
Hydrogen Atom Abstraction and Reduction Study of 21-Thiaporphyrin and 21,23-Dithiaporphyrin
 and
and
Abstract
:
1. Introduction

2. Results and Discussion
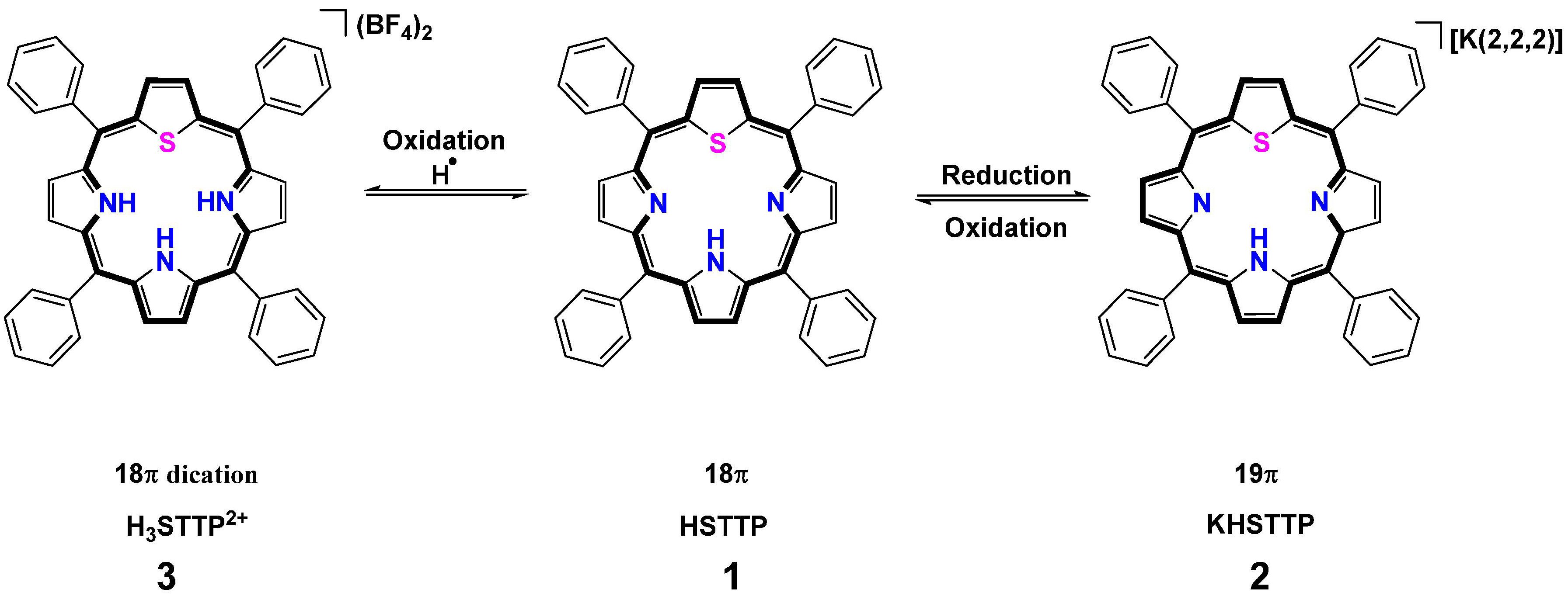
2.1. H-Atoms Abstraction and Reduction in 21-Thiaporphyrin
2.1.1. Syntheses
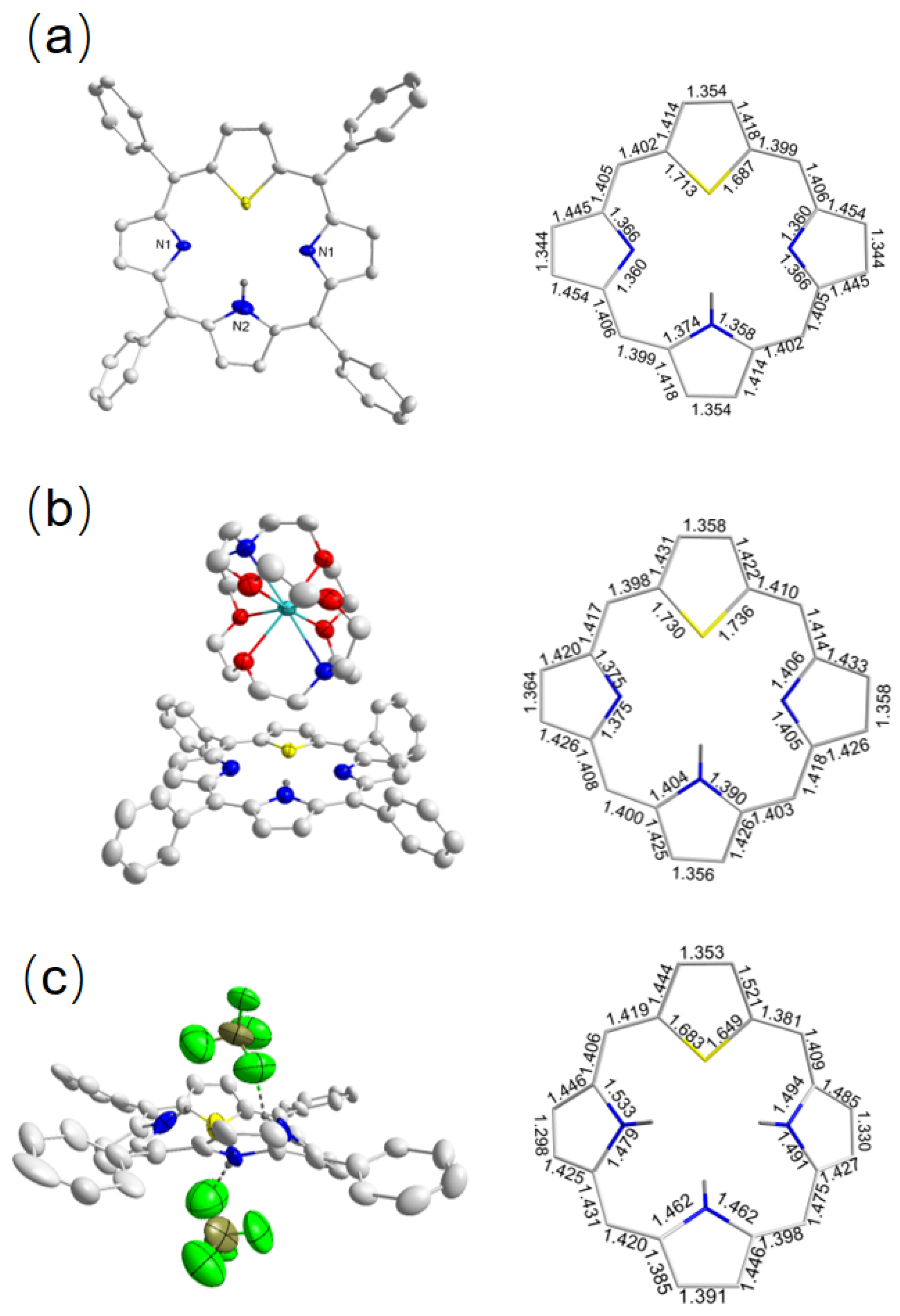
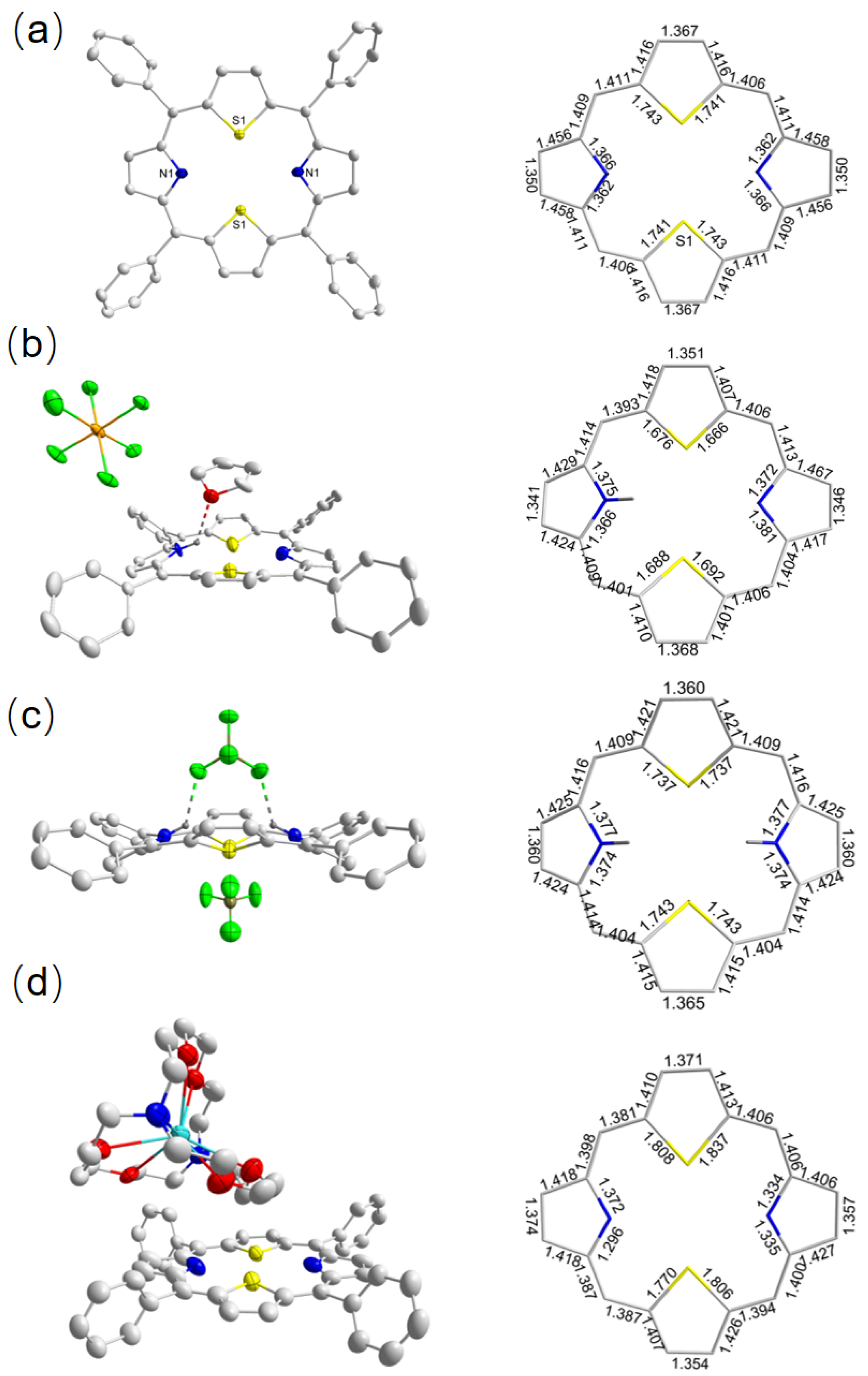
2.1.2. Crystallographic Details
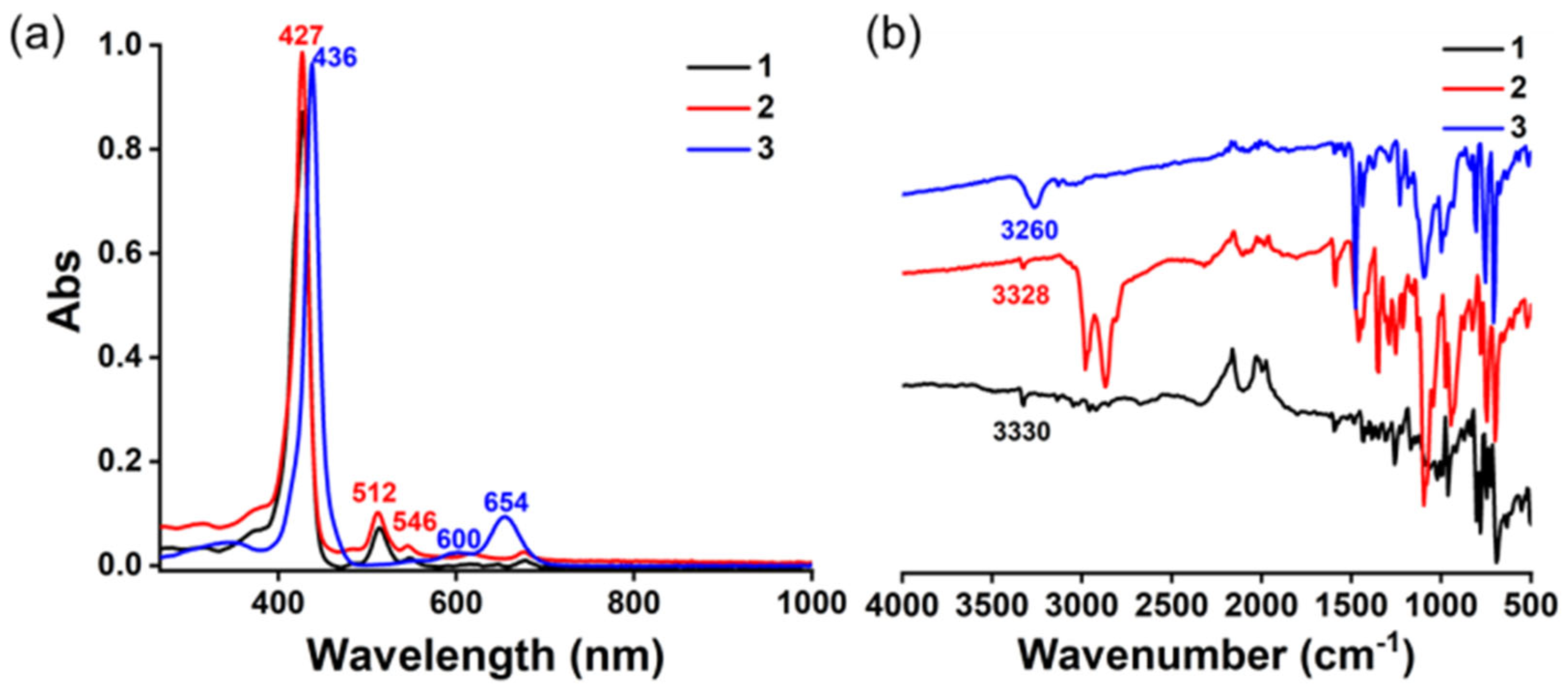
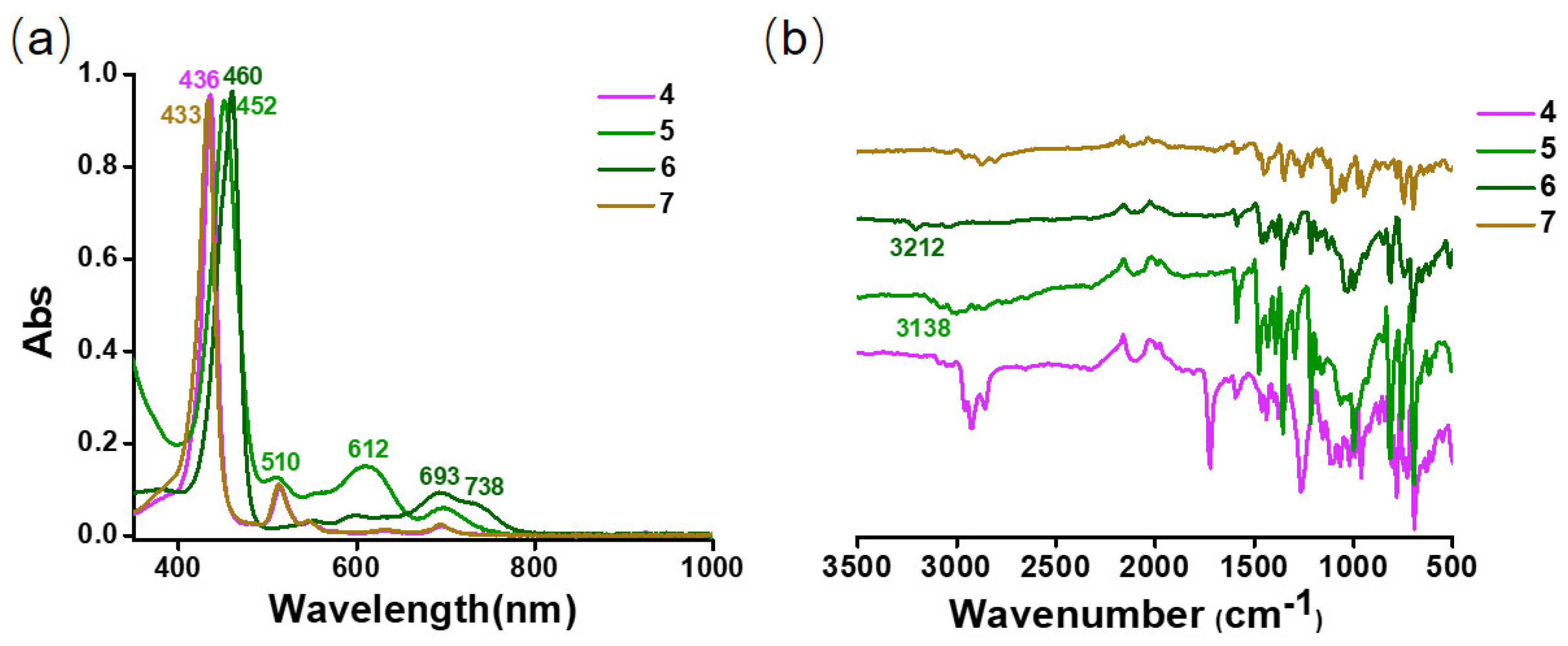
2.1.3. Spectra Characterization
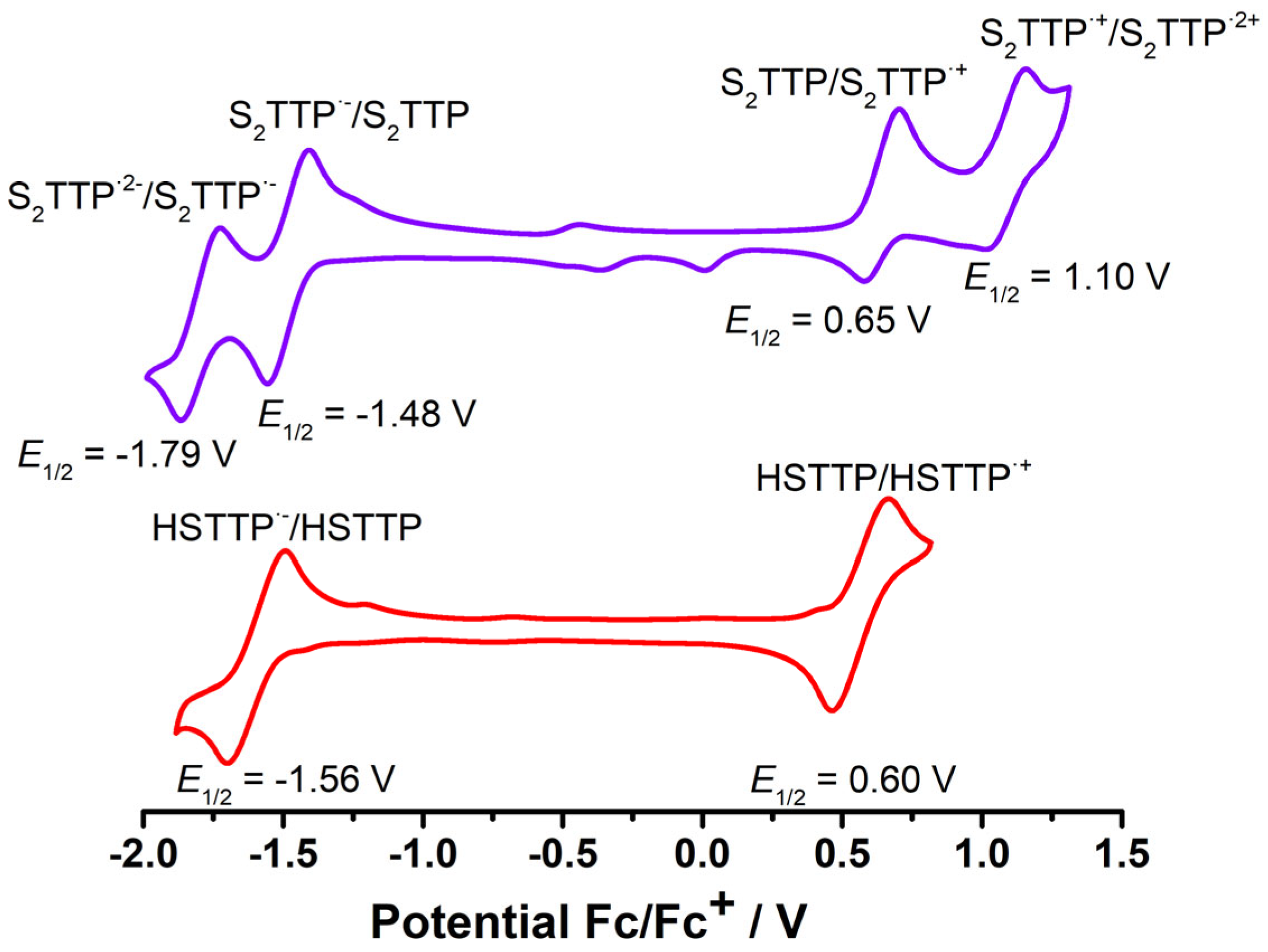
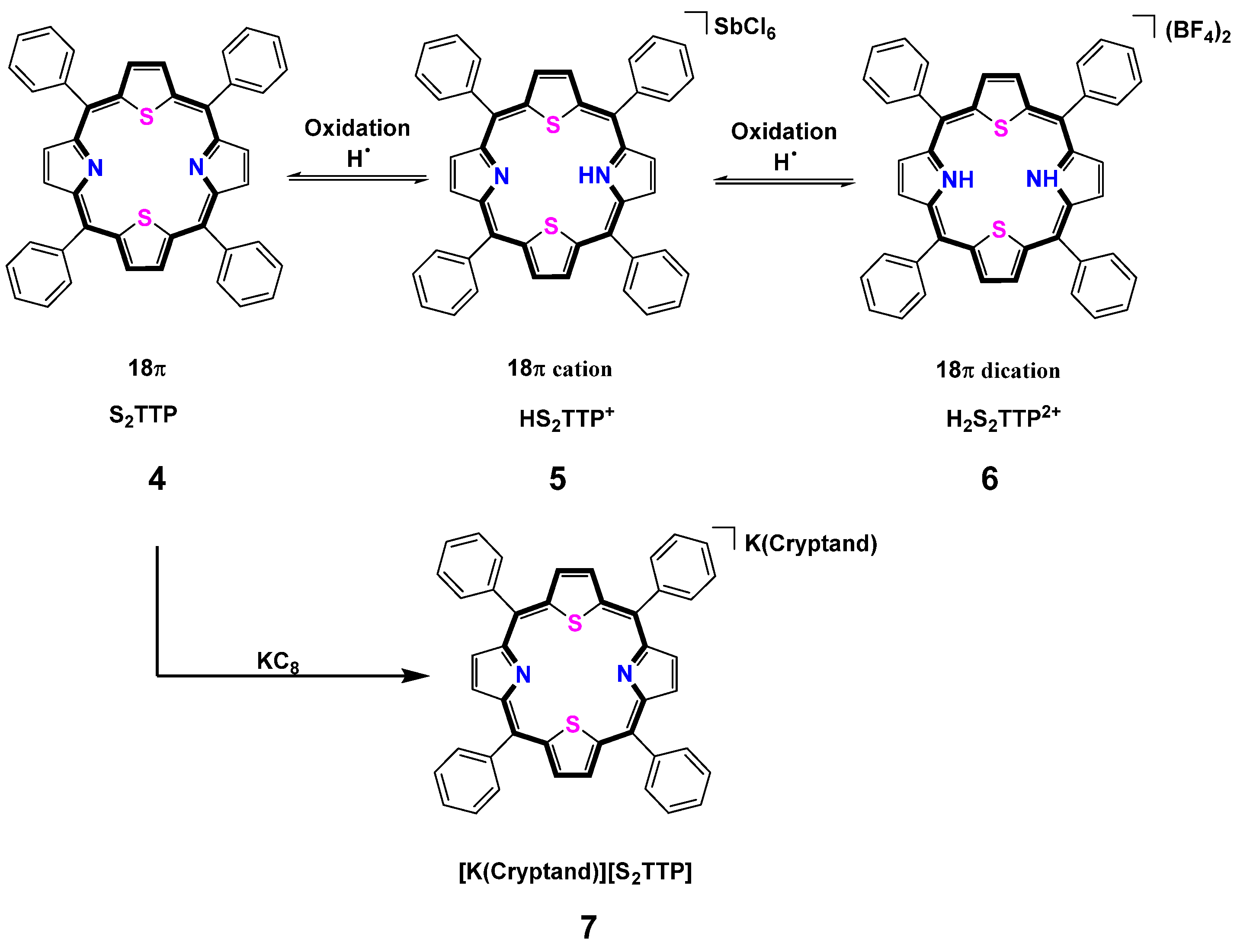
2.2. Stepwise H-Atom Abstraction and Reduction in 21,23-Thiaporphyrin
2.2.1. Syntheses
2.2.2. Crystallographic Details
2.2.3. Spectra Characterization
2.3. ZINDO/S Calculation
3. Materials and Methods
3.1. Instruments
3.2. Materials
3.3. EPR Spectroscopy
3.4. Single-Crystal X-ray Structure Determinations
3.5. Synthesis of [Na(Cryptand)](HSTTP) (2)
3.6. Synthesis of [H3STTP](BF4)2 (3)
3.7. Synthesis of [HS2TTP]SbCl6 (5)
3.8. Synthesis of [H2S2TTP](BF4)2 (6)
3.9. Synthesis of [K(Cryptand)](S2TTP) (7)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hiroto, S.; Miyake, Y.; Shinokubo, H. Synthesis and functionalization of porphyrins through organometallic methodologies. Chem. Rev. 2017, 117, 2910–3043. [Google Scholar] [CrossRef]
- Ishizuka, T.; Grover, N.; Kingsbury, C.J.; Kotani, H.; Senge, M.O.; Kojima, T. Nonplanar porphyrins: Synthesis, properties, and unique functionalities. Chem. Soc. Rev. 2022, 51, 7560–7630. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, H.; Ryu, D.Y.; Jang, W.-D. Porphyrin-based supramolecular polymers. Chem. Soc. Rev. 2023, 52, 1947–1974. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.; Grzeszczuk, M.; Latos-Grazynski, L.; Lisowski, J. Studies of the reduction of the nickel(II) complex of 5,10,15,20-tetraphenyl-21-thiaporphyrin to form corresponding nickel(I) complexes. Inorg. Chem. 1989, 28, 3546–3552. [Google Scholar] [CrossRef]
- Latos-Grazynski, L.; Lisowski, J.; Chmielewski, P.; Grzeszczuk, M.; Olmstead, M.M.; Balch, A.L. Palladium complexes of 21-thiaporphyrin: Syntheses and characterization. Inorg. Chem. 1994, 33, 192–197. [Google Scholar] [CrossRef]
- Chmielewski, P.; Latos-Grazynski, L.; Pacholska, E. Low-valent nickel thiaporphyrins nuclear magnetic resonance and electron paramagnetic resonance studies. Inorg. Chem. 1994, 33, 1992–1999. [Google Scholar] [CrossRef]
- Wang, F.; Zhong, Z.-B.; Jia, R.-X.; Xing, K.; Cao, R.; Bai, F.; Duan, P.-C. Three oxidation states of cobalt(I/II/III) complexes by thiaporphyrin. Inorg. Chem. 2024, 63, 7233–7240. [Google Scholar] [CrossRef] [PubMed]
- Lash, T.D. Recent developments in the Chemistry of heteroporphyrins and heterocarbaporphyrins. In Advance in Heterocyclic chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Elsevier: Oxford, UK, 2022; Volume 138, pp. 243–334. [Google Scholar] [CrossRef]
- Pareek, Y.; Ravikanth, M. Thiaporphyrins: From building blocks to multiporphyrin arrays. RSC Adv. 2014, 4, 7851–7880. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef]
- Shivatare, S.S.; Shivatare, V.S.; Wong, C.-H. Glycoconjugates: Synthesis, functional studies, and therapeutic developments. Chem. Rev. 2022, 122, 15603–15671. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D.; Fagnoni, M. Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C–H bonds elaboration. Chem. Rev. 2022, 122, 1875–1924. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D. Hydrogen atom transfer (HAT): A versatile strategy for substrate activation in photocatalyzed organic synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.E.R.; Bols, M.L.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Iron and copper active sites in zeolites and their correlation to metalloenzymes. Chem. Rev. 2018, 118, 2718–2768. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, H.; Watanabe, E.; Yoshida, Z. Porphyrin Acids. Tetrahedron 1973, 29, 3241–3245. [Google Scholar] [CrossRef]
- Hirayama, N.; Takehara, A.; Sasada, Y.; Watanabe, E.; Ogoshi, H.; Yoshida, Z. X-ray crystal structure of octaethylporphinium(monocation) tri-iodide. J. Chem. Soc. Chem. Commun. 1974, 330–331. [Google Scholar] [CrossRef]
- Honda, T.; Kojima, T.; Fukuzumi, S. Crystal structures and properties of a monoprotonated porphyrin. Chem. Commun. 2009, 4994–4996. [Google Scholar] [CrossRef]
- Hrung, C.P.; Tsutsui, M.; Culten, D.L.; Meyer, E.F., Jr.; Morimoto, C.N. Synthesis, characterization, and structure of tri-μ-halogeno-hexacarbonyldirhenate(I) salts of monocationic porphyrin acids. J. Am. Chem. Soc. 1978, 100, 6068–6076. [Google Scholar] [CrossRef]
- Karaman, R.; Bruice, T.C. Unusual behavior of 5,10,15,20-tetraphenylporphine diacid toward oxygen Broensted bases. Inorg. Chem. 1992, 31, 2455–2459. [Google Scholar] [CrossRef]
- Almarsson, Ö.; Blaskó, A.; Bruice, T.C. Studies on a hydrocarbon capped free base tetraphenylporphyrin and its conjugate acids -first observation of a monoprotonated tetrapehylporphyrin {CapTPP(H3+)CF3CO2−}. Tetrahedron 1993, 49, 10239–10252. [Google Scholar] [CrossRef]
- De Luca, G.; Romeo, A.; Scolaro, L.M.; Ricciardi, G.; Rosa, A. Evidence for tetraphenylporphyrin monoacids. Inorg. Chem. 2007, 46, 5979–5988. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, J.; Grzeszczuk, M.; Latos-Grazynski, L. Spectrochemical and electrochemical studies of 21-thiatetra (p-tolyl)porphyrin and its copper (II) complexes. Inorg. Chim. Acta. 1989, 161, 153–163. [Google Scholar] [CrossRef]
- Tagawa, K.; Mori, S.; Okujima, T.; Takase, M.; Uno, H. Protonation behavior of thiaporphyrin and thiabenzoporphyrin. Tetrahedron 2017, 73, 794–801. [Google Scholar] [CrossRef]
- Chimielewski, P.J.; Latos-Grazynski, L.; Olmstead, M.M.; Balch, A.L. Nickel Complexes of 21-oxaporphyrin and 21, 23-dioxaporphyrin. Chem. Eur. J. 1997, 3, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Petrukhina, M.A. Planar, curved and twisted molecular nanographenes: Reduction-induced alkali metal coordination. Coord. Chem. Rev. 2023, 486, 215144. [Google Scholar] [CrossRef]
- Zabula, A.V.; Spisak, S.N.; Filatov, A.S.; Rogachev, A.Y.; Petrukhina, M.A. Record alkali metal intercalation by highly charged corannulene. Acc. Chem. Res. 2018, 51, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Stawski, W.; Zhu, Y.; Wei, Z.; Petrukhina, M.A.; Anderson, H.L. Crystallographic evidence for global aromaticity in the di-anion and tetra-anion of a cyclophane hydrocarbon. Chem. Sci. 2023, 14, 14109–14114. [Google Scholar] [CrossRef] [PubMed]
- Stawski, W.; Zhu, Y.; Roncevic, I.; Wei, Z.; Petrukhina, M.A.; Anderson, H.L. A general alkene aminoarylation enabled by N-centred radical reactivity of sulfonamides. Nat. Chem. 2024, 16, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Kuzmin, A.V.; Khasanov, S.S.; Shestakov, A.F.; Yudanova, E.I.; Otsuka, A.; Yamochi, H.; Kitagawa, H.; Lyubovskaya, R.N. Solid state structure, and optical and magnetic properties, of free base tetra(4-pyridyl)porphyrin {H2T(4-Py)P}•– radical anions. J. Org. Chem. 2018, 83, 1861–1866. [Google Scholar] [CrossRef]
- Konarev, D.V.; Kuzmin, A.V.; Khasanov, S.S.; Shestakov, A.F.; Nazarov, D.I.; Otsuka, A.; Yamochi, H.; Kitagawa, H.; Lyubovskaya, R.N. Radical anions of free-base tetraphenyl- and tetrakis(pentafluorophenyl)porphyrins: Effect of substituentson the properties and charge dsproportionation in {Cryptand[2.2.2](Cs+)}(H2TPP•–). Eur. J. Inorg. Chem. 2020, 2020, 2615–2623. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. A critical evaluation of DFT, including time-dependent DFT, applied to bioinorganic chemistry. J. Biol. Inorg. Chem. 2006, 11, 702–711. [Google Scholar] [CrossRef]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Abe, M.; Hilmey, D.G.; Stilts, C.E.; Sukumaran, D.K.; Detty, M.R. 21-Telluraporphyrins. 1. Impact of 21,23-heteroatom interactions on electrochemical redox potentials, 125Te NMR Spectra, and Absorption Spectra. Organometallics 2002, 21, 2986–2992. [Google Scholar] [CrossRef]
- Hicks, J.; Juckel, M.; Paparo, A.; Dange, D.; Jones, C. Multigram syntheses of magnesium(I) compounds using alkali metal halide supported alkali metals as dispersible reducing agents. Organometallics 2018, 37, 4810–4813. [Google Scholar] [CrossRef]
- Bill, E.; Jul, X. Program for Simulation of Molecular Magnetic Data; Max-Planck Institute for Chemical Energy Conversion: Mülheim/Ruhr, Germany, 2008. [Google Scholar]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. 2015, 71, 9–18. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
|---|---|---|---|---|---|---|---|---|
| Angle (°) | Tilt a | S1 4.62(5) N1 11.39(6) N2 4.12(6) | S1 4.75(8) N1 2.39(1) N2 4.07(1) N3 4.33(1) | S1 27.72(3) N1 33.40(4) N2 34.88(4) N3 32.81(4) | S1 4.98(4) N1 12.99(6) | S1 16.79(1) S2 15.93(1) N1 21.28(2) N2 9.31(2) | S1 22.20(6) S2 29.77(2) N1 26.75(2) | S1 1.780(2) S2 3.350(2) N1 3.921(2) N2 2.391(3) |
| C-N-C | 105.91(1) 120.52(5) | 110.63(5) 111.04(5) 111.81(5) | 99.177(1) 102.52(1) 100.92(1) | 106.32(2) | 109.81(5) 104.72(5) | 110.39(3) | 119.70(2) 121.13(2) | |
| C-S-C | 88.46(1) | 85.71(2) | 93.00(8) | 91.98(1) | 91.98(4) 92.60(4) | 93.06(2) 92.58(2) | 84.15(5) 82.61(4) | |
| Length (Å) b | 1.70 1.36 | 1.73 1.38 | 1.68 1.48 | 1.74 1.36 | 1.68 1.37 | 1.74 1.38 | 1.81 1.33 | |
| Nonbonded Length(Å) N-NN(S)-S | 4.40(3) 3.69(6) | 4.29(9) 3.77(7) | 4.21(2) 3.92(1) | 4.64(3) 3.06(7) | 4.51(4) 3.32(4) | 4.81(4) 3.31(2) | 4.63(2) 3.06(7) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.-R.; Xing, K.; Liu, T.; Cao, R.; Dang, L.-L.; Bai, F.; Duan, P.-C. Hydrogen Atom Abstraction and Reduction Study of 21-Thiaporphyrin and 21,23-Dithiaporphyrin. Molecules 2024, 29, 3424. https://doi.org/10.3390/molecules29143424
Ren X-R, Xing K, Liu T, Cao R, Dang L-L, Bai F, Duan P-C. Hydrogen Atom Abstraction and Reduction Study of 21-Thiaporphyrin and 21,23-Dithiaporphyrin. Molecules. 2024; 29(14):3424. https://doi.org/10.3390/molecules29143424
Chicago/Turabian StyleRen, Xiao-Rui, Kang Xing, Teng Liu, Ronghui Cao, Li-Long Dang, Feng Bai, and Peng-Cheng Duan. 2024. "Hydrogen Atom Abstraction and Reduction Study of 21-Thiaporphyrin and 21,23-Dithiaporphyrin" Molecules 29, no. 14: 3424. https://doi.org/10.3390/molecules29143424
APA StyleRen, X.-R., Xing, K., Liu, T., Cao, R., Dang, L.-L., Bai, F., & Duan, P.-C. (2024). Hydrogen Atom Abstraction and Reduction Study of 21-Thiaporphyrin and 21,23-Dithiaporphyrin. Molecules, 29(14), 3424. https://doi.org/10.3390/molecules29143424