
A Nickel/Organoboron-Catalyzed Coupling of Aryl Bromides with Sodium Sulfinates: The Synthesis of Sulfones under Visible Light

Abstract
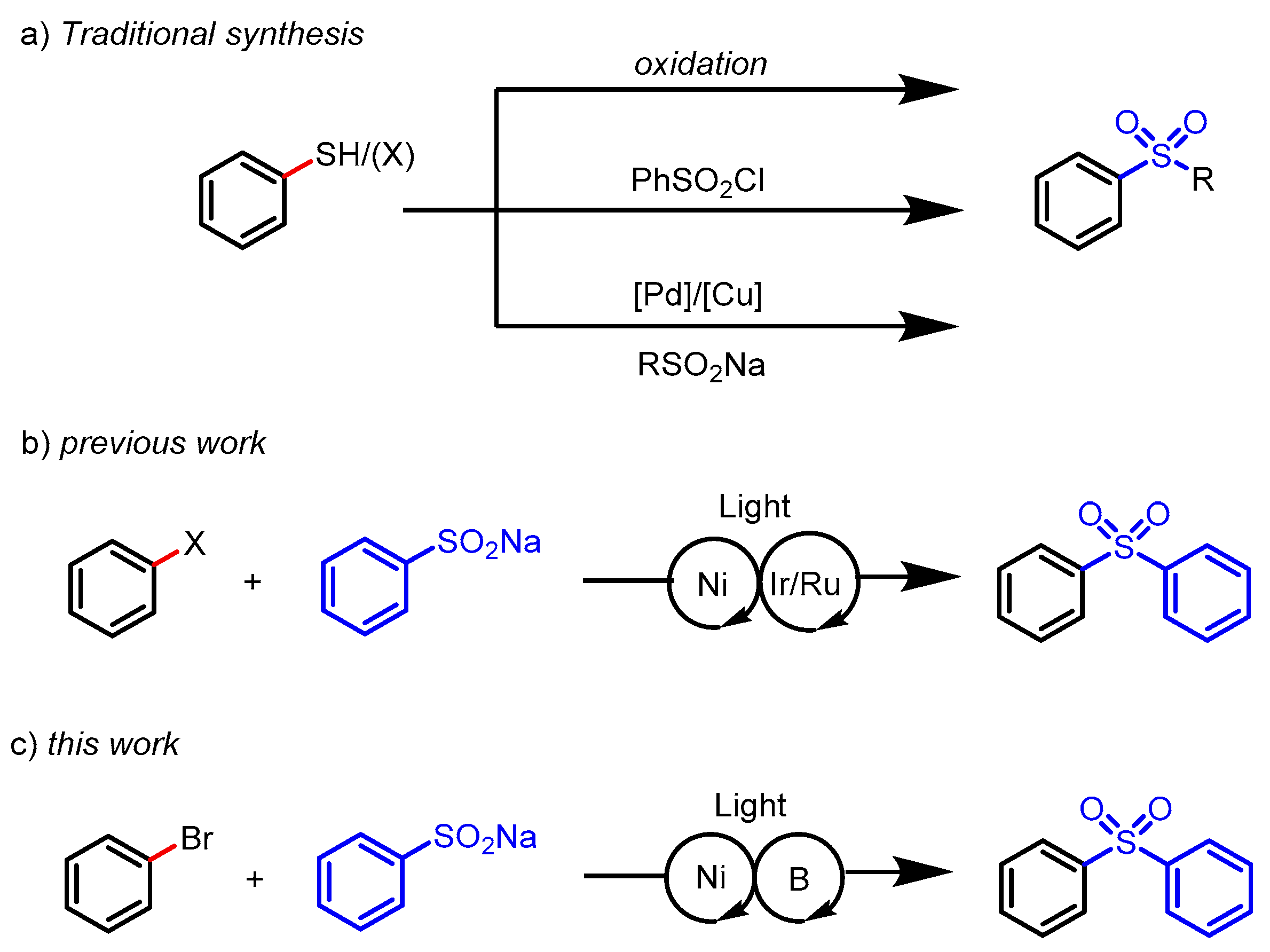
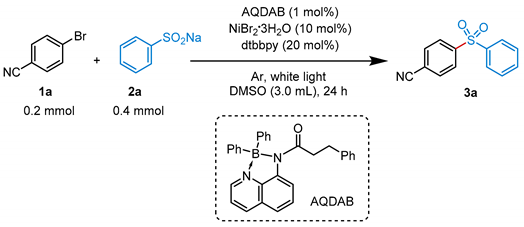
:1. Introduction
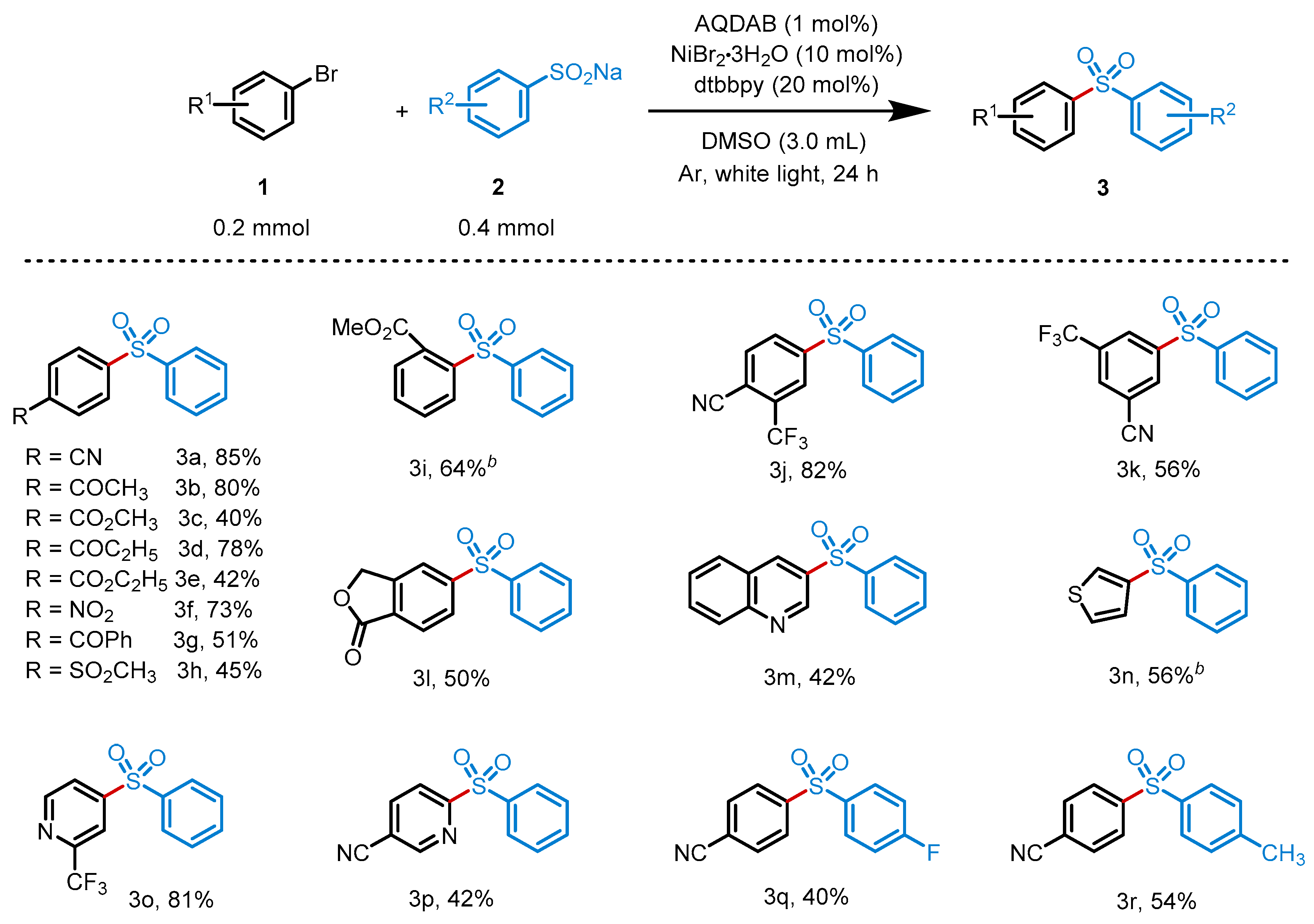
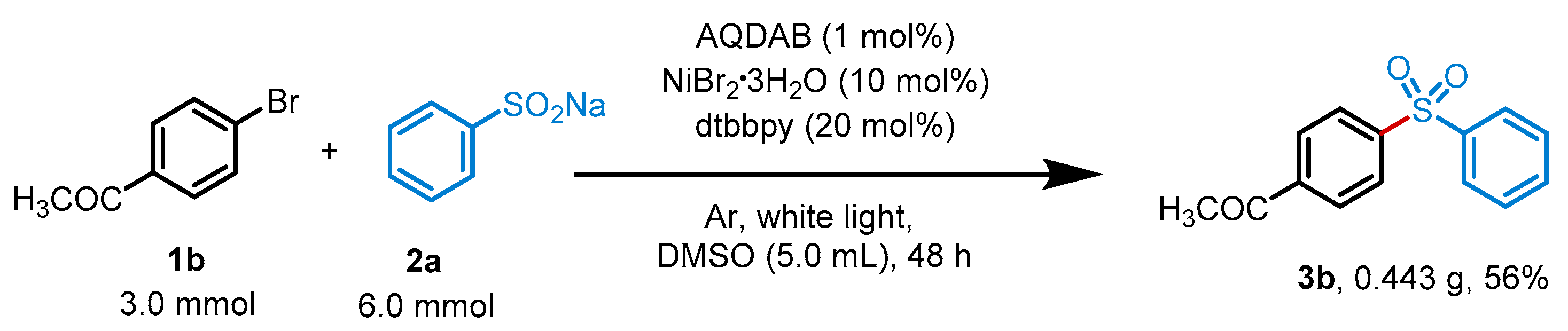
2. Results
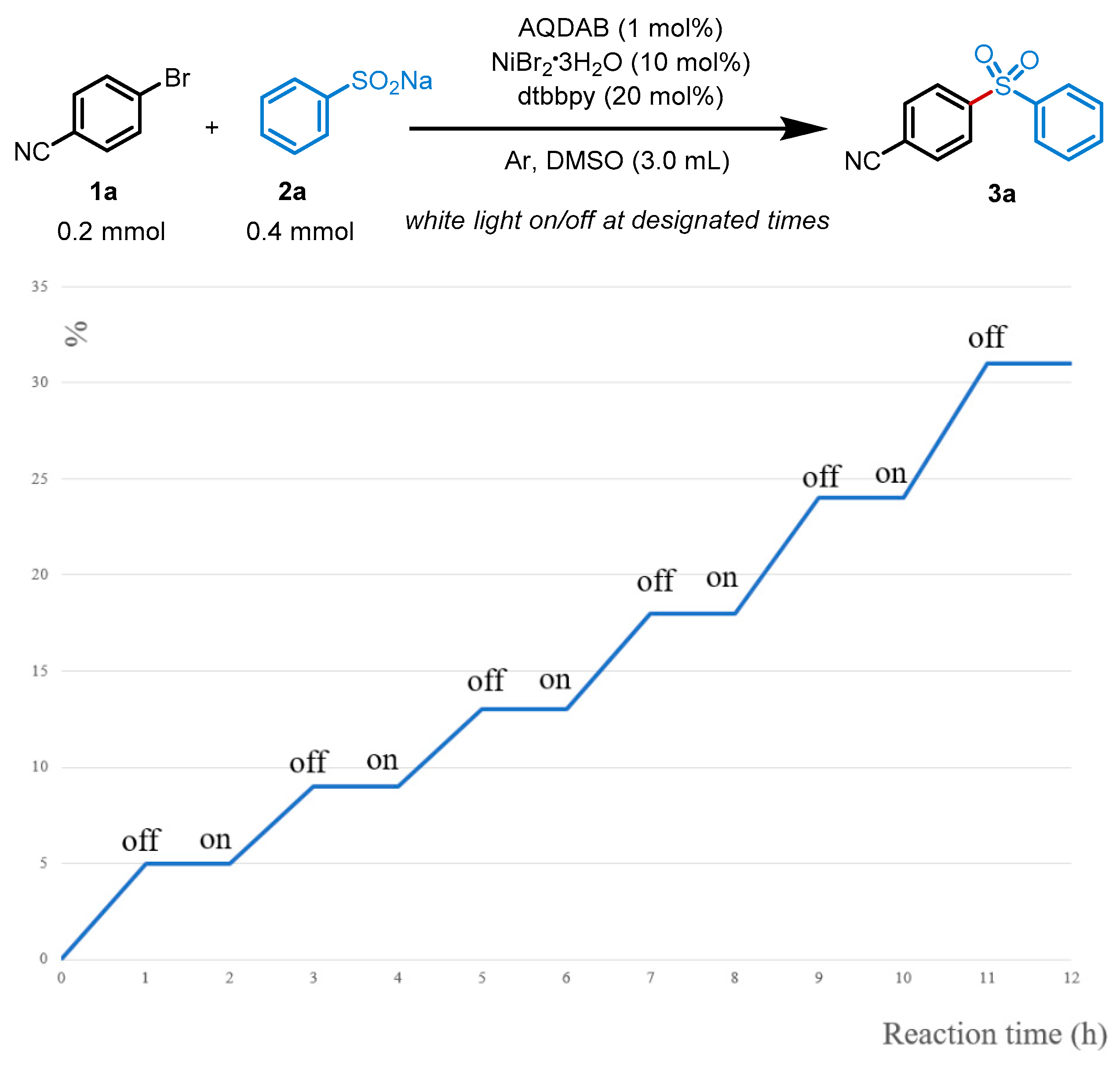
3. Discussion
4. Materials, Methods, Reaction Procedure, and Analytical Data
4.1. Methods and Materials
4.2. The Preparation of Sulfone Compounds
4.2.1. General Procedure
4.2.2. Analytical Data of Products 3a–3r
- (3d) 1-(4-(phenylsulfonyl)phenyl)propan-1-one (CAS: 69567-00-6)
- (3p) 6-(phenylsulfonyl)nicotinonitrile (205514-29-0)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Cheng, Y.; Xu, Z.; Xu, P.; Qu, H.; Fang, Y.; Xu, T.; Wen, L. Evaluation of polyamidoamine (PAMAM) dendrimers as drug carriers of anti-bacterial drugs using sulfamethoxazole (SMZ) as a model drug. Eur. J. Med. Chem. 2007, 42, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, H.; Seino, Y.; Kimura, M.; Kido, J. A m-Terphenyl-Modifed Sulfone Derivative as a Host Material for High-Efficiency Blue and Green Phosphorescent OLEDs. Chem. Mater. 2012, 24, 1404–1406. [Google Scholar] [CrossRef]
- Kakimoto, M.-A.; Grunzinger, S.J.; Hayakawa, T. Hyperbranched poly(ether sulfone)s: Preparation and application to ion-exchange membranes. Polym. J. 2010, 42, 697–705. [Google Scholar] [CrossRef]
- Sandrini, G.; Färkkilä, M.; Burgess, G.; Forster, E.; Haughie, S. Eletriptan vs sumatriptan. A double-blind, placebo-controlled, multiple migraine attack study. Neurology 2002, 59, 1210–1217. [Google Scholar] [CrossRef]
- Le, Y.; Ji, H.; Chen, J.-F.; Shen, Z.; Yun, J.; Pu, M. Nanosized bicalutamide and its molecular structure in solvents. Int. J. Pharm. 2009, 370, 175–180. [Google Scholar] [CrossRef]
- Beaudegnies, R.; Edmunds, A.J.F.; Fraser, T.E.M.; Hall, R.G.; Hawkes, T.R.; Mitchell, G.; Schaetzer, J.; Wendeborn, S.; Wibley, J. Herbicidal 4-hydroxyphenylpyruvate dioxygenase inhibitors—A review of the triketone chemistry story from a Syngenta perspective. Bioorganic Med. Chem. 2009, 17, 4134–4152. [Google Scholar] [CrossRef]
- Tanetani, Y.; Kaku, K.; Kawai, K.; Fujioka, T.; Shimizu, T. Action mechanism of a novel herbicide, pyroxasulfone. Pestic. Biochem. Physiol. 2009, 95, 47–55. [Google Scholar] [CrossRef]
- Takahashi, H.; Ohki, A.; Kanzaki, M.; Tanaka, A.; Sato, Y.; Matthes, B.; Böger, P.; Wakabayashi, K. Very-Long-Chain Fatty Acid Biosynthesis is Inhibited by Cafenstrole, N,N-Diethyl-3-mesitylsulfonyl-1H-1,2,4-triazole-1-carboxamide and Its Analogs. Z. Naturforschung C 2001, 56, 781–786. [Google Scholar] [CrossRef]
- Liu, N.-W.; Liang, S.; Manolikakes, G. Recent Advances in the Synthesis of Sulfones. Synthesis 2016, 48, 1939–1973. [Google Scholar] [CrossRef]
- Pritzius, A.B.; Breit, B. Asymmetric Rhodium-Catalyzed Addition of Thiols to Allenes: Synthesis of Branched Allylic Thioethers and Sulfones. Angew. Chem. Int. Ed. 2015, 54, 3121–3125. [Google Scholar] [CrossRef] [PubMed]
- Répichet, S.; Le Roux, C.; Hernandez, P.; Dubac, J.; Desmurs, J.-R. Bismuth(III) Trifluoromethanesulfonate: An Efficient Catalyst for the Sulfonylation of Arenes. J. Org. Chem. 1999, 64, 6479–6482. [Google Scholar] [CrossRef]
- Yao, B.; Zhang, Y. Sulfonylation of arenes with sulfonamides. Tetrahedron Lett. 2008, 49, 5385–5388. [Google Scholar] [CrossRef]
- Baskin, J.M.; Wang, Z. An Efficient Copper Catalyst for the Formation of Sulfones from Sulfinic Acid Salts and Aryl Iodides. Org. Lett. 2002, 4, 4423–4425. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.-Y.; Sun, L.; Lu, Z.-P.; Yu, D.-G. Photoredox sheds new light on nickel catalysis: From carbon-carbon to carbon-heteroatom bond formation. Org. Chem. Front. 2016, 3, 522–526. [Google Scholar] [CrossRef]
- Fan, L.; Jia, J.; Hou, H.; Lefebvre, Q.; Rueping, M. Decarboxylative Aminomethylation of Aryl- and Vinylsulfonates through Combined Nickel- and Photoredox-Catalyzed Cross-Coupling. Chem. A Eur. J. 2016, 22, 16437–16440. [Google Scholar] [CrossRef] [PubMed]
- Terrett, J.A.; Cuthbertson, J.D.; Shurtleff, V.W.; MacMillan, D.W.C. Switching on elusive organometallic mechanisms with photoredox catalysis. Nature 2015, 524, 330–334. [Google Scholar] [CrossRef]
- Corcoran, E.B.; Pirnot, M.T.; Lin, S.; Dreher, S.D.; DiRocco, D.A.; Davies, I.W.; Buchwald, S.L.; MacMillan, D.W.C. Aryl amination using ligand-free Ni(II) salts and photoredox catalysis. Science 2016, 353, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Tellis, J.C.; Primer, D.N.; Molander, G.A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 2014, 345, 433–436. [Google Scholar] [CrossRef]
- Zuo, Z.; Ahneman, D.T.; Chu, L.; Terrett, J.A.; Doyle, A.G.; MacMillan, D.W.C. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. [Google Scholar] [CrossRef]
- Welin, E.R.; Le, C.; Arias-Rotondo, D.M.; McCusker, J.K.; MacMillan, D.W.C. Photosensitized, energy transfer-mediated organometallic catalysis through electronically excited nickel(II). Science 2017, 355, 380–385. [Google Scholar] [CrossRef]
- Oderinde, M.S.; Frenette, M.; Robbins, D.W.; Aquila, B.; Johannes, J.W. Photoredox Mediated Nickel Catalyzed Cross-Coupling of Thiols with Aryl and Heteroaryl Iodides via Thiyl Radicals. J. Am. Chem. Soc. 2016, 138, 1760–1763. [Google Scholar] [CrossRef]
- Oderinde, M.S.; Jones, N.H.; Juneau, A.; Frenette, M.; Aquila, B.; Tentarelli, S.; Robbins, D.W.; Johannes, J.W. Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides. Angew. Chem. Int. Ed. 2016, 55, 13219–13223. [Google Scholar] [CrossRef]
- Yue, H.; Zhu, C.; Rueping, M. Cross-Coupling of Sodium Sulfinates with Aryl, Heteroaryl, and Vinyl Halides by Nickel/Photoredox Dual Catalysis. Angew. Chem. Int. Ed. 2018, 57, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-W.; Hofman, K.; Herbert, A.; Manolikakes, G. Visible-Light Photoredox/Nickel Dual Catalysis for the Cross-Coupling of Sulfinic Acid Salts with Aryl Iodides. Org. Lett. 2018, 20, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Afonso, M.-J.; Lu, Z.-P.; Kelly, C.-B.; Lang, S.-B.; Dykstra, R.; Gutierrez, O.; Molander, G.-A. Engaging sulfinate salts via Ni/photoredox dual catalysis enables facile Csp2-SO2R coupling. Chem. Sci. 2018, 9, 3186–3191. [Google Scholar] [CrossRef] [PubMed]
- Zu, W.; Day, C.; Wei, L.; Jia, X.; Xu, L. Dual aminoquinolate diarylboron and nickel catalysed metallaphotoredox platform for carbon-oxygen bond construction. Chem. Commun. 2020, 56, 8273–8276. [Google Scholar] [CrossRef]
- Zhu, Y.; Zu, W.; Tian, Q.; Cao, Z.; Wei, Y.; Xu, L. A nickel/organoboron catalyzed metallaphotoredox platform for C(sp2)–P and C(sp2)–S bond construction. Org. Chem. Front. 2022, 9, 1070–1076. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, Z.-T.; Young, D.J.; Chai, L.-L.; Wu, Q.; Li, H.-X. Visible-light mediated cross-coupling of aryl halides with sodium sulfinates via carbonyl-photoredox/nickel dual catalysis. Org. Chem. Front. 2022, 9, 1437–1444. [Google Scholar] [CrossRef]
- Nandi, G.C. An efficient Cu-catalyzed microwave-assisted synthesis of diaryl sulfones. Synth. Commun. 2017, 47, 319–323. [Google Scholar] [CrossRef]
- Liu, N.-W.; Liang, S.; Margraf, N.; Shaaban, S.; Luciano, V.; Drost, M.; Manolikakes, G. Nickel-Catalyzed Synthesis of Diaryl Sulfones from Aryl Halides and Sodium Sulfinates. Eur. J. Org. Chem. 2018, 2018, 1208–1210. [Google Scholar] [CrossRef]
- Zhu, D.L.; Wu, Q.; Li, H.Y.; Li, H.X.; Lang, J.P. Hantzsch Ester as a Visible-Light Photoredox Catalyst for Transition-Metal-Free Coupling of Arylhalides and Arylsulfinates. Eur. J. Org. Chem. 2020, 26, 3484–3488. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Variation from Standard Conditions | Yield b | |
|---|---|---|---|
| 1 | Control experiments | None | 85% |
| 2 | Without light or under sunlight | n.d. | |
| 3 | Heating the reaction at 40 °C while shielding any light for 24 h or 48 h | n.d. | |
| 4 | Without AQDAB | trace | |
| 5 | Without NiBr2·3H2O | n.d. | |
| 6 | Without dtbbpy | 30% | |
| 7 | The screening of Ni catalysts | NiBr2·dme | 35% |
| 8 | NiBr2 | 71% | |
| 9 | NiCl2 | 58% | |
| 10 | NiCl2·dme | 40% | |
| 11 | Ni(acac)2 | trace | |
| 12 | NiCl2·6H2O | 48% | |
| 13 | The screening of ligands | dtbbpy (10 mol%) | 55% |
| 14 | dpy (20 mol%) | 70% | |
| 15 | dmedpy (20 mol%) | 55% | |
| 16 | 1,10-phen (20 mol%) | 26% | |
| 17 | The screening of solvents | (CH2OH)2 | n.d. |
| 18 | CH3OH | n.d. | |
| 19 | THF | n.d. | |
| 20 | CH3CN | n.d. | |
| 21 | DMF | 40% | |
| 22 | NMP | 48% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, S.; Tian, W.; Lv, Q.; Miao, Z.; Xu, L. A Nickel/Organoboron-Catalyzed Coupling of Aryl Bromides with Sodium Sulfinates: The Synthesis of Sulfones under Visible Light. Molecules 2024, 29, 3418. https://doi.org/10.3390/molecules29143418
Ding S, Tian W, Lv Q, Miao Z, Xu L. A Nickel/Organoboron-Catalyzed Coupling of Aryl Bromides with Sodium Sulfinates: The Synthesis of Sulfones under Visible Light. Molecules. 2024; 29(14):3418. https://doi.org/10.3390/molecules29143418
Chicago/Turabian StyleDing, Siyi, Weina Tian, Qiaohuan Lv, Zongcheng Miao, and Liang Xu. 2024. "A Nickel/Organoboron-Catalyzed Coupling of Aryl Bromides with Sodium Sulfinates: The Synthesis of Sulfones under Visible Light" Molecules 29, no. 14: 3418. https://doi.org/10.3390/molecules29143418