenes by a three-component reaction as potential antioxidants. Mol. Divers. 2017, 21, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Yahagi, H.; Uesawa, Y.; Sugita, Y. 3-(E)-Styryl-2H-chromene derivatives as potent and selective monoamine oxidase B inhibitors. Bioorg. Chem. 2018, 77, 436–442. [Google Scholar] [CrossRef]
- Razdan, R.K.; Pars, H.G.; Granchelli, F.E.; Harris, L.S. Steroidal Analog of a Tetrahydrocannabinol. J. Med. Chem. 1968, 11, 377–378. [Google Scholar] [CrossRef]
- Pars, H.G.; Granchelli, F.E.; Keller, J.K.; Razdan, R.K. Physiologically Active Nitrogen Analogs of Tetrahydrocannabinols. Tetrahydrobenzopyrano[3,4-d]Pyridines. J. Am. Chem. Soc. 1966, 88, 3664–3665. [Google Scholar] [CrossRef]
- Pars, H.G.; Granchelli, F.E.; Razdan, R.K.; Keller, J.K.; Teiger, D.G.; Rosenberg, F.J.; Harris, L.S. Drugs derived from cannabinoids. 1. Nitrogen analogs, benzopyranopyridines and benzopyranopyrroles. J. Med. Chem. 1976, 19, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.D.; Altenbach, R.J.; Basha, F.Z.; Carroll, W.A.; Drizin, I.; Kerwin, J.F., Jr.; Wendt, M.D.; Haight, A.R.; Zhang, W. Benzopyranopyrrole and Benzopyranopyridine Alpha-1 Adrenergic Compounds. WO Patent WO9824791A1, 11 June 1998. [Google Scholar]
- Brown, R.E.; Puchalski, C.; Shavel, J., Jr. Novel Substituted Benzopyranopyridine. U.S. Patent 3,962,266, 8 June 1976. [Google Scholar]
- Connor, D.T.; Unangst, P.C.; Schwender, C.F.; Sorenson, R.J.; Carethers, M.E.; Puchalski, C.; Brown, R.E. Synthesis of 1,2,3,4-tetrahydro-5H-[1]benzopyrano[3,4-c]pyridin-5-ones. II. Substitution at the 3-position with 2-aminoethyl and 2-aminopropyl side chains. J. Heterocycl. Chem. 1984, 21, 1561–1564. [Google Scholar] [CrossRef]
- Connor, D.T.; Unangst, P.C.; Schwender, C.F.; Sorenson, R.J.; Carethers, M.E.; Brown, R.E.; Puchalski, C. Synthesis of 1,2,3,4-tetrahydro-5H-[1]benzopyrano[3,4-c]pyridin-5-ones. I. 3-unsubstituted compounds. J. Heterocycl. Chem. 1984, 21, 1557–1559. [Google Scholar] [CrossRef]
- Radulovic, N.; Stojanovic, G.; Vukicevic, R.; Dekic, V.; Dekic, B.; Palic, R. New 3,4-Annelated Coumarin Derivatives: Synthesis, Antimicrobial Activity, Antioxidant Capacity, and Molecular Modeling. Monatshefte Chem./Chem. Mon. 2006, 137, 1477–1486. [Google Scholar] [CrossRef]
- Dawane, B.S.; Konda, S.G.; Bodade, R.G.; Bhosale, R.B. An efficient one-pot synthesis of some new 2,4-diaryl pyrido[3,2-c]coumarins as potent antimicrobial agents. J. Heterocycl. Chem. 2010, 47, 237–241. [Google Scholar] [CrossRef]
- Nunez-Vergara, L.J.; Squella, J.A.; Navarrete-Encina, P.A.; Vicente-Garcia, E.; Preciado, S.; Lavilla, R. Chromenopyridines: Promising Scaffolds for Medicinal and Biological Chemistry. Curr. Med. Chem. 2011, 18, 4761–4785. [Google Scholar] [CrossRef] [PubMed]
- Delost, M.D.; Smith, D.T.; Anderson, B.J.; Njardarson, J.T. From Oxiranes to Oligomers: Architectures of U.S. FDA Approved Pharmaceuticals Containing Oxygen Heterocycles. J. Med. Chem. 2018, 61, 10996–11020. [Google Scholar] [CrossRef]
- Mishra, S.; Ghosh, R. K2CO3-Mediated, One-Pot, Multicomponent Synthesis of Medicinally Potent Pyridine and Chromeno[2,3-b]Pyridine Scaffolds. Synth. Commun. 2012, 42, 2229–2244. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Kireev, A.S.; Yakovenko, A.A.; Antipin, M.Y.; Magedov, I.V.; Kornienko, A. Convenient one-step synthesis of a medicinally relevant benzopyranopyridine system. Tetrahedron Lett. 2006, 47, 9309–9312. [Google Scholar] [CrossRef]
- Banerjee, S.; Wang, J.; Pfeffer, S.; Ma, D.; Pfeffer, L.M.; Patil, S.A.; Li, W.; Miller, D.D. Design, Synthesis and Biological Evaluation of Novel 5H-Chromenopyridines as Potential Anti-Cancer Agents. Molecules 2015, 20, 17152–17165. [Google Scholar] [CrossRef] [PubMed]
- Molla, A.; Ranjan, S.; Rao, M.S.; Dar, A.H.; Shyam, M.; Jayaprakash, V.; Hussain, S. Borax Catalysed Domino Synthesis of Highly Functionalised Spirooxindole and Chromenopyridine Derivatives: X-Ray Structure, Hirshfeld Surface Analysis and Molecular Docking Studies. ChemistrySelect 2018, 3, 8669–8677. [Google Scholar] [CrossRef]
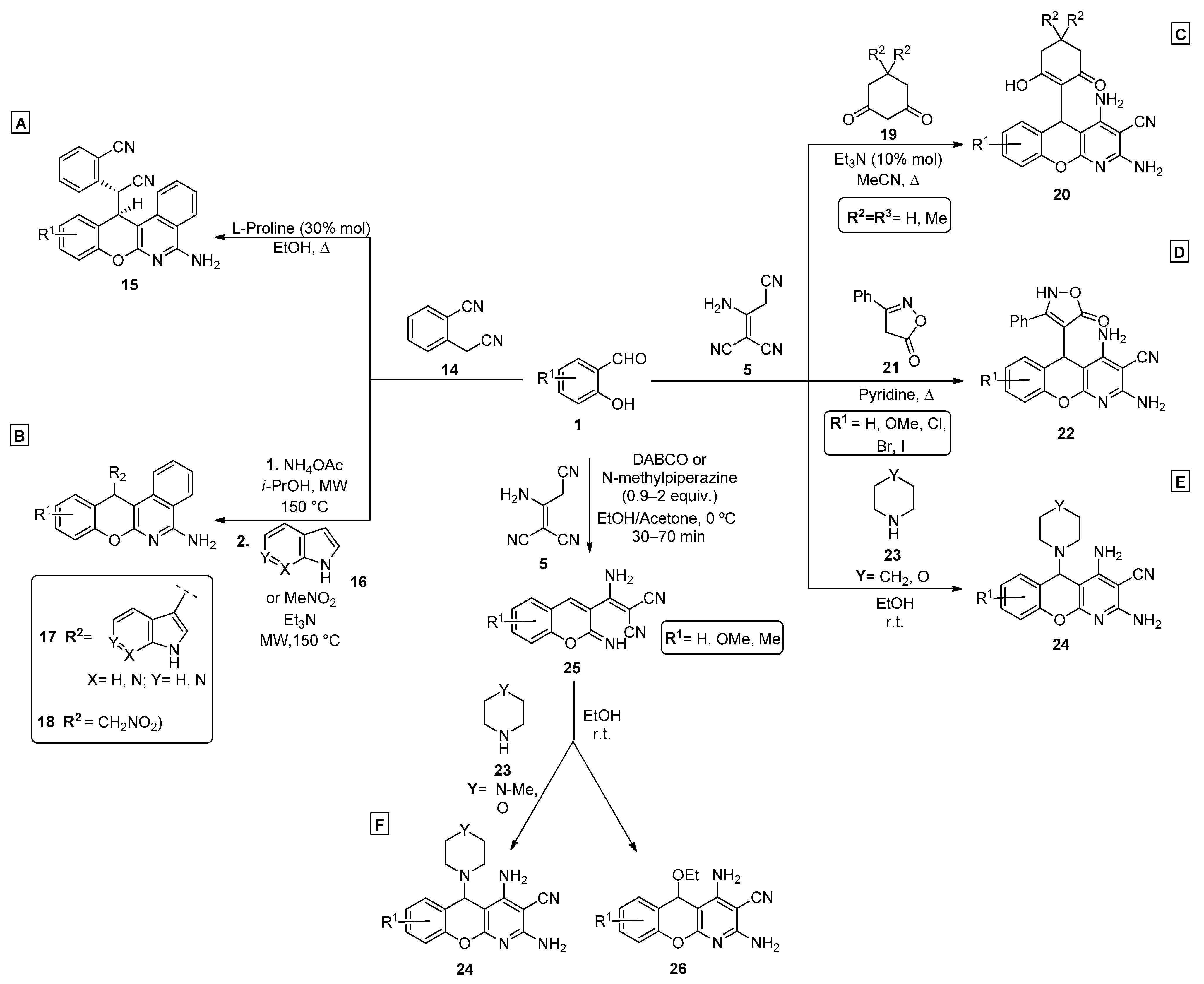
- Vereshchagin, A.N.; Elinson, M.N.; Anisina, Y.E.; Ryzhkov, F.V.; Goloveshkin, A.S.; Bushmarinov, I.S.; Zlotin, S.G.; Egorov, M.P. Pot, atom and step economic (PASE) synthesis of 5-isoxazolyl-5H-chromeno[2,3-b]pyridine scaffold. Mendeleev Commun. 2015, 25, 424–426. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Anisina, Y.E.; Ryzhkov, F.V.; Novikov, R.A.; Egorov, M.P. PASE Pseudo-Four-Component Synthesis and Docking Studies of New 5-C-Substituted 2,4-Diamino-5H-Chromeno[2,3-b]Pyridine-3-Carbonitriles. ChemistrySelect 2017, 2, 4593–4597. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Anisina, Y.E.; Ryzhkov, F.V.; Goloveshkin, A.S.; Novikov, R.A.; Egorov, M.P. Synthesis, structural, spectroscopic and docking studies of new 5C-substituted 2,4-diamino-5H-chromeno[2,3-b]pyridine-3-carbonitriles. J. Mol. Struct. 2017, 1146, 766–772. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Goloveshkin, A.S.; Ushakov, I.E.; Egorov, M.P. PASE facile and efficient multicomponent approach to the new type of 5-C-substituted 2,4-diamino-5H-chromeno[2,3-b]pyridine scaffold. Mendeleev Commun. 2018, 28, 372–374. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Goloveshkin, A.S.; Ushakov, I.E.; Egorov, M.P. Multicomponent transformation of salicylaldehydes, 2-aminoprop-1-ene-1,1,3-tricarbonitrile, and pyrazolin-5-ones into substituted 2,4-diamino-5-(5-hydroxy-3-methyl-1H-pyrazol-4-yl)-5H-chromeno[2,3-b]pyridine-3-carbonitriles. Russ. Chem. Bull. 2018, 67, 1695–1703. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Krymov, S.K.; Fakhrutdinov, A.N.; Egorov, M.P. Selective multicomponent ‘one-pot’ approach to the new 5-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)chromeno[2,3-b]pyridine scaffold in pyridine–ethanol catalyst/solvent system. Monatshefte Chem./Chem. Mon. 2019, 150, 1073–1078. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Fakhrutdinov, A.N.; Goloveshkin, A.S.; Egorov, M.P. Pot-, Atom- and Step-Economic (PASE) Multicomponent Approach to the 5-(Dialkylphosphonate)-Substituted 2,4-Diamino-5H-Chromeno[2,3-b]Pyridine Scaffold. Eur. J. Org. Chem. 2019, 2019, 4171–4178. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Krymov, S.K.; Fakhrutdinov, A.N.; Goloveshkin, A.S.; Egorov, M.P. Pot, atom and step economic (PASE) assembly of salicylaldehydes, malononitrile dimer and 4-hydroxypyridine-2(1H)-ones into medicinally relevant 5H-chromeno[2,3-b]pyridine scaffold. Mol. Divers. 2019, 24, 617–626. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Karpenko, K.A.; Elinson, M.N.; Dorofeeva, E.O.; Goloveshkin, A.S.; Egorov, M.P. Pseudo six-component stereoselective synthesis of 2,4,6-triaryl-3,3,5,5-tetracyanopiperidines. Mendeleev Commun. 2018, 28, 384–386. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Fakhrutdinov, A.N.; Goloveshkin, A.S.; Egorov, M.P. A facile and efficient multicomponent approach to 5-[5-hydroxy-3-(trifluoromethyl)-1H-pyrazol-4-yl]-5H-chromeno[2,3-b]pyridines. J. Fluor. Chem. 2018, 213, 31–36. [Google Scholar] [CrossRef]
- Festa, A.A.; Storozhenko, O.A.; Golantsov, N.E.; Subramani, K.; Novikov, R.A.; Zaitseva, S.O.; Baranov, M.S.; Varlamov, A.V.; Voskressensky, L.G. Homophtalonitrile for Multicomponent Reactions: Syntheses and Optical Properties of o-Cyanophenyl- or Indol-3-Yl-SubstitutedChromeno[2,3-c]Isoquinolin-5-Amines. ChemistryOpen 2019, 8, 23–30. [Google Scholar] [CrossRef]
- Festa, A.A.; Storozhenko, O.A.; Bella Ndoutoume, D.R.; Varlamov, A.V.; Voskressensky, L.G. Sequential three-component reaction of homophthalonitrile, salicylaldehydes and nitromethane. Mendeleev Commun. 2017, 27, 451–453. [Google Scholar] [CrossRef]
- Shaabani, A.; Hajishaabanha, F.; Mofakham, H.; Maleki, A. A new one-pot three-component synthesis of 2,4-diamino-5H-chromeno[2,3-b] pyridine-3-carbonitrile derivatives. Mol. Divers. 2010, 14, 179–182. [Google Scholar] [CrossRef]
- Lopes, D.; Oliveira-Pinto, S.; Pontes, O.; Sampaio-Marques, B.; Costa, M.D.; Carvalho, L.; Gonçalves, C.S.; Costa, B.M.; Maciel, P.; Ludovico, P.; et al. Unravelling the anticancer potential of functionalized chromeno[2,3-b]pyridines for breast cancer treatment. Bioorg. Chem. 2020, 100, 103942. [Google Scholar] [CrossRef]
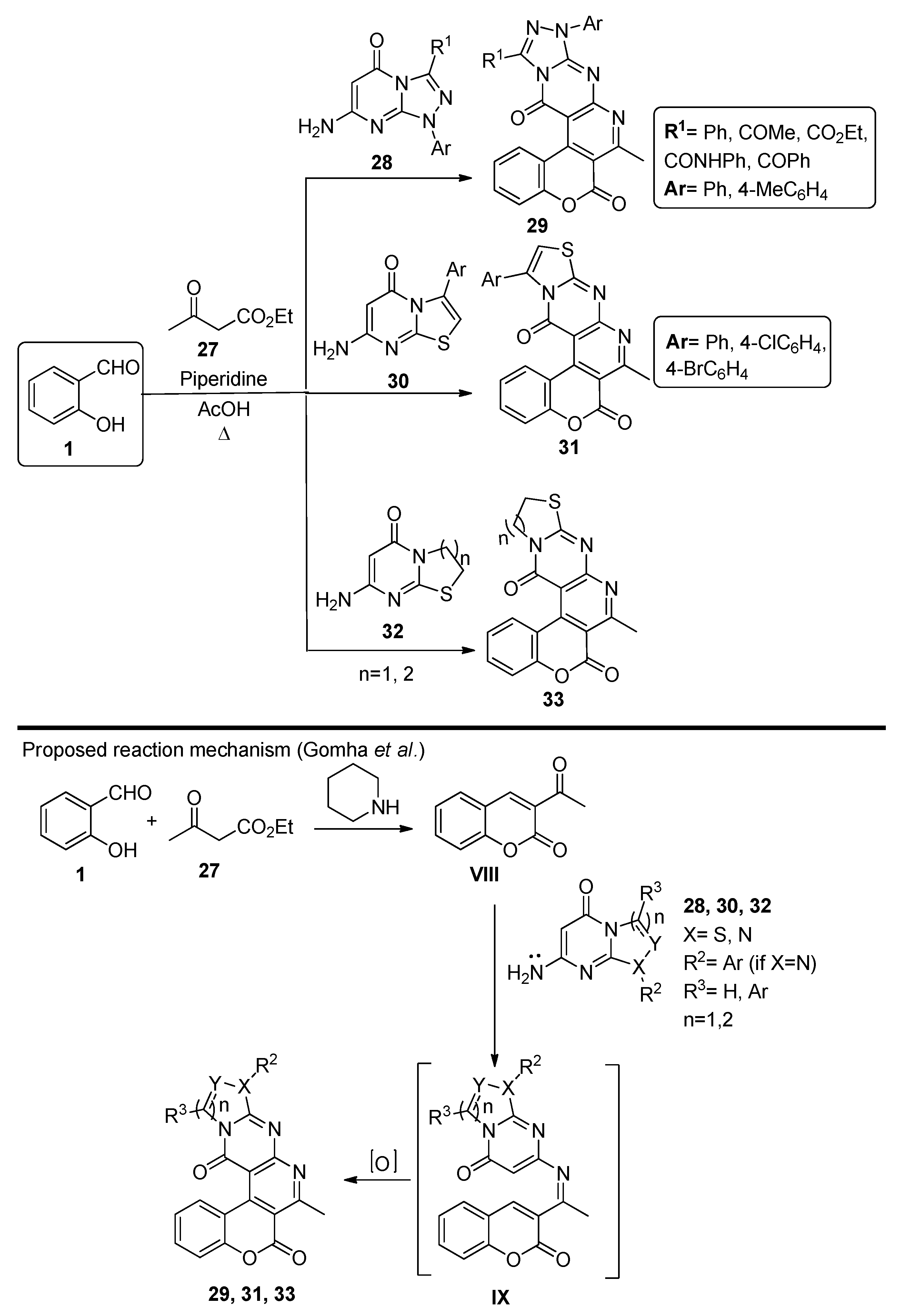
- Gomha, S.M.; Riyadh, S.M. Multicomponent Synthesis of Novel Penta-Heterocyclic Ring Systems Incorporating a Benzopyranopyridine Scaffold. Synthesis 2014, 46, 258–262. [Google Scholar] [CrossRef]
- Navarrete-Encina, P.A.; Salazar, R.; Vega-Retter, C.; Pérez, K.; Squella, J.A.; Nuñez-Vergara, L.J. On the one pot syntheses of chromeno[4,3-b]pyridine-3-carboxylate and chromeno[3,4-c]pyridine-3-carboxylate and dihydropyridines. J. Braz. Chem. Soc. 2010, 21, 413–418. [Google Scholar] [CrossRef]
- Povarov, L.S.; Grigos, V.I.; Mikhailov, B.M. Reaction of benzylideneaniline with some unsaturated compounds. Russ. Chem. Bull. 1963, 12, 1878–1880. [Google Scholar] [CrossRef]
- Adolfsson, D.E.; Tyagi, M.; Singh, P.; Deuschmann, A.; Ådén, J.; Gharibyan, A.L.; Jayaweera, S.W.; Lindgren, A.E.G.; Olofsson, A.; Almqvist, F. Intramolecular Povarov Reactions for the Synthesis of Chromenopyridine Fused 2-Pyridone Polyheterocycles Binding to α-Synuclein and Amyloid-β Fibrils. J. Org. Chem. 2020, 85, 14174–14189. [Google Scholar] [CrossRef]
- Dimitriadou, E.; Raftopoulou, M.; Kasapidou, P.M.; Tsoleridis, C.A.; Stephanidou-Stephanatou, J.; Hadjipavlou-Litina, D.J.; Kontogiorgis, C.; Pritsa, A.; Papadopoulos, A. Ultrasound promoted synthesis of chromeno[2,3-b]pyridines and their evaluation as lipid peroxidation inhibitors. Arkivoc 2014, 2014, 372–384. [Google Scholar] [CrossRef]
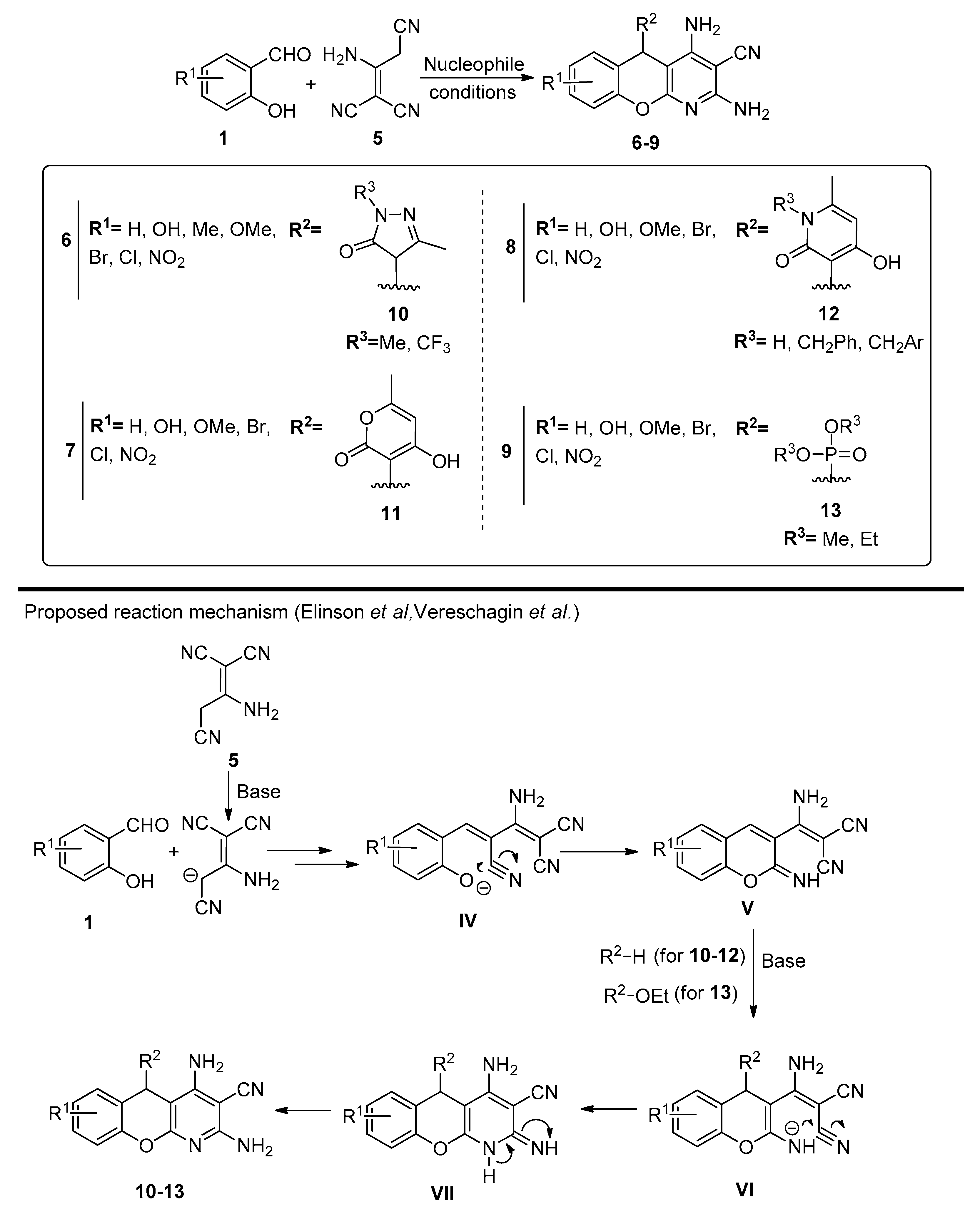
- Ibrahim, M.A.; El-Gohary, N.M.; Ibrahim, S.S.; Said, S. Synthesis of Some Novel Heteroannelated Chromones by Basic Rearrangement of 6-Methylchromone-3-Carbonitrile. Chem. Heterocycl. Compd. 2015, 50, 1624–1633. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; El-Gohary, N.M. Studies on the Chemical Transformations of 6-Methylchromone-3-Carbonitrile under Nucleophilic Conditions. J. Heterocycl. Chem. 2016, 53, 859–864. [Google Scholar] [CrossRef]
- Savych, I.; Ejaz, S.A.; Shah, S.J.A.; Iaroshenko, V.O.; Villinger, A.; Sosnovskikh, V.Y.; Iqbal, J.; Abbasi, A.; Langer, P. Reactions of 3-Acylchromones with Heterocyclic Ketene Aminals: One-Pot Synthesis and Phosphatase Inhibitory Activity of Fused Pyridine Derivatives. Eur. J. Org. Chem. 2017, 2017, 186–202. [Google Scholar] [CrossRef]
- Siddiqui, Z.N.; Praveen, S.; Farooq, F. Novel benzopyranopyridine derivatives of 2-amino-3-formylchromone. Chem. Pap. 2010, 64, 818–824. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Farag, A.A.M.; Roushdy, N.; El-Gohary, N.M. Synthesis, optical and photoelectrical characterizations of the novel 10-chloro-6H,8H-dichromeno[2,3-b:3′,4′-e]pyridine-6,8-dione (CDPD) and its photodiode application. Opt. Mater. 2016, 51, 70–77. [Google Scholar] [CrossRef]
- Ponduri, R.; Kumar, P.; Vadali, L.R.A.O.; Modugu, N.R. Water-PEG-400 Mediated an Efficient One-Pot Eco-Friendly Synthesis of Functionalized Isoxazole Substituted Chromeno[2,3-b]pyridine-3-carboxylate Derivatives. ChemistrySelect 2018, 3, 7766–7770. [Google Scholar] [CrossRef]
- Dolatkhah, Z.; Nasiri-Aghdam, M.; Bazgir, A. A Three-Component Synthesis of Benzochromenodiazocines and Chromenopyridines. Tetrahedron Lett. 2013, 54, 1960–1962. [Google Scholar] [CrossRef]
- Zhang, C.H.; Huang, R.; Hu, X.M.; Lin, J.; Yan, S.J. Three-Component Site-Selective Synthesis of Highly Substituted 5H-Chromeno-[4,3-b]Pyridines. J. Org. Chem. 2018, 83, 4981–4989. [Google Scholar] [CrossRef]
- Lozinski, O.A.; Shokol, T.V.; Zubatyuk, R.I.; Shishkin, O.V.; Khilya, V.P. An alternative approach to the synthesis of 5H-chromeno[4,3-b]pyridin-5-one system using the cleavage of 5H,9H-pyrano[2′,3′:5,6]chromeno[4,3-b]pyridine-5,9-diones with binucleophiles. Chem. Heterocycl. Comp. 2018, 54, 96–99. [Google Scholar] [CrossRef]
- Ali, K.A.; Abdel Hafez, N.A.; Elsayed, M.A.; Ibrahim, A.A. Microwave-assisted synthesis and heterocyclic functionalization of chromenopyridines on calixarene scaffold. J. Heterocycl. Chem. 2020, 57, 1838–1844. [Google Scholar] [CrossRef]
- Thapa, U.; Thapa, P.; Karki, R.; Yun, M.; Choi, J.H.; Jahng, Y.; Lee, E.; Jeon, K.H.; Na, Y.; Ha, E.M.; et al. Synthesis of 2,4-diaryl chromenopyridines and evaluation of their topoisomerase I and II inhibitory activity, cytotoxicity, and structure-activity relationship. Eur. J. Med. Chem. 2011, 46, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Lee, E.S. 2,4-Diaryl-5,6-Dihydro-1,10-Phenanthrolines with Furyl or Thienyl Moiety at 4-Position: Synthesis, Topoisomerase I and II Inhibitory Activity, and Cytotoxicity. Bull. Korean Chem. Soc. 2012, 33, 1769–1772. [Google Scholar] [CrossRef]
- Thapa, P.; Jun, K.Y.; Kadayat, T.M.; Park, C.; Zheng, Z.; Thapa Magar, T.B.; Bist, G.; Shrestha, A.; Na, Y.; Kwon, Y.; et al. Design and synthesis of conformationally constrained hydroxylated 4-phenyl-2-aryl chromenopyridines as novel and selective topoisomerase II-targeted antiproliferative agents. Bioorg. Med. Chem. 2015, 23, 6454–6466. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.B.; Park, C.; Jeon, K.H.; Lee, E.; Park, S.E.; Jun, K.Y.; Kadayat, T.M.; Thapa, P.; Karki, R.; Na, Y.; et al. A Series of Novel Terpyridine-Skeleton Molecule Derivants Inhibit Tumor Growth and Metastasis by Targeting Topoisomerases. J. Med. Chem. 2015, 58, 1100–1122. [Google Scholar] [CrossRef] [PubMed]
- Magar, T.B.T.; Seo, S.H.; Kadayat, T.M.; Jo, H.; Shrestha, A.; Bist, G.; Katila, P.; Kwon, Y.; Lee, E.S. Synthesis and SAR study of new hydroxy and chloro-substituted 2,4-diphenyl 5H-chromeno[4,3-b]pyridines as selective topoisomerase IIα-targeting anticancer agents. Bioorg. Med. Chem. 2018, 26, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Behbehani, H.; Dawood, K.M.; Aryan, F.A.; Ibrahim, H.M. Green Protocol for the Novel Synthesis of Thiochromeno[4,3-b]Pyridine and Chromeno[4,3-b]Pyridine Derivatives Utilizing a High-Pressure System. ACS Omega 2021, 6, 34065–34074. [Google Scholar] [CrossRef] [PubMed]
- El-Essawy, F.; El-Etrawy, A.S. Synthesis of New Chromeno[4,3-b]pyrazolo[4,3-e]pyridines Derivatives with Antimicrobial Evaluation. J. Heterocycl. Chem. 2013, 51, 191–195. [Google Scholar] [CrossRef]
- Rong, L.; Han, H.; Jiang, H.; Zhang, Q.; Tu, S. Efficient one-pot synthesis of 4-aryl-3-cyano-2,5-dihydro-1H-indeno[1,2-b]pyridin-2-one and 4-aryl-3-cyano-1,2,5,6-tetrahydrobenzo[h] quinolin-2-one derivatives under solvent-free conditions. Synth. Commun. 2009, 39, 1027–1034. [Google Scholar] [CrossRef]
- Kok, T.; Wapenaar, H.; Wang, K.; Neochoritis, C.G.; Zarganes-Tzitzikas, T.; Proietti, G.; Eleftheriadis, N.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Cool, R.H.; et al. Discovery of chromenes as inhibitors of macrophage migration inhibitory factor. Bioorg. Med. Chem. 2018, 26, 999–1005. [Google Scholar] [CrossRef]
- Patel, M.A.; Bhila, V.G.; Patel, N.H.; Patel, A.K.; Brahmbhatt, D.I. Synthesis, characterization and biological evaluation of some pyridine and quinoline fused chromenone derivatives. Med. Chem. Res. 2012, 21, 4381–4388. [Google Scholar] [CrossRef]
- Pal, S.; Khan, M.N.; Karamthulla, S.; Choudhury, L.H. Molecular iodine catalyzed one-pot multicomponent reactions for the synthesis of dihydrochromeno[4,3-b]pyrazolo[4,3-e]pyridin-6(7H)-ones. RSC Adv. 2013, 3, 15705–15711. [Google Scholar] [CrossRef]
- Patel, A.A.; Lad, H.B.; Pandya, K.R.; Patel, C.V.; Brahmbhatt, D.I. Synthesis of a new series of 2-(2-oxo-2H-chromen-3-yl)-5H-chromeno[4,3-b]pyridin-5-ones by two facile methods and evaluation of their antimicrobial activity. Med. Chem. Res. 2013, 22, 4745–4754. [Google Scholar] [CrossRef]
- Khan, A.T.; Das, D.K. Michael Initiated Ring Closure (MIRC) reaction on in situ generated benzylidenecyclohexane-1,3-diones for the construction of chromeno[3,4-b]quinoline derivatives. Tetrahedron Lett. 2012, 53, 2345–2351. [Google Scholar] [CrossRef]
- Khan, A.T.; Das, D.K.; Islam, K.; Das, P. A simple and expedient synthesis of functionalized pyrido[2,3-c] coumarin derivatives using molecular iodine catalyzed three-component reaction. Tetrahedron Lett. 2012, 53, 6418–6422. [Google Scholar] [CrossRef]
- Chen, Z.; Gu, J.; Su, W. An Efficient Protocol for Multicomponent Synthesis of 1H-Chromeno[4,3-b]Pyridin-5(4H)-Ones Derivatives. J. Chem. Res. 2013, 37, 327–330. [Google Scholar] [CrossRef]
- Oshiro, P.B.; Bregadiolli, B.A.; da Silva-Filho, L.C. A facile one-step synthesis of chromeno[4,3-b]pyridine derivatives promoted by niobium pentachloride. J. Heterocycl. Chem. 2020, 57, 2795–2800. [Google Scholar] [CrossRef]
- Paul, S.; Das, A.R. An efficient green protocol for the synthesis of coumarin fused highly decorated indenodihydropyridyl and dihydropyridyl derivatives. Tetrahedron Lett. 2012, 53, 2206–2210. [Google Scholar] [CrossRef]
- Motamedi, R. Solvent-free synthesis of novel 5-oxo-5H-chromeno [4,3-b]pyridine derivatives. Chem. Heterocyc. Compd. 2013, 48, 1839–1843. [Google Scholar] [CrossRef]
- Symeonidis, T.S.; Litinas, K.E. Synthesis of methyl substituted [5,6]- and [7,8]-fused pyridocoumarins via the iodine-catalyzed reaction of aminocoumarins with n-butyl vinyl ether. Tetrahedron Lett. 2013, 54, 6517–6519. [Google Scholar] [CrossRef]
- Ganguly, N.C.; Chandra, S. One-pot access to pyridocoumarins via Povarov-hydrogen transfer cascade under auto-tandem catalysis of iodine in aqueous micelles. Tetrahedron Lett. 2014, 55, 1564–1568. [Google Scholar] [CrossRef]
- Epstein, O.; Bryan, M.C.; Cheng, A.C.; Derakhchan, K.; Dineen, T.A.; Hickman, D.; Hua, Z.; Human, J.B.; Kreiman, C.; Marx, I.E.; et al. Lead Optimization and Modulation of HERG Activity in a Series of Aminooxazoline Xanthene β-Site Amyloid Precursor Protein Cleaving Enzyme (BACE1) Inhibitors. J. Med. Chem. 2014, 57, 9796–9810. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Brown, J.; Judd, T.C.; Lopez, P.; Qian, W.; Powers, T.S.; Chen, J.J.; Bartberger, M.D.; Chen, K.; Dunn, R.T.; et al. An Orally Available BACE1 Inhibitor That Affords Robust CNS Aβ Reduction without Cardiovascular Liabilities. ACS Med. Chem. Lett. 2015, 6, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; La, D.S.; Cheng, A.C.; Whittington, D.A.; Patel, V.F.; Chen, K.; Dineen, T.A.; Epstein, O.; Graceffa, R.; Hickman, D.; et al. Structure- and Property-Based Design of Aminooxazoline Xanthenes as Selective, Orally Efficacious, and Cns Penetrable BACE Inhibitors for the Treatment of Alzheimers Disease. J. Med. Chem. 2012, 55, 9156–9169. [Google Scholar] [CrossRef]
- Dineen, T.A.; Chen, K.; Cheng, A.C.; Derakhchan, K.; Epstein, O.; Esmay, J.; Hickman, D.; Kreiman, C.E.; Marx, I.E.; Wahl, R.C.; et al. Inhibitors of β-Site Amyloid Precursor Protein Cleaving Enzyme (BACE1): Identification of (S)-7-(2-Fluoropyridin-3-yl)-3-((3-methyloxetan-3-yl)ethynyl)-5′H-Spiro[Chromeno[2,3-b]pyridine-5,4′-oxazol]-2′-amine (AMG-8718). J. Med. Chem. 2014, 57, 9811–9831. [Google Scholar] [CrossRef]


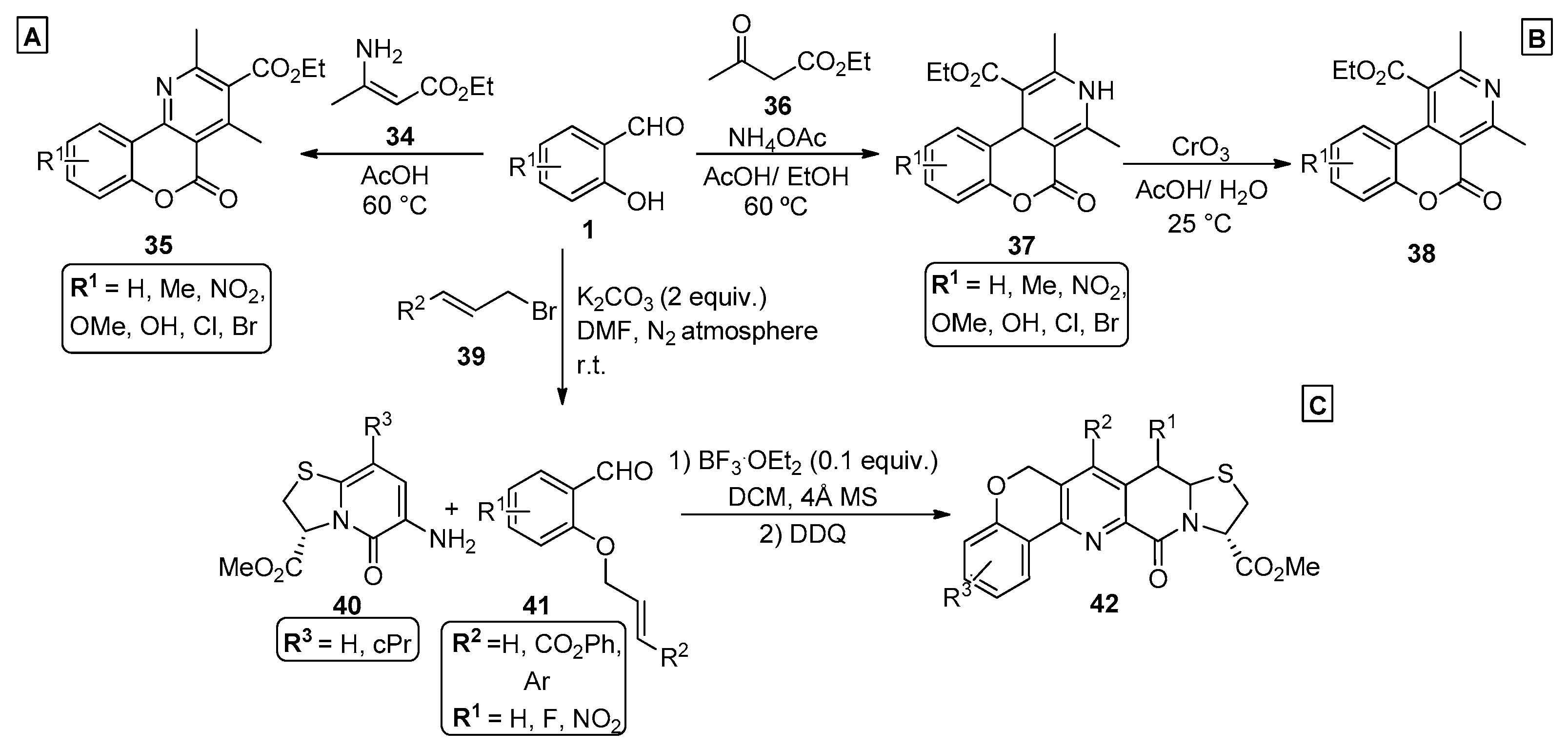


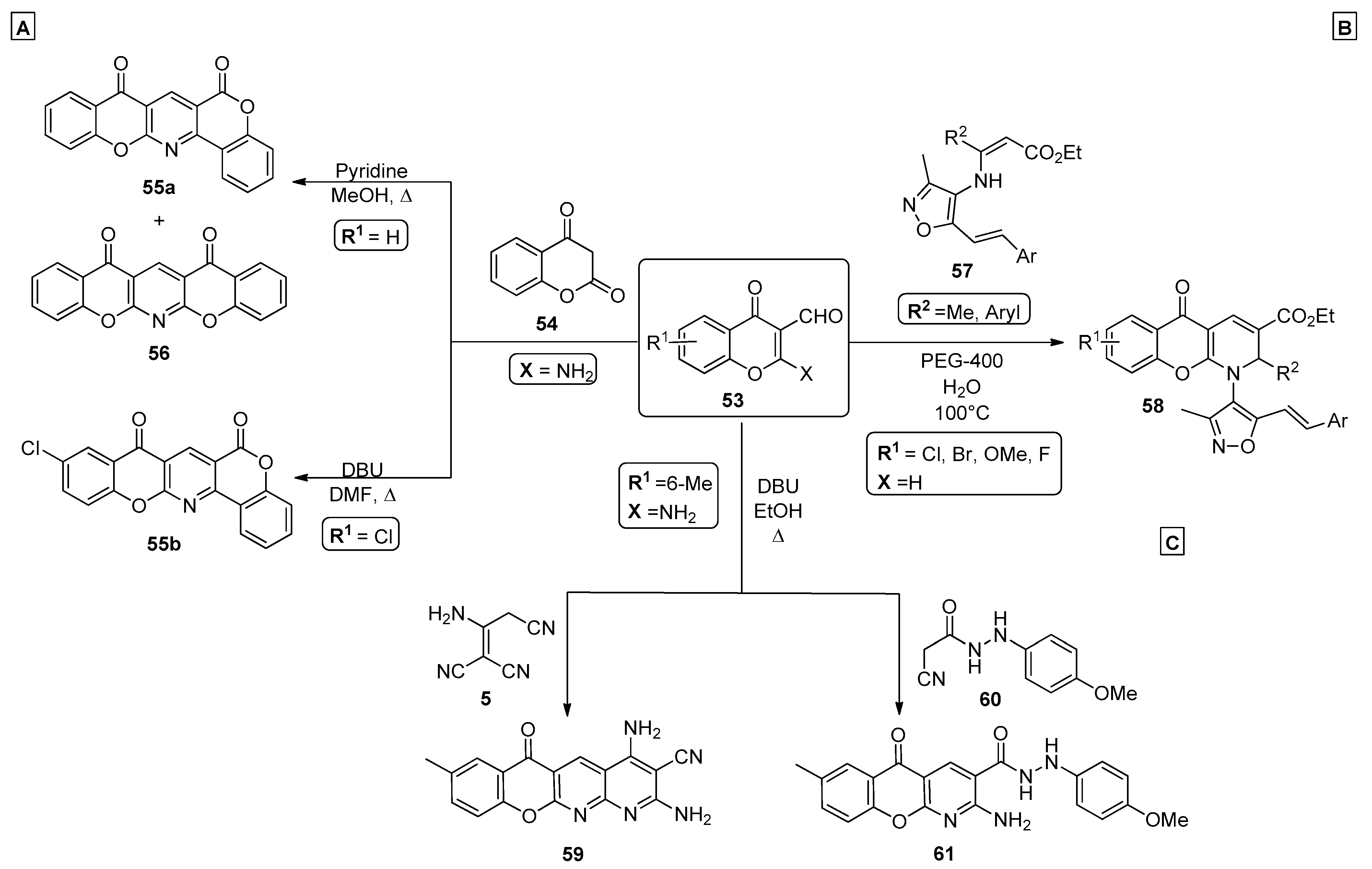


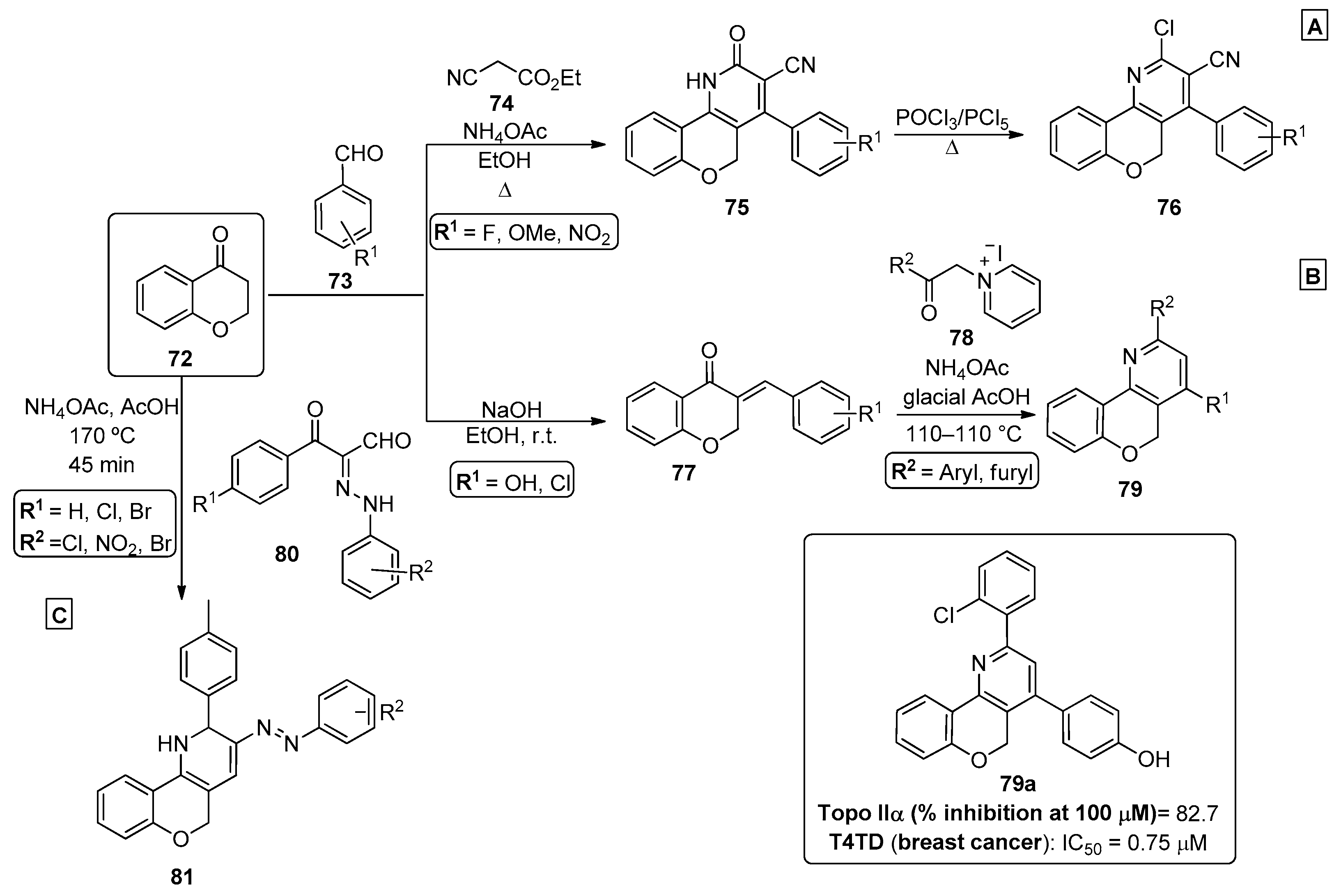

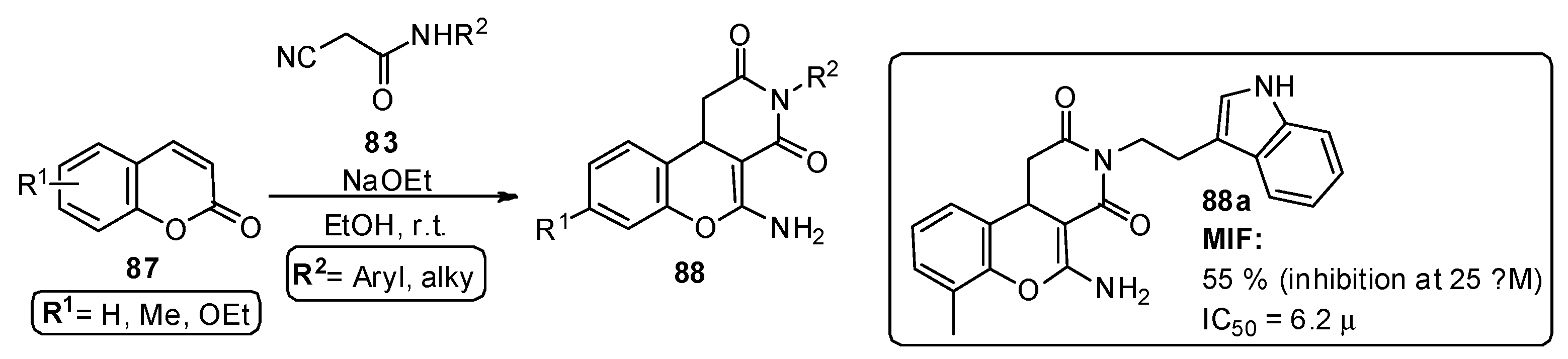


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedroso de Lima, F.; Costa, M.; Sousa, A.; Proença, M.F. The Chromenopyridine Scaffold: A Privileged Platform in Drug Design. Molecules 2024, 29, 3004. https://doi.org/10.3390/molecules29133004
Pedroso de Lima F, Costa M, Sousa A, Proença MF. The Chromenopyridine Scaffold: A Privileged Platform in Drug Design. Molecules. 2024; 29(13):3004. https://doi.org/10.3390/molecules29133004
Chicago/Turabian StylePedroso de Lima, Fábio, Marta Costa, Ana Sousa, and Maria Fernanda Proença. 2024. "The Chromenopyridine Scaffold: A Privileged Platform in Drug Design" Molecules 29, no. 13: 3004. https://doi.org/10.3390/molecules29133004