

Polymethylenetetrazole: Synthesis, Characterization, and Energetic Properties
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
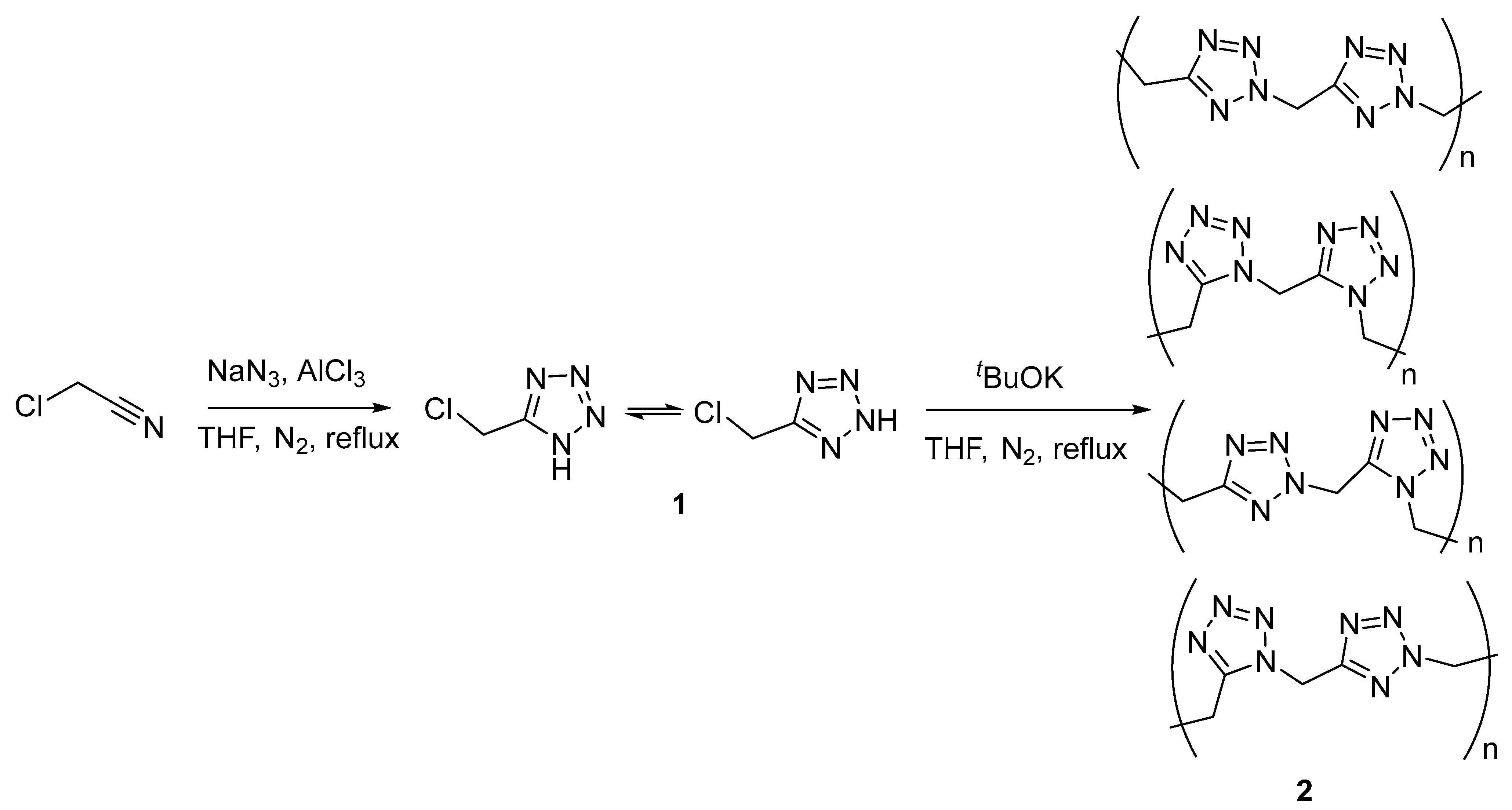
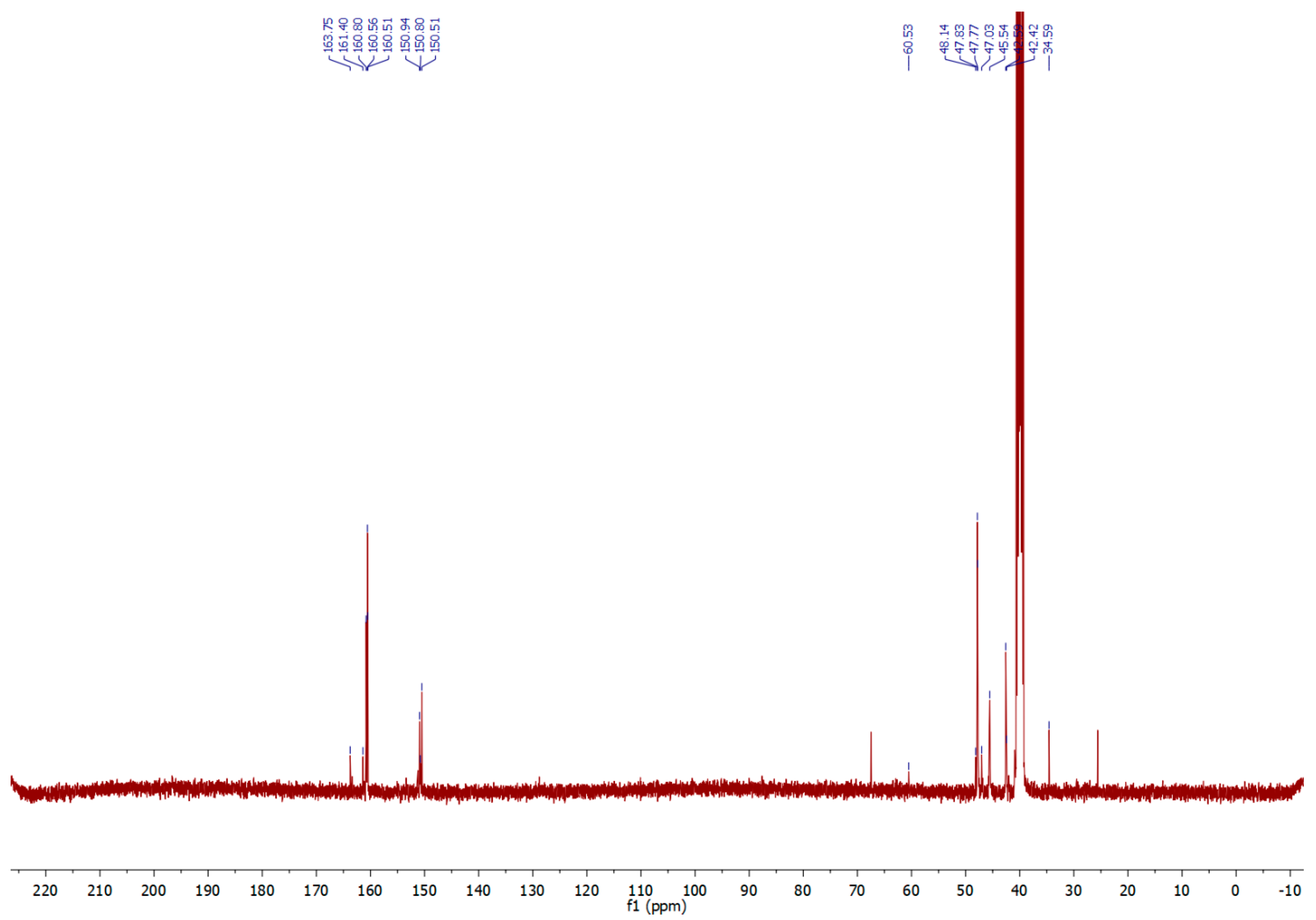
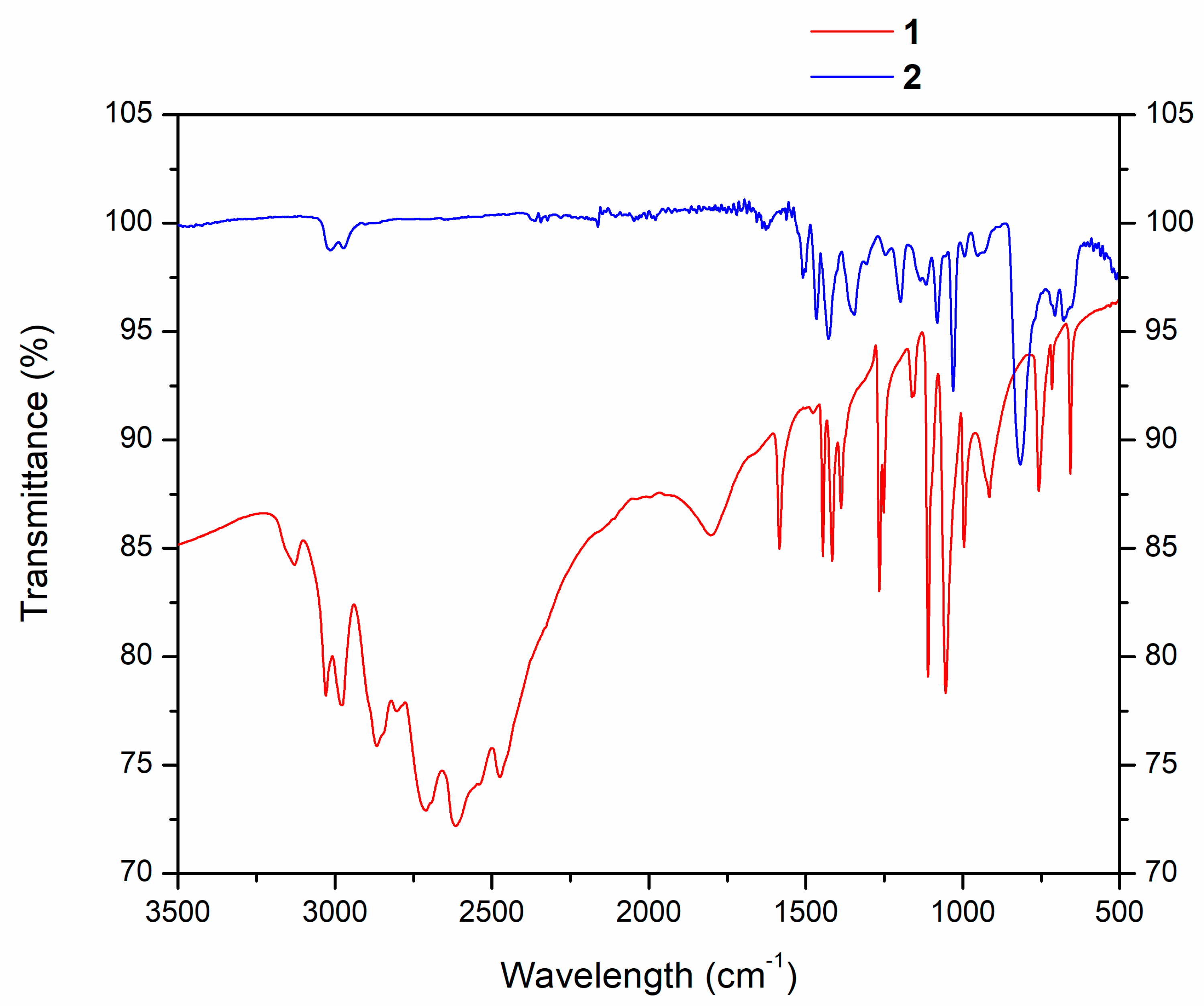
2.1. Synthesis and Analysis of NMR and IR Spectra
2.2. XRD Analysis
2.3. Molecular Weight Determination
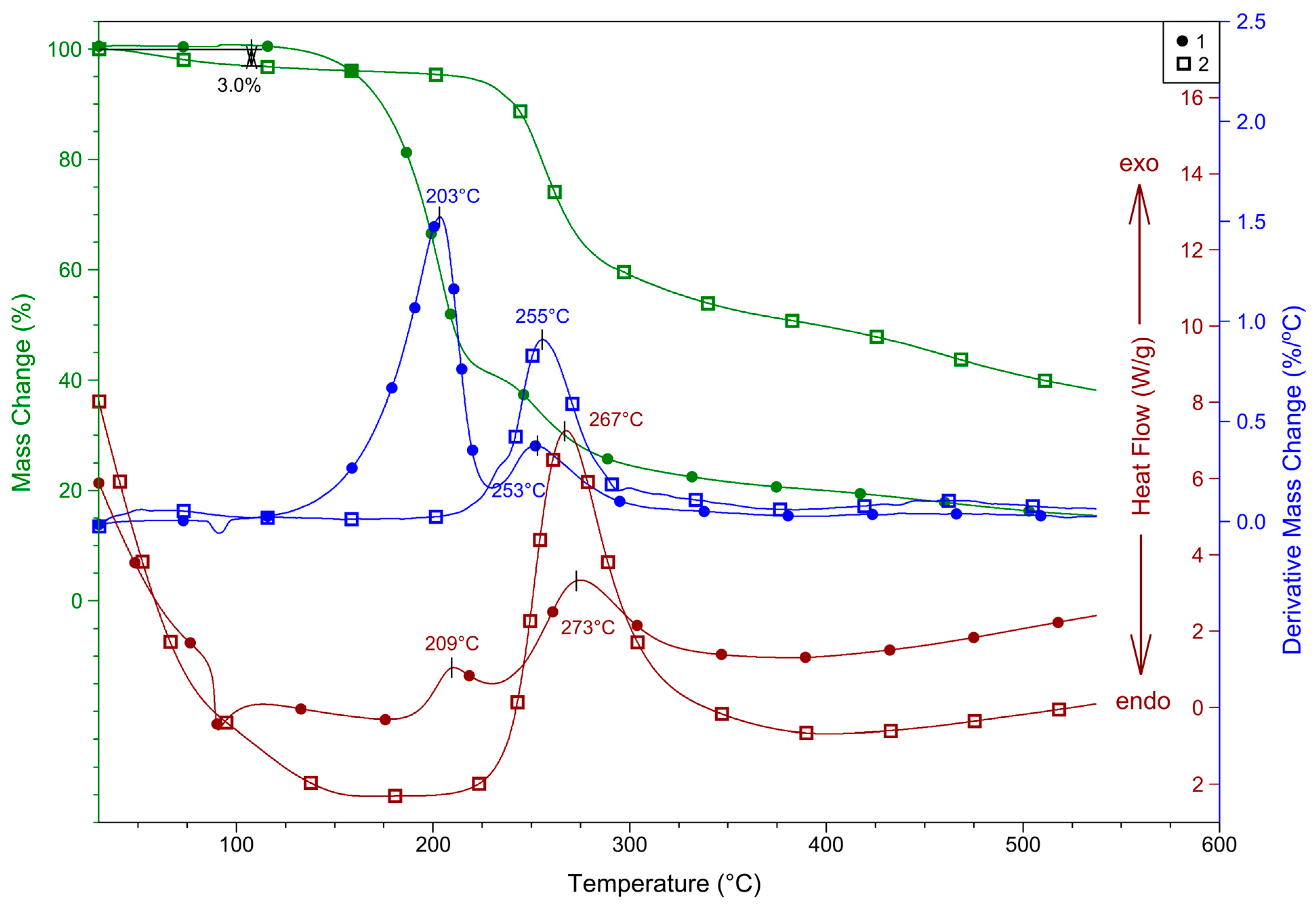
2.4. Thermal Properties of 1 and 2
2.5. Energetic Properties of PMT
3. Materials and Methods
3.1. Materials and Measurements
3.2. Synthesis
3.2.1. 5-Chloromethyl-1H-tetrazole (1)
3.2.2. Polymethylenetetrazole (2)
3.3. Determination of Weight Average Molecular Weight with Static Light Scattering
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fried, L.E.; Manaa, M.R.; Pagoria, P.F.; Simpson, R.L. Design and Synthesis of Energetic Materials. Annu. Rev. Mater. Res. 2001, 31, 291–321. [Google Scholar] [CrossRef]
- Gao, H.; Shreeve, J.M. The Many Faces of FOX-7: A Precursor to High-Performance Energetic Materials. Angew. Chem. 2015, 127, 6433–6436. [Google Scholar] [CrossRef]
- Klapötke, T.M. New Nitrogen-Rich High Explosives. In High Energy Density Materials. Structure and Bonding; Klapötke, T.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 125, pp. 85–121. [Google Scholar] [CrossRef]
- He, P.; Zhang, J.-G.; Yin, X.; Wu, J.-T.; Wu, L.; Zhou, Z.-N.; Zhang, T.-L. Energetic Salts Based on Tetrazole N-Oxide. Chem. Eur. J. 2016, 22, 7670–7685. [Google Scholar] [CrossRef] [PubMed]
- Klapötke, T.M.; Sabaté, C.M.; Stierstorfer, J. Neutral 5-nitrotetrazoles: Easy initiation with low pollution. New J. Chem. 2009, 33, 136–147. [Google Scholar] [CrossRef]
- Fischer, N.; Karaghiosoff, K.; Klapötke, T.M.; Stierstorfer, J. New Energetic Materials featuring Tetrazoles and Nitramines—Synthesis, Characterization and Properties. Z. Anorg. Allg. Chem. 2010, 636, 735–749. [Google Scholar] [CrossRef]
- Hammerl, A.; Klapötke, T.M.; Mayer, P.; Weigand, J.J.; Holl, G. Synthesis, Structure, Molecular Orbital Calculations and Decomposition Mechanism for Tetrazolylazide CHN7, its Phenyl Derivative PhCN7 and Tetrazolylpentazole CHN9. Propellants, Explos. Pyrotech. 2005, 30, 17–26. [Google Scholar] [CrossRef]
- Talawar, M.B.; Chhabra, J.S.; Agrawal, A.P.; Asthana, S.N.; Rao, K.U.B.; Singh, H. Synthesis, characterization, thermolysis and performance evaluation of mercuric-5-nitro tetrazole (MNT). J. Hazard. Mater. A 2004, 113, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Klapötke, T.M.; Stierstorfer, J.; Wiedemann, C. 1-Nitratoethyl-5-nitriminotetrazole derivatives—Shaping future high explosives. Polyhedron 2011, 30, 2374–2386. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Sabaté, C.M.; Stierstorfer, J. Hydrogen-bonding Stabilzation in Energetic Perchlorate Salts: 5-Amino-1H-tetrazolium Perchlorate and its Adduct with 5-Amino-1H-tetrazole. Z. Anorg. Allg. Chem. 2008, 634, 1867–1874. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Karaghiosoff, K.; Mayer, P.; Penger, A.; Welch, J.M. Synthesis and Characterization of 1,4-Dimethyl-5-Aminotetrazolium 5-Nitrotetrazolate. Propellants, Explos. Pyrotech. 2006, 31, 188–195. [Google Scholar] [CrossRef]
- Zhilin, A.Y.; Ilyushin, M.A.; Tselinskii, I.V.; Kozlov, A.S.; Kuz’mina, N.E. Synthesis and Properties of Tetraamminebis(1-methyl-5-aminotetrazole-N3, N4)cobalt(III) Perchlorate. Russ. J. Appl. Chem. 2002, 75, 1849–1851. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, J.; Zhang, T.; Yang, L.; Zhang, J.; Hu, X. Synthesis, structural investigation, thermal decomposition mechanism and sensitivity properties of an energetic compound [Cd(DAT)6](ClO4)2 (DAT = 1,5-diaminotetrazole). J. Hazard. Mater. 2008, 160, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.V.; Coburn, M.D.; Meyer, T.J.; Wetzler, M. Green primary explosives: 5-Nitrotetrazolato-N2-ferrate hierarchies. Proc. Natl. Acad. Sci. USA 2006, 103, 10322–10327. [Google Scholar] [CrossRef] [PubMed]
- Loebbecke, S.; Schuppler, H.; Schweikert, W. Thermal analysis of the extremely Nitrogen-Rich Solids BTT and DAAT. J. Therm. Anal. Calorim. 2003, 72, 453–463. [Google Scholar] [CrossRef]
- Wang, R.; Guo, Y.; Zeng, Z.; Twamley, B.; Shreeve, J.M. Furazan-Functionalized Tetrazolate-Based Salts: A New Family of Insensitive Energetic Materials. Chem. Eur. J. 2009, 15, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Staples, R.J.; Shreeve, J.M. Engineering bistetrazoles: (E)-5,5′-(ethene-1,2-diyl)bis(1H-tetrazol-1-ol) as a new planar high-energy-density material. Mater. Adv. 2022, 3, 6062. [Google Scholar] [CrossRef]
- Chinnam, A.K.; Singh, J.; Staples, R.J.; Shreeve, J.M. Manipulating sensitivities of planar oxadiazole-based high performing energetic materials. J. Heterocycl. Chem. 2024, 61, 506–513. [Google Scholar] [CrossRef]
- Thaltiri, V.; Singh, J.; Staples, R.J.; Shreeve, J.M. A domino reaction from a sensitive azide: The impact of positional isomerism on chemical reactivity featuring ortho azido/nitro substituted derivatives. J. Mater. Chem. A 2024, 12, 9546. [Google Scholar] [CrossRef]
- Cao, W.-L.; Tariq, Q.-N.; Li, Z.-M.; Yang, J.-Q.; Zhang, J.-G. Recent advances on the nitrogen-rich 1,2,4-oxadiazole-azoles-based energetic materials. Def. Technol. 2022, 18, 344–367. [Google Scholar] [CrossRef]
- Tang, J.; Yang, H.-W.; Cheng, G.-B. Nitrogen-rich tetracyclic-based heterocyclic energetic materials. Energetic Mater. Front. 2023, 4, 110–122. [Google Scholar] [CrossRef]
- Kizhnyaev, V.N.; Pokatilov, F.A.; Vereshchagin, L.I. Branched Tetrazole-Containing Polymers. Polym. Sci. Ser. A 2007, 49, 28–34. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies. Tables and Charts, 3rd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2001; p. 198. [Google Scholar]
- Matveeva, N.A.; Suschko, N.I.; Makarevich, N.I.; Gaponik, P.N.; Ivashkevich, O.A.; Koren’, A.O. IR absorption spectra of 5-substituted tetrazoles. J. Appl. Spectrosc. 1992, 57, 845–852. [Google Scholar] [CrossRef]
- Anderson, M.; Wittgren, B.; Wahlund, K.-G. Accuracy in Multiangle Light Scattering Measurements for Molar Mass and Radius Estimations. Model Calculations and Experiments. Anal. Chem. 2003, 75, 4279–4291. [Google Scholar] [CrossRef]
- Quigley, A.; Williams, D.R. The second virial coefficient as a predictor of protein aggregation propensity: A self-interaction chromatography study. Eur. J. Pharm. Biopharm. 2015, 96, 282–290. [Google Scholar] [CrossRef]
- Sukhanov, G.T.; Bosov, K.K.; Filippova, Y.V.; Sukhanova, A.G.; Krupnova, I.A.; Pivovarova, E.V. New 5-Aminotetrazole-Based Energetic Polymers: Synthesis, Structure and Properties. Materials 2022, 15, 6936. [Google Scholar] [CrossRef] [PubMed]
- NATO Standardization Agreement (STANAG) on Explosives; Impact Sensitivity Tests, No 4489, Ed. 1; NATO: Brussels, Belgium, 1999.
- NATO Standardization Agreement (STANAG) on Explosives; Friction Sensitivity Tests, No 4487, Ed. 2; NATO: Brussels, Belgium, 2009.
- Behringer, H.; Kohl, K. Über Tetrazole, I. Mitteil.: Die Synthese des 5-[β-Amino-äthyl]-tetrazols und einiger Tetrazolyl-(5)-methylderivate. Chem. Berichte 1956, 89, 2648–2653. [Google Scholar] [CrossRef]
- Moderhack, D. 2-Substituierte 5-Tetrazolcarbaldehyde. Chem. Berichte 1975, 108, 887–896. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenjo, L.; Oklješa, A.; Tomšič, M.; Barta Holló, B.; Nešić, J.; Tóth, E.; Podlipnik, Č. Polymethylenetetrazole: Synthesis, Characterization, and Energetic Properties. Molecules 2024, 29, 3389. https://doi.org/10.3390/molecules29143389
Brenjo L, Oklješa A, Tomšič M, Barta Holló B, Nešić J, Tóth E, Podlipnik Č. Polymethylenetetrazole: Synthesis, Characterization, and Energetic Properties. Molecules. 2024; 29(14):3389. https://doi.org/10.3390/molecules29143389
Chicago/Turabian StyleBrenjo, Ljubica, Aleksandar Oklješa, Matija Tomšič, Berta Barta Holló, Jovica Nešić, Elvira Tóth, and Črtomir Podlipnik. 2024. "Polymethylenetetrazole: Synthesis, Characterization, and Energetic Properties" Molecules 29, no. 14: 3389. https://doi.org/10.3390/molecules29143389
APA StyleBrenjo, L., Oklješa, A., Tomšič, M., Barta Holló, B., Nešić, J., Tóth, E., & Podlipnik, Č. (2024). Polymethylenetetrazole: Synthesis, Characterization, and Energetic Properties. Molecules, 29(14), 3389. https://doi.org/10.3390/molecules29143389