
Transition Metal Chelation Effect in MOF-253 Materials: Guest Molecule Adsorption Dynamics and Proposed Formic Acid Synthesis Investigated by Atomistic Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
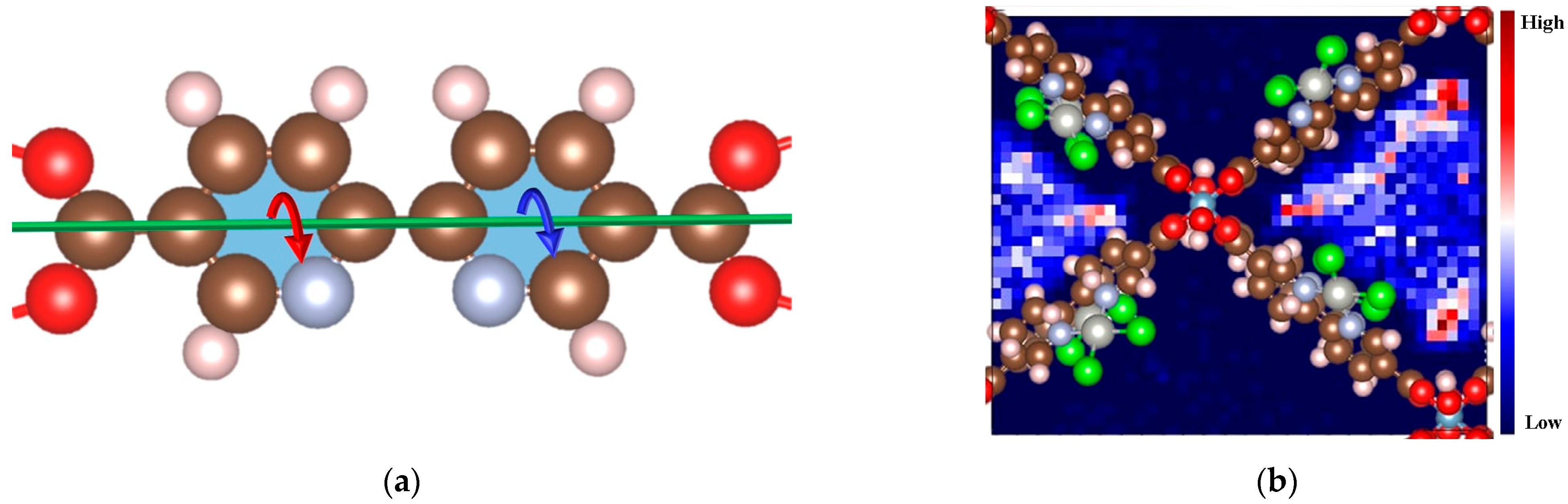
2.1. The Dynamics Phenomenon of Linker in MOF-253
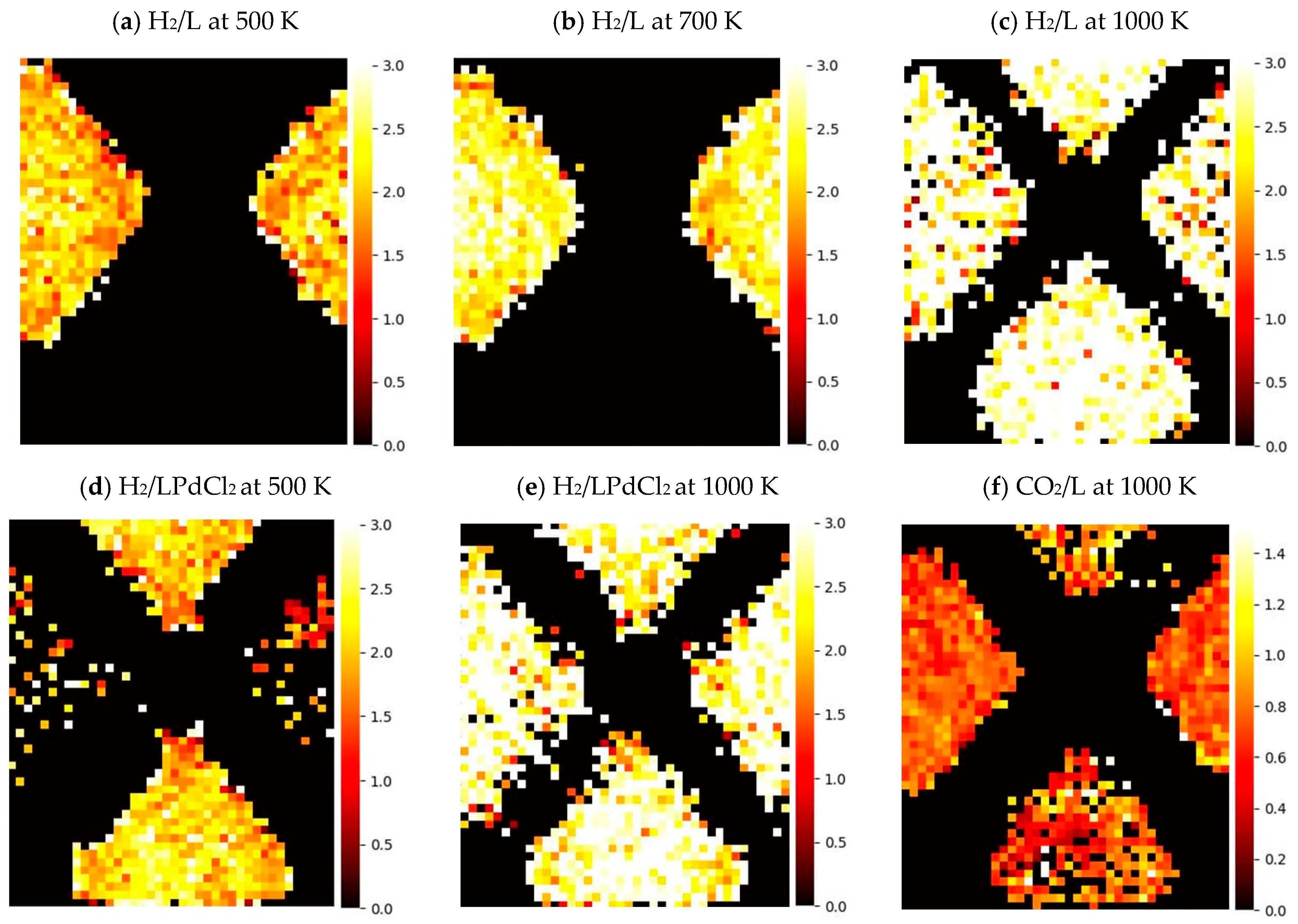
2.2. The Adsorption of the Guest Molecules in MOF-253
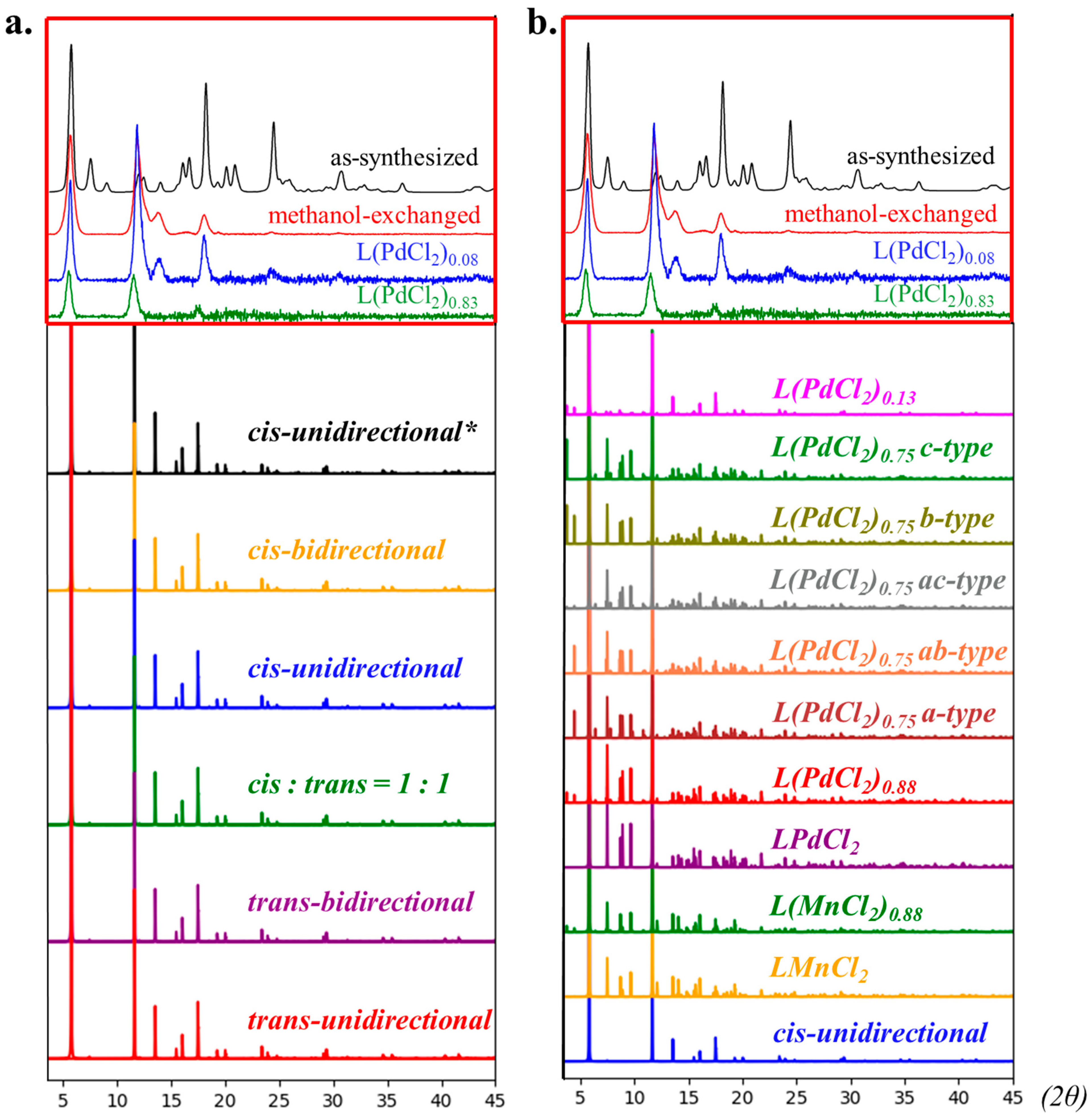
2.3. The Examination of the PXRD Simulation
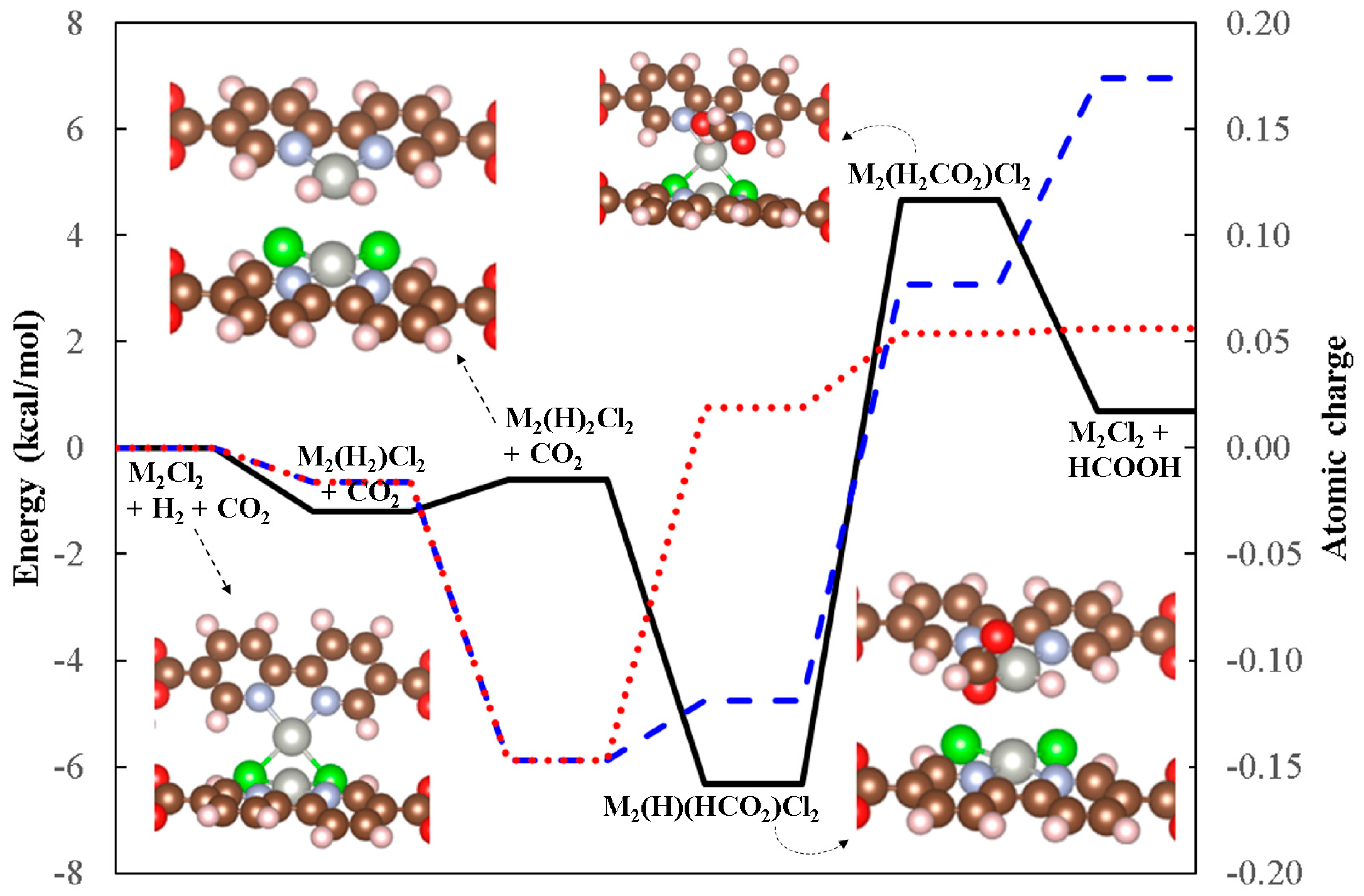
2.4. The Catalytic Mechanism for Another LPdCl2 Isomer
3. Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forgan, R.S. Modulated self-assembly of metal-organic frameworks. Chem. Sci. 2020, 11, 4546–4562. [Google Scholar] [CrossRef] [PubMed]
- Shakya, D.M.; Ejegbavwo, O.A.; Rajeshkumar, T.; Senanayake, S.D.; Brandt, A.J.; Farzandh, S.; Acharya, N.; Ebrahim, A.M.; Frenkel, A.I.; Rui, N.; et al. Selective Catalytic Chemistry at Rhodium(II) Nodes in Bimetallic Metal–Organic Frameworks. Angew. Chem. Int. Ed. 2019, 58, 16533–16537. [Google Scholar] [CrossRef] [PubMed]
- Szécsényi, Á.; Li, G.; Gascon, J.; Pidko, E.A. Unraveling reaction networks behind the catalytic oxidation of methane with H2O2 over a mixed-metal MIL-53(Al,Fe) MOF catalyst. Chem. Sci. 2018, 9, 6765–6773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Zhou, H.-C. Synthesis of MOFs for heterogeneous catalysis via linker design. Polyhedron 2018, 154, 189–201. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Q.; Jiang, H.-L. Metal–organic frameworks meet metal nanoparticles: Synergistic effect for enhanced catalysis. Chem. Soc. Rev. 2017, 46, 4774–4808. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Yan, R.; Shu, Y.; Cao, Q.; Zhang, L. Strategies for the application of metal–organic frameworks in catalytic reactions. RSC Adv. 2022, 12, 10114–10125. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Dong, L.-Z.; Liu, J.; Li, S.-L.; Lan, Y.-Q. From molecular metal complex to metal-organic framework: The CO2 reduction photocatalysts with clear and tunable structure. Coord. Chem. Rev. 2019, 390, 86–126. [Google Scholar] [CrossRef]
- Seo, J.S.; Whang, D.; Lee, H.; Jun, S.I.; Oh, J.; Jeon, Y.J.; Kim, K. A homochiral metal–organic porous material for enantioselective separation and catalysis. Nature 2000, 404, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Kent, C.A.; Mehl, B.P.; Ma, L.; Papanikolas, J.M.; Meyer, T.J.; Lin, W. Energy Transfer Dynamics in Metal−Organic Frameworks. J. Am. Chen. Soc. 2010, 132, 12767–12769. [Google Scholar] [CrossRef]
- Wang, C.; Xie, Z.; deKrafft, K.E.; Lin, W. Doping Metal–Organic Frameworks for Water Oxidation, Carbon Dioxide Reduction, and Organic Photocatalysis. J. Am. Chen. Soc. 2011, 133, 13445–13454. [Google Scholar] [CrossRef]
- Chen, F.; Drake, H.F.; Feng, L.; Powell, J.A.; Wang, K.-Y.; Yan, T.-H.; Zhou, H.-C. Metal–Organic Frameworks as Versatile Platforms for Organometallic Chemistry. Inorganics 2021, 9, 27. [Google Scholar] [CrossRef]
- Kaes, C.; Katz, A.; Hosseini, M.W. Bipyridine: The Most Widely Used Ligand. A Review of Molecules Comprising at Least Two 2,2‘-Bipyridine Units. Chem. Rev. 2000, 100, 3553–3590. [Google Scholar] [CrossRef] [PubMed]
- Bloch, E.D.; Britt, D.; Lee, C.; Doonan, C.J.; Uribe-Romo, F.J.; Furukawa, H.; Long, J.R.; Yaghi, O.M. Metal Insertion in a Microporous Metal−Organic Framework Lined with 2,2′-Bipyridine. J. Am. Chen. Soc. 2010, 132, 14382–14384. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Cohen, S.M. A robust, catalytic metal–organic framework with open 2,2′-bipyridine sites. Chem. Commun. 2014, 50, 4810–4812. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Peng, X.; Zeng, H. Synthesis, structure and luminescence property of Eu(III) metal–organic framework based on 2,2′-bipyridine-5,5′-dicarboxylic acid. Inorg. Chem. Commun. 2014, 46, 39–42. [Google Scholar] [CrossRef]
- Vizuet, J.P.; Lewis, A.L.; McCandless, G.T.; Balkus, K.J. Synthesis and characterization of a holmium 2,2′-bipyridine-5,5′-dicarboxylate MOF: Towards the construction of a suitable holmium carrier. Polyhedron 2019, 159, 12–17. [Google Scholar] [CrossRef]
- Wu, Y.; Wei, L.; Wang, H.; Chen, L.; Zhang, Q. First principles study of enhanced CO2 adsorption on MOF-253 by salt-insertion. Comput. Mater. Sci. 2016, 111, 79–85. [Google Scholar] [CrossRef]
- Deng, X.; Qin, Y.; Hao, M.; Li, Z. MOF-253-Supported Ru Complex for Photocatalytic CO2 Reduction by Coupling with Semidehydrogenation of 1,2,3,4-Tetrahydroisoquinoline (THIQ). Inorg. Chem. 2019, 58, 16574–16580. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, Y.; Fu, J.; Zeng, X.; Chen, Z.; Li, Z. Construction of a supported Ru complex on bifunctional MOF-253 for photocatalytic CO2 reduction under visible light. Chem. Commun. 2015, 51, 2645–2648. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xie, J.; Zhang, J. MOF-253 immobilized Pd and Cu as recyclable and efficient green catalysts for Sonogashira reaction. Arab. J. Chem. 2022, 15, 103962. [Google Scholar] [CrossRef]
- Hsieh, M.-C.; Krishnan, R.; Tsai, M.-K. Formic Acid Generation from CO2 Reduction by MOF-253 Coordinated Transition Metal Complexes: A Computational Chemistry Perspective. Catalysts 2022, 12, 890. [Google Scholar] [CrossRef]
- Gonzalez-Nelson, A.; Coudert, F.-X.; van der Veen, M.A. Rotational Dynamics of Linkers in Metal–Organic Frameworks. Nanomaterials 2019, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.L.; Tranchemontagne, D.; Yaghi, O.M.; Garcia-Garibay, M.A. Amphidynamic Character of Crystalline MOF-5: Rotational Dynamics of Terephthalate Phenylenes in a Free-Volume, Sterically Unhindered Environment. J. Am. Chen. Soc. 2008, 130, 3246–3247. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kolokolov, D.I.; da Silva, I.; Stepanov, A.G.; Blake, A.J.; Dailly, A.; Manuel, P.; Tang, C.C.; Yang, S.; Schröder, M. Porous Metal–Organic Polyhedral Frameworks with Optimal Molecular Dynamics and Pore Geometry for Methane Storage. J. Am. Chen. Soc. 2017, 139, 13349–13360. [Google Scholar] [CrossRef] [PubMed]
- Inukai, M.; Fukushima, T.; Hijikata, Y.; Ogiwara, N.; Horike, S.; Kitagawa, S. Control of Molecular Rotor Rotational Frequencies in Porous Coordination Polymers Using a Solid-Solution Approach. J. Am. Chen. Soc. 2015, 137, 12183–12186. [Google Scholar] [CrossRef] [PubMed]
- Vogelsberg, C.S.; Uribe-Romo, F.J.; Lipton, A.S.; Yang, S.; Houk, K.N.; Brown, S.; Garcia-Garibay, M.A. Ultrafast rotation in an amphidynamic crystalline metal organic framework. Prod. Nat. Acad. Sci. USA 2017, 114, 13613–13618. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Wang, Z.; Yeary, P.; Feng, X.; Lin, W. Self-adaptive Metal-Organic Framework Assembles Di-iron Active Sites to Mimic Monooxygenases. J. Am. Chen. Soc. 2023; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Rüger, R.; Franchini, M.; Trnka, T.; Yakovlev, A.; Lenthe, E.v.; Philipsen, P.; Vuren, T.v.; Klumpers, B.; Soini, T. Amsterdam Modeling Suite, AMS2020.101; SCM: Amsterdam, The Netherlands, 2020.
- Coupry, D.E.; Addicoat, M.A.; Heine, T. Extension of the Universal Force Field for Metal–Organic Frameworks. J. Chem. Theory Comput. 2016, 12, 5215–5225. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chen. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Lenthe, E.v.; Snijders, J.G.; Baerends, E.J. The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, M.-C.; Liang, W.-L.; Chang, C.-C.; Tsai, M.-K. Transition Metal Chelation Effect in MOF-253 Materials: Guest Molecule Adsorption Dynamics and Proposed Formic Acid Synthesis Investigated by Atomistic Simulations. Molecules 2024, 29, 3211. https://doi.org/10.3390/molecules29133211
Hsieh M-C, Liang W-L, Chang C-C, Tsai M-K. Transition Metal Chelation Effect in MOF-253 Materials: Guest Molecule Adsorption Dynamics and Proposed Formic Acid Synthesis Investigated by Atomistic Simulations. Molecules. 2024; 29(13):3211. https://doi.org/10.3390/molecules29133211
Chicago/Turabian StyleHsieh, Meng-Chi, Wei-Lun Liang, Chun-Chih Chang, and Ming-Kang Tsai. 2024. "Transition Metal Chelation Effect in MOF-253 Materials: Guest Molecule Adsorption Dynamics and Proposed Formic Acid Synthesis Investigated by Atomistic Simulations" Molecules 29, no. 13: 3211. https://doi.org/10.3390/molecules29133211
APA StyleHsieh, M.-C., Liang, W.-L., Chang, C.-C., & Tsai, M.-K. (2024). Transition Metal Chelation Effect in MOF-253 Materials: Guest Molecule Adsorption Dynamics and Proposed Formic Acid Synthesis Investigated by Atomistic Simulations. Molecules, 29(13), 3211. https://doi.org/10.3390/molecules29133211