
MgO Modified by X2, HX, or Alkyl Halide (X = Cl, Br, or I) Catalytic Systems and Their Activity in Chemoselective Transfer Hydrogenation of Acrolein into Allyl Alcohol



Abstract
:
1. Introduction
2. Results
2.1. Reaction of MgO with Bromine or Iodine in Vapor Phase or in Solution with Various Alcohols






2.2. Vapor Phase Reaction of Various Alkyl Halides with MgO
2.3. Characterization of MgO-X2, MgO-HX, and MgO-RX Systems
2.3.1. Powder X-ray Diffraction Measurements
2.3.2. Strength of Acidic and Basic Site Measurements of Catalysts
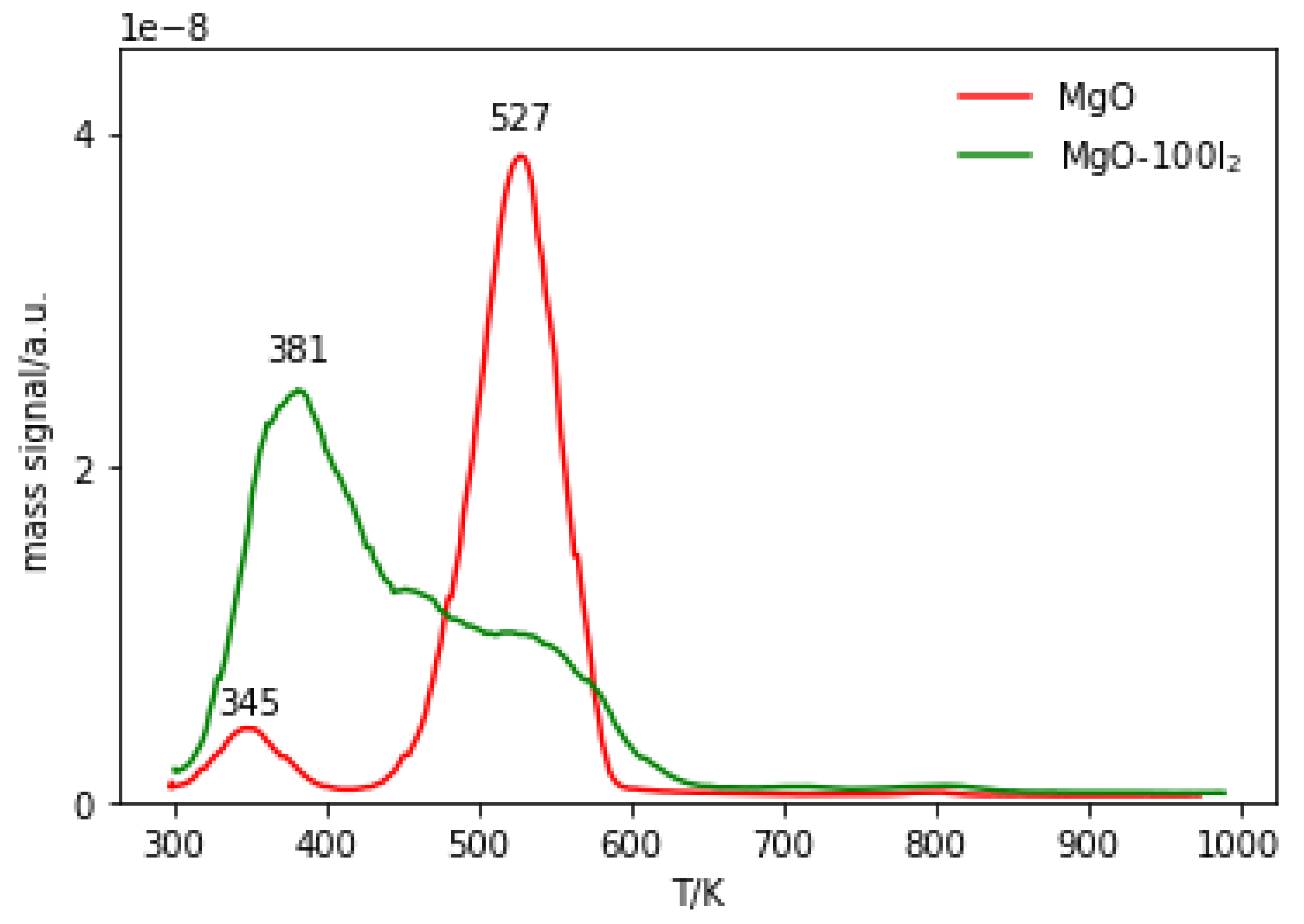
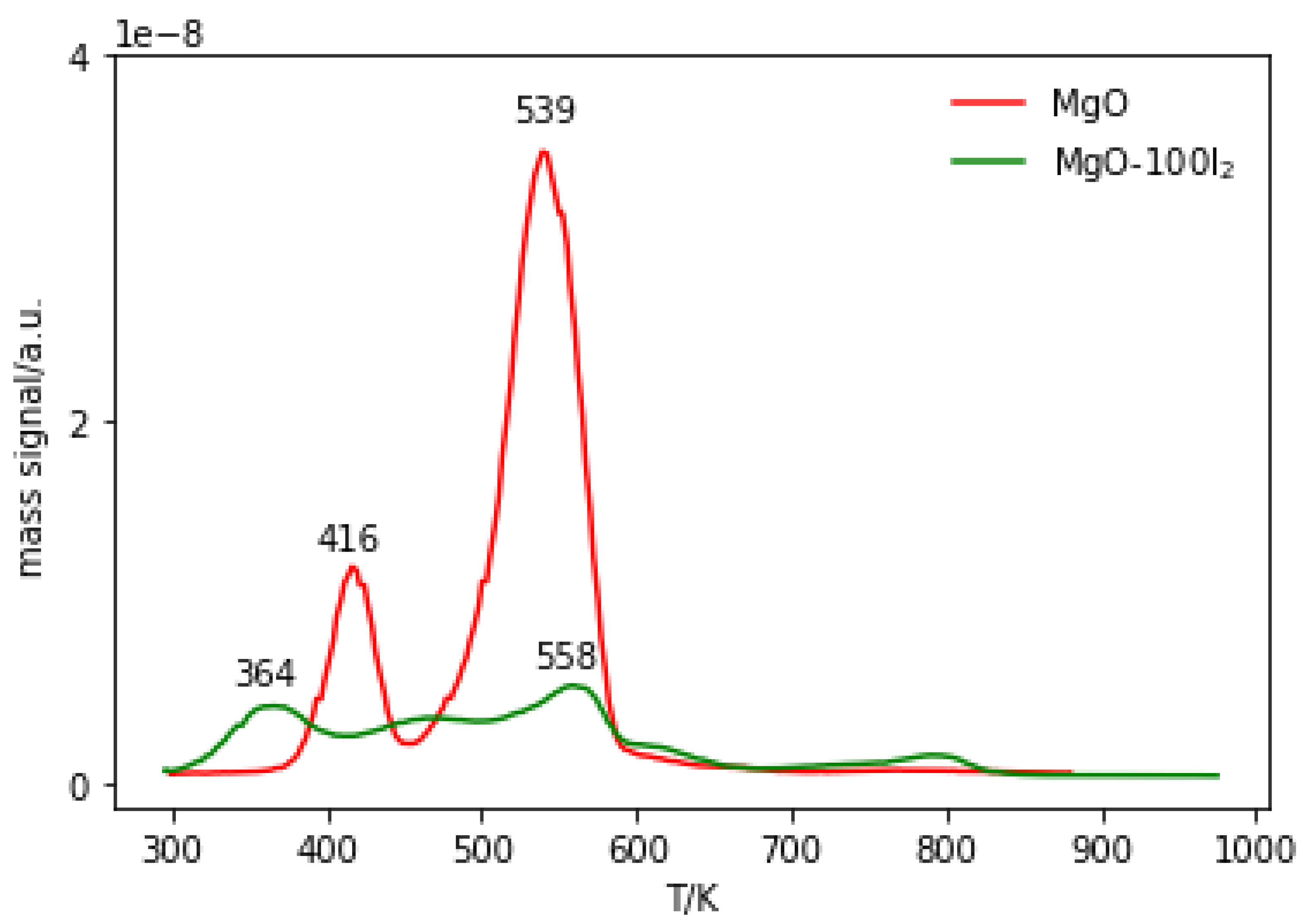
2.3.3. Temperature-Programmed Desorption (TPD) of Probe Molecules


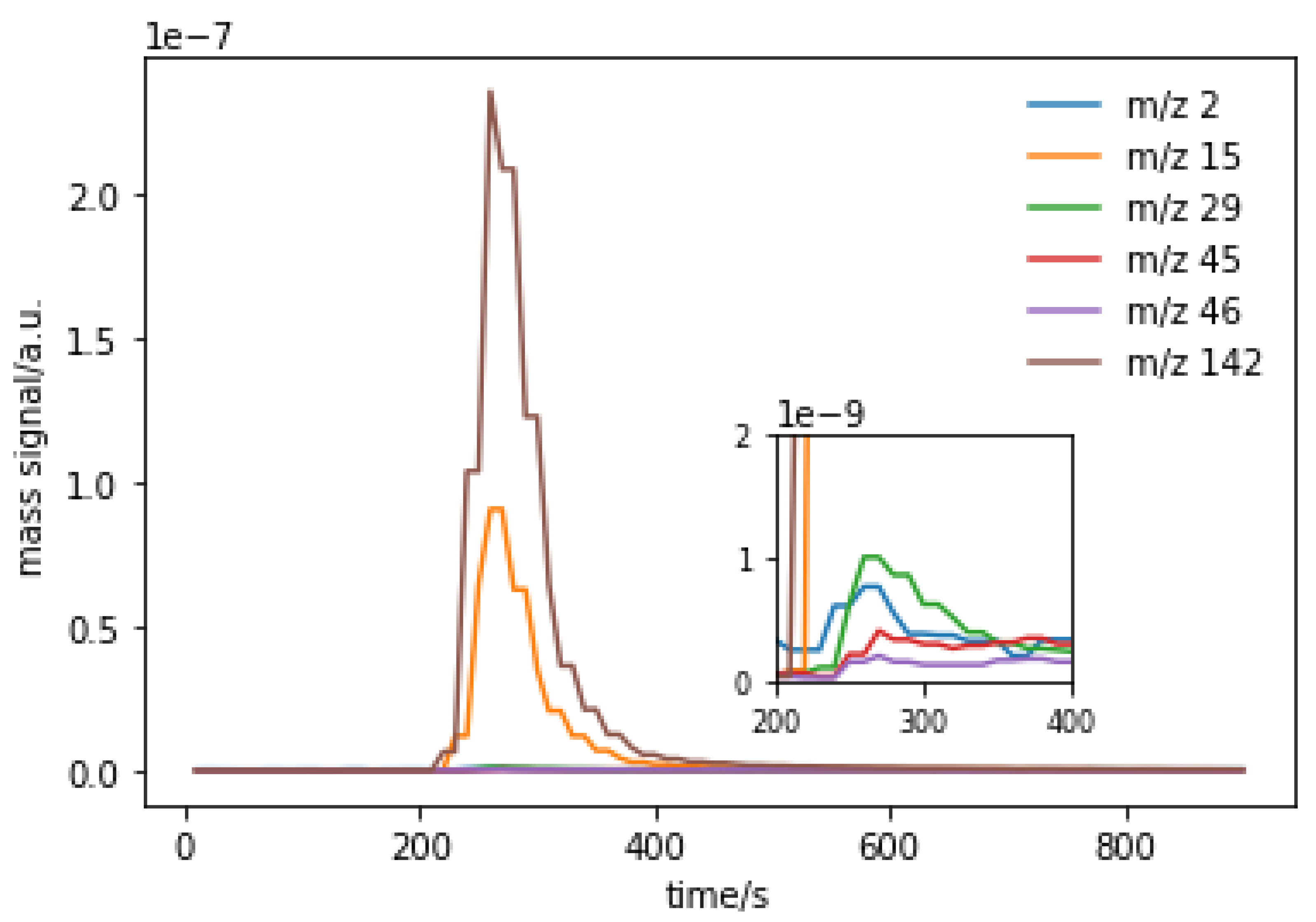
2.3.4. Temperature-Programmed Desorption (TPD) and Mass Spectrometry (MS) of Reaction Products Derived from Methyl Iodide and MgO
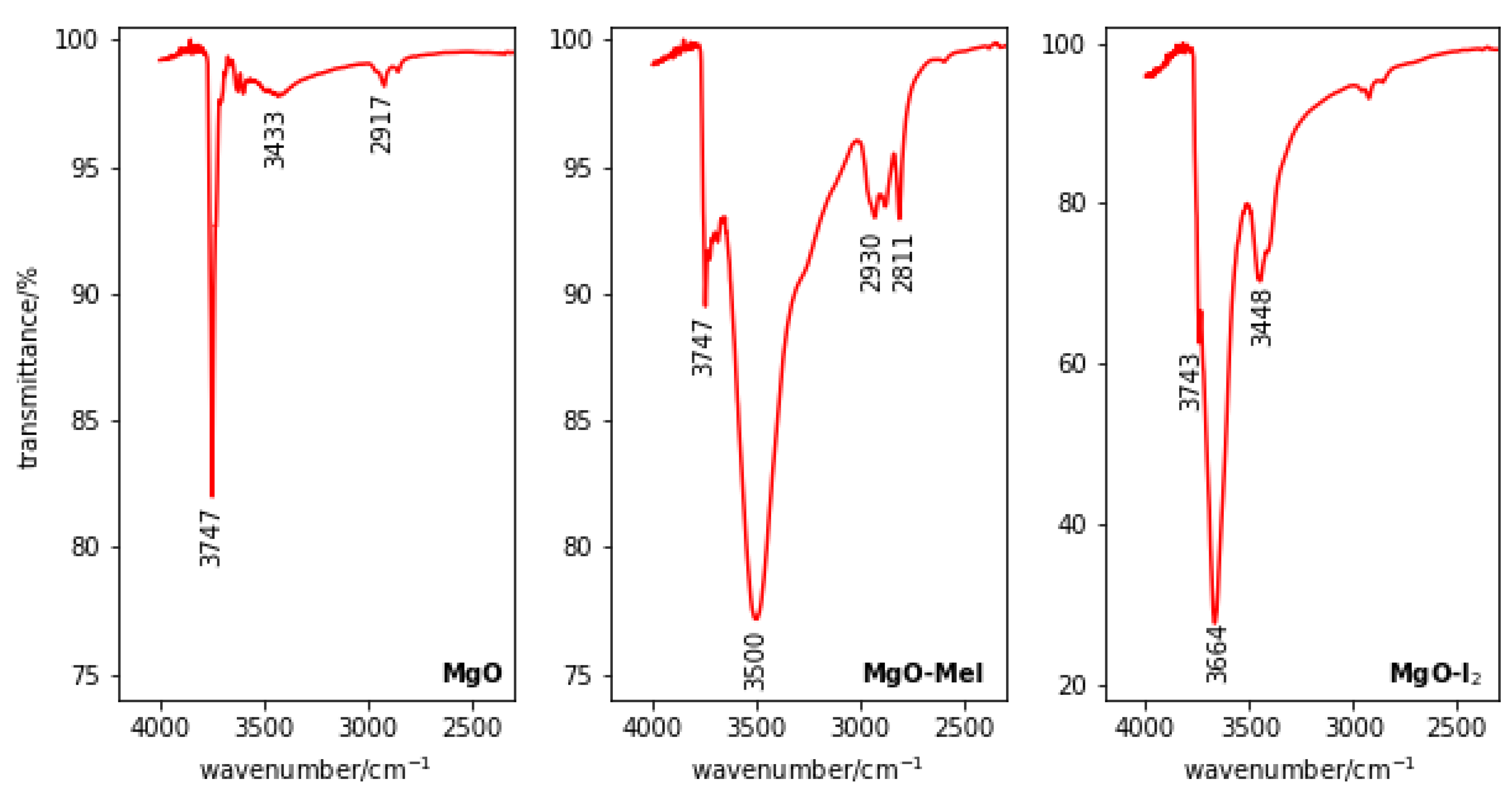
2.3.5. Fourier Transform Infrared Spectroscopy (FTIR)
2.4. Catalytic Activity of MgO-X2, MgO-HX, and MgO-RX Systems in Transfer Hydrogenation Reaction between Ethanol and Acrolein





3. Materials and Methods
3.1. Solvents and Organic and Inorganic Reagents
- alkyl chlorides: MeCl, CH2Cl2, CHCl3, CCl4, n-BuCl, i-BuCl, s-BuCl, and t-BuCl;
- alkyl bromides: MeBr, CH2Br2, CHBr3, n-BuBr, i-BuBr, s-BuBr, and t-BuBr;
- alkyl iodides: MeI, CH2I2, CHI3, EtI, n-BuI, i-BuI, s-BuI, and t-BuI;
- other alkyl halides: BrCH2Cl, ClCH2I, BrCH2I, and BrCH2CH2CH2Cl.
3.2. Synthesis of Magnesium Oxide
3.3. Reaction of MgO with Br2, I2, or HX in Alcohols
3.4. Reaction of MgO with RX, IBr, or ICl3 Vapors (with the Exception of MeCl, MeBr, and CHI3)
3.5. Reaction of MgO with MeCl or MeBr Vapors
3.6. Reaction of MgO with CHI3 Vapors
3.7. Analytical Determinations
3.7.1. Qualitative Tests for I− and IO3− Ions
3.7.2. Quantitative Test for I2 on MgO
3.7.3. Quantitative Test for Br2 on MgO
3.7.4. Quantitative Test for I− Ions on MgO
3.7.5. Quantitative Test for Cl− and Br− Ions on MgO
3.7.6. Analysis of Organic Reaction Products
3.8. Characterization of MgO, MgO-X2 (X = Br and I), MgO-HX, and MgO-RX Catalysts
- -
- MeI adsorption on MgO at 373 K for 10 min, desorption at 293 K for 10 min;
- -
- I2 adsorption on MgO at 473 K for 15 min; desorption at 473 K for 15 min.
3.9. Catalytic Activity Measurements of MgO-X (X = Cl, Br and I) Catalysts in Transfer Hydrogenation of Acrolein with Alcohols
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corma, A.; Iborra, S. Optimization of Earth Alkaline Metal Oxide and Hydroxide Catalysts for Based-Catalyzed Reactions. Adv. Catal. 2006, 49, 239–302. [Google Scholar]
- Hattori, H. Solid Base Catalysts: Generation, Characterization, and Catalytic Behavior of Basic Sites. J. Jpn. Petrol. Inst. 2004, 47, 67–81. [Google Scholar] [CrossRef]
- Tanabe, K.; Hőlderich, W.F. Industrial application of solid acid-base catalysts. Appl. Catal. A Gen. 1999, 181, 399–434. [Google Scholar] [CrossRef]
- Montero, J.M.; Gai, P.; Wilson, K.; Lee, A.F. Structure-sensitive biodiesel synthesis over MgO nanocrystals. Green Chem. 2009, 11, 265–268. [Google Scholar] [CrossRef]
- Xu, C.; Enache, D.I.; Lloyd, R.; Knight, D.W.; Bartley, J.K.; Hutchins, G.J. MgO Catalysed Triglyceride Transesterification for Biodiesel Synthesis. Catal. Lett. 2010, 138, 1–7. [Google Scholar] [CrossRef]
- Chuah, G.K.; Jaenicke, S.; Zhu, Y.Z.; Liu, S.H. Meerwein-Ponndorf-Verley Reduction Over Heterogeneous Catalysts. Curr. Org. Chem. 2006, 10, 1639–1654. [Google Scholar] [CrossRef]
- Ruiz, J.R.; Jiménez-Sanchidrián, C. Heterogeneous Catalysis in the Meerwein-Ponndorf-Verley Reduction of Carbonyl Compounds. Curr. Org. Chem. 2007, 11, 1113–1125. [Google Scholar] [CrossRef]
- Gliński, M. Catalytic hydrogen transfer over magnesia: Vapour and liquid phase reduction of various aralkyl ketones. Appl. Catal. A Gen. 2008, 349, 133–139. [Google Scholar] [CrossRef]
- Gliński, M. Catalytic Transfer Hydrogenation of Cycloalkanones on MgO. Vapour and Liquid Phase Modes of Reaction. Polish J. Chem. 2009, 83, 187–194. [Google Scholar]
- Gliński, M. Structure-reactivity relationship in transfer hydrogenation of aliphatic ketones over magnesium oxide. React. Kinet. Catal. Lett. 2009, 97, 275–279. [Google Scholar] [CrossRef]
- Gliński, M. Highly diasteroselective transfer hydrogenation of 4-t-butylcyclohexanone in the presence of magnesium oxide. React. Kinet. Mech. Cat. 2010, 99, 93–98. [Google Scholar]
- Kijeński, J.; Malinowski, S. Influence of sodium on the physico-chemical and catalytic properties of magnesium oxide. JCS Faraday Trans. I 1978, 74, 250–261. [Google Scholar] [CrossRef]
- Kijeński, J.; Gliński, M.; Quiroz, C.W.A. The direct synthesis of alkenylaromatics during catalytic transfer reduction (CTR) of aralkyl ketones with isopropyl alcohol over MgO of enhanced acidity. Appl. Catal. A Gen. 1997, 150, 77–84. [Google Scholar] [CrossRef]
- Mishakov, I.V.; Heroux, D.S.; Chesnokov, V.V.; Koscheev, S.G.; Mel’gunov, M.S.; Bedilo, A.F.; Buyanov, R.A.; Klabunde, K.J. Reaction of nanocrystalline MgO with 1-iodobutane. J. Catal. 2005, 229, 344–351. [Google Scholar] [CrossRef]
- Szőllősi, G.; Bartók, M. Vapour-phase heterogeneous catalytic transfer hydrogenation of alkyl methyl ketones on MgO: Prevention of the deactivation of MgO in the presence of carbon tetrachloride. Appl. Catal. A Gen. 1998, 169, 263–269. [Google Scholar] [CrossRef]
- Szőllősi, G.; Bartók, M. Role of basic and acidic centers of MgO and modified MgO in catalytic transfer hydrogenation of ketones studied by infrared spectroscopy. J. Mol. Struct. 1999, 482, 13–17. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Catalytic Hydrogen Tranfer Over Magnesia. XXI. Liquid phase reduction of Cyclopentanone Over CH4-xClx/MgO (n = 1-4) Catalysts. Polish, J. Chem. 2008, 88, 1117–1119. [Google Scholar]
- Matsuda, T.; Tanabe, J.; Hayashi, N.; Sasaki, Y.; Miura, H.; Sugiyama, K. Properties of Magnesium Oxides Prepared from Various Salts and Their Catalytic Activity in 1-Butene Isomerization. Bull. Soc. Chem. Jpn. 1982, 55, 990–994. [Google Scholar] [CrossRef]
- Kibblewhite, J.F.J.; Tench, A.J. Reaction of halogens with oxide surfaces. JCS Faraday Trans. I 1974, 70, 72–83. [Google Scholar] [CrossRef]
- Flockhart, B.D.; Lieuw, K.Y.; Pink, R.C. Electron-transfer at alumina surfaces: 4. Reduction of iodine. J Catal. 1974, 32, 20–24. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Reaction of iodine with metal oxides. Can. J. Chem. 2011, 89, 1370–1374. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Liquid phase hydrogen transfer over MgO-I2 and MgO-RI catalysts. React. Kinet. Catal. Lett. 2008, 95, 107–112. [Google Scholar] [CrossRef]
- Sun, N.; Klabunde, K.J. Nanocrystal Metal Oxide−Chlorine Adducts: Selective Catalysts for Chlorination of Alkanes. J. Am. Chem. Soc. 1999, 121, 5587–5588. [Google Scholar] [CrossRef]
- Stoimenov, P.K.; Zaikovski, V.; Klabunde, K.J. Novel Halogen and Interhalogen Adducts of Nanoscale Magnesium Oxide. J. Am. Chem. Soc. 2003, 125, 12907–12913. [Google Scholar] [CrossRef] [PubMed]
- Chesnokov, V.V.; Bedilo, A.F.; Heroux, D.S.; Mishakov, I.V.; Klabunde, K.J. Oxidative dehydrogenation of butane over nanocrystalline MgO, Al2O3, and VOx/MgO catalysts in the presence of small amounts of iodine. J. Catal. 2003, 218, 438–446. [Google Scholar] [CrossRef]
- Mishakov, I.V.; Bedilo, A.F.; Richards, R.M.; Chesnokov, V.V.; Volodin, A.M.; Zaikovskii, V.I.; Buyanov, R.A.; Klabunde, K.J. Nanocrystalline MgO as a Dehydrohalogenation Catalyst. J. Catal. 2002, 206, 40–48. [Google Scholar] [CrossRef]
- Grünert, W.; Brückner, A.; Hofmeister, H.; Claus, P. Structural Properties of Ag/TiO2 Catalysts for Acrolein Hydrogenation. J. Phys. Chem. B 2004, 108, 5709–5717. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Reactivity of Alcohols in Chemoselective Transfer Hydrogenation of Acrolein over Magnesium Oxide as the Catalyst. Catal. Lett. 2011, 141, 293–299. [Google Scholar] [CrossRef]
- Che, M.; Naccache, C.; Imielik, B. Electron spin resonance studies on titanium dioxide and magnesium oxide—Electron donor properties. J. Catal. 1972, 24, 328–335. [Google Scholar] [CrossRef]
- Joseph, R.; Pallan, P.; Sudalai, A.; Ravindranathan, T. Direct conversion of alcohols into the corresponding iodides. Tetrahedron Lett. 1995, 36, 609–612. [Google Scholar] [CrossRef]
- Mori, N.; Togo, H. Facile oxidative conversion of alcohols to esters using molecular iodine. Tetrahedron 2005, 61, 5915–5925. [Google Scholar] [CrossRef]
- Verma, R.M.; Bose, S. Determination of formates by oxidation with iodine. Anal. Chim. Acta 1962, 27, 176–178. [Google Scholar] [CrossRef]
- Collucia, S.; Lavagnino, S.; Marchese, L. The hydroxylated surface of MgO powders and the formation of surface sites. Mater. Chem. Phys. 1988, 18, 445–464. [Google Scholar] [CrossRef]
- Iwanek, E.; Ulkowska, U.; Gliński, M. Surface studies of magnesium oxide-based catalysts modified with X2 or MgX2 (X = Br, I). Surf. Interface Anal. 2015, 47, 1001–1008. [Google Scholar] [CrossRef]
- Iwanek, E.; Ulkowska, U.; Gliński, M. Magnesium oxide modified with various iodine-containing compounds––Surface studies. Surf. Interface Anal. 2017, 49, 945–952. [Google Scholar] [CrossRef]
- Tanabe, K.; Misono, M.; Ono, Y.; Hattori, H. Acid and base centers: Structure and acid-base property. Stud. Surf. Sci Catal. 1989, 51, 27–213. [Google Scholar]
- Busca, G. Bases and Basic Materials in Chemical and Environmental Processes. Liquid versus Solid Basicity. Chem. Rev. 2010, 110, 2217–2249. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Brause, W.; Radeglia, R. H-Brückenwechselwirkungen zwischen OH-Protonendonatoren und Cyanverbindungen. Korrelationen. J. Prakt. Chem. 1970, 312, 1174. [Google Scholar] [CrossRef]
- Sokoll, R.; Hobert, H.; Schmuck, J. Thermal desorption and infrared studies of amines adsorbed on SiO2, Al2O3, Fe2O3, MgO, and CaO I. Diethylamine and triethylarnine. J. Catal. 1990, 121, 153–164. [Google Scholar] [CrossRef]
- Cunningham, J.; Penny, A.L. Reactions involving electron transfer at semiconductor surfaces. III. Dissociation of methyl iodide over zinc oxide. J. Phys. Chem. 1972, 76, 2353–2361. [Google Scholar] [CrossRef]
- Gliński, M.; Ulkowska, U. Description of the structure-chemoselectivity relationship in the transfer hydrogenation of α,β-unsaturated aldehydes and ketones with alcohols in the presence of magnesium oxide. Appl. Catal. A Gen. 2018, 554, 117–124. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TR [K] | Concentration [μmol g−1 MgO] | |
|---|---|---|
| Br2 | Br− | |
| 295 | 43 | 125 |
| 373 | 5 | 120 |
| 473 | 0 | 115 |
| 673 | 0 | 117 |
| 873 | 0 | 115 |
| Alcohol | B.p. [K] | [I−] [mmol g−1] 1,2,3 | ([I−]/[Mg2+]) 4surf | ([I−]/[Mg2+]) 5bulk |
|---|---|---|---|---|
| CH3OH | 337.9 | 10.8 | 5.8 | 0.44 |
| C2H5OH | 351.5 | 10.4 | 5.6 | 0.42 |
| 2-C3H7OH | 355.7 | 6.8 | 3.6 | 0.27 |
| 2-C5H11OH | 392.5 | 12.1 | 6.5 | 0.49 |
| 3-C5H11OH | 388.5 | 9.9 | 5.3 | 0.40 |
| t-C4H9OH | 355.7 | 6.5 | 3.5 | 0.26 |
| Alcohol | Main Reaction Products |
|---|---|
| MeOH | No organic reaction products were detected |
| EtOH | 1,1-diethoxyethane, acetaldehyde, ethyl acetate |
| 2-PrOH | 2-iodopropane, acetone, di i-propyl ether |
| 2-PeOH | 2-pentanone, 2-iodopentane, di 2-pentyl ether |
| 3-PeOH | 3-pentanone, 3-iodopentane, di 3-pentyl ether |
| t-BuOH | methylpropene, 2,4,4-trimethyl-1-pentene t-butyl i-butyl ether |
| Time [h] | [I2] [μmol g−1] | [I−] 1 [μmol g−1] | [I2] [mmol g−1] | [I−] 2 [mmol g−1] |
|---|---|---|---|---|
| 0.0 | 200 | 0 | 3.00 | 0.00 |
| 0.5 | 98 | 211 | 2.64 | 0.66 |
| 1.0 | 73 | 263 | 2.60 | 0.69 |
| 3.0 | 41 | 331 | 2.57 | 0.85 |
| 6.0 | 23 | 368 | 2.47 | 1.08 |
| 10.0 | 0 | 404 | 2.42 | 1.13 |
| 16.0 | 0 | 397 | 2.37 | 1.24 |
| 24.0 | 0 | 402 | 2.34 | 1.32 |
| 30.0 | -- | -- | 2.32 | 1.34 3 |
| Time [h] | [Br2] [μmol g−1] | [Br−] 1 [μmol g−1] | [Br2] [mmol g−1] | [Br−] 2 [mmol g−1] |
|---|---|---|---|---|
| 0.0 | 200 | 0 | 3.00 | 0.00 |
| 0.2 | 65 | 270 | -- | -- |
| 0.5 | 5 | 382 | 2.51 | 0.88 |
| 1.0 | 0 | 394 | 2.30 | 1.36 |
| 3.0 | 0 | 392 | 2.01 | 1.94 |
| 6.0 | 0 | 395 | 1.47 | 3.00 |
| 16.0 | -- | -- | 0.34 | 5.25 |
| 24.0 | -- | -- | 0.06 | 5.80 |
| 30.0 | -- | -- | 0.02 | 5.94 3 |
| RCl | [Cl−] [mmol g−1] | SBET [m2 g−1] | Color |
|---|---|---|---|
| CH3Cl | 0.17 | 104 | Colorless |
| CH2Cl2 | 0.21 | 90 | Colorless |
| CHCl3 | 0.27 | 94 | Colorless |
| CCl4 | 0.15 | 89 | Colorless |
| EtCl | 0.17 | 32 | Colorless |
| n-BuCl | 0.24 | 95 | Dark yellow |
| i-BuCl | 0.18 | -- | Colorless |
| s-BuCl | 0.84 | 69 | Colorless |
| t-BuCl | 19.31 | 9 | Pink |
| t-AmCl | 12.22 | -- | Pink |
| RBr | [Br−] [mmol g−1] | SBET [m2 g−1] | Color |
|---|---|---|---|
| CH3Br | 0.21 | 97 | Colorless |
| CH2Br2 | 0.17 | 91 | Pale yellow |
| CHBr3 | 0.23 1 | 105 | Beige |
| EtBr | 0.18 | 58 | Pale yellow |
| n-BuBr | 0.28 | 72 | Dark yellow |
| i-BuBr | 0.22 | -- | Pale yellow |
| s-BuBr | 3.49 | 4 | Pale pink |
| t-BuBr | 12.42 | 1 | Violet |
| RI | [I−] [mmol g−1] | SBET [m2 g−1] | Color |
|---|---|---|---|
| CH3I | 0.25 1,2,3,4 | 85 | Pale yellow |
| CH2I2 | 0.08 5 | -- | Yellow |
| CHI3 | 0.10 6 | -- | Yellow |
| EtI | 0.23 7 | 89 | Pale yellow |
| n-BuI | 0.28 | 88 | Pale yellow |
| i-BuI | 0.27 | 91 | Yellow |
| s-BuI | 0.56 | 41 | Yellow |
| t-BuI | 2.33 8 | 15 | Yellow-red |
| RX2 or IXn | [X−] [mmol g−1] | Color | ||
|---|---|---|---|---|
| [Cl−] | [Br−] | [I−] | ||
| ClCH2Br | 0.06 | 0.19 | -- | Colorless |
| ClCH2I | 0.20 | -- | 0.04 | Yellowish |
| BrCH2I | -- | 0.17 | - 1 | Yellow |
| Cl(CH2)3Br | 0.10 | 0.18 | -- | Colorless |
| Br2 | -- | 0.12 2 | -- | Yellowish |
| I2 | -- | -- | 0.01 3 | Yellow |
| IBr | -- | 0.19 | 0 | Yellow–brown |
| ICl3 | 0.10 | -- | 0 | Colorless |
| Modifier | SBET [m2∙g−1] | Strength of Sites | |
|---|---|---|---|
| Acidic | Basic | ||
| -- | 100 | H0 > 4.8 | 7.2 ≤ H- < 33.0 |
| Br2 | 98 | 0.8 < H0 ≤ 4.8 | 7.2 ≤ H- < 15.0 |
| I2 | 100 | −3.0 < H0 ≤ 4.8 | 7.2 ≤ H- < 9.3 |
| HCl | 95 | −5.6 < H0 ≤ 4.8 | 7.2 ≤ H- < 18.4 |
| HBr | 96 | −3.0 < H0 ≤ 4.8 | 7.2 ≤ H- < 18.4 |
| HI | 98 | −3.0 < H0 ≤ 4.8 | 7.2 ≤ H- < 18.4 |
| Modifier | SBET [m2∙g−1] | Strength of Sites | |
|---|---|---|---|
| Acidic | Basic | ||
| CH3Cl | 104 | 0.8 < H0 ≤ 4.8 | 7.2≤ H- < 22.3 |
| CH2Cl2 | 90 | 0.8 < H0 ≤ 4.8 | 7.2 ≤ H- < 22.3 |
| CHCl3 | 94 | −3.0 < H0 ≤ 4.8 | 7.2 ≤ H- < 22.3 |
| CCl4 | 98 | H0 > 4.8 | 7.2 ≤ H- < 33.0 |
| RX | SBET [m2∙g−1] | Strength of Sites | |
|---|---|---|---|
| Acidic | Basic | ||
| MeCl | 104 | 0.8 < H0 ≤ 4.8 | 7.2 ≤ H- < 22.3 |
| t-BuCl | 9 | 0.8 < H0 ≤ 4.8 | H- < 7.2 |
| MeBr | 97 | 0.8 < H0 ≤ 4.8 | 7.2 ≤ H- < 15.0 |
| EtBr | 58 | 0.8 < H0 ≤ 4.8 | 7.2 ≤ H- < 18.4 |
| MeI | 91 | −3.0 < H0 ≤ 4.8 | 7.2 ≤ H- < 9.3 |
| EtI | 19 | 0.8 < H0 ≤ 4.8 | 7.2 ≤ H- < 15.0 |
| [I2] [μmol∙g−1] | T [K] | Conversion [%] | Moles from 100 Moles of Acrolein | |||
|---|---|---|---|---|---|---|
| UOL | SAL | SOL | Others | |||
| 0 | 473 | 15 | 15 | 0 | 0 | 0 |
| 523 | 32 | 31 | 1 | 0 | 0 | |
| 573 | 51 | 47 | 2 | 1 | 1 | |
| 50 | 473 | 45 | 45 | 0 | 0 | 0 |
| 523 | 68 | 65 | 0 | 2 | 1 | |
| 573 | 69 | 69 | 1 | 5 | 1 | |
| 100 | 473 | 67 | 65 | 0 | 1 | 1 |
| 523 | 78 | 73 | 0 | 4 | 1 | |
| 573 | 82 | 67 | 1 | 9 | 1 | |
| 200 | 473 | 73 | 57 | 0 | 3 | 13 |
| 523 | 78 | 48 | 1 | 6 | 23 | |
| 573 | 82 | 47 | 1 | 10 | 24 | |
| 100 1 | 473 | 67 | 64 | 0 | 2 | 1 |
| 523 | 78 | 67 | 1 | 5 | 5 | |
| 573 | 83 | 66 | 1 | 9 | 7 | |
| [Br2] [μmol∙g−1] | T [K] | Conversion [%] | Moles from 100 Moles of Acrolein | |||
|---|---|---|---|---|---|---|
| UOL | SAL | SOL | Others | |||
| 0 | 473 | 15 | 15 | 0 | 0 | 0 |
| 523 | 32 | 31 | 1 | 0 | 0 | |
| 573 | 51 | 47 | 2 | 1 | 1 | |
| 100 | 473 | 63 | 63 | 0 | 0 | 0 |
| 523 | 77 | 75 | 0 | 1 | 1 | |
| 573 | 82 | 74 | 1 | 5 | 2 | |
| 200 | 473 | 72 | 72 | 0 | 0 | 0 |
| 523 | 81 | 76 | 0 | 2 | 3 | |
| 573 | 91 | 46 | 0 | 13 | 32 | |
| 300 | 473 | 63 | 58 | 0 | 1 | 4 |
| 523 | 82 | 41 | 0 | 9 | 32 | |
| 573 | 86 | 37 | 0 | 11 | 38 | |
| HX | T [K] | Conversion [%] | Moles from 100 Moles of Acrolein | |||
|---|---|---|---|---|---|---|
| UOL | SAL | SOL | Others | |||
| HCl | 473 | 35 | 34 | 0 | 0 | 1 |
| 523 | 56 | 50 | 0 | 1 | 5 | |
| 573 | 80 | 58 | 1 | 6 | 15 | |
| HBr | 473 | 64 | 62 | 0 | 1 | 1 |
| 523 | 69 | 60 | 1 | 1 | 7 | |
| 573 | 75 | 54 | 0 | 3 | 18 | |
| HI | 473 | 62 | 60 | 0 | 1 | 1 |
| 523 | 73 | 70 | 0 | 2 | 1 | |
| 573 | 74 | 63 | 1 | 5 | 5 | |
| 1 | 473 | 18 | 18 | 9 | 0 | 0 |
| 523 | 34 | 32 | 2 | 0 | 0 | |
| 573 | 53 | 46 | 4 | 2 | 1 | |
| RX | T [K] | Conversion [%] | Moles from 100 Moles of Acrolein | |||
|---|---|---|---|---|---|---|
| UOL | SAL | SOL | Others | |||
| -- | 473 | 15 | 15 | 0 | 0 | 0 |
| 523 | 32 | 31 | 1 | 0 | 0 | |
| 573 | 51 | 47 | 2 | 1 | 1 | |
| MeI | 473 | 69 | 65 | 1 | 2 | 1 |
| 523 | 76 | 58 | 2 | 15 | 1 | |
| 573 | 81 | 53 | 4 | 21 | 3 | |
| EtCl | 473 | 45 | 44 | 0 | 0 | 1 |
| 523 | 65 | 57 | 0 | 2 | 6 | |
| 573 | 86 | 43 | 0 | 4 | 38 | |
| EtBr | 473 | 75 | 73 | 0 | 1 | 1 |
| 523 | 83 | 77 | 0 | 3 | 3 | |
| 573 | 87 | 62 | 0 | 11 | 14 | |
| EtI | 473 | 73 | 70 | 0 | 2 | 1 |
| 523 | 77 | 65 | 1 | 10 | 1 | |
| 573 | 80 | 60 | 2 | 15 | 3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gliński, M.; Ulkowska, U.; Kaszkur, Z.; Łomot, D.; Winiarek, P. MgO Modified by X2, HX, or Alkyl Halide (X = Cl, Br, or I) Catalytic Systems and Their Activity in Chemoselective Transfer Hydrogenation of Acrolein into Allyl Alcohol. Molecules 2024, 29, 3180. https://doi.org/10.3390/molecules29133180
Gliński M, Ulkowska U, Kaszkur Z, Łomot D, Winiarek P. MgO Modified by X2, HX, or Alkyl Halide (X = Cl, Br, or I) Catalytic Systems and Their Activity in Chemoselective Transfer Hydrogenation of Acrolein into Allyl Alcohol. Molecules. 2024; 29(13):3180. https://doi.org/10.3390/molecules29133180
Chicago/Turabian StyleGliński, Marek, Urszula Ulkowska, Zbigniew Kaszkur, Dariusz Łomot, and Piotr Winiarek. 2024. "MgO Modified by X2, HX, or Alkyl Halide (X = Cl, Br, or I) Catalytic Systems and Their Activity in Chemoselective Transfer Hydrogenation of Acrolein into Allyl Alcohol" Molecules 29, no. 13: 3180. https://doi.org/10.3390/molecules29133180
APA StyleGliński, M., Ulkowska, U., Kaszkur, Z., Łomot, D., & Winiarek, P. (2024). MgO Modified by X2, HX, or Alkyl Halide (X = Cl, Br, or I) Catalytic Systems and Their Activity in Chemoselective Transfer Hydrogenation of Acrolein into Allyl Alcohol. Molecules, 29(13), 3180. https://doi.org/10.3390/molecules29133180