
Deciphering the Role of Trehalose in Chroococcidiopsis sp. 029’s High-Desiccation Resistance: Sequence Determination, Structural Modelling and Simulative Analysis of the 30S Ribosomal Subunit
 ,
,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sequence Identification
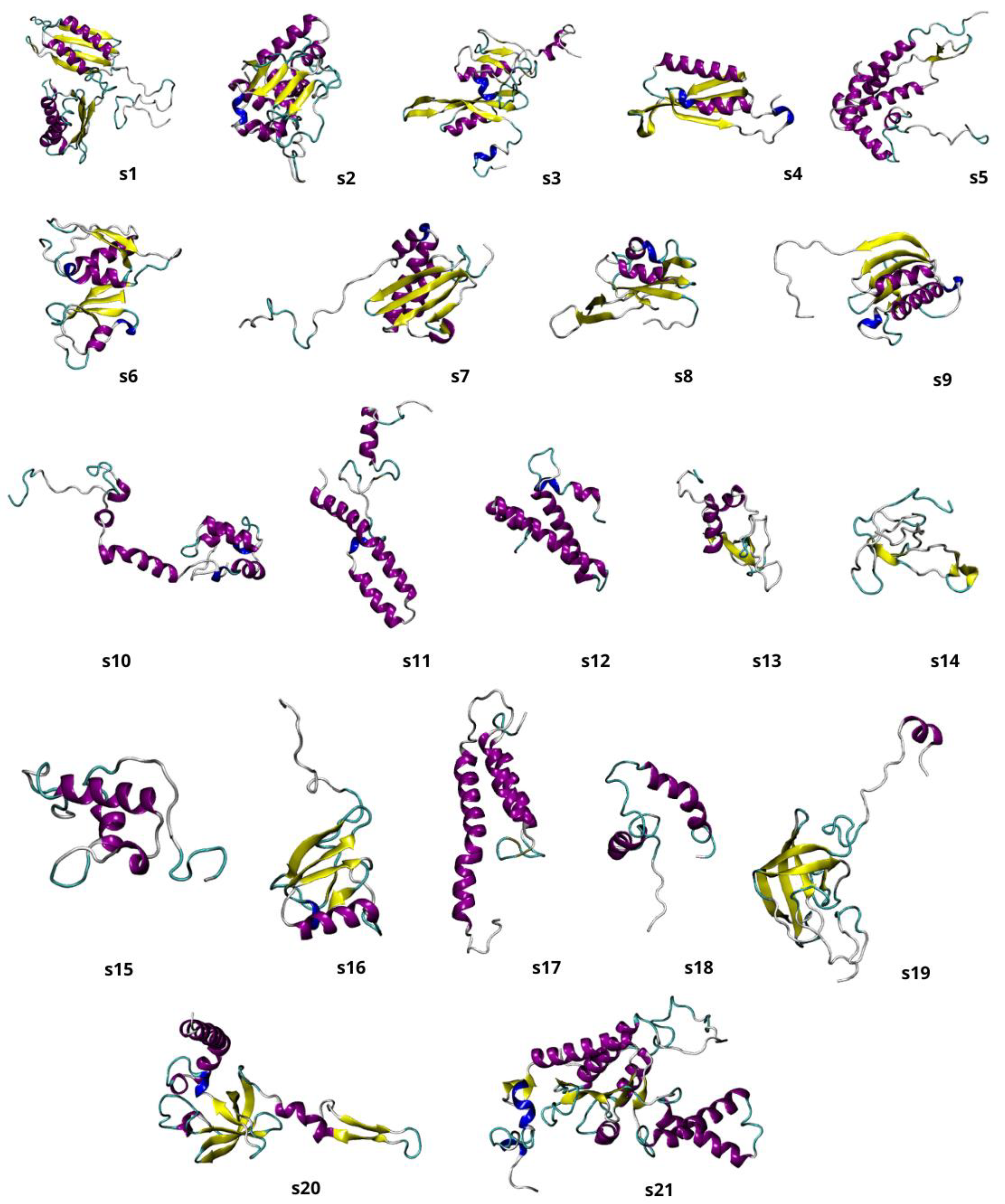
2.2. Models Building
2.3. RMSD, RMSF, Gyration Radius, and Secondary Structure Analyses
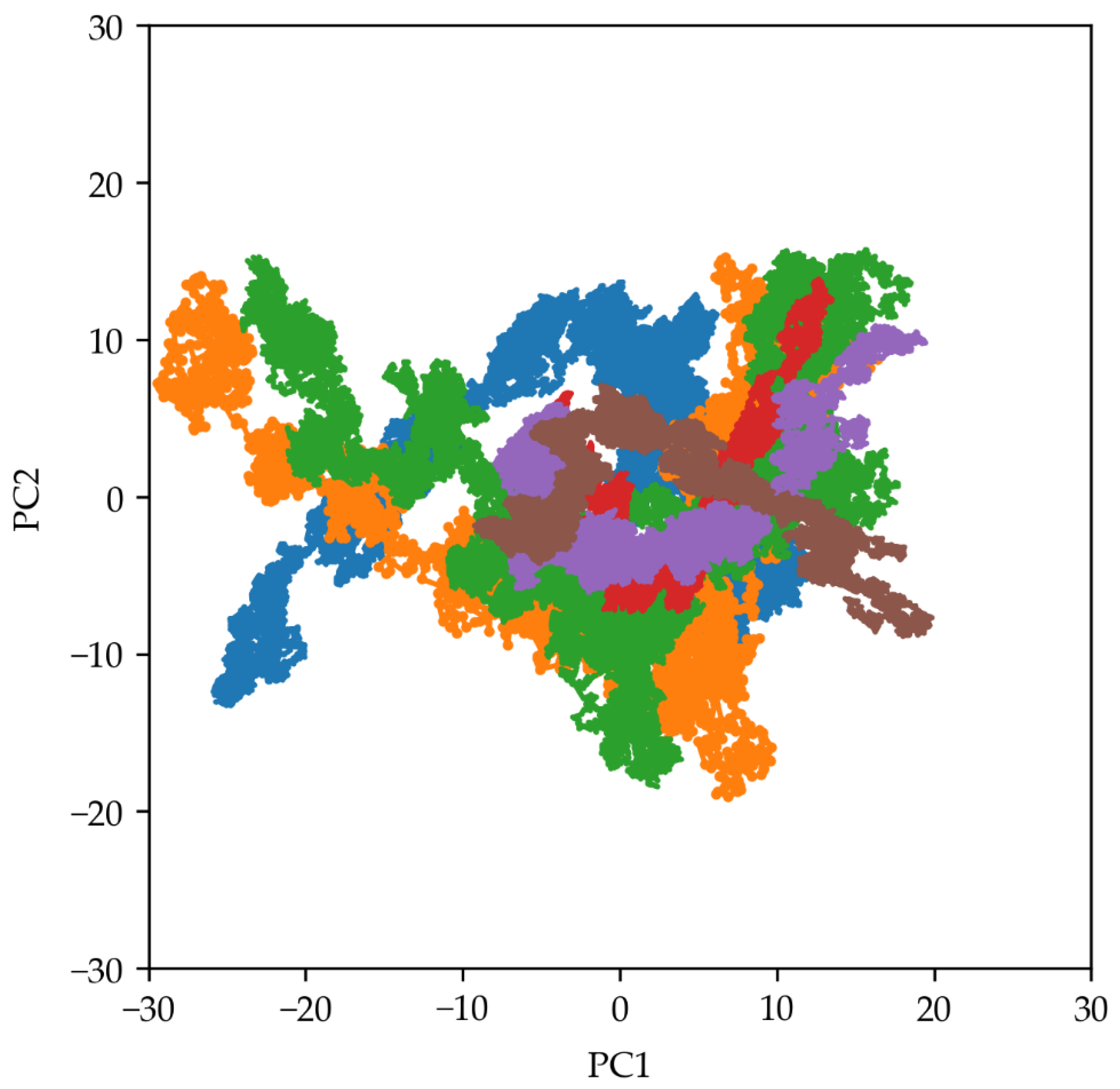
2.4. Principal Components Analysis
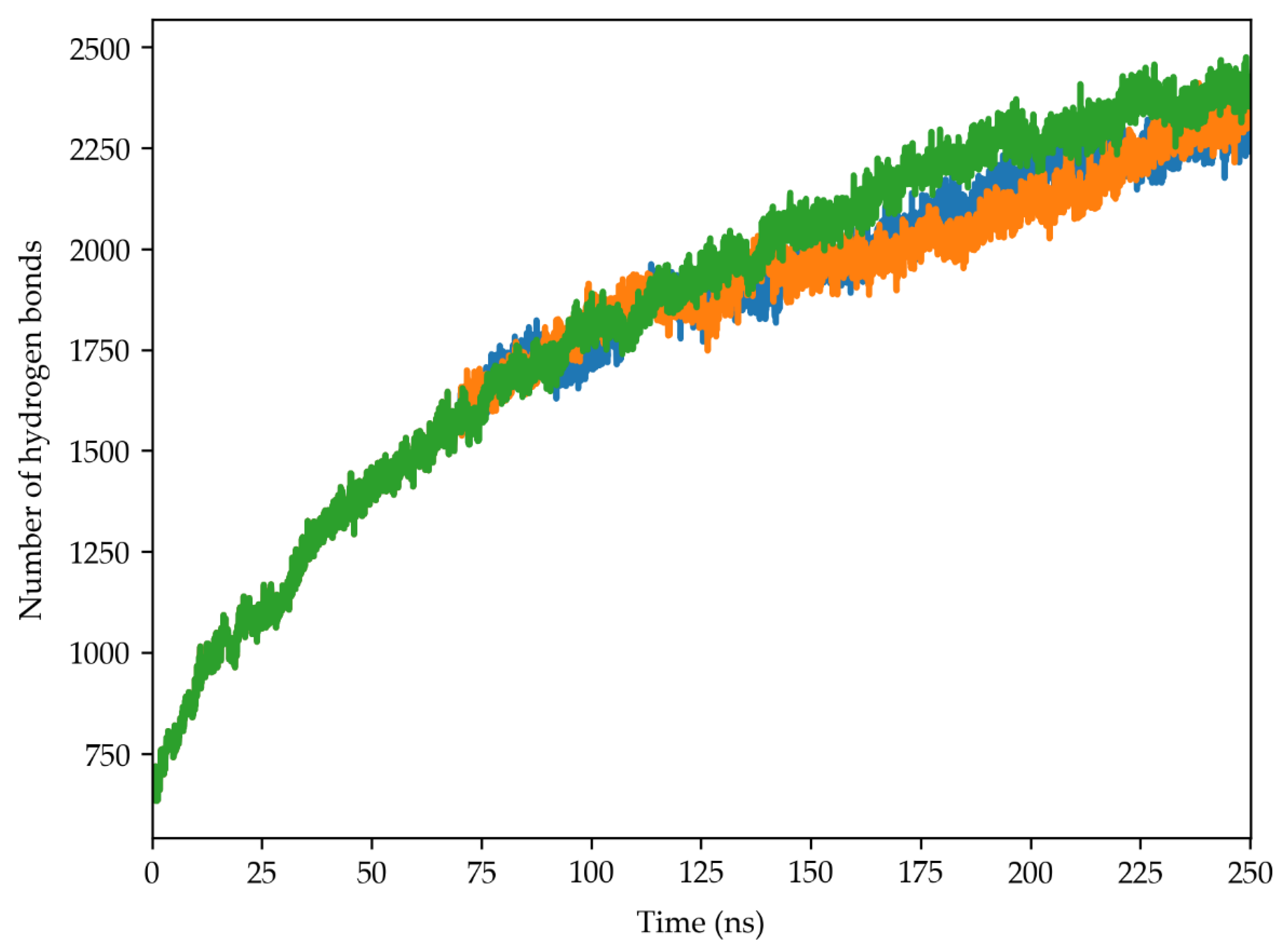
2.5. Hydrogen Bond Analysis
2.6. Radial Distribution Function
3. Discussion
4. Materials and Methods
4.1. Computational Methods
4.1.1. Identification of Genes Encoding the Elements of the 30S Ribosomal Subunit
4.1.2. Molecular Modelling of the 21 Proteins and the 16S rRNA Molecule
4.1.3. Molecular Dynamics Simulations
4.1.4. Simulation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alpert, P. Constraints of tolerance: Why are desiccation-tolerant organisms so small or rare? J. Exp. Biol. 2006, 209, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Potts, M. Desiccation Tolerance of Prokaryotes. Microbiol. Rev. 1994, 58, 755–805. [Google Scholar] [CrossRef] [PubMed]
- Crowe, J.H.; Carpenter, J.F.; Crowe, L.M. The role of vitrification in anhydrobiosis. Annu. Rev. Physiol. 1998, 60, 73–1103. [Google Scholar] [CrossRef] [PubMed]
- Billi, D. Subcellular integrities in Chroococcidiopsis sp. CCMEE 029 survivors after prolonged desiccation revealed by molecular probes and genome stability assays. Extremophiles 2009, 13, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Fagliarone, C.; Mosca, C.; Ubaldi, I.; Verseux, C.; Baqué, M.; Wilmotte, A.; Billi, D. Avoidance of protein oxidation correlates with the desiccation and radiation resistance of hot and cold desert strains of the cyanobacterium Chroococcidiopsis. Extremophiles 2017, 21, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Fagliarone, C.; Napoli, A.; Chiavarini, S.; Baqué, M.; De Vera, J.-P.; Billi, D. Biomarker preservation and survivability under extreme dryness and Mars-like UV flux of a desert cyanobacterium capable of trehalose and sucrose accumulation. Front. Astron. Space Sci. 2020, 7, 31. [Google Scholar] [CrossRef]
- Mosca, C.; Napoli, A.; Fagliarone, C.; Fujimori, A.; Moeller, R.; Billi, D. Role of DNA repair pathways in the recovery of a dried, radioresistant cyanobacterium exposed to high-LET radiation: Implications for the habitability of Mars. Int. J. Astrobiol. 2022, 21, 380–391. [Google Scholar] [CrossRef]
- Verseux, C.; Baqué, M.; Lehto, K.; de Vera, J.-P.P.; Rothschild, L.J.; Billi, D. Sustainable life support on Mars—The potential roles of cyanobacteria. Int. J. Astrobiol. 2016, 15, 65–92. [Google Scholar] [CrossRef]
- Gallego Fernandez, B.; Rothschild, L.J.; Fagliarone, C.; Chiavarini, S.; Billi, D. Feasibility as feedstock of the cyanobacterium Chroococcidiopsis sp. 029 cultivated with urine-supplemented Moon and Mars regolith simulants. Algal Res. 2023, 71, 10304. [Google Scholar] [CrossRef]
- Billi, D. Desert cyanobacteria under space and planetary simulations: A tool for searching for life beyond Earth and supporting human space exploration. Int. J. Astrobiol. 2019, 18, 483–489. [Google Scholar] [CrossRef]
- Billi, D.; Baqué, M.; Verseux, C.; Rothschild, L.J.; de Vera, J.-P. Desert cyanobacteria—Potential for space and Earth applications. In Adaption of Microbial Life to Environmental Extremes; Stan-Lotter, H., Fendrihan, S., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [CrossRef]
- Lu, X.-J.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Corradini, D.; Strekalova, E.G.; Stanley, H.E.; Gallo, P. Microscopic mechanism of protein cryopreservation in an aqueous solution with trehalose. Sci. Rep. 2013, 3, 1218. [Google Scholar] [CrossRef] [PubMed]
- Lins, R.D.; Pereira, C.S.; Hünenberger, P.H. Trehalose-protein interaction in aqueous solution. Proteins 2004, 55, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 19 July 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Mládek, A.; Banáš, P.; Cheatham, T.E.; Jurečka, P. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; De Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C. Efficient algorithms for Langevin and DPD dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrafesa, D.; Napoli, A.; Iacovelli, F.; Romeo, A.; Tucci, F.G.; Billi, D.; Falconi, M. Deciphering the Role of Trehalose in Chroococcidiopsis sp. 029’s High-Desiccation Resistance: Sequence Determination, Structural Modelling and Simulative Analysis of the 30S Ribosomal Subunit. Molecules 2024, 29, 3486. https://doi.org/10.3390/molecules29153486
Pietrafesa D, Napoli A, Iacovelli F, Romeo A, Tucci FG, Billi D, Falconi M. Deciphering the Role of Trehalose in Chroococcidiopsis sp. 029’s High-Desiccation Resistance: Sequence Determination, Structural Modelling and Simulative Analysis of the 30S Ribosomal Subunit. Molecules. 2024; 29(15):3486. https://doi.org/10.3390/molecules29153486
Chicago/Turabian StylePietrafesa, Davide, Alessandro Napoli, Federico Iacovelli, Alice Romeo, Fabio Giovanni Tucci, Daniela Billi, and Mattia Falconi. 2024. "Deciphering the Role of Trehalose in Chroococcidiopsis sp. 029’s High-Desiccation Resistance: Sequence Determination, Structural Modelling and Simulative Analysis of the 30S Ribosomal Subunit" Molecules 29, no. 15: 3486. https://doi.org/10.3390/molecules29153486