
Triiron Complexes Featuring Azadiphosphine Related to the Active Site of [FeFe]-Hydrogenases: Their Redox Behavior and Protonation


Abstract

1. Introduction
2. Results and Discussion
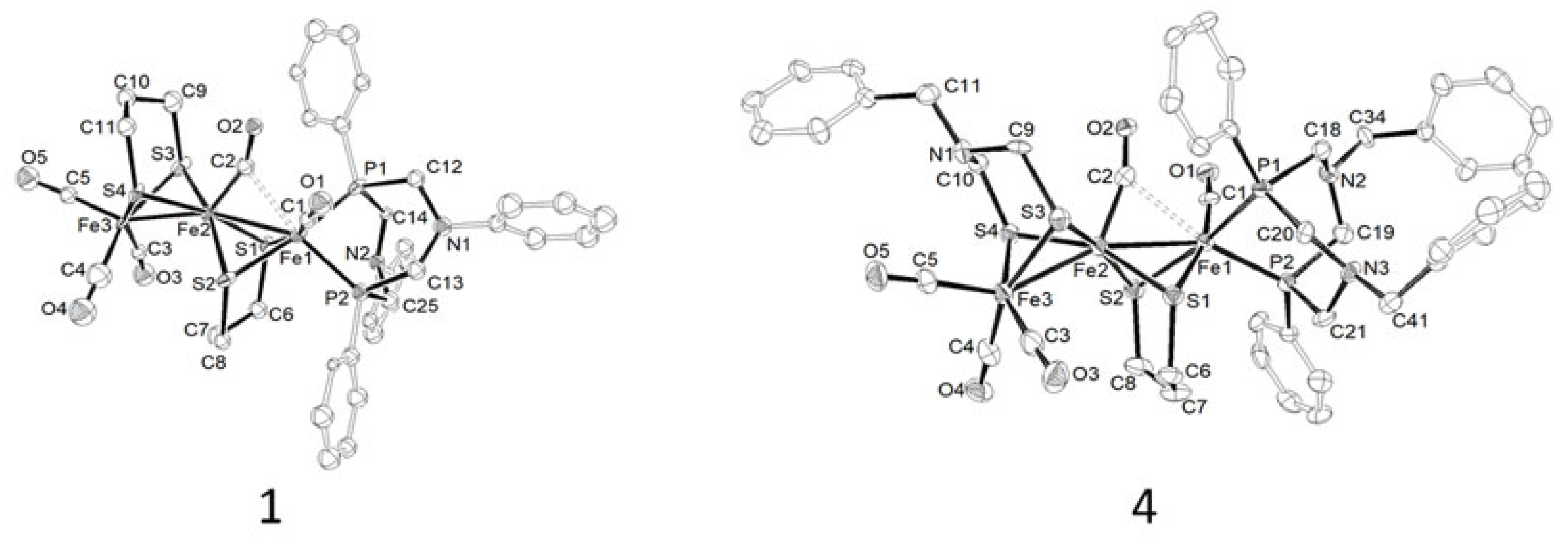
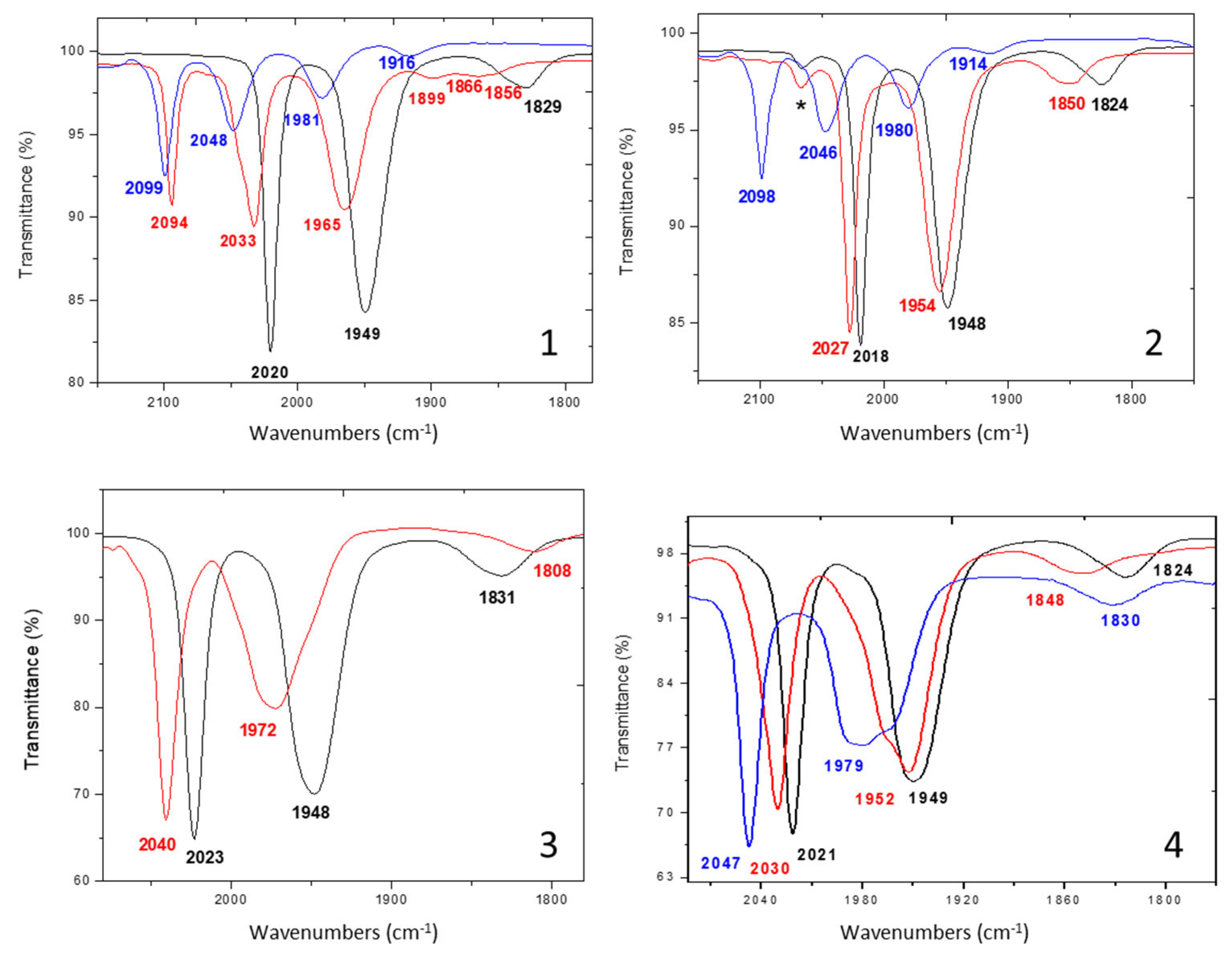
2.1. Synthesis, Spectroscopic, and Structural Characterization of Complexes [Fe3(CO)5(κ2-PPh2NR2)(μ-pdt)2] (R = Ph (1), Bn (2)), [Fe3(CO)5(κ2-PPh2NR2)(μ-adtBn)(μ-pdt)] (R = Ph (3), Bn (4))
2.2. Electrochemical Behavior of Complexes in the Absence of Protons
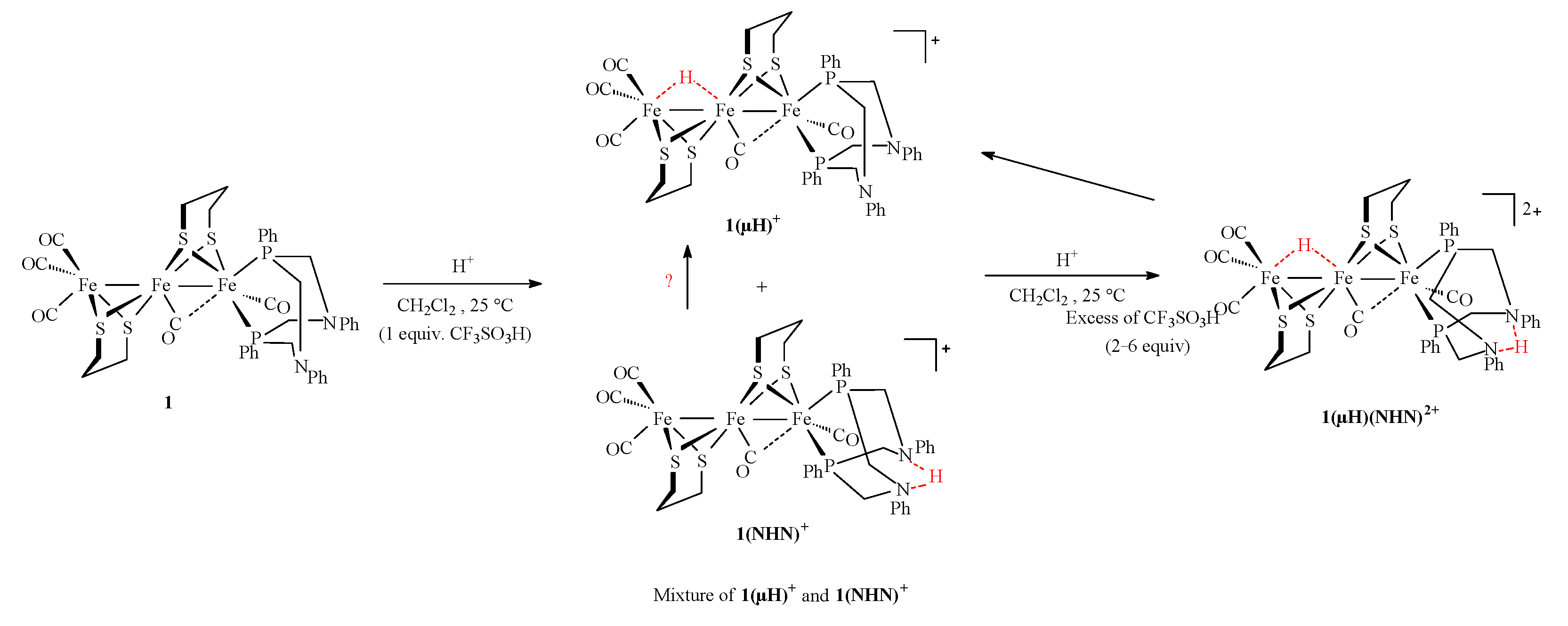
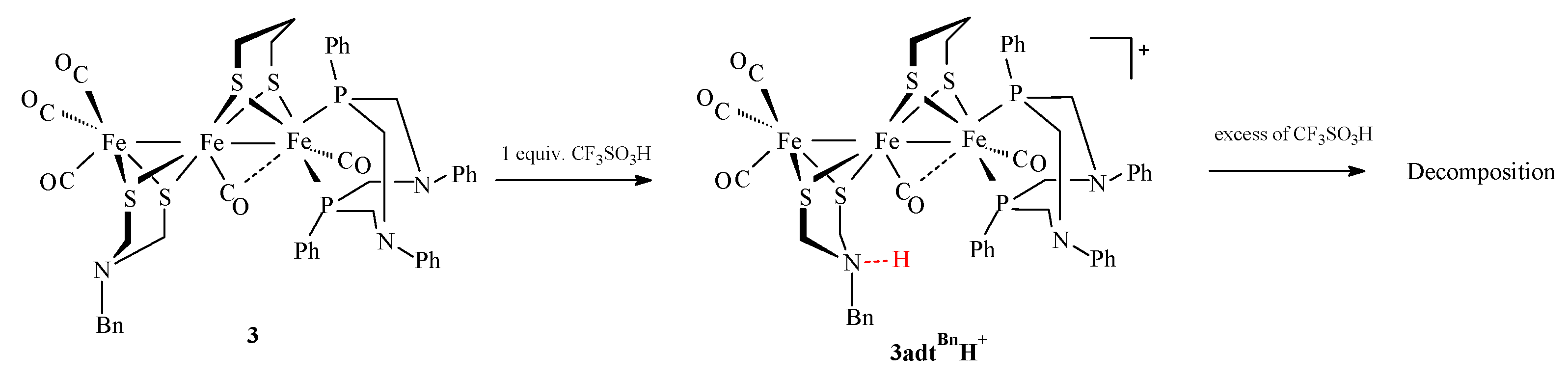
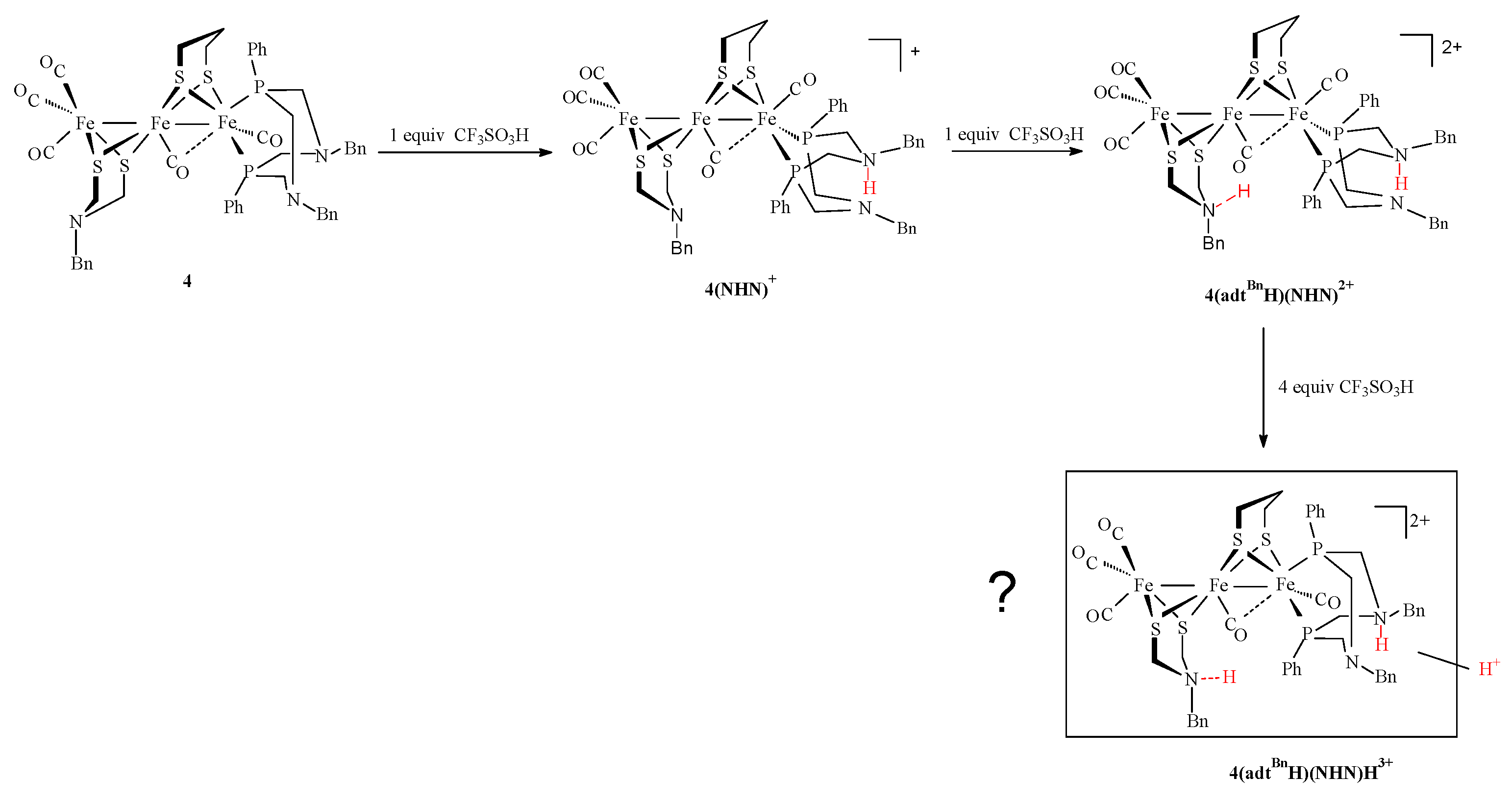
2.3. Behavior of Complexes in the Presence of Protons
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Preparation of complexes [Fe3(CO)5(κ2-PPh2NR2)(μ-pdt)2] (R = Ph (1), Bn (2)), [Fe3(CO)5(κ2-PPh2NR2)(μ-adtBn)(μ-pdt)] (R = Ph (3), Bn (4))
- Data for [Fe3(CO)5(κ2-PPh2NPh2)(μ-pdt)2] (1)m = 0.26 g, yield = 52%IR (CH2Cl2, cm−1): ῡ(CO) = 2020 (s), 1949 (s), 1829 (w).31P{1H} NMR (CDCl3, ppm): δ = 56.6 (d, 2JPP = 80 Hz), 40. 6 (d, 2JPP = 80 Hz).1H NMR (CDCl3, ppm): δ = 7.89–6.95 (m, 20H, Ph), 4.55–3.79 (m, 8H, CH2, PPh2NPh2), 2.15–1.09 (m, 12H, CH2, pdt).Anal. Calcd (%) for C39H40Fe3N2O5P2S4(%) Theoretical: C = 48.07; H = 4.14; N = 2.87(%) Experimental: C = 48.13; H = 4.09; N = 2.74
- Data for [Fe3(CO)5(κ2-PPh2NBn2)(μ-pdt)2] (2)m = 0.27 g, yield = 53%IR (CH2Cl2, cm−1): ῡ(CO) = 2018 (s), 1948 (s), 1824 (w).31P{1H} NMR (CDCl3, ppm): δ = 53.3 (d, 2JPP = 81 Hz), 40.8 (d, 2JPP = 81 Hz).1H NMR (CDCl3, ppm): δ = 7.65–7.19 (m, 20H, Ph), 4.09 (q(AB), 2JHH = 13 Hz, 2H, NCH2Ph), 3.84 (q(AB), 2JHH = 13 Hz, 2H, NCH2Ph), 3.77–2.88 (m, 8H, PCH2N), 2.37–1.07 (m, 12H, CH2, pdt).Anal. Calcd (%) for C41H44Fe3N2O5P2S4(%) Theoretical: C = 49.12; H = 4.42; N = 2.79(%) Experimental: C = 48.75; H = 4.41; N = 2.50
- Data for [Fe3(CO)5(κ2-PPh2NPh2)(μ-adtBn)(μ-pdt)] (3)m = 0.24 g, yield = 55%.IR (CH2Cl2, cm−1): ῡ(CO) = 2023 (s), 1948 (s), 1831 (w).31P{1H} NMR (CDCl3, ppm): δ = 57.0 (d, 2JPP = 80 Hz), 40.6 (d, 2JPP = 80 Hz).1H NMR (CDCl3, ppm): δ = 7.89–6.95 (m, 25H, Ph), 4.54–3.77 (m, 8H, PPh2NPh2), 3.45 (d(AB), 2JHH = 14Hz, 2H, NCH2Ph), 3.15 (d, 2JHH = 11Hz, 1H, SCH2N), 3.00 (d, 2JHH = 11Hz, 1H, SCH2N), 2.51 (d, 2JHH = 11Hz, 1H, SCH2N), 1.86 (d, 2JHH = 11Hz, 1H, SCH2N), 2.12–1.28 (m, CH2, 6H, pdt).Anal. Calcd (%) for 3, 2 CH2Cl2: C47H49Cl4Fe3N3O5P2S4(%) Theoretical: C = 45.69; H = 4.00; N = 3.40(%) Experimental: C = 46.17; H = 4.07; N = 3.39
- Data for [Fe3(CO)5(κ2-PPh2NBn2)(μ-adt)(μ-pdt)] (4)m = 0.24 g, yield = 53%IR (CH2Cl2, cm−1): ῡ(CO) = 2021 (s), 1949 (s), 1824 (w).31P{1H} NMR (CDCl3, ppm): δ = 53.7 (d, 2JPP = 81.5 Hz), 40.8 (d, 2JPP = 81.5 Hz).1H NMR (CDCl3, ppm): δ = 7.64–7.13 (m, 25H, Ph), 4.04 (q(AB), 2JHH = 13 Hz, 2H, NCH2Ph), 3.83 (q(AB), 2JHH = 13 Hz, 1H, NCH2Ph), 3.75–3.27 (m, 8H, PCH2N), 3.42 (s, 2H, NCH2Ph), 3.12 (d, 2JHH = 11.5 Hz, 1H, SCH2N), 2.91 (d, 2JHH = 11.5 Hz, 1H, SCH2N), 2.50 (d, 2JHH = 11.5 Hz, 1H, SCH2N), 2.32–1.26 (m, 6H, pdt), 1.78 (d, 2JHH = 11.5 Hz, 1H, SCH2N).Anal. Calcd (%) for C47H49Fe3N3O5P2S4(%) Theoretical: C = 51.62; H = 4.52; N = 3.84.(%) Experimental: C = 50.83; H = 4.47; N = 3.68.
Protonation Experiments
References
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Stripp, S.T.; Duffus, B.R.; Fourmond, V.; Léger, C.; Leimkühler, S.; Hirota, S.; Hu, Y.; Jasniewski, A.; Ogata, H.; Ribbe, M.W. Second and outer coordination sphere effects in nitrogenase, hydrogenase, formate dehydrogenase, and CO dehydrogenase. Chem. Rev. 2022, 122, 11900–11973. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.A.; Bikbaev, K.; Pang, Y.; Lorent, C.; Wiemann, C.; Breuer, N.; Zebger, I.; DeBeer, S.; Span, I.; Bjornsson, R.; et al. Binding of exogenous cyanide reveals new active-site states in [FeFe] hydrogenases. Chem. Sci. 2023, 14, 2826–2838. [Google Scholar] [CrossRef] [PubMed]
- Birrell, J.A.; Rodríguez-Maciá, P.; Reijerse, E.J.; Martini, M.A.; Lubitz, W. The catalytic cycle of [FeFe] hydrogenase: A tale of two sites. Coord. Chem. Rev. 2021, 449, 214191. [Google Scholar] [CrossRef]
- Tai, H.; Hirota, S.; Stripp, S.T. Proton transfer mechanisms in bimetallic hydrogenases. Acc. Chem. Res. 2021, 54, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Mebs, S.; Senger, M.; Duan, J.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Winkler, M.; Stripp, S.T.; Haumann, M. Bridging hydride at reduced H-cluster species in [FeFe]-hydrogenases revealed by infrared spectroscopy, isotope editing, and quantum chemistry. J. Am. Chem. Soc. 2017, 139, 12157–12160. [Google Scholar] [CrossRef]
- Lorent, C.; Katz, S.; Duan, J.; Julia Kulka, C.; Caserta, G.; Teutloff, C.; Yadav, S.; Apfel, U.-P.; Winkler, M.; Happe, T.; et al. Shedding light on proton and electron dynamics in [FeFe] hydrogenases. J. Am. Chem. Soc. 2020, 142, 5493–5497. [Google Scholar] [CrossRef]
- Mebs, S.; Duan, J.; Wittkamp, F.; Stripp, S.T.; Happe, T.; Apfel, U.-P.; Winkler, M.; Haumann, M. Differential protonation at the catalytic six-iron cofactor of [FeFe]-hydrogenases revealed by 57Fe nuclear resonance X-ray scattering and quantum mechanics/molecular mechanics analyses. Inorg. Chem. 2019, 58, 4000–4013. [Google Scholar] [CrossRef]
- Silakov, A.; Wenk, B.; Reijerse, E.; Lubitz, W. 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: Evidence for a nitrogen in the dithiol bridge. Phys. Chem. Chem. Phys. 2009, 11, 6592–6599. [Google Scholar] [CrossRef]
- Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T.R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J.M.; Reijerse, E.; Lubitz, W.; et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 499, 66–70. [Google Scholar] [CrossRef]
- Boncella, A.E.; Sabo, E.T.; Santore, R.M.; Carter, J.; Whalen, J.; Hudspeth, J.D.; Morrison, C.N. The expanding utility of iron-sulfur clusters: Their functional roles in biology, synthetic small molecules, maquettes and artificial proteins, biomimetic materials, and therapeutic strategies. Coord. Chem. Rev. 2022, 451, 214229. [Google Scholar] [CrossRef]
- Laun, K.; Baranova, I.; Duan, J.; Kertess, L.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Senger, M.; Stripp, S.T. Site-selective protonation of the one-electron reduced cofactor in [FeFe]-hydrogenase. Dalton Trans. 2021, 50, 3641–3650. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Mebs, S.; Duan, J.; Shulenina, O.; Laun, K.; Kertess, L.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Winkler, M.; et al. Protonation/reduction dynamics at the [4Fe–4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases. Phys. Chem. Chem. Phys. 2018, 20, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.-P. [FeFe]-hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2021, 50, 1668–1784. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.; Laun, K.; Wittkamp, F.; Duan, J.; Haumann, M.; Happe, T.; Winkler, M.; Apfel, U.-P.; Stripp, S.T. Proton-coupled reduction of the catalytic [4Fe-4S] cluster in [FeFe]-hydrogenases. Angew. Chem. Int. Ed. 2017, 56, 16503–16506. [Google Scholar] [CrossRef] [PubMed]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.-P. [FeFe]-hydrogenases: Recent developments and future perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Senger, M.; Berggren, G.; Stripp, S.T. Current state of [FeFe]-hydrogenase research: Biodiversity and spectroscopic investigations. ACS Catal. 2020, 10, 7069–7086. [Google Scholar] [CrossRef]
- Senger, M.; Eichmann, V.; Laun, K.; Duan, J.; Wittkamp, F.; Knör, G.; Apfel, U.-P.; Happe, T.; Winkler, M.; Heberle, J.; et al. How [FeFe]-hydrogenase facilitates bidirectional proton transfer. J. Am. Chem. Soc. 2019, 141, 17394–17403. [Google Scholar] [CrossRef] [PubMed]
- Wiedner, E.S.; Appel, A.M.; Raugei, S.; Shaw, W.J.; Bullock, R.M. Molecular catalysts with diphosphine ligands containing pendant amines. Chem. Rev. 2022, 122, 12427–12474. [Google Scholar] [CrossRef] [PubMed]
- Hobballah, A.; Lounissi, S.; Motei, R.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. Synthesis, Characterization and electrochemical reductive properties of complexes [Fe2(CO)4(κ2-PPh2NR2)(μ-dithiolato)] related to the H-cluster of [FeFe]-H2ases. Eur. J. Inorg. Chem. 2021, 2021, 205–216. [Google Scholar] [CrossRef]
- Carroll, M.E.; Barton, B.E.; Rauchfuss, T.B.; Carroll, P.J. Synthetic models for the active site of the [FeFe]-hydrogenase: Catalytic proton reduction and the structure of the doubly protonated intermediate. J. Am. Chem. Soc. 2012, 134, 18843–18852. [Google Scholar] [CrossRef] [PubMed]
- Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Shubina, E.S. Ligand versus metal protonation of an iron hydrogenase active site mimic. Chem. Rev. 2016, 116, 8545–8587. [Google Scholar] [CrossRef] [PubMed]
- Chambers, G.M.; Johnson, S.I.; Raugeri, S.; Bullock, R.M. Anion control of tautomeric equilibria: Fe–H vs. N–H influenced by NH/F hydrogen bonding. Chem. Sci. 2019, 10, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Muir, K.W. Di-iron aza diphosphido complexes: Mimics for the active site of Fe-only hydrogenase, and effects of changing the coordinating atoms of the bridging ligand in [Fe2{µ-(ECH2)2NR}(CO)6]. Inorg. Chem. 2004, 43, 8203–8205. [Google Scholar] [CrossRef] [PubMed]
- Eilers, G.; Schwartz, L.; Stein, M.; Zampella, G.; de Gioia, L.; Ott, S.; Lomoth, R. Ligand versus metal protonation of an iron hydrogenase active site mimic. Chem. Eur. J. 2007, 13, 7075–7084. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.E.; Olsen, M.T.; Rauchfuss, T.B. Aza- and oxadithiolates are probable proton relays in functional models for the [FeFe]-hydrogenases. J. Am. Chem. Soc. 2008, 130, 16834–16835. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.T.; Rauchfuss, T.B.; Wilson, S.R. Role of the azadithiolate cofactor in models for [FeFe]-hydrogenase: Novel structures and catalytic implications. J. Am. Chem. Soc. 2010, 132, 17733–17740. [Google Scholar] [CrossRef] [PubMed]
- Hobballah, A.; Arrigoni, F.; Elleouet, C.; Greco, C.; Laurans, M.; Pétillon, F.Y.; Schollhammer, P. Triiron clusters derived from dinuclear complexes related to the active site of [Fe–Fe] hydrogenases: Steric effect of the dithiolate bridge on redox properties, a DFT analysis. Inorg. Chem. Front. 2021, 8, 3659–3674. [Google Scholar] [CrossRef]
- Rahaman, A.; Ghosh, S.; Unwin, D.G.; Basak-Modi, S.; Holt, K.B.; Kabir, S.E.; Nordlander, E.; Richmond, M.G.; Hogarth, G. Bioinspired hydrogenase models: The mixed-valence triiron complex [Fe3(CO)7(μ-edt)2] and phosphine derivatives [Fe3(CO)7−x(PPh3)x(μ-edt)2] (x = 1, 2) and [Fe3(CO)5(κ2-diphosphine)(μ-edt)2] as proton reduction catalysts. Organometallics 2014, 33, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Zsolnai, L.; Huttner, G. Zweikernige und dreikernige carbonyleisenkomplexe mit 1.2- und 1.3-dithiolatobrückenIiganden, Dinuclear and trinuclear carbonyliron complexes containing 1,2- and 1,3-dithiolato bridging ligands. Z. Naturforsch. 1982, 37b, 1430–1436. [Google Scholar] [CrossRef]
- Ghosh, S.; Hogarth, G.; Holt, K.B.; Kabir, S.E.; Rahaman, A.; Unwin, D.G. Bio-inspired hydrogenase models: Mixed valence triion complexes as proton reduction catalysts. Chem. Commun. 2011, 47, 11222–11224. [Google Scholar] [CrossRef] [PubMed]
- Lounissi, S.; Capon, J.-F.; Gloaguen, F.; Matoussi, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Diiron species containing a cyclic PPh2NPh2 diphosphine related to the [FeFe]H2ases active site. Chem. Commun. 2011, 47, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gao, W.; Åkermark, T.; Li, M.; Åkermark, B. An air-stable Fe3S4 complex with properties similar to those of the HOX air state of the diiron hydrogenases. Eur. J. Inorg. Chem. 2012, 2012, 4259–4263. [Google Scholar] [CrossRef]
- Beaume, L.; Clémancey, M.; Blondin, G.; Greco, C.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. New systematic route to mixed-valence triiron clusters derived from dinuclear models of the active site of [Fe−Fe]-hydrogenases. Organometallics 2014, 33, 6290–6293. [Google Scholar] [CrossRef]
- Ezzaher, S.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Pichon, R.; Kervarec, N. Evidence for the formation of terminal hydrides by protonation of an asymmetric iron hydrogenase active site mimic. Inorg. Chem. 2007, 46, 3426–3428. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, F.; Mohamed Bouh, S.; De Gioia, L.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; Zampella, G. Influence of the dithiolate bridge on the oxidative processes of diiron models related to the active site of [FeFe] hydrogenases. Chem. Eur. J. 2017, 23, 4364–4372. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, F.; Mohamed Bouh, S.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; De Gioia, L.; Zampella, G. Electrochemical and theoretical investigations of the oxidatively induced reactivity of the complex [Fe2(CO)4(κ2-dmpe)(µ-adtBn)] related to the active site of [FeFe] hydrogenases. Chem. Eur. J. 2018, 24, 15036–15051. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Eilers, G.; Eriksson, L.; Gogoll, A.; Lomoth, R.; Ott, S. Iron hydrogenase active site mimic holding a proton and a hydride. Chem. Commun. 2006, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Brammer, L. Metals and hydrogen bonds. Dalton Trans. 2003, 3145–3157. [Google Scholar] [CrossRef]
- Duan, J.; Mebs, S.; Laun, K.; Wittkamp, F.; Heberle, J.; Hofmann, E.; Apfel, U.-P.; Winkler, M.; Senger, M.; Haumann, M.; et al. Geometry of the catalytic active site in [FeFe]-hydrogenase is determined by hydrogen bonding and proton transfer. ACS Catal. 2019, 9, 9140–9149. [Google Scholar] [CrossRef]
- Wilson, D.; Newell, R.H.; McNevin, M.J.; Muckerman, J.T.; Rakowski DuBois, M.; DuBois, D.L. Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J. Am. Chem. Soc. 2006, 128, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Takács, J.; Markó, L.; Párkányi, L. Synthesis and some reactions of Fe(CO)2(dppe)(SR)2 and Fe3(CO)6(SAr)6 complexes. The crystal structure of cis,cis,cis-Fe(CO)2(dppe)(SPh)2. J. Organomet. Chem. 1989, 361, 109–116. [Google Scholar] [CrossRef]
- Laurence, J.D.; Li, H.; Rauchfuss, T.B. Beyond Fe-only hydrogenases: N-functionalized 2-aza-1,3-dithiolates Fe2[(SCH2)2NR](CO)x (x = 5, 6). Chem. Commun. 2001, 1482–1483. [Google Scholar] [CrossRef]
- Arrigoni, F.; De Gioia, L.; Elleouet, C.; Pétillon, F.; Schollhammer, P.; Talarmin, J.; Zampella, G. Normal vs. inverted ordering of reduction potentials in [FeFe]-hydrogenases biomimetics: Effect of the dithiolate bulk chemistry. Chem.–A Eur. J. 2023, 29, e202300569. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | IR (CH2Cl2, cm−1), ῡ(CO) | 31P{1H} NMR (CDCl3, δ: ppm, J: Hz) |
|---|---|---|
| 1 | 2020 (s), 1949 (s), 1829 (w) | 56.6 (d, 2JPP = 80.0), 40.6 (d, 2JPP = 80.0) |
| 2 | 2018 (s), 1948 (s), 1824 (w) | 53.3 (d, 2JPP = 81.0), 40.8 (d, 2JPP = 81.0) |
| 3 | 2023 (s), 1948 (s), 1831 (w) | 57.0 (d, 2JPP = 80.0), 40.6 (d, 2JPP = 80.0) |
| 4 | 2021 (s), 1949 (s), 1824 (w) | 53.7 (d, 2JPP = 81.5), 40.8 (d, 2JPP = 81.5) |
| 4 | |||
|---|---|---|---|
| Distance (Å) | Angles (°) | ||
| Fe1-Fe2 | 2.5459(19) | Fe1-Fe2-Fe3 | 155.85(7) |
| Fe2-Fe3 | 2.5693(19) | Fe2-C2-O2 | 160.8(8) |
| Fe1-COterminal | 1.767(11) | Fe1-C2-O2 | 125.7(7) |
| Fe3-COterminal | 1.732(12)–1.797(10) | Fe2-C2-Fe1 | 73.5(3) |
| Fe1-CObridging | 2.403(9) | Fe1-Fe2-C2 | 64.8(3) |
| Fe2-CObridging | 1.765(10) | P1-Fe1-P2 | 81.30(10) |
| Fe1-P1 | 2.206(2) | ||
| Fe1-P2 | 2.179(3) | ||
| Complex | Oxidation | Reduction |
|---|---|---|
| 1 | E1/2ox1 = −0.28 V; Epox2 = 0.61 V | E1/2red1 = −1.90 V; Epred2 = −2.20 V |
| 2 | E1/2ox1 = −0.34 V; Epox2 = 0.57 V | E1/2red1 = −1.95 V; Epred2 = −2.25 V |
| 3 | E1/2ox1 = −0.28 V; Epox2 = 0.48 V | Epred1 = −1.90 V; Epred2 = −2.16 V |
| 4 | E1/2ox1 = −0.34 V; Epox2 = 0.47 V | Epred1 = −1.96 V; Epred2 = −2.22 V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hobballah, A.; Elleouet, C.; Schollhammer, P. Triiron Complexes Featuring Azadiphosphine Related to the Active Site of [FeFe]-Hydrogenases: Their Redox Behavior and Protonation. Molecules 2024, 29, 3270. https://doi.org/10.3390/molecules29143270
Hobballah A, Elleouet C, Schollhammer P. Triiron Complexes Featuring Azadiphosphine Related to the Active Site of [FeFe]-Hydrogenases: Their Redox Behavior and Protonation. Molecules. 2024; 29(14):3270. https://doi.org/10.3390/molecules29143270
Chicago/Turabian StyleHobballah, Ahmad, Catherine Elleouet, and Philippe Schollhammer. 2024. "Triiron Complexes Featuring Azadiphosphine Related to the Active Site of [FeFe]-Hydrogenases: Their Redox Behavior and Protonation" Molecules 29, no. 14: 3270. https://doi.org/10.3390/molecules29143270
APA StyleHobballah, A., Elleouet, C., & Schollhammer, P. (2024). Triiron Complexes Featuring Azadiphosphine Related to the Active Site of [FeFe]-Hydrogenases: Their Redox Behavior and Protonation. Molecules, 29(14), 3270. https://doi.org/10.3390/molecules29143270