
Asymmetric Synthesis and Applications of Chiral Organoselenium Compounds: A Review


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Asymmetric Synthesis of Organoselenium Compounds
2.1. Chiral Catalyst or Ligand-Controlled Method
2.1.1. Asymmetric Cycloaddition
2.1.2. Selenolactonizations
2.1.3. Selenoaminations and Amidations
2.1.4. Asymmetric Addition Reaction
2.1.5. Asymmetric Selenization of Aldehydes/Ketones
2.1.6. Asymmetric Ring-Opening Reaction
2.1.7. Asymmetric Substitution Reaction
2.1.8. Asymmetric Decarboxylation Reaction
2.2. Substrate Control Enantioselectivity
3. Application of Chiral Organoselenium Compounds in the Asymmetric Catalysis
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berzelius, J.J. Undersökning af en ny Mineral-kropp, funnen i de orenare sorterna af det i Falun tillverkade svaflet. Afh. Fys. Kemi Mineral. 1818, 6, 42. [Google Scholar]
- Winkel, L.H.E.; Johnson, C.A.; Lenz, M.; Grundl, T.; Leupin, O.X.; Amini, M.; Charlet, L. Environmental Selenium Research: From Microscopic Processes to Global Understanding. Environ. Sci. Technol. 2012, 46, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Lenardão, E.J.; Santi, C.; Sancineto, L. Bioactive Organoselenium Compounds and Therapeutic Perspectives. In New Frontiers in Organoselenium Compounds; Springer: Cham, Switzerland, 2018; pp. 99–143. [Google Scholar]
- Chuai, H.; Zhang, S.-Q.; Bai, H.; Li, J.; Wang, Y.; Sun, J.; Wen, E.; Zhang, J.; Xin, M. Small molecule selenium-containing compounds: Recent development and therapeutic applications. Eur. J. Med. Chem. 2021, 223, 113621. [Google Scholar] [CrossRef] [PubMed]
- Löwig, C.J. Ueber Schwefelwasserstoff- und Selenwasserstoffäther. Ann. Phys. 1836, 113, 550–553. [Google Scholar] [CrossRef]
- Mugesh, G.; Mont, W.-W.d.; Sies, H. Chemistry of Biologically Important Synthetic Organoselenium Compounds. Chem. Rev. 2001, 101, 2125–2180. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P. Transition-Metal-Catalyzed C-S, C-Se, and C-Te Bond Formation via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef] [PubMed]
- Rathke, B. Beiträge zur Kenntniss des Selens. Justus Liebigs Ann. Chem. 1869, 152, 181–220. [Google Scholar] [CrossRef]
- Foster, D.G.; Brown, S.F. Organic Selenium Compounds. Some Derivatives of Aromatic Seleno-Ethers. J. Am. Chem. Soc. 1928, 50, 1182–1188. [Google Scholar] [CrossRef]
- Rathke, B. Ueber das Selenmercaptan. Ann. Chem. 1869, 152, 211. [Google Scholar]
- Corrigan, J.F. A Guide to Chalcogen−Nitrogen Chemistry By Tristram Chivers. J. Am. Chem. Soc. 2007, 129, 5782. [Google Scholar] [CrossRef]
- Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 1986; p. 463. [Google Scholar]
- Mont, W.-W.d. Book Review: The Chemistry of Organic Selenium and Tellurium Compounds. Angew. Chem. Int. Ed. 1988, 27, 444–445. [Google Scholar] [CrossRef]
- Back, G.T. Organoselenium Chemistry: A Practical Approach; OUP Oxford: Oxford, UK, 1999; p. 295. [Google Scholar]
- Takouachet, R.; Benali-Cherif, R.; Bendeif, E.-E.; Jelsch, C.; Cherif, F.Y.; Rahmouni, A.; Benali-Cherif, N. The supramolecular behavior and molecular recognition of adeninium cations on anionic hydrogen selenite/diselenite frameworks: A structural and theoretical analysis. J. Mol. Struct. 2021, 1229, 129836. [Google Scholar] [CrossRef]
- Djeribi, M.; Nagazi, I.; Cocetta, V.; Dege, N.; Issaoui, N.; Zanetti, L.; Carraro, M.; Ayed, B. Synthesis of novel supramolecular selenomolybdate as anticancer agents: An experimental and DFT computational analysis. J. Mol. Struct. 2024, 1306, 137880. [Google Scholar] [CrossRef]
- Yu, L.; Bin, L.; Chang-Cheng, Y.; Takehiko, W.; Yoshihisa, I. Molecular Recognition Studies on Supramolecular Systems. 32.1 Molecular Recognition of Dyes by Organoselenium-Bridged Bis(β-cyclodextrin)s. J. Org. Chem. 2001, 66, 225–232. [Google Scholar] [CrossRef]
- Cester Bonati, F.; Andreoni, L.; Cattani, S.; Baccini, C.; Anzellotti, S.; Cera, G.; Silvi, S.; Secchi, A. Selenoureido Calix[6]arenes: A Novel Platform for Pseudorotaxane Synthesis. Eur. J. Org. Chem. 2024, 27, e202400237. [Google Scholar] [CrossRef]
- Vyhivskyi, O.; Laikov, D.N.; Finko, A.V.; Skvortsov, D.A.; Zhirkina, I.V.; Tafeenko, V.A.; Zyk, N.V.; Majouga, A.G.; Beloglazkina, E.K. Ullmann-type C–Se Cross-Coupling in the Hydantoin Family: Synthesis, Mechanistic Studies, and Tests of Biological Activity. J. Org. Chem. 2020, 85, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, H.; Zhang, X.; Ye, J.; Liu, J.; Xu, Q.; Lautens, M. Organoselenium-Catalyzed Mild Dehydration of Aldoximes: An Unexpected Practical Method for Organonitrile Synthesis. Org. Lett. 2014, 16, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jian, Y.; Hu, W.; Zhao, S.; Shi, Z.-J.; Selander, N.; Zhou, T. Ni and Fe catalyzed cascade radical reactions of oxime esters with diselenides. Org. Chem. Front. 2022, 9, 3480–3485. [Google Scholar] [CrossRef]
- Tang, J.; Singh, T.; Li, X.; Liu, L.; Zhou, T. Selenium-Directed ortho-C–H Borylation by Iridium Catalysis. J. Org. Chem. 2020, 85, 11959–11967. [Google Scholar] [CrossRef]
- Xia, J.; Li, T.; Lu, C.; Xu, H. Selenium-Containing Polymers: Perspectives toward Diverse Applications in Both Adaptive and Biomedical Materials(Review). Macromolecules 2018, 51, 7435–7455. [Google Scholar] [CrossRef]
- Chen, Z.; Li, M.; Gu, Q.; Peng, X.; Qiu, W.; Xie, W.; Liu, D.; Jiao, Y.; Liu, K.; Zhou, J.; et al. Highly Efficient Purely Organic Phosphorescence Light-Emitting Diodes Employing a Donor-Acceptor Skeleton with a Phenoxaselenine Donor. Adv. Sci. 2023, 10, e2207003. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Hu, C.; Li, H.; Huang, H.; Ding, L.; Zhang, J.; Wu, J.; Du, C.; He, Z.; Chen, X. Molecule-Enhanced Electrocatalysis of Sustainable Oxygen Evolution Using Organoselenium Functionalized Metal-Organic Nanosheets. J. Am. Chem. Soc. 2023, 145, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Brutchey, R. Diorganyl Dichalcogenides as Useful Synthons for Colloidal Semiconductor Nanocrystals. Acc. Chem. Res. 2015, 48, 2918–2926. [Google Scholar] [CrossRef]
- Patra, A.; Wijsboom, Y.H.; Leitus, G.; Bendikov, M. Tuning the Band Gap of Low-Band-Gap Polyselenophenes and Polythiophenes: The Effect of the Heteroatom. Chem. Mater. 2011, 23, 896–906. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B.T. Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology. Chem. Rev. 2004, 104, 6255–6286. [Google Scholar] [CrossRef] [PubMed]
- Radomska, D.; Czarnomysy, R.; Radomski, D.; Bielawski, K. Selenium Compounds as Novel Potential Anticancer Agents. Int. J. Mol. Sci. 2021, 22, 1009. [Google Scholar] [CrossRef]
- Ali, W.; Benedetti, R.; Handzlik, J.; Zwergel, C.; Battistelli, C. The innovative potential of selenium-containing agents for fighting cancer and viral infections. Drug. Discov. 2021, 26, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Frost, D.V. The two faces of selenium—Can selenophobia be cured? CRC Crit. Rev. Toxicol. 1972, 1, 467–514. [Google Scholar] [CrossRef]
- Zhu, C.; Huang, Y. Asymmetric synthesis of chiral organoselenium compounds. Curr. Org. Chem. 2006, 10, 1905–1920. [Google Scholar] [CrossRef]
- Lai, S.; Liang, X.; Zeng, Q. Recent Progress in Synthesis and Application of Chiral Organoselenium Compounds. Chem. Eur. J. 2024, 30, e202304067. [Google Scholar] [CrossRef] [PubMed]
- Stadel, J.T.; Back, T.G. Asymmetric Synthesis with Organoselenium Compounds—The Past Twelve Years. Chem. Eur. J. 2024, 30, e202304074. [Google Scholar] [CrossRef] [PubMed]
- Sonego, J.M.; de Diego, S.I.; Szajnman, S.H.; Gallo-Rodriguez, C.; Rodriguez, J.B. Organoselenium Compounds: Chemistry and Applications in Organic Synthesis. Chem.—Eur. J. 2023, 29, e202300030. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C-S, C-Se, and C-Te Bond Formations via Cross-Coupling and Atom-Economic Addition Reactions. Achievements and Challenges. Chem. Rev. 2022, 122, 16110–16293. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, S.-i.; Takahashi, K.; Kato, H.; Yamazaki, H. Aymmetric Methoxyselenenylation of alkenes with chiral freeocenylselenium reagents. J. Organomet. Chem. 1997, 62, 7711–7716. [Google Scholar] [CrossRef]
- Chen, F.; Tan, C.K.; Yeung, Y.-Y. C2-symmetric cyclic selenium-catalyzed enantioselective bromoaminocyclization. J. Am. Chem. Soc. 2013, 135, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Hashimoto, T.; Maruoka, K. A Chiral Electrophilic Selenium Catalyst for Highly Enantioselective Oxidative Cyclization. J. Am. Chem. Soc. 2016, 138, 5206–5209. [Google Scholar] [CrossRef]
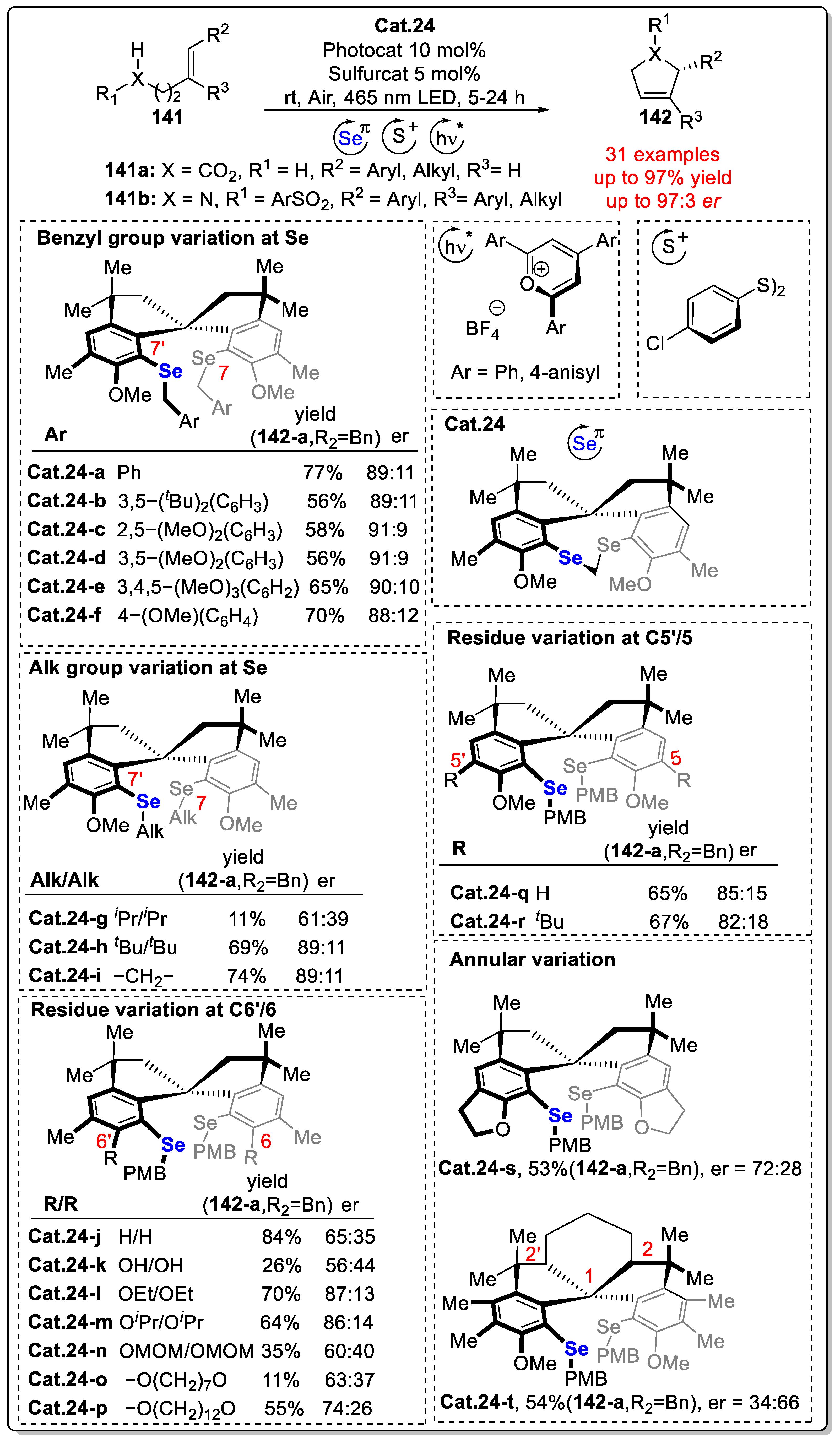
- Lei, T.; Graf, S.; Schöll, C.; Krätzschmar, F.; Gregori, B.; Appleson, T.; Breder, A. Asymmetric Photoaerobic Lactonization and Aza-Wacker Cyclization of Alkenes Enabled by Ternary Selenium–Sulfur Multicatalysis. ACS Catal. 2023, 13, 16240–16248. [Google Scholar] [CrossRef]
- Peters, B.B.C.; Andersson, P.G. The Implications of the Brønsted Acidic Properties of Crabtree-Type Catalysts in the Asymmetric Hydrogenation of Olefins. J. Am. Chem. Soc. 2022, 144, 16252–16261. [Google Scholar] [CrossRef]
- Tan, M.; Peters, B.B.C.; Andersson, P.G.; Zhou, T. Recent advances in the metal-catalyzed asymmetric alkene hydrogenation of cyclic conjugated carbonyl compounds. Org. Chem. Front. 2024, 11, 2934–2953. [Google Scholar] [CrossRef]
- Zhao, S.; Peters, B.B.C.; Zhang, H.; Xue, R.; Yang, Y.; Wu, L.; Huang, T.; He, L.; Andersson, P.G.; Zhou, T. Asymmetric and Chemoselective Iridium Catalyzed Hydrogenation of Conjugated Unsaturated Oxime Ethers. Chem. Eur. J. 2024, 30, e202401333. [Google Scholar] [CrossRef] [PubMed]
- Verendel, J.J.; Pàmies, O.; Diéguez, M.; Andersson, P.G. Asymmetric Hydrogenation of Olefins Using Chiral Crabtree-type Catalysts: Scope and Limitations. Chem. Rev. 2014, 114, 2130–2169. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ponra, S.; Li, X.; Peters, B.B.C.; Massaro, L.; Zhou, T.; Andersson, P.G. Catalytic enantioselective synthesis of fluoromethylated stereocenters by asymmetric hydrogenation. Chem. Sci. 2022, 13, 8590–8596. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Zhou, T.-G.; Li, X.-W.; Shao, X.-R.; Xu, P.-L.; Wu, W.-L.; Hartwig, J.F.; Shi, Z.-J. A Chiral Nitrogen Ligand for Enantioselective, Iridium-Catalyzed Silylation of Aromatic C−H Bonds. Angew. Chem. Int. Ed. 2017, 56, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Erdelmeier, I.; Tailhan-Lomont, C.; Yadan, J.-C. Copper(I)-Assisted Mild and Convenient Synthesis of New Se−N Heterocycles: Access to a Promising Class of GPx Mimics. J. Org. Chem. 2000, 65, 8152–8157. [Google Scholar] [CrossRef] [PubMed]
- Balkrishna, S.J.; Bhakuni, B.S.; Chopra, D.; Kumar, S. Cu-Catalyzed Efficient Synthetic Methodology for Ebselen and Related Se-N Heterocycles. Org. Lett. 2010, 12, 5394–5397. [Google Scholar] [CrossRef] [PubMed]
- Thanna, S.; Goins, C.M.; Knudson, S.E.; Slayden, R.A.; Ronning, D.R.; Sucheck, S.J. Thermal and Photoinduced Copper-Promoted C–Se Bond Formation: Synthesis of 2-Alkyl-1,2-benzisoselenazol-3(2H)-ones and Evaluation against Mycobacterium tuberculosis. J. Org. Chem. 2017, 82, 3844–3854. [Google Scholar] [CrossRef]
- Shinichi, Y.; Mayumi, T.; Shoko, Y. Asymmetric [2 + 1] Cycloaddition Reactions of 1-Seleno-2-silylethene. J. Org. Chem. 1996, 61, 4046–4050. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kalyani, D.; Collins, W.R. Preparative and mechanistic studies toward the rational development of catalytic, enantioselective selenoetherification reactions. J. Am. Chem. Soc. 2010, 132, 15752–15765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, S.; Jacobsen, E.N. Enantioselective Selenocyclization via Dynamic Kinetic Resolution of Seleniranium Ions by Hydrogen-Bond Donor Catalysts. J. Am. Chem. Soc. 2014, 136, 16485–16488. [Google Scholar] [CrossRef]
- Niu, W.; Yeung, Y.-Y. Catalytic and Highly Enantioselective Selenolactonization. Org. Lett. 2015, 17, 1660–1663. [Google Scholar] [CrossRef]
- See, J.Y.; Yang, H.; Zhao, Y.; Wong, M.W.; Ke, Z.; Yeung, Y.-Y. Desymmetrizing Enantio- and Diastereoselective Selenoetherification through Supramolecular Catalysis. ACS Catal. 2018, 8, 850–858. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, Y.-Y.; Du, Y.-L.; Gong, L.-Z. Organocatalytic asymmetric selenofunctionalization of tryptamine for the synthesis of hexahydropyrrolo[2,3-b]indole derivatives. Beilstein J. Org. Chem. 2013, 9, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Yu, L.; Huo, Y.; Li, Y.; Xue, X.; Chen, Z. Chiral Lewis Base Catalyzed Enantioselective Selenocyclization of 1,1-Disubstituted Alkenes: Asymmetric Synthesis of Selenium-Containing 4H-3,1-Benzoxazines. Org. Lett. 2022, 24, 4093–4098. [Google Scholar] [CrossRef] [PubMed]
- Pluim, H.; Wynberg, H. Alkaloid catalysed asymmetric synthesis. The addition of selenophenols to 2-cyclohexen-1-ones and conversion to optically active allylic alcohols. Tetrahedron Lett. 1979, 20, 1251–1254. [Google Scholar] [CrossRef]
- Miyake, Y.; Oda, M.; Oyamada, A.; Takada, H.; Ohe, K.; Uemura, S. Asymmetric imidation of organic selenides into selenimides. J. Organomet. Chem. 2000, 611, 475–487. [Google Scholar] [CrossRef]
- Burlant, W.J.; Gould, E.S. Metalation of Dibenzoselenophene. J. Am. Chem. Soc. 1954, 76, 5775–5776. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Young, M.W.; Lauer, R.F. Reactions of selenoxides: Thermal syn-elimination and H218O exchange. Tetrahedron Lett. 1973, 14, 1979–1982. [Google Scholar] [CrossRef]
- Tamagaki, S.; Oae, S.; Sakaki, K. Preparation and properties of selenium imides. Tetrahedron Lett. 1975, 16, 649–652. [Google Scholar] [CrossRef]
- Davis, F.A.; Stringer, O.D.; McCauley, J.P. Asymmetric oxidation of achiral selenides to optically active selenoxides: Stereochemistry of the allyl selenoxide-selenenate [2,3] sigmatropic rearrangement. Tetrahedron 1985, 41, 4747–4757. [Google Scholar] [CrossRef]
- Marcos, V.; Alemán, J.; Ruano, J.L.G.; Marini, F.; Tiecco, M. Asymmetric Synthesis of α-Alkyl α-Selenocarbonyl Compounds Catalyzed by Bifunctional Organocatalysts. Org. Lett. 2011, 13, 3052–3055. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Wang, H.; Huang, D.; Shi, Y. Enantioselective oxysulfenylation and oxyselenenylation of olefins catalyzed by chiral Bronsted acids. Tetrahedron 2012, 68, 2728–2735. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, N.; Wang, Z.; Yan, H. Enantioselective addition of selenosulfonates to α,β-unsaturated ketones. Org. Biomol. Chem. 2018, 16, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, F.; Huang, S.; Zhao, Z.; Mao, H.; Qin, W. Organocatalytic Enantioselective Selenosulfonylation of a C–C Double Bond To Form Two Stereogenic Centers in an Aqueous Medium. J. Org. Chem. 2019, 84, 8100–8111. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, Q.; Bian, Z.; Wang, J. Rhodium-Catalyzed Enantioselective Hydroselenation of Heterobicyclic Alkenes. Org. Lett. 2020, 22, 2781–2785. [Google Scholar] [CrossRef]
- Oroz, P.; Navo, C.D.; Avenoza, A.; Busto, J.H.; Corzana, F.; Jiménez-Osés, G.; Peregrina, J.M. Toward Enantiomerically Pure β-Seleno-α-amino Acids via Stereoselective Se-Michael Additions to Chiral Dehydroalanines. Org. Lett. 2020, 23, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Compañón, I.; Guerreiro, A.; Mangini, V.; Castro-López, J.; Escudero-Casao, M.; Avenoza, A.; Busto, J.H.; Castillón, S.; Jiménez-Barbero, J.; Asensio, J.L.; et al. Structure-Based Design of Potent Tumor-Associated Antigens: Modulation of Peptide Presentation by Single-Atom O/S or O/Se Substitutions at the Glycosidic Linkage. J. Am. Chem. Soc. 2019, 141, 4063–4072. [Google Scholar] [CrossRef]
- Li, E.; Chen, J.; Huang, Y. Enantioselective Seleno-Michael Addition Reactions Catalyzed by a Chiral Bifunctional N-Heterocyclic Carbene with Noncovalent Activation. Angew. Chem. Int. Ed. 2022, 61, e202202040. [Google Scholar] [CrossRef]
- Slocumb, H.S.; Nie, S.; Dong, V.M.; Yang, X.-H. Enantioselective Selenol-ene Using Rh-Hydride Catalysis. J. Am. Chem. Soc. 2022, 144, 18246–18250. [Google Scholar] [CrossRef]
- Yang, X.; Davison, R.T.; Nie, S.; Cruz, F.A.; McGinnis, T.M.; Dong, V.M. Catalytic Hydrothiolation: Counterion-Controlled Regioselectivity. J. Am. Chem. Soc. 2019, 141, 3006–3013. [Google Scholar] [CrossRef]
- Lin, X.; Tan, Z.; Yang, W.; Yang, W.; Liu, X.; Feng, X. Chiral cobalt(II) complex catalyzed asymmetric [2,3]-sigmatropic rearrangement of allylic selenides with α-diazo pyrazoleamides. CCS Chem. 2021, 3, 1423. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, H.M.; Yin, L. Copper(I)-Catalyzed Conjugate Addition/Enantioselective Protonation with Selenols and α-Substituted α,β-Unsaturated Thioamides. Angew. Chem. 2023, 135, e202301422. [Google Scholar] [CrossRef]
- Nakamura, S.; Aoki, T.; Ogura, T.; Wang, L.; Toru, T. Highly Enantioselective Reaction of α-Selenoorganolithium Compounds with Chiral Bis(oxazoline)s and Preparation of Enantioenriched Benzylidencyclohexanes. J. Org. Chem. 2005, 69, 8916–8923. [Google Scholar] [CrossRef] [PubMed]
- Tiecco, M.; Carlone, A.; Sternativo, S.; Marini, F.; Bartoli, G.; Melchiorre, P. Organocatalytic Asymmetric α-Selenenylation of Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 6882–6885. [Google Scholar] [CrossRef] [PubMed]
- Sunden, H.; Rios, R.; Cordova, A. Organocatalytic highly enantioselective α-selenenylation of aldehydes. Tetrahedron Lett. 2007, 48, 7865–7869. [Google Scholar] [CrossRef]
- Hess, L.C.; Posner, G.H. Asymmetric, organocatalytic, three-step synthesis of alpha-hydroxy-(E)-beta,gamma-unsaturated esters. Org. Lett. 2010, 12, 2120–2122. [Google Scholar] [CrossRef] [PubMed]
- Genna, D.T.; Hencken, C.P.; Siegler, M.A.; Posner, G.H. α-Chloro-β,γ-ethylenic Esters: Enantiocontrolled Synthesis and Substitutions. Org. Lett. 2010, 12, 4694–4697. [Google Scholar] [CrossRef]
- Armstrong, A.; Emmerson, D.P.G. Enantioselective Synthesis of α-Alkyl,α-Vinyl Amino Acids via [2,3]-Sigmatropic Rearrangement of Selenimides. Org. Lett. 2011, 13, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.; Emmerson, D.P.G.; Milner, H.J.; Sheppard, R.J. [2,3]-Sigmatropic rearrangement of allylic selenimides: Strategy for the synthesis of peptides, peptidomimetics, and N-aryl vinyl glycines. J. Org. Chem. 2014, 79, 3895–3907. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Z. Cr-Catalyzed Diastereo- and Enantioselective Synthesis of β-Hydroxy Sulfides and Selenides. ACS Catal. 2022, 12, 11152–11158. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, C.; Yuan, F.; Huang, Y.; Pan, Y. Enantioselective ring-opening reaction of meso-epoxides with ArSeH catalyzed by heterometallic Ti-Ga-Salen system. Org. Lett. 2005, 7, 1927–1930. [Google Scholar] [CrossRef]
- You, Y.; Wu, Z.-J.; Wang, Z.-H.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Enantioselective Synthesis of 3,3-Disubstituted Oxindoles Bearing Two Different Heteroatoms at the C3 Position by Organocatalyzed Sulfenylation and Selenenylation of 3-Pyrrolyl-oxindoles. J. Org. Chem. 2015, 80, 8470–8477. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Qiu, J.; Li, C.; Yuan, L.; Yin, H.; Chen, F.-X. Lewis Acid-Catalyzed Asymmetric Selenocyanation of β-Ketoesters with N-Selenocyanatosaccharin. J. Org. Chem. 2020, 85, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Xie, P.; Zhao, F.; Zheng, C.; Gu, Q.; You, S. Rh(III)-Catalyzed Atroposelective C–H Selenylation of 1-Aryl Isoquinolines. ACS Catal. 2024, 14, 6009–6015. [Google Scholar] [CrossRef]
- Waetzig, S.R.; Tunge, J.A. Synthesis of allyl selenides by palladium-catalyzed decarboxylative coupling. Chem. Commun. 2008, 3311–3313. [Google Scholar] [CrossRef] [PubMed]
- Ruano, J.L.G.; Frateschi, L.; Capperucci, A. Synthesis of Enantiomerically Pure anti-1,2-Diaryl and syn-1,2-Alkylaryl vic-Selenoamines. J. Org. Chem. 2012, 77, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhuang, Z.; Liao, W. Asymmetric Synthesis of Dihydronaphthoquinones Containing Adjacent Stereocenters via a Sulfa-Michael Addition Triggered Ring-Expansion Approach. J. Org. Chem. 2015, 80, 4627–4637. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, A.; Mitsudera, H.; Omata, Y.; Matsuura, K.; Shirai, M.; Kakehi, A. Magnesium cation-induced anti-aldol selective tandem Michael/aldol reaction. Tetrahedron 2002, 58, 9817–9826. [Google Scholar] [CrossRef]
- Kamimura, A.; Okawa, H.; Morisaki, Y.; Ishikawa, S.; Uno, H. Asymmetric Thio-Michael/Nucleophilic Addition Domino Reaction with Chiral N-Sulfinimines. J. Org. Chem. 2007, 72, 3569–3572. [Google Scholar] [CrossRef]
- Yang, X.F.; Hou, X.L.; Dai, L.X. A stereoselective Michael–imino aldol tandem reaction triggered by thiolate anions. Tetrahedron Lett. 2000, 41, 4431–4434. [Google Scholar] [CrossRef]
- Jiang, M.; Yang, H.; Fu, H. Visible-Light Photoredox Synthesis of Chiral α-Selenoamino Acids. Org. Lett. 2016, 18, 1968–1971. [Google Scholar] [CrossRef] [PubMed]
- Satheesh, V.; Srivastava, H.K.; Kumar, S.V.; Sengoden, M.; Punniyamurthy, T. Stereospecific Al-Catalysed Tandem C−N/C−Se Bond Formation of Isoselenocyanates with Aziridines: Synthesis and DFT Study. Adv. Synth. Catal. 2019, 361, 55–58. [Google Scholar] [CrossRef]
- Li, F.; Wang, D.; Chen, H.; He, Z.; Zhou, L.; Zeng, Q. Transition metal-free coupling reactions of benzylic trimethylammonium salts with di(hetero)aryl disulfides and diselenides. Chem. Commun. 2020, 56, 13029–13032. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, F.; Zeng, Q. Synthesis of Dibenzylic Diselenides from Elemental Selenium and Benzylic Quaternary Ammonium Salts. Eur. J. Org. Chem. 2021, 2021, 5605–5608. [Google Scholar] [CrossRef]
- Braga, A.L.; Paixão, M.W.; Lüdtke, D.S.; Silveira, C.C.; Rodrigues, O.E.D. Synthesis of New Chiral Aliphatic Amino Diselenides and their Application as Catalysts for the Enantioselective Addition of Diethylzinc to Aldehydes. Org. Lett. 2003, 5, 2635–2638. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Liu, Y.; Zhao, X. Chiral Selenide-Catalyzed Enantioselective Construction of Saturated Trifluoromethylthiolated Azaheterocycles. Org. Lett. 2017, 19, 3434–3437. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhao, X. Indane-Based Chiral Aryl Chalcogenide Catalysts: Development and Applications in Asymmetric Electrophilic Reactions. Acc. Chem. Res. 2022, 55, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Qin, T.; Zhao, X. Catalytic Regio- and Enantioselective Oxytrifluoromethylthiolation of Aliphatic Internal Alkenes by Neighboring Group Assistance. Org. Lett. 2018, 20, 6384–6388. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Ji, J.; Luo, J.; Zhao, X. Chiral Selenide-Catalyzed Enantioselective Allylic Reaction and Intermolecular Difunctionalization of Alkenes: Efficient Construction of C-SCF3 Stereogenic Molecules. J. Am. Chem. Soc. 2018, 140, 4782–4786. [Google Scholar] [CrossRef]
- Luo, J.; Cao, Q.; Cao, X.; Zhao, X. Selenide-catalyzed enantioselective synthesis of trifluoromethylthiolated tetrahydronaphthalenes by merging desymmetrization and trifluoromethylthiolation. Nat. Commun. 2018, 9, 527. [Google Scholar] [CrossRef]
- Qin, T.; Jiang, Q.; Ji, J.; Luo, J.; Zhao, X. Chiral selenide-catalyzed enantioselective synthesis of trifluoromethylthiolated 2,5-disubstituted oxazolines. Org. Biomol. Chem. 2019, 17, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, X. Enantioselective Construction of Chiral Sulfides via Catalytic Electrophilic Azidothiolation and Oxythiolation of N-Allyl Sulfonamides. ACS Catal. 2019, 9, 6896–6902. [Google Scholar] [CrossRef]
- Guo, R.; Liu, Z.; Zhao, X. Efficient Synthesis of P-Chirogenic Compounds Enabled by Chiral Selenide-Catalyzed Enantioselective Electrophilic Aromatic Halogenation. CCS Chem. 2021, 3, 2617–2628. [Google Scholar] [CrossRef]
- Cheng, P.-T.; Tseng, Y.-H.; Chein, R.-J. Organoselenium-Catalyzed Asymmetric Cyclopropanations of (E)-Chalcones. Org. Lett. 2021, 23, 8104–8108. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kurose, N.; Kawanami, S.; Nojiri, A.; Arai, Y.; Koizumi, T.; Shiro, M. Nucleophilic Substitution Reaction of Optically Pure Chloroselenurane with Active Methylene Compounds. Formation of Optically Pure Selenonium Ylides. Chem. Lett. 1995, 24, 379–380. [Google Scholar] [CrossRef]
- Kamigata, N.; Nakamura, Y.; Kikuchi, K.; Ikemoto, I.; Shimizu, T.; Matsuyama, H. Synthesis and stereochemistry of an optically active selenonium ylide. X-Ray molecular structure of (+)Se-{4′-[(-)-menthyloxycarbonyl]phenyl}(methyl)-selenonium 4,4-dimethyl-2,6-dioxocyclohexylide. J. Chem. Soc. Perkin Trans. 1 1992, 1721–1728. [Google Scholar] [CrossRef]
- Cook, A.F.; Moffatt, J.G. Carbodiimide-sulfoxide reactions. VII. Synthesis of stabilized sulfonium ylides. J. Am. Chem. Soc. 1968, 90, 740–746. [Google Scholar] [CrossRef]
- Ratts, K.W. NMR spectra of s-ylids. Sulfur bonding and magnetic nonbquivalence. Tetrahedron Lett. 1966, 7, 4707–4712. [Google Scholar] [CrossRef]
- Liang, Y.; Jiao, H.; Zhang, H.; Wang, Y.-Q.; Zhao, X. Chiral Chalcogenide-Catalyzed Enantioselective Electrophilic Hydrothiolation of Alkenes. Org. Lett. 2022, 24, 7210–7215. [Google Scholar] [CrossRef]
- Wirth, T. Asymmetric Reaction of Arylalkenes with Diselenides. Angew. Chem. Int. Ed. 1995, 34, 1726–1728. [Google Scholar] [CrossRef]
- Fujita, K.-i.; Murata, K.; Iwaoka, M.; Tomoda, S. Design of optically active selenium reagents having a chiral tertiary amino group and their application to asymmetric inter-and intramolecular oxyselenenylations. Tetrahedron 1997, 53, 2029–2048. [Google Scholar] [CrossRef]
- Xu, X.; Qin, T.; Huang, N.; Liao, L.; Zhao, X. Catalytic Enantioselective Electrophilic Difunctionalization of Unsaturated Sulfones. Org. Lett. 2024, 26, 4514–4519. [Google Scholar] [CrossRef] [PubMed]
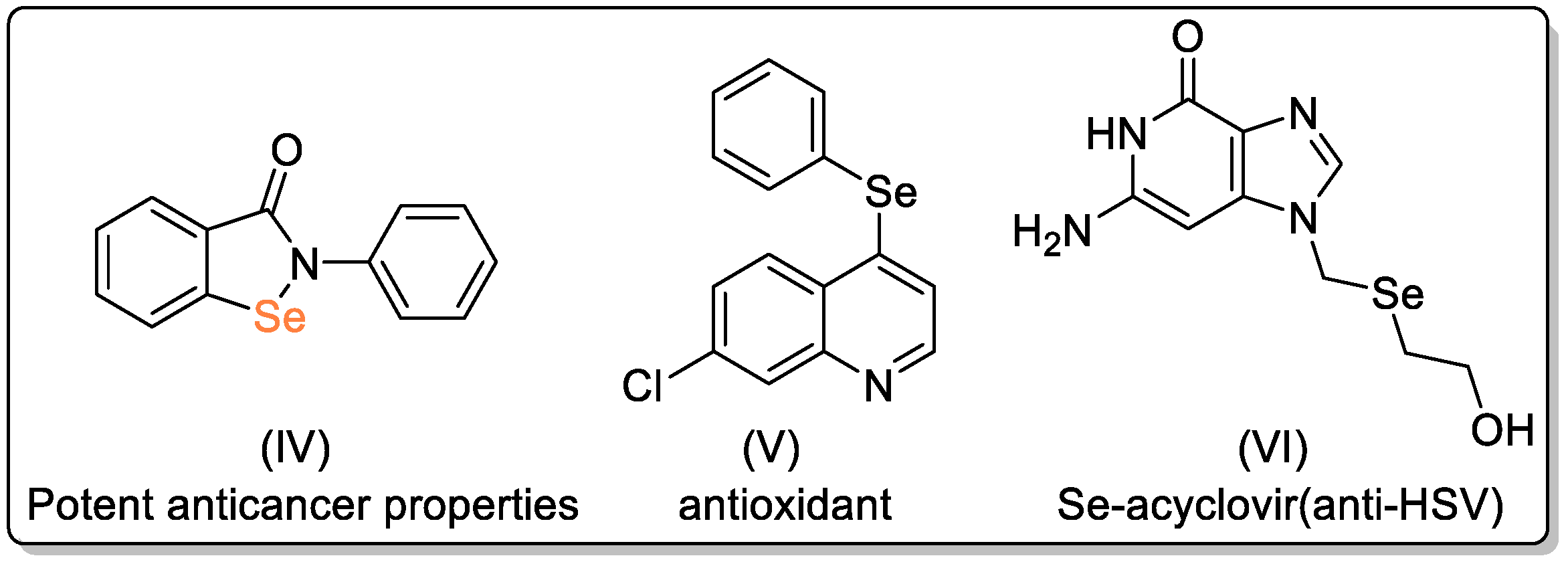
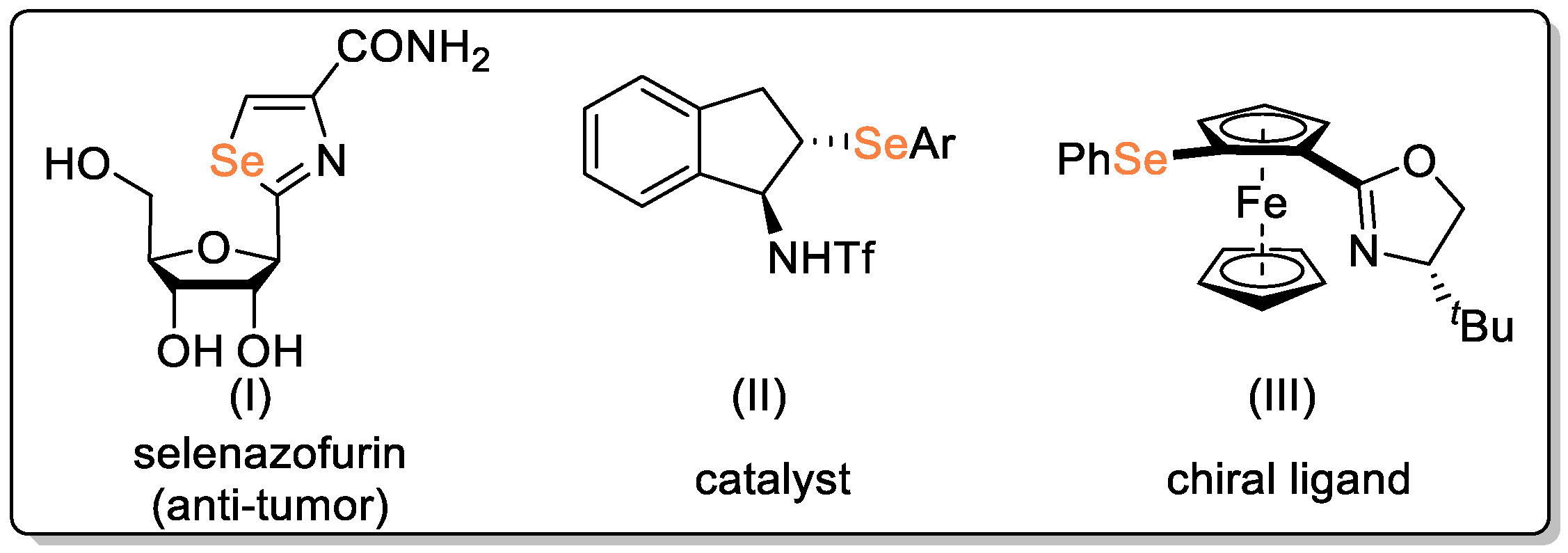

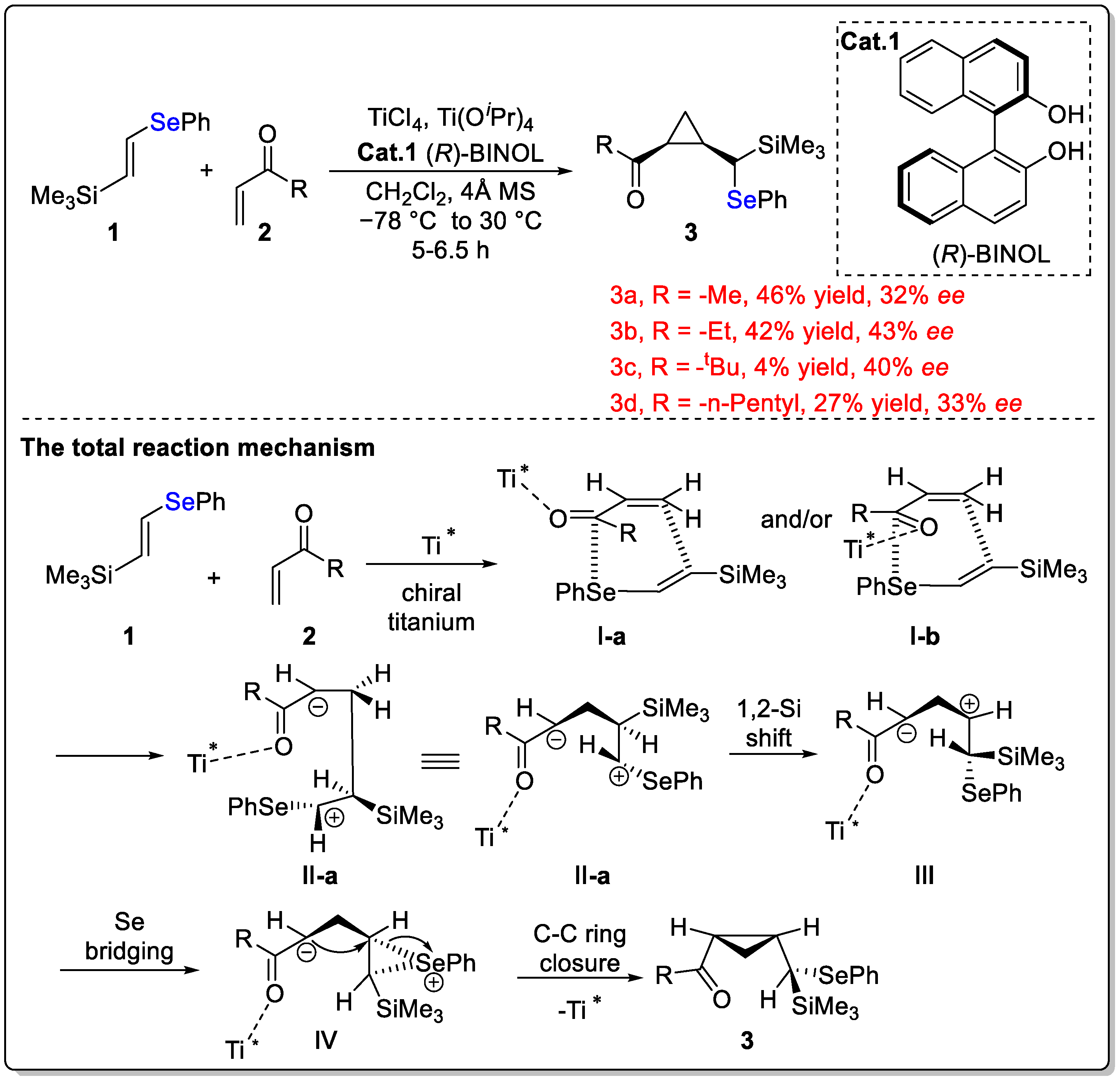
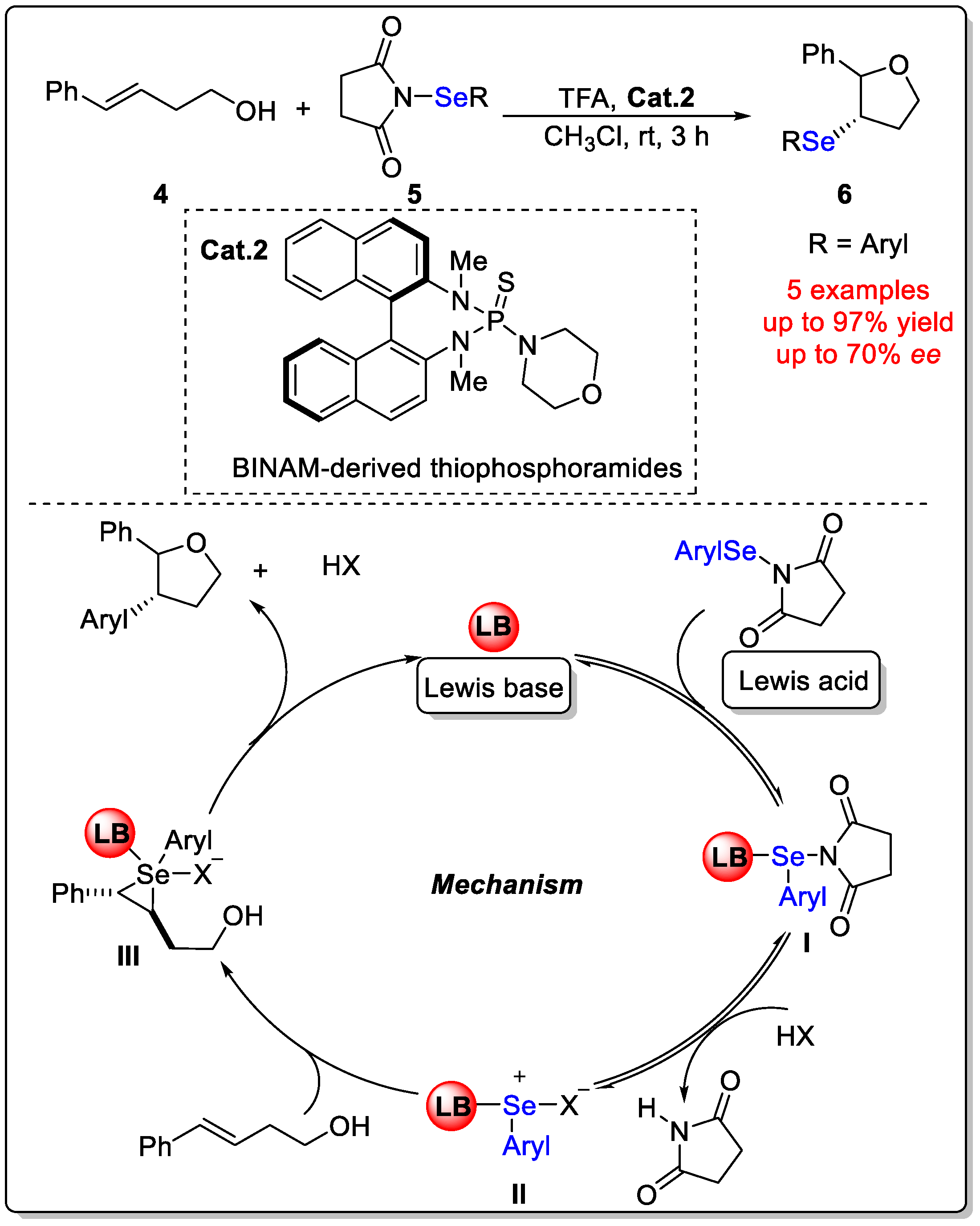
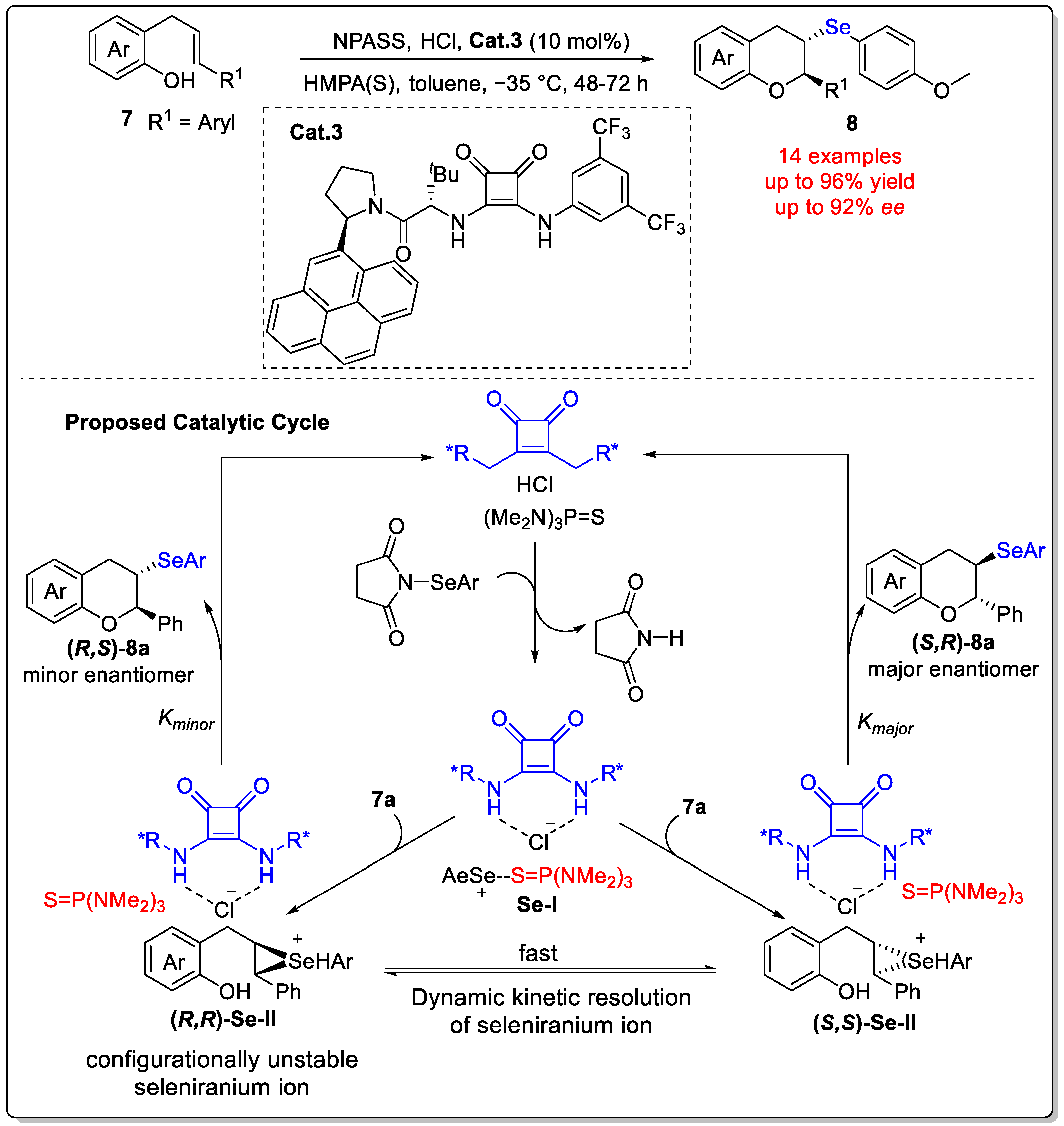

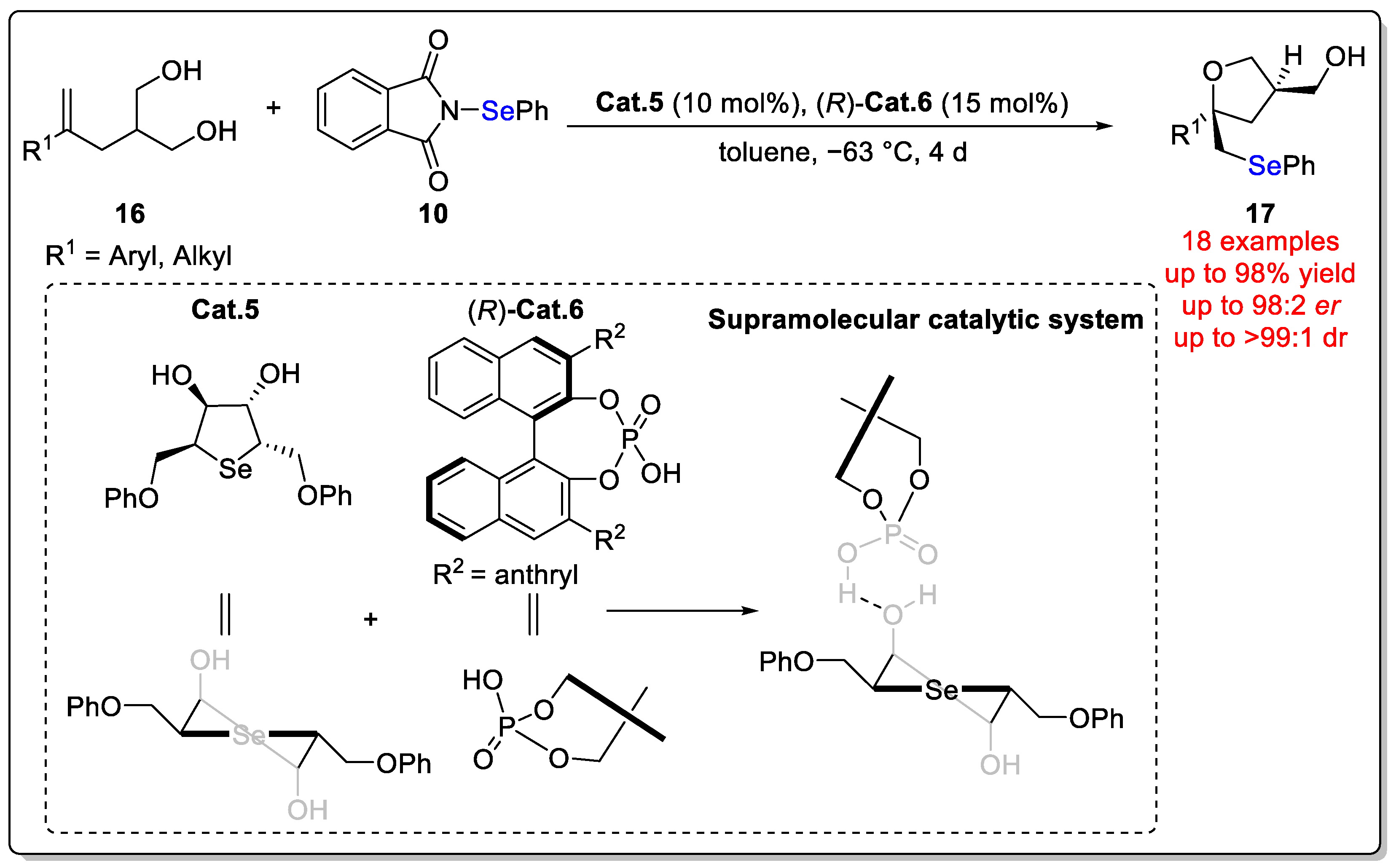
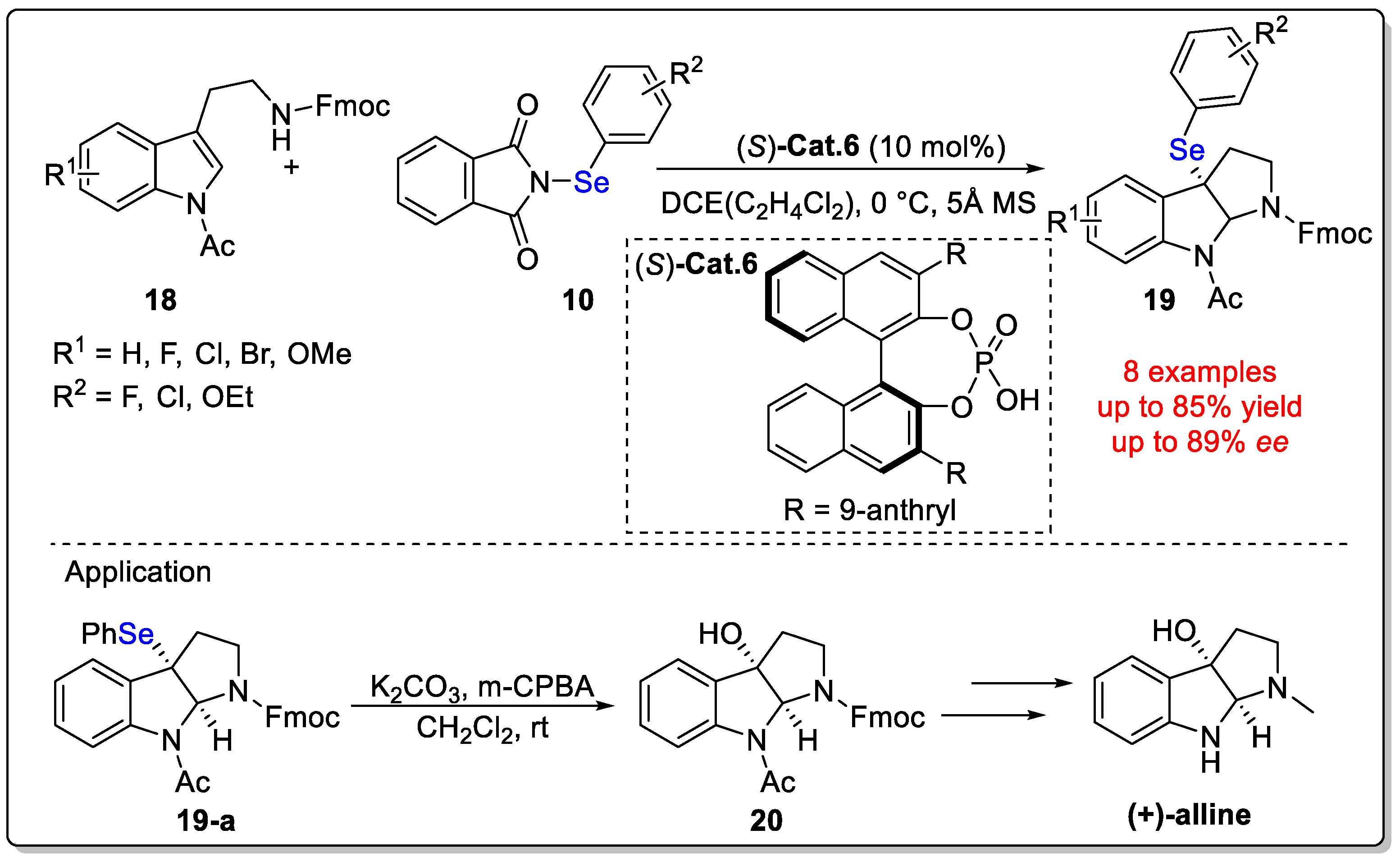
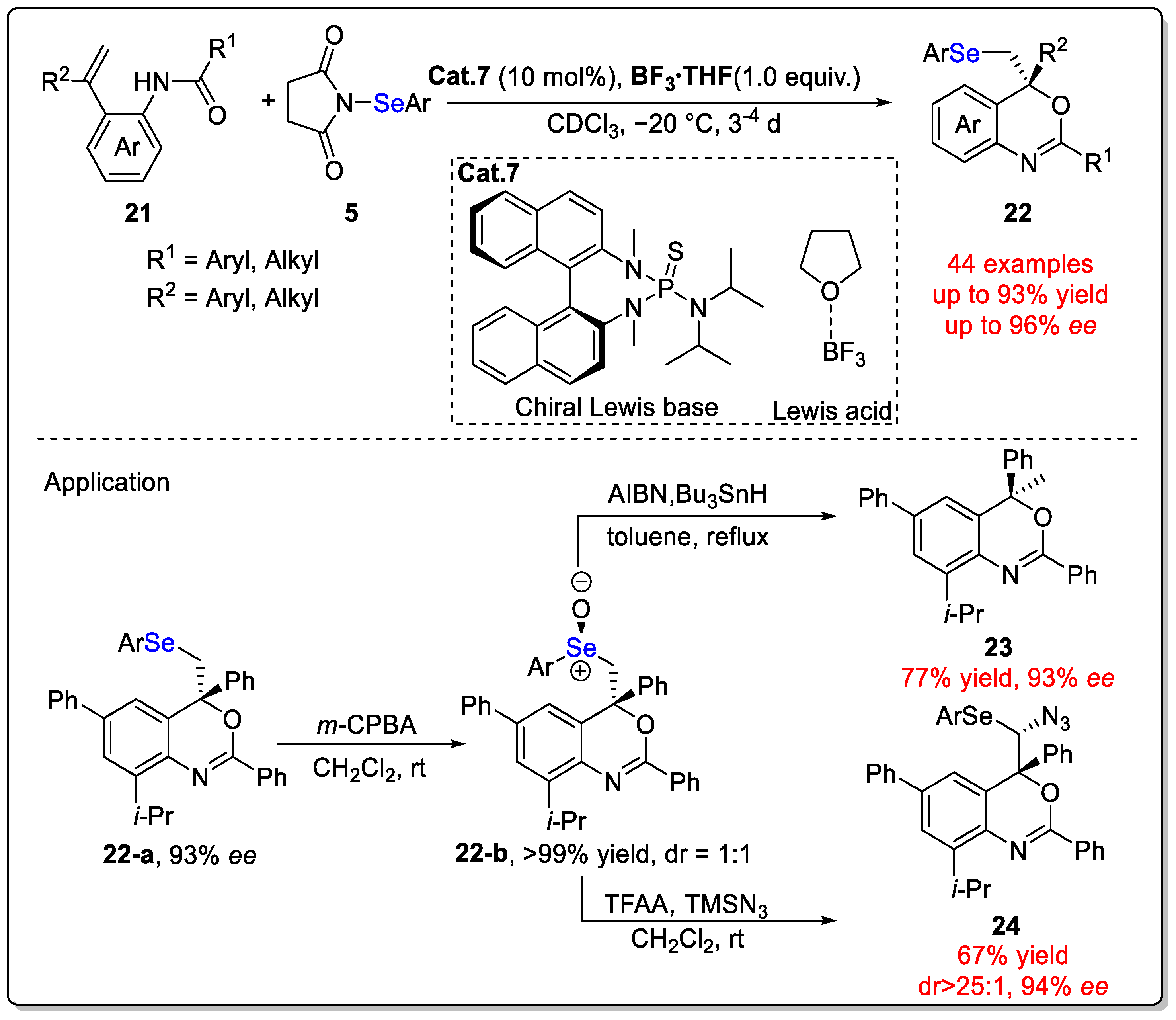


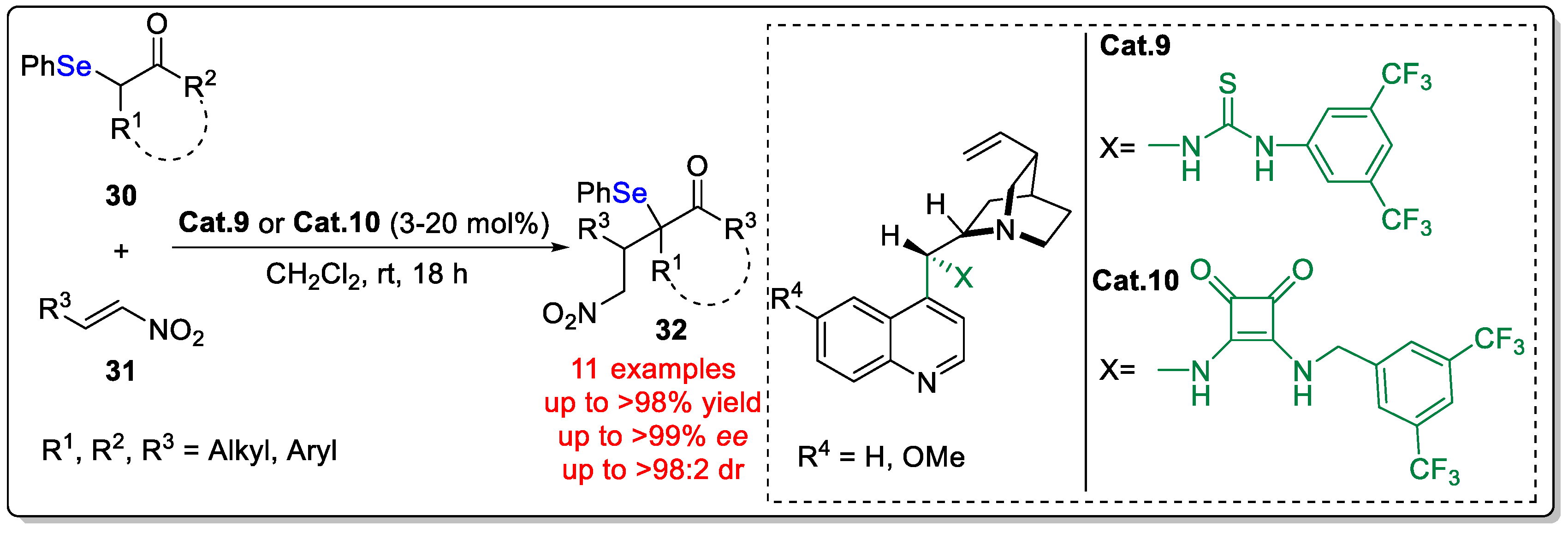
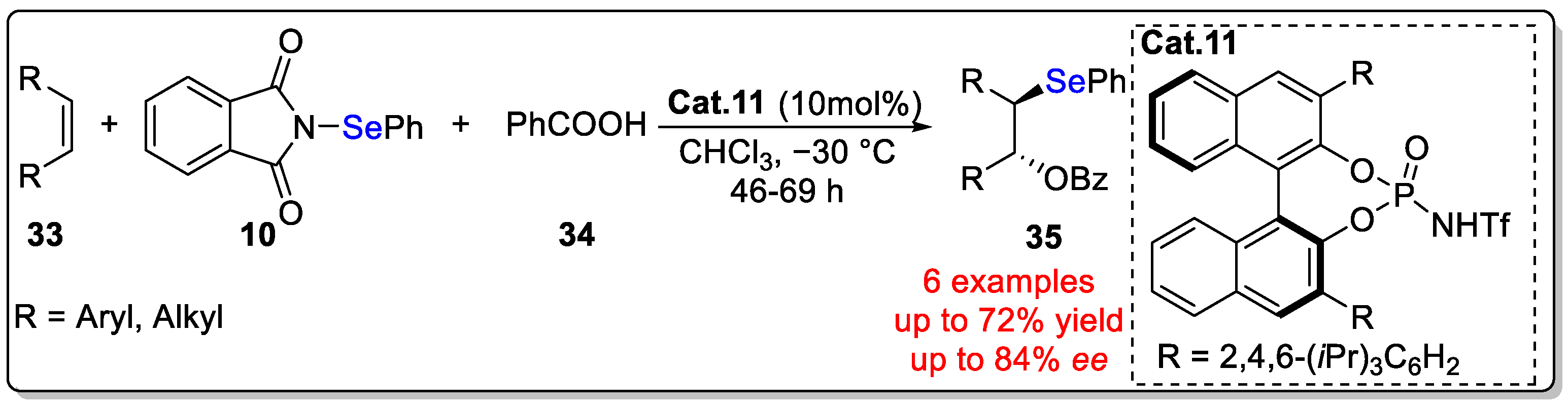
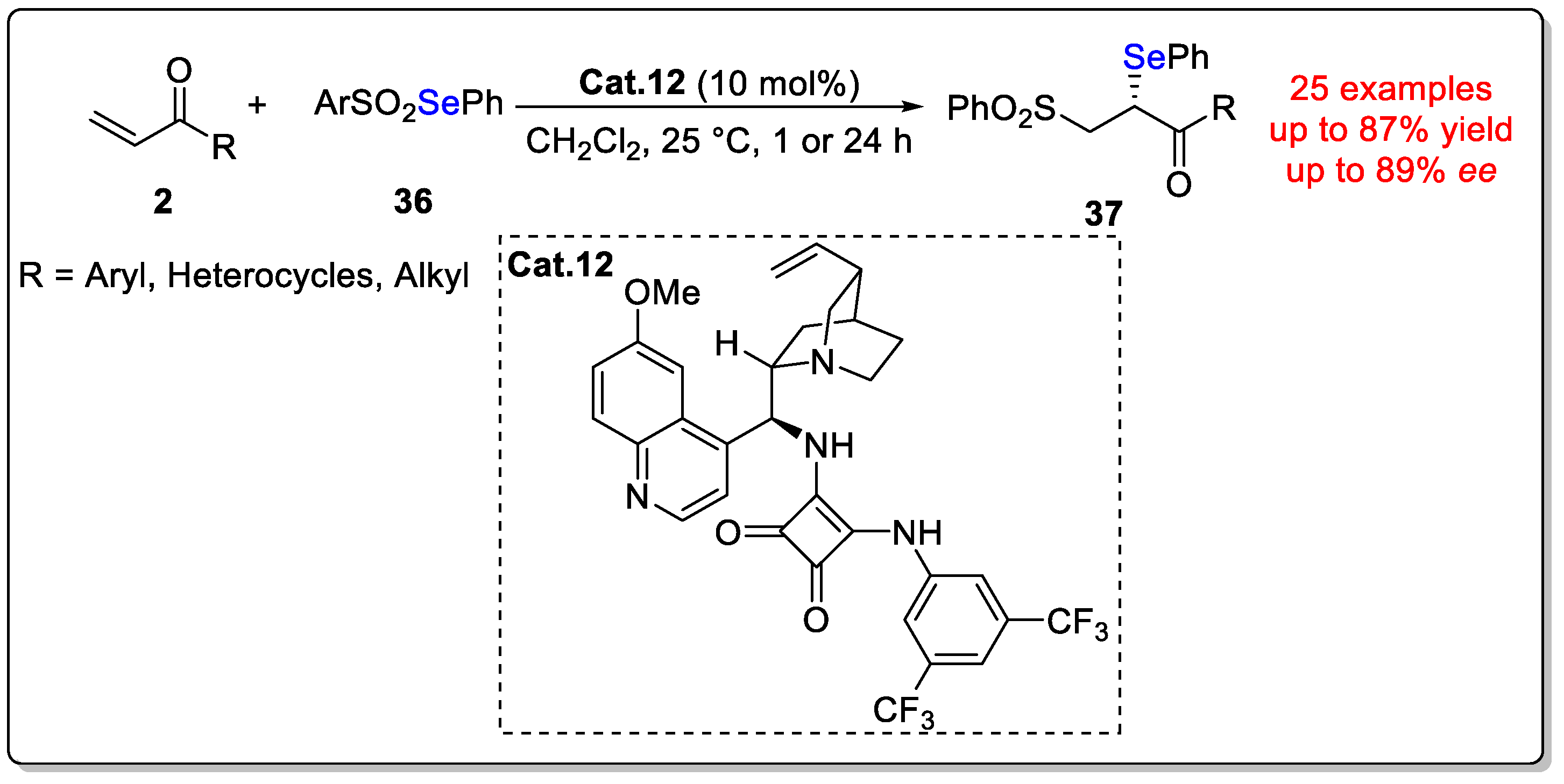
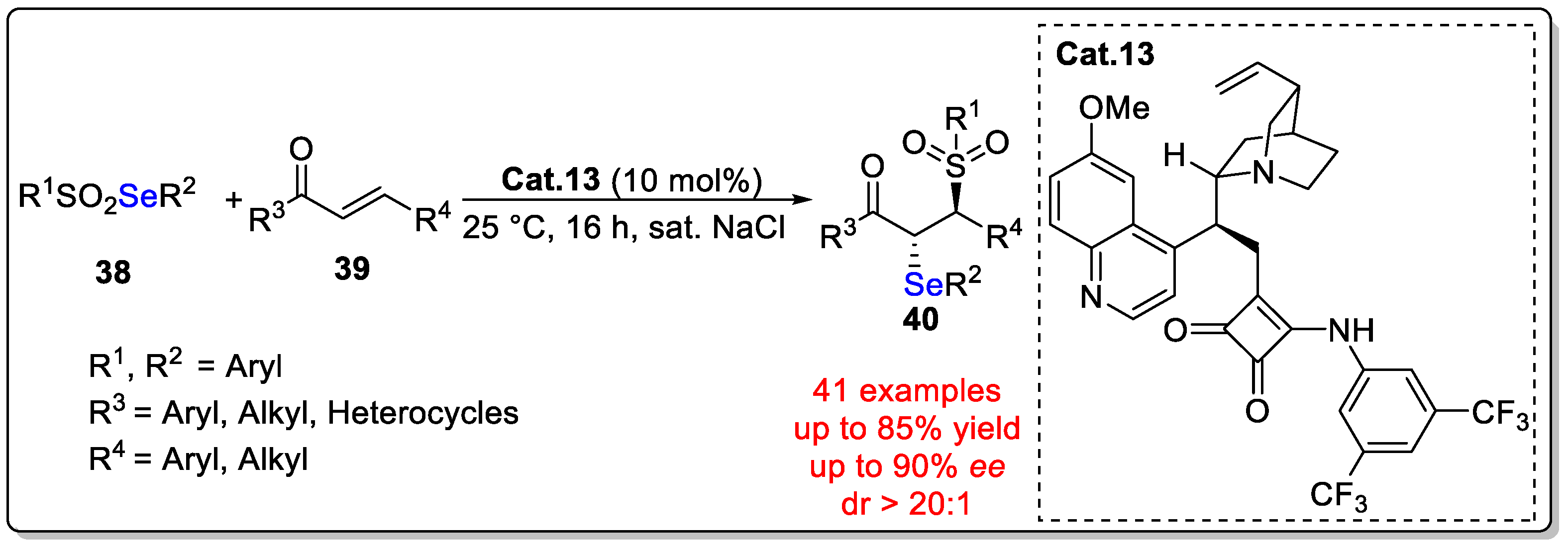


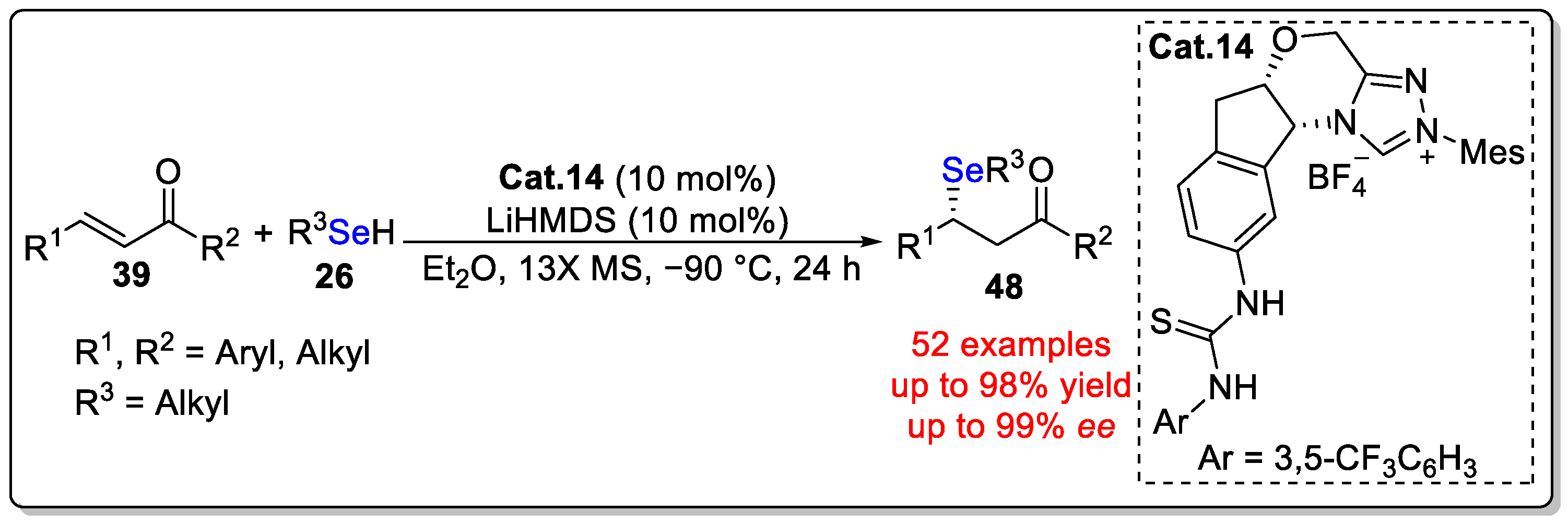


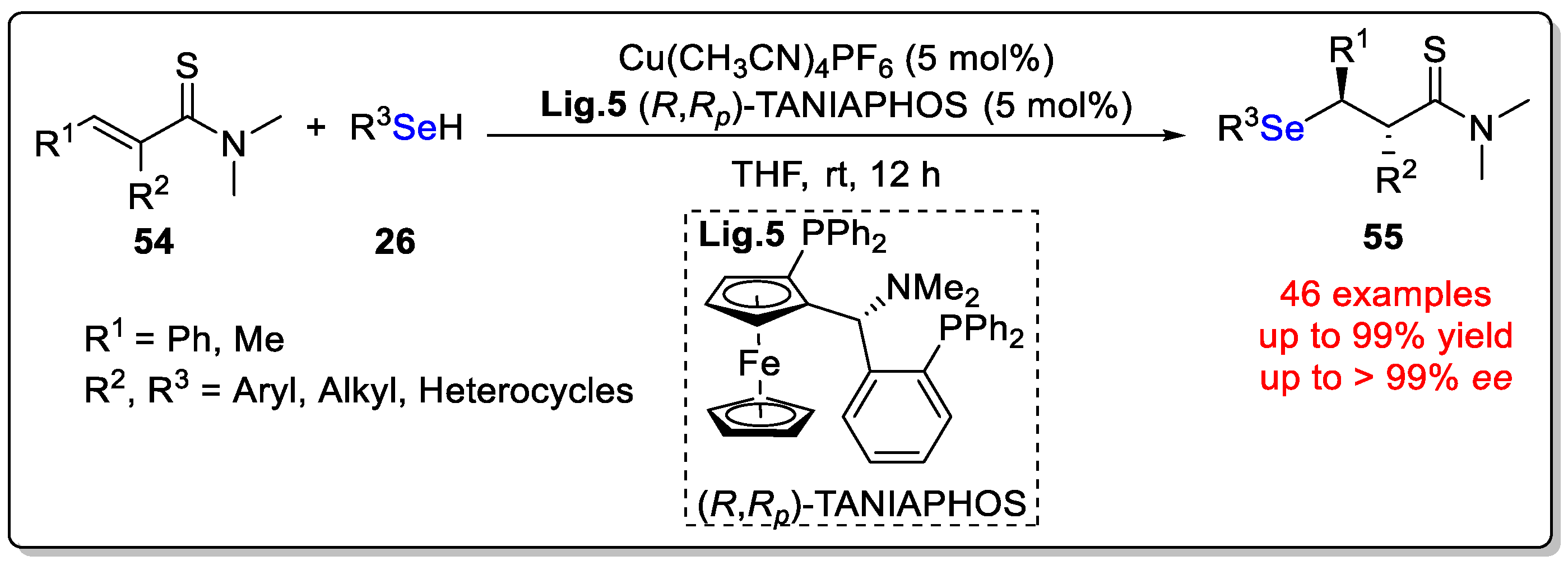

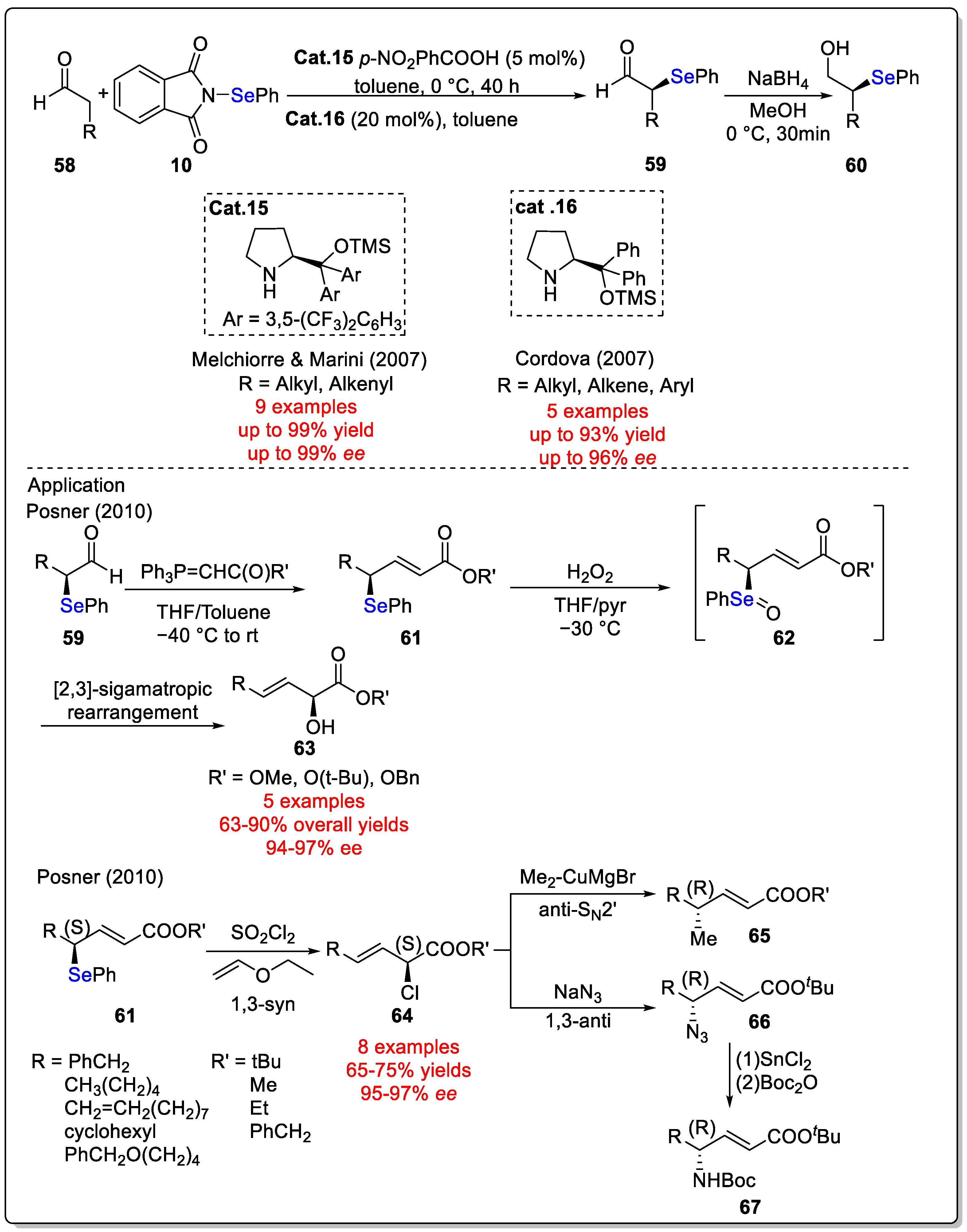
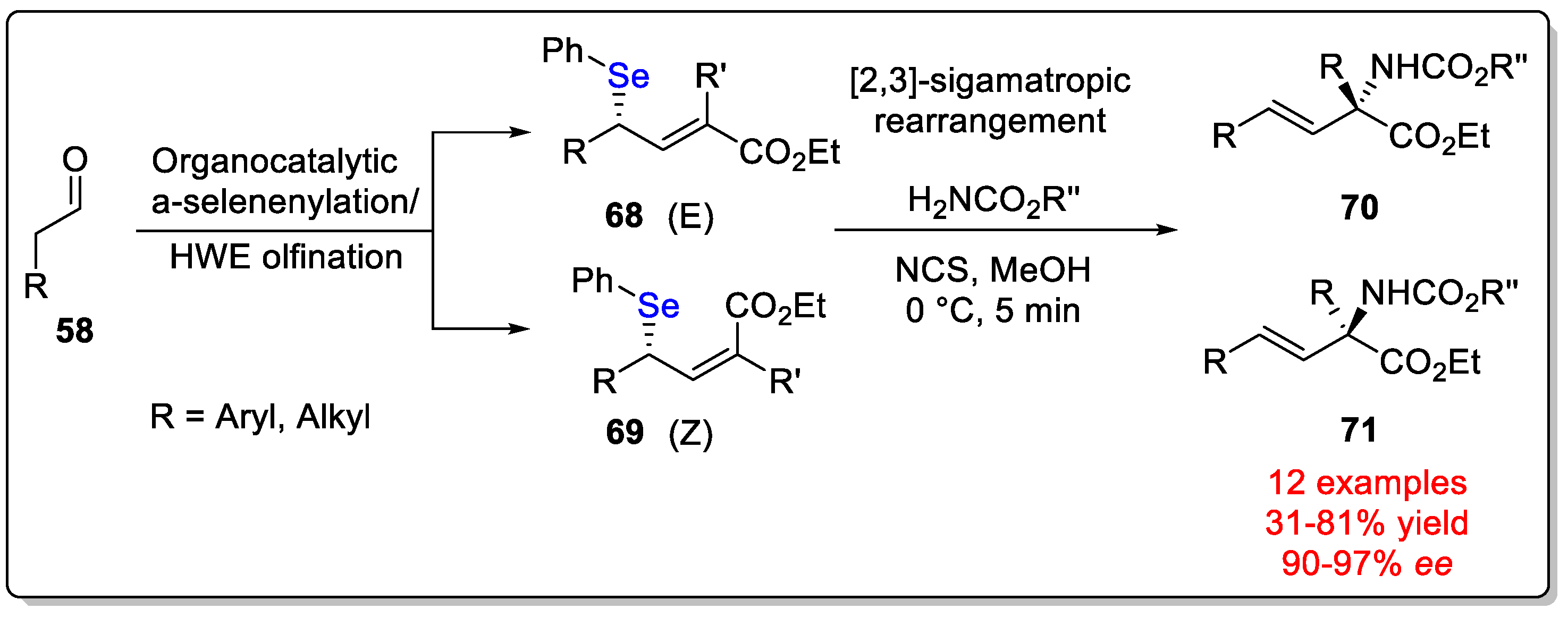

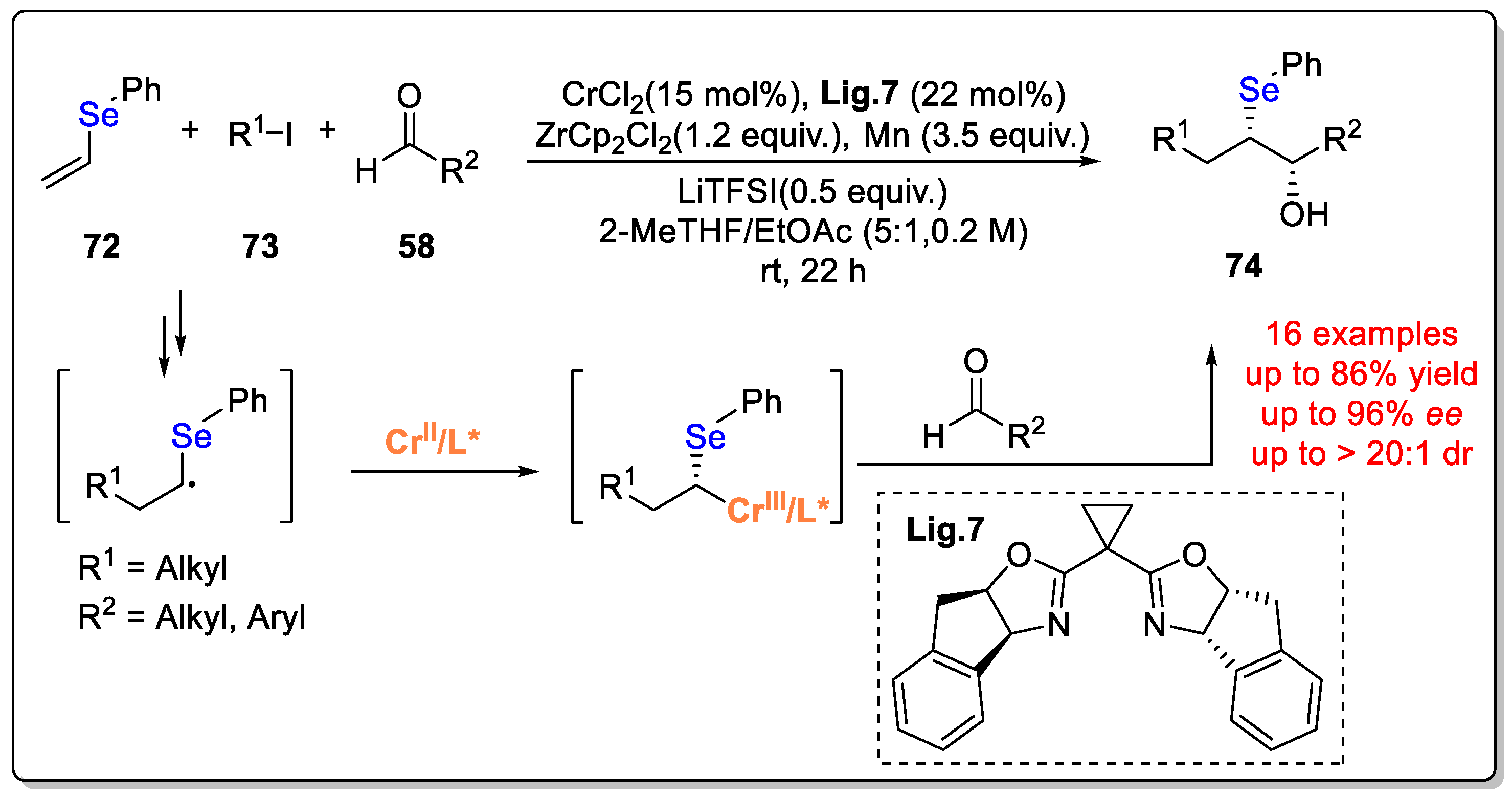
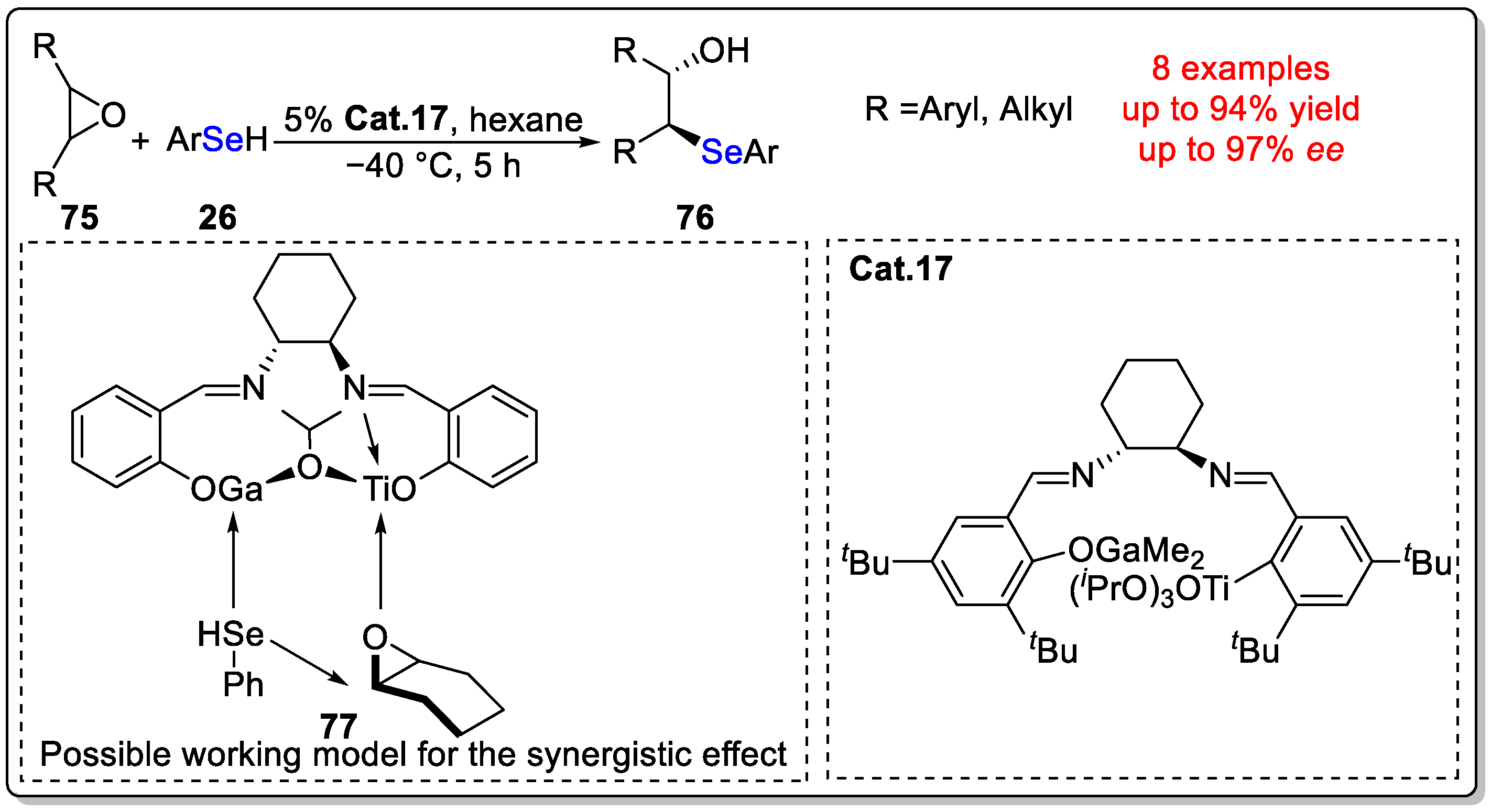
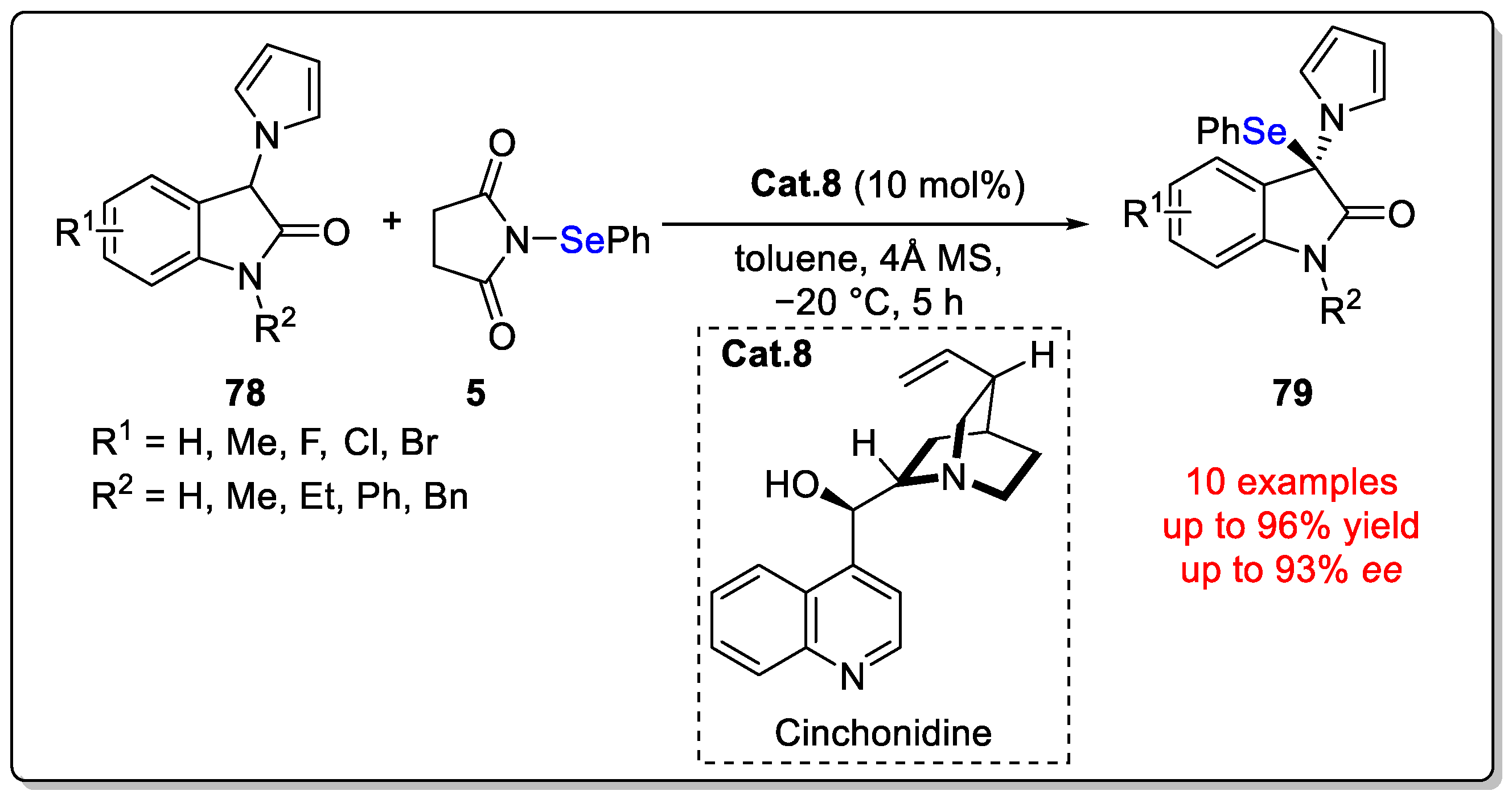
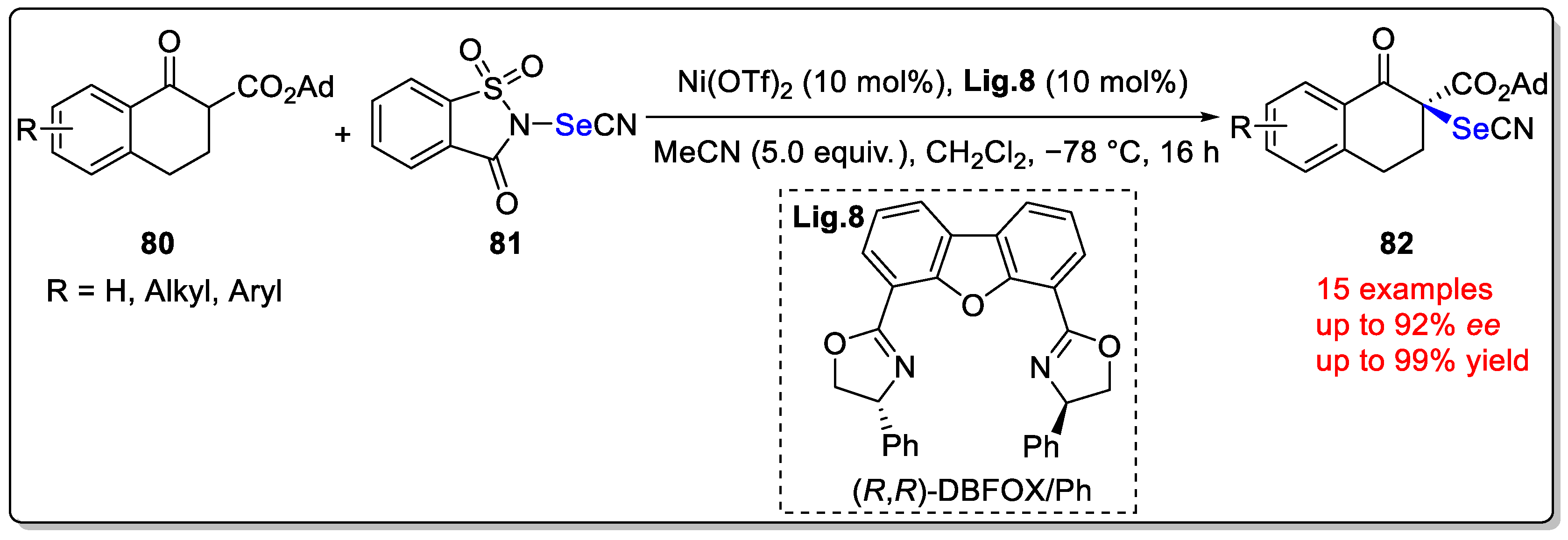
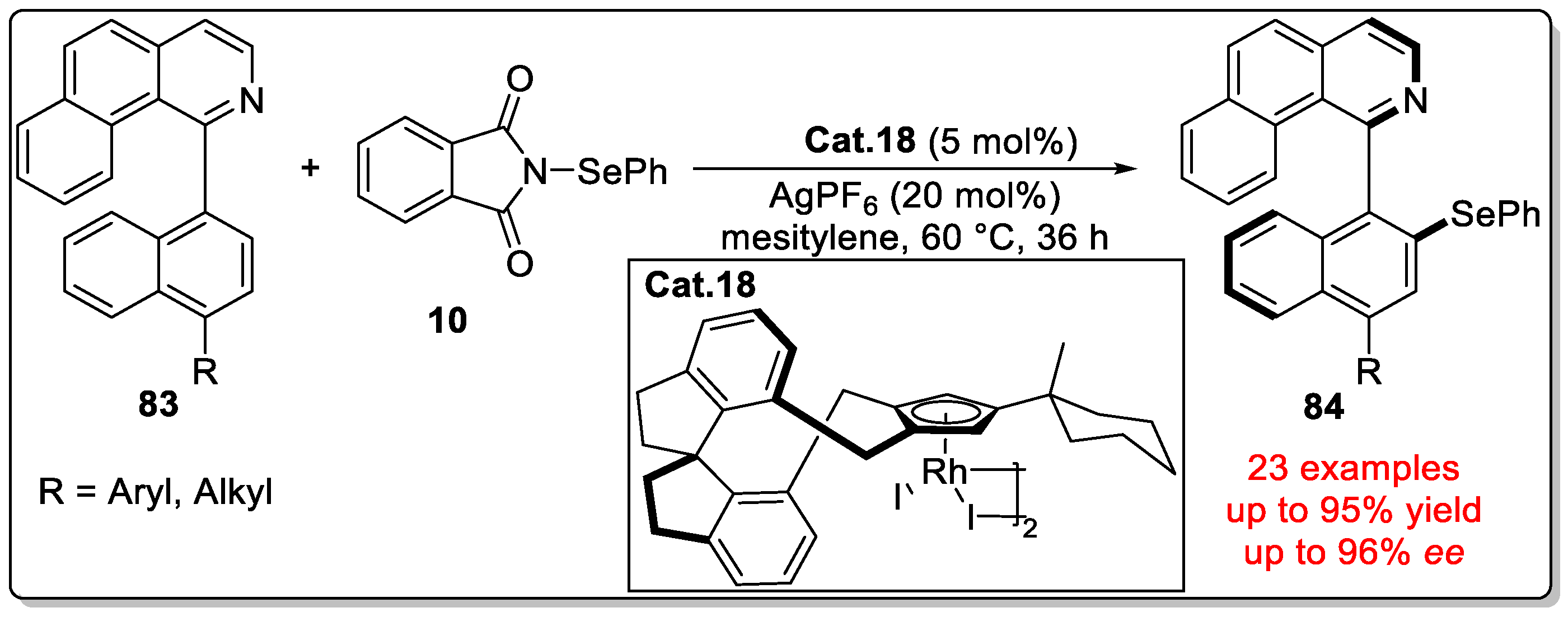

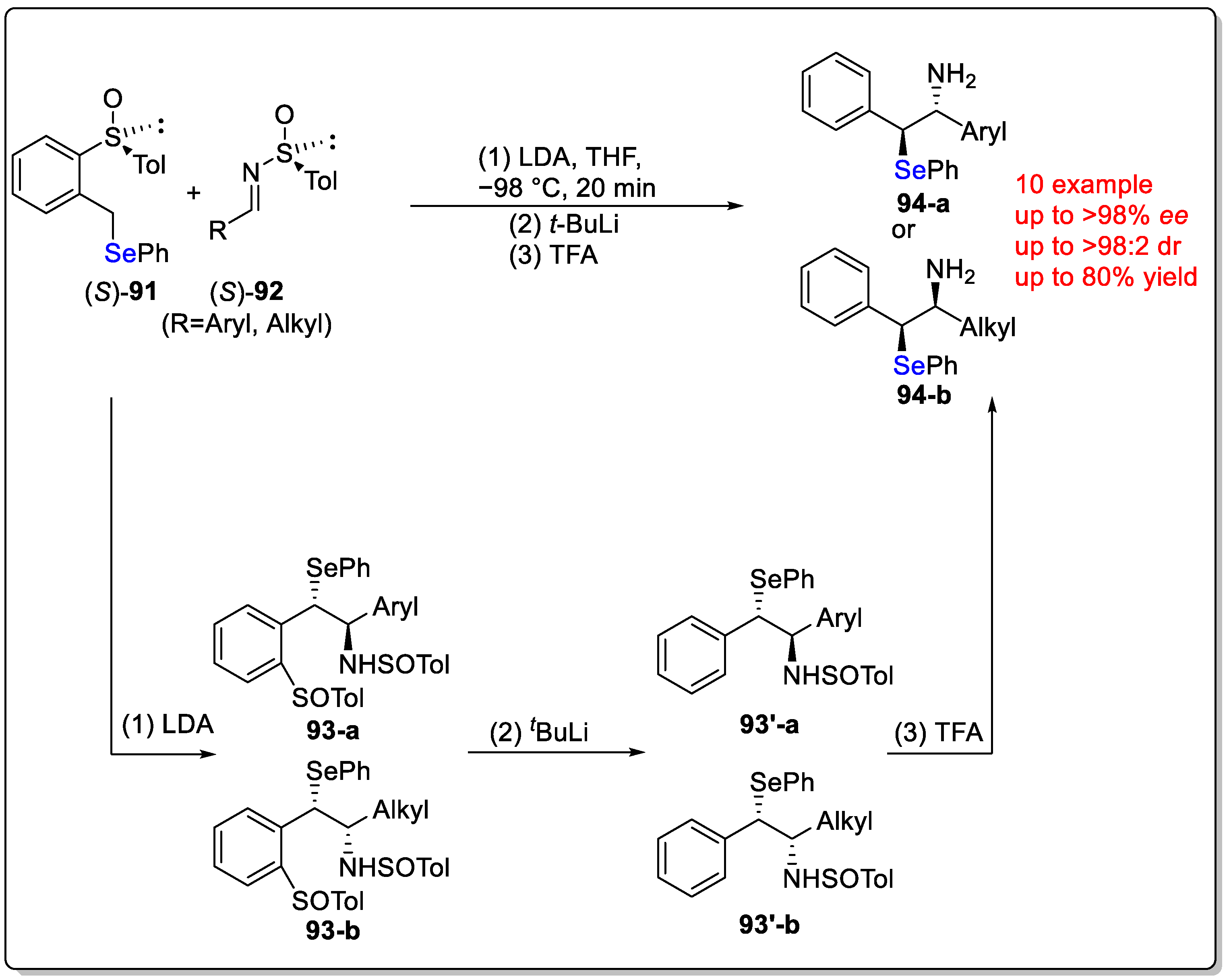

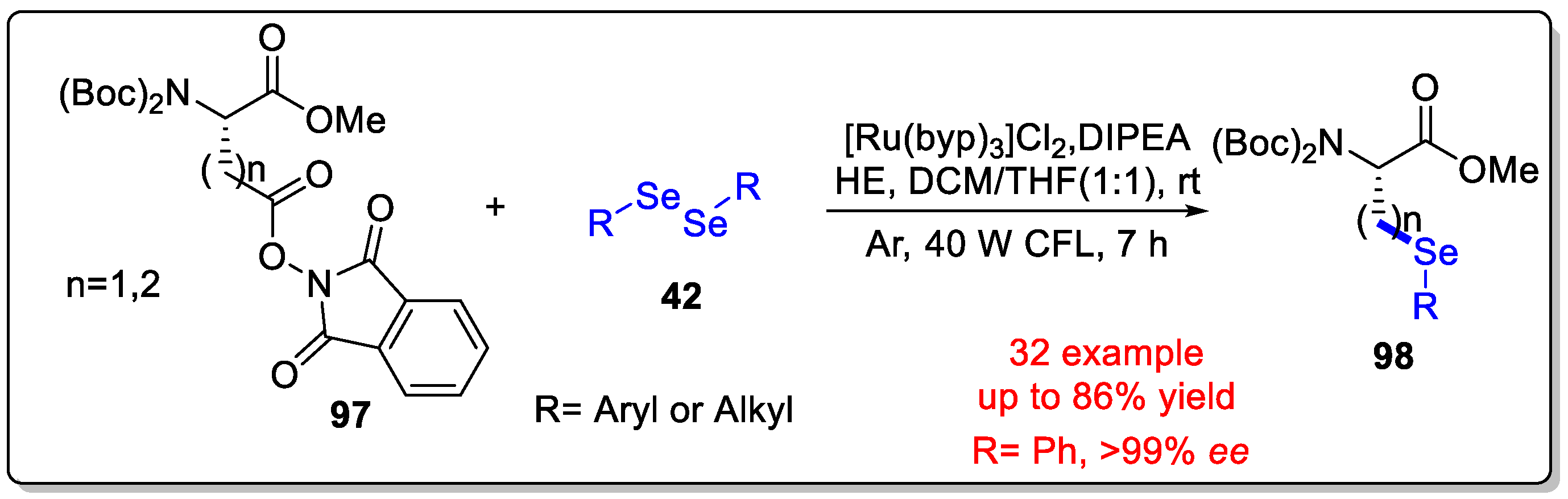

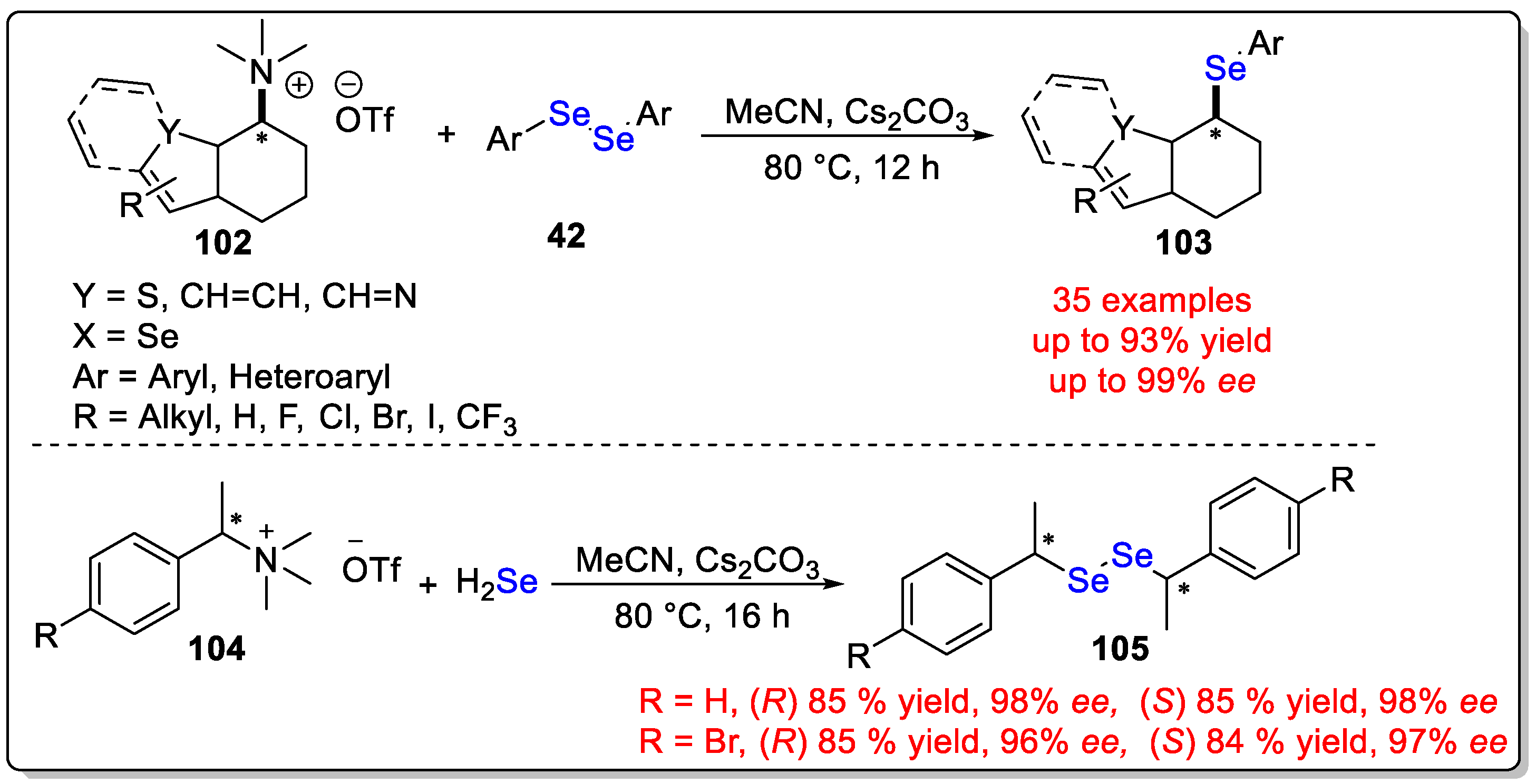
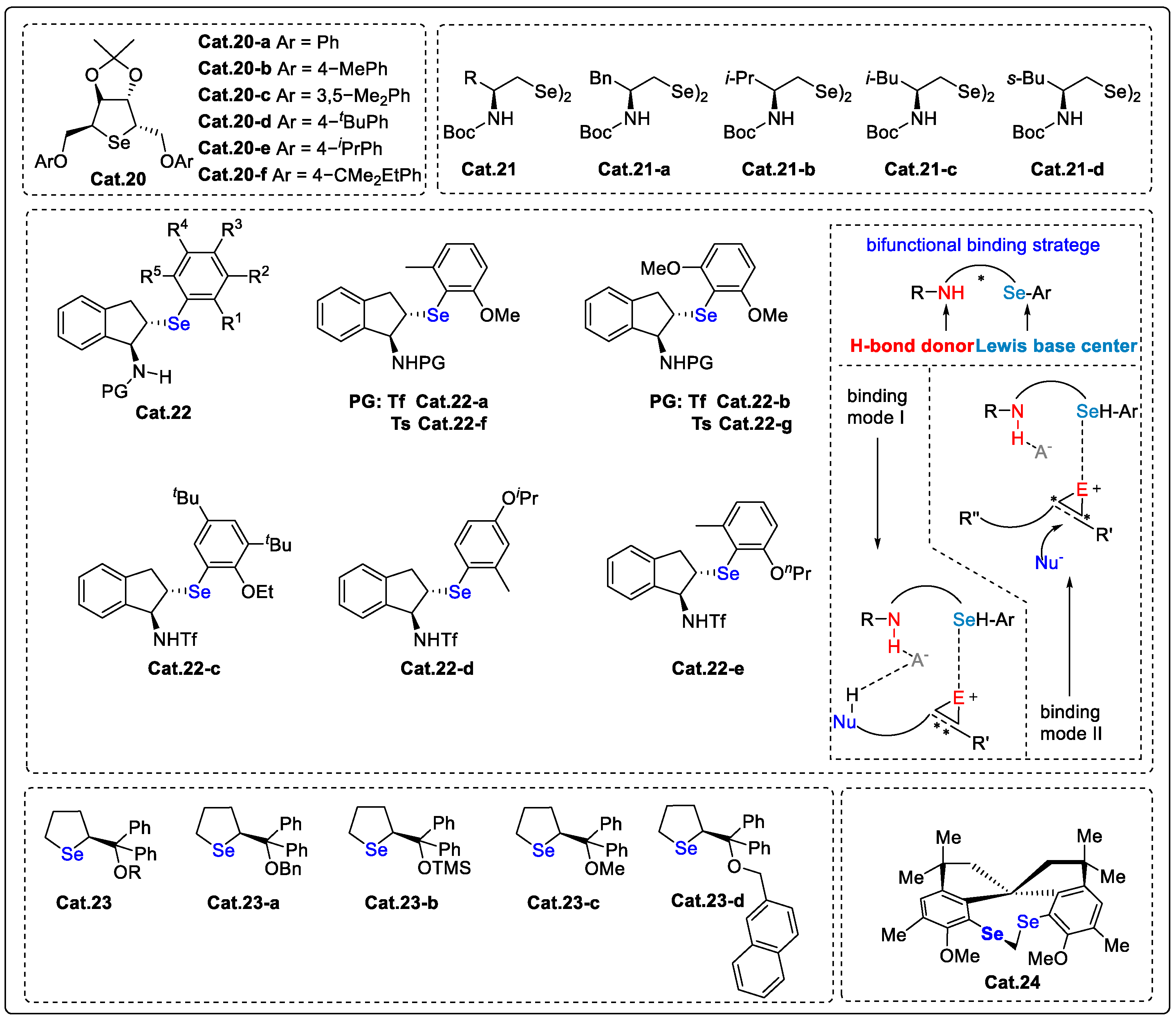
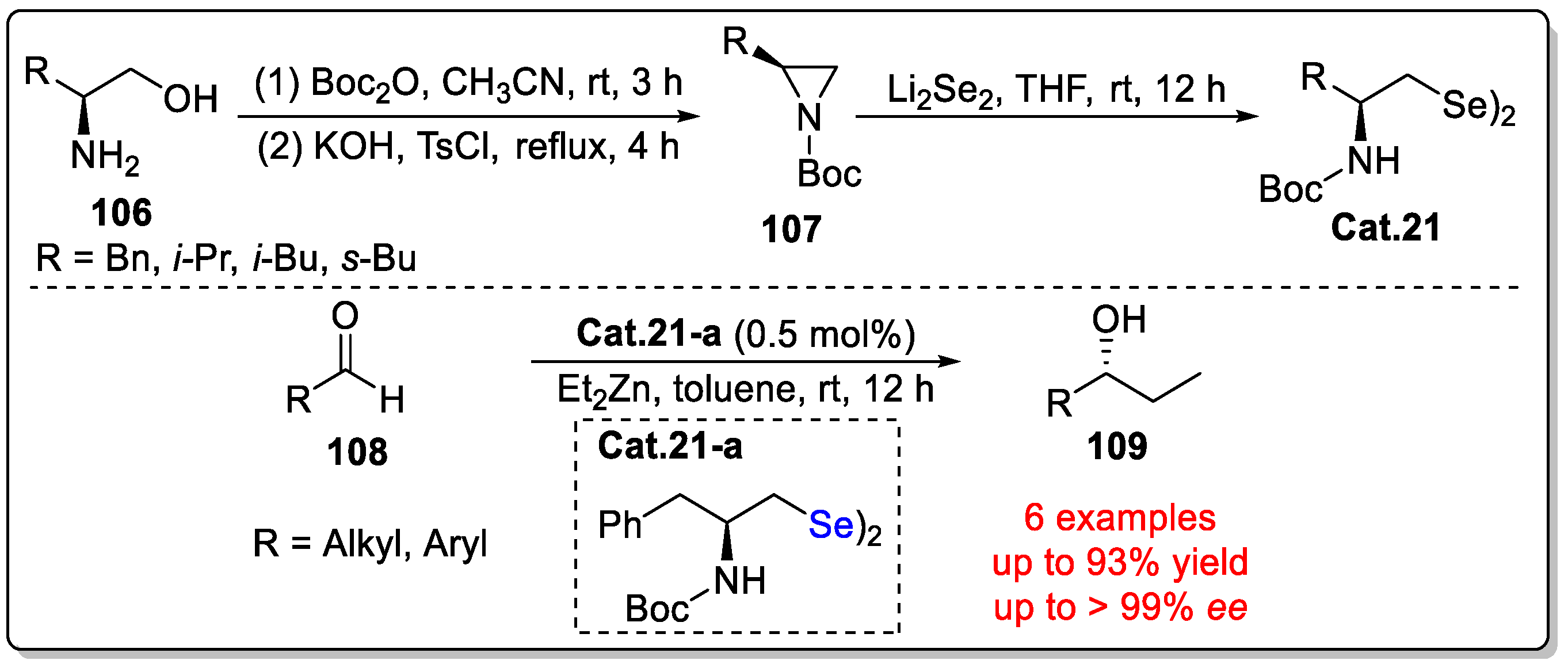

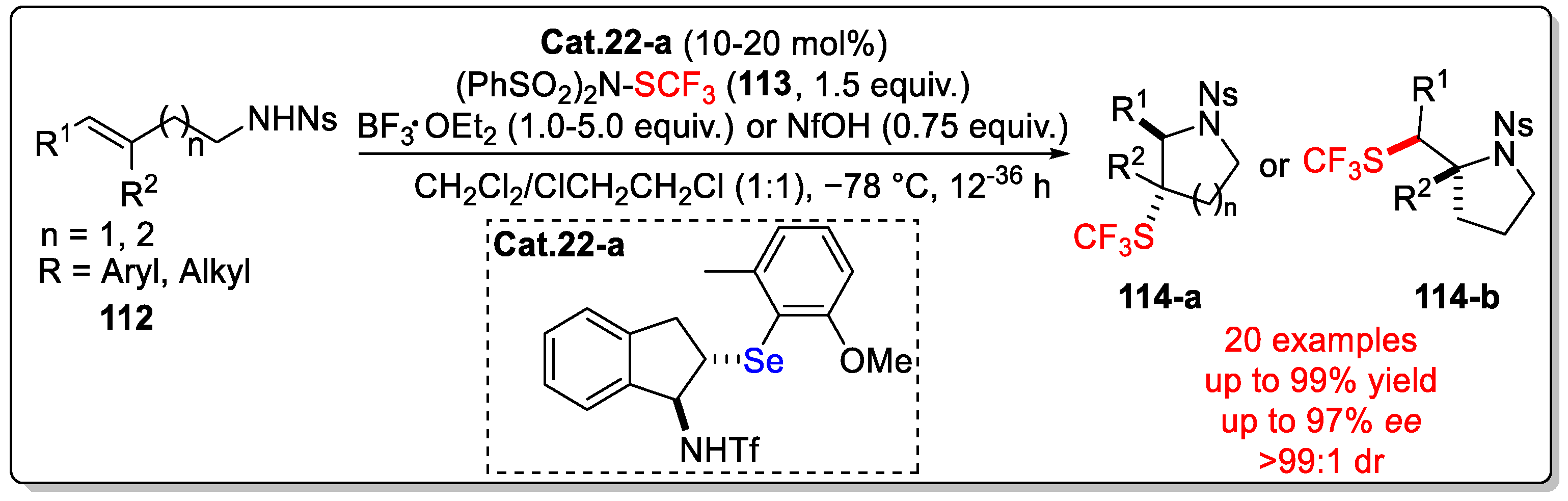

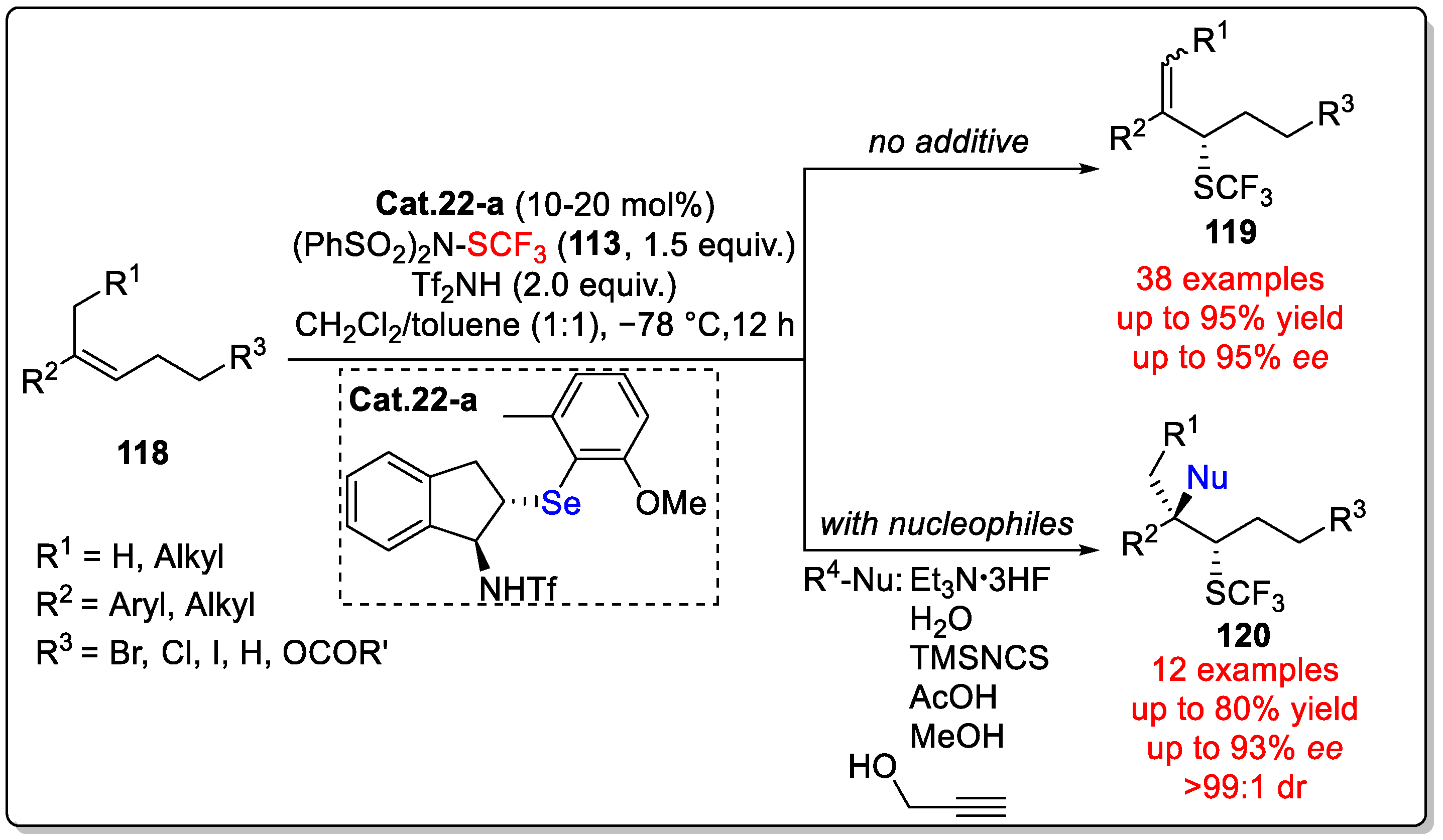
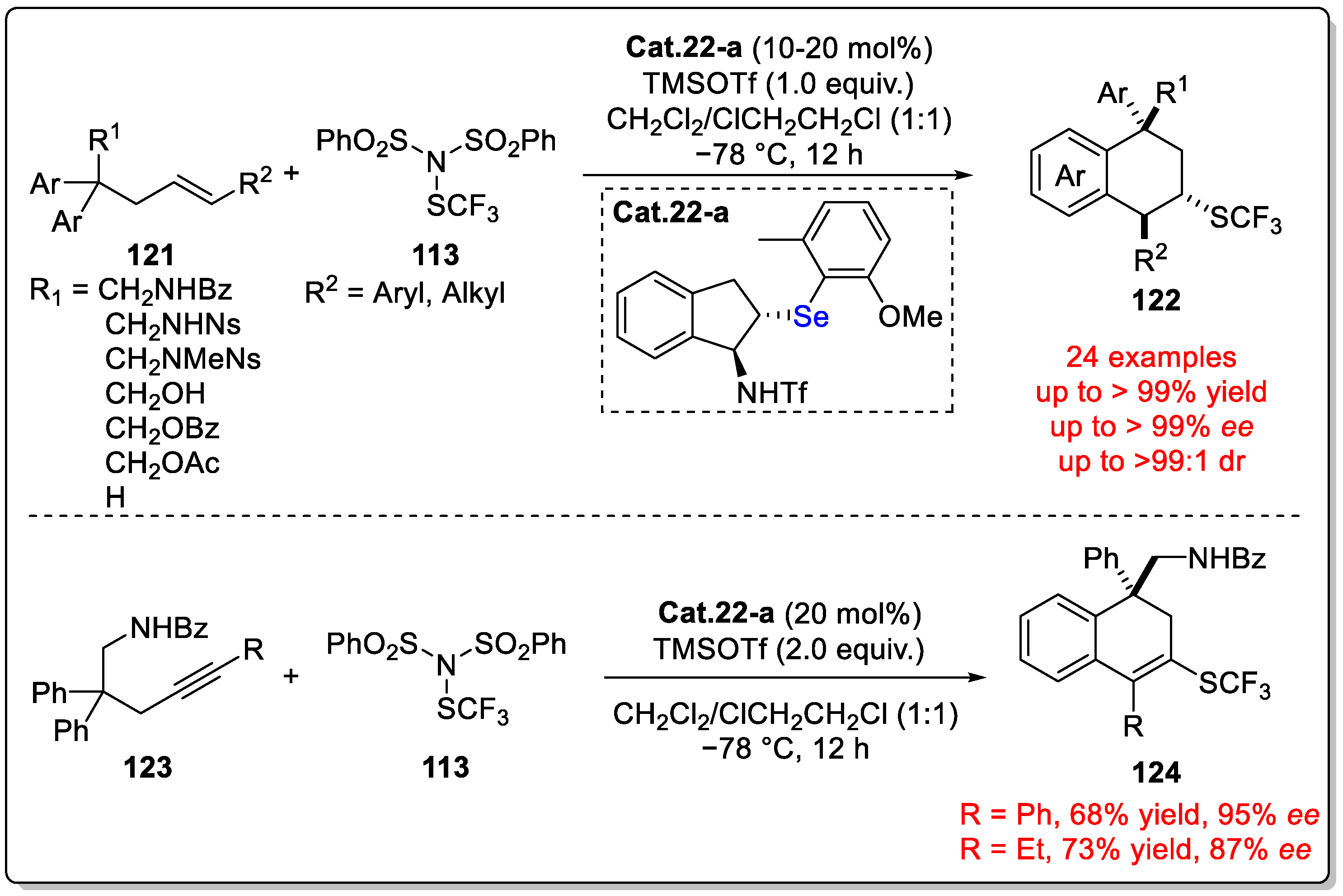

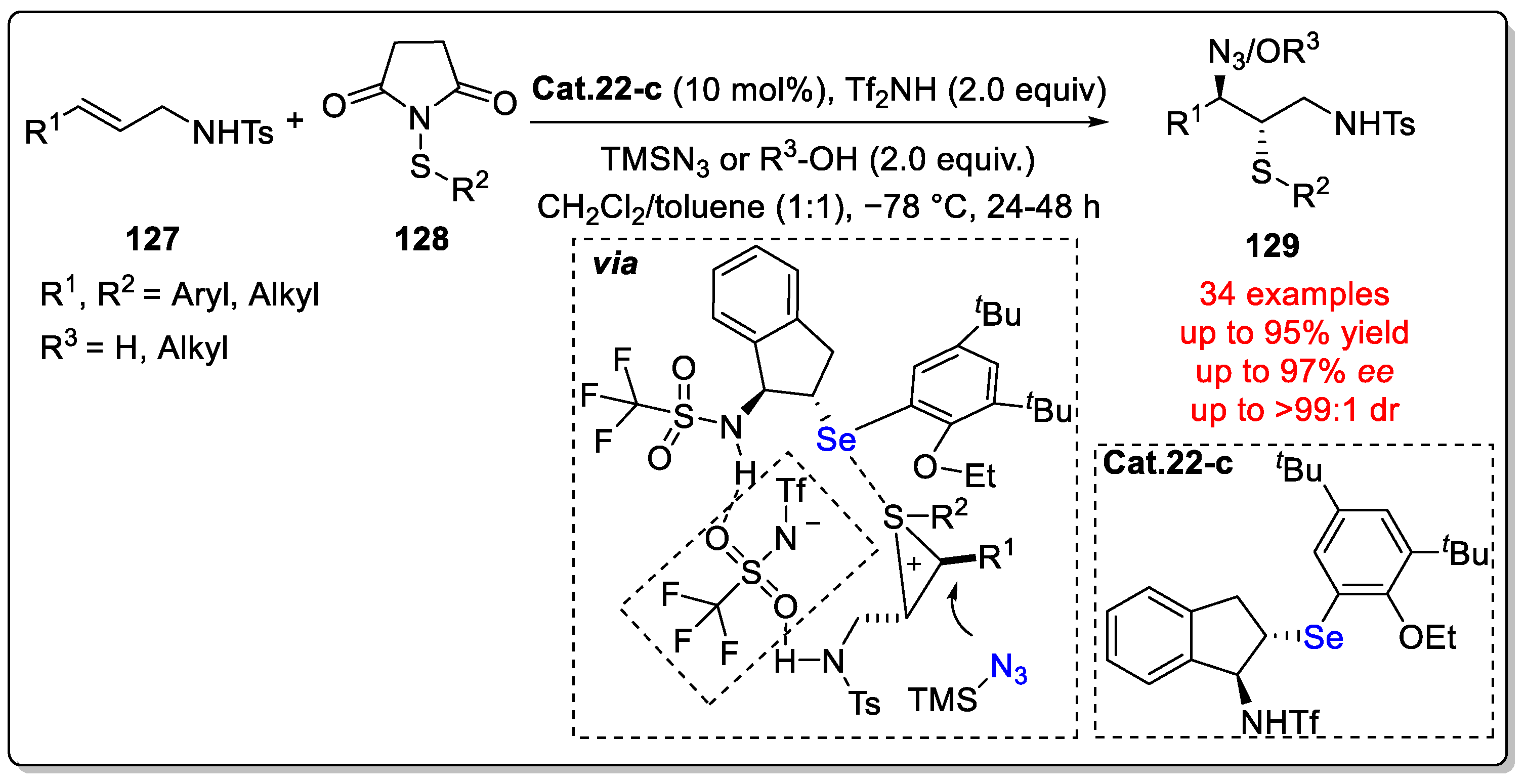
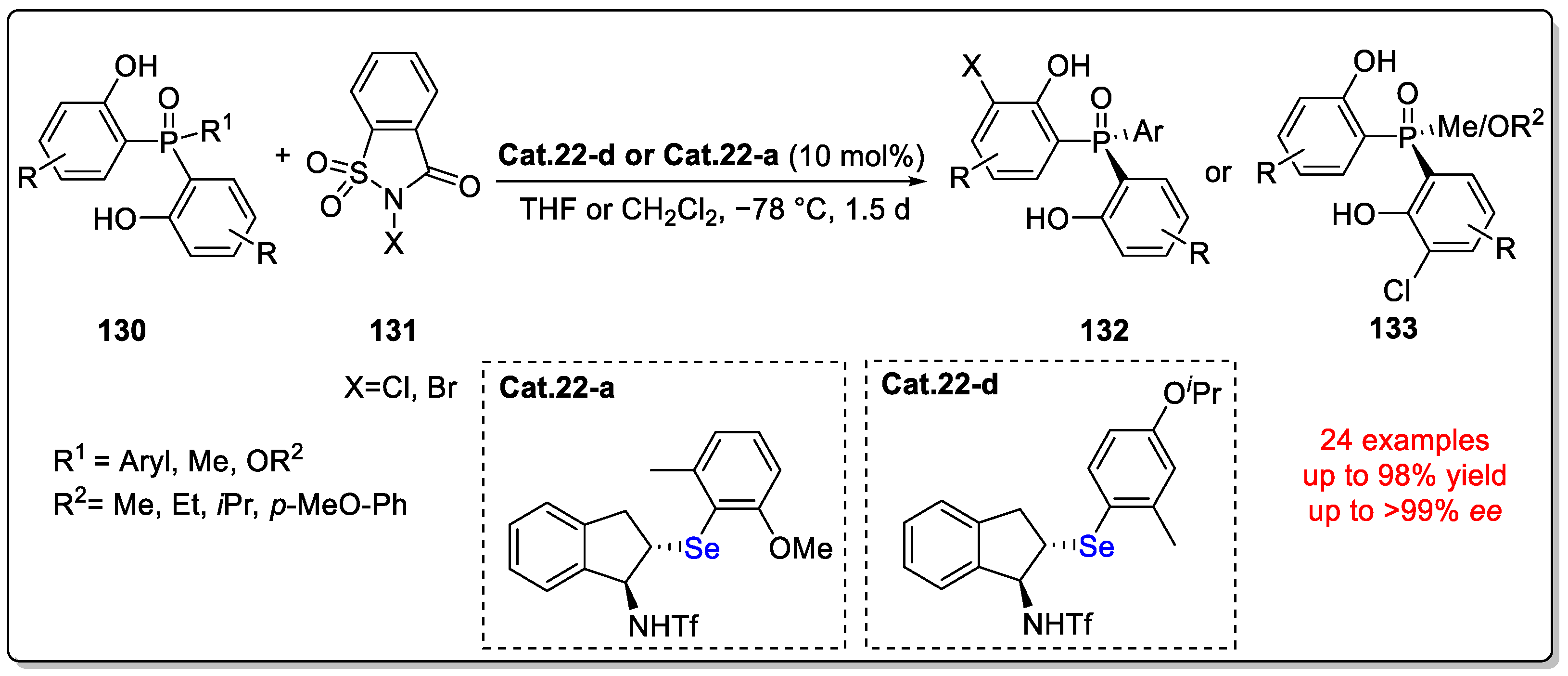
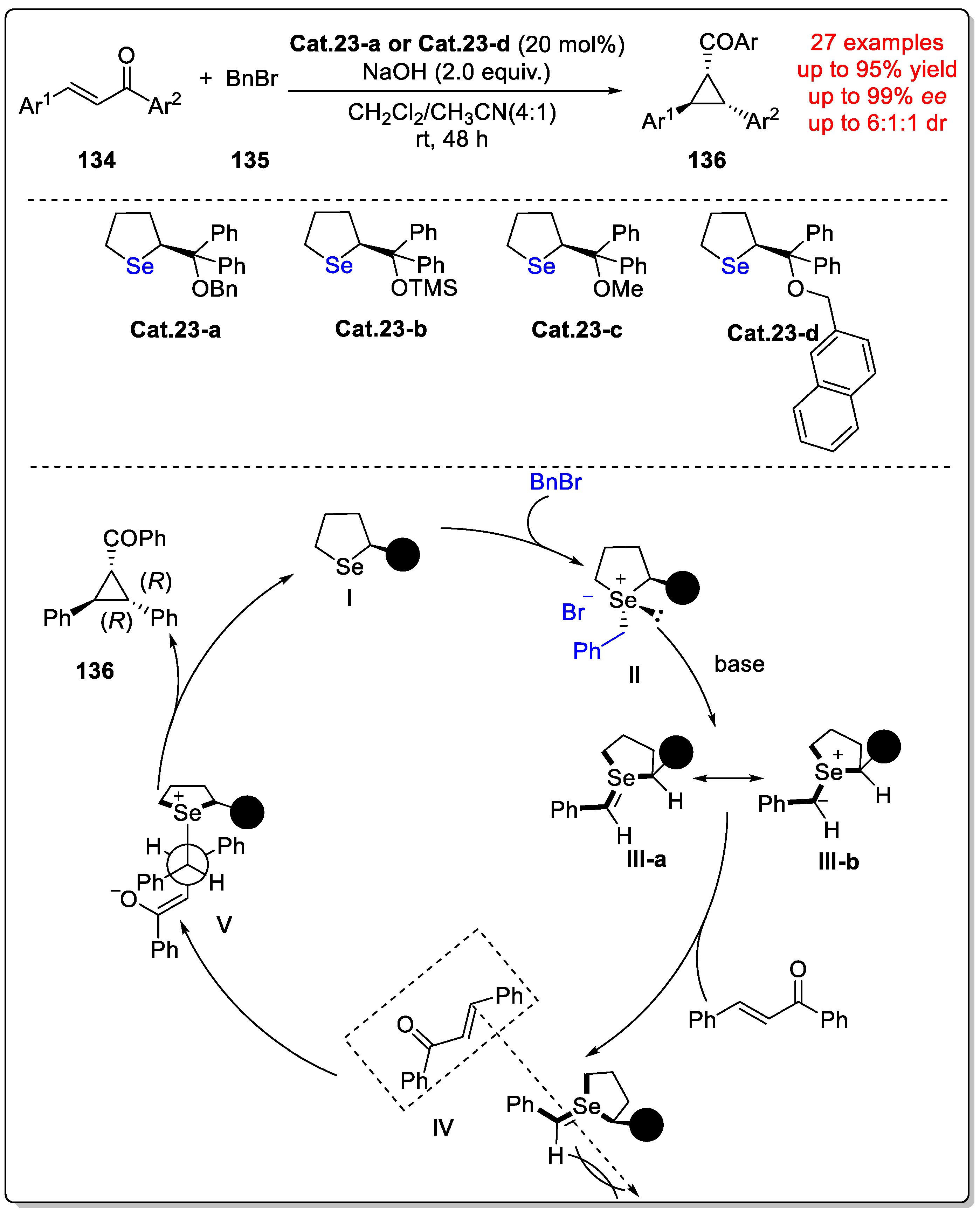


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jian, Y.; Singh, T.; Andersson, P.G.; Zhou, T. Asymmetric Synthesis and Applications of Chiral Organoselenium Compounds: A Review. Molecules 2024, 29, 3685. https://doi.org/10.3390/molecules29153685
Jian Y, Singh T, Andersson PG, Zhou T. Asymmetric Synthesis and Applications of Chiral Organoselenium Compounds: A Review. Molecules. 2024; 29(15):3685. https://doi.org/10.3390/molecules29153685
Chicago/Turabian StyleJian, Yanyu, Thishana Singh, Pher G. Andersson, and Taigang Zhou. 2024. "Asymmetric Synthesis and Applications of Chiral Organoselenium Compounds: A Review" Molecules 29, no. 15: 3685. https://doi.org/10.3390/molecules29153685
APA StyleJian, Y., Singh, T., Andersson, P. G., & Zhou, T. (2024). Asymmetric Synthesis and Applications of Chiral Organoselenium Compounds: A Review. Molecules, 29(15), 3685. https://doi.org/10.3390/molecules29153685