

Effects of Surface Charge Distribution and Electrolyte Ions on the Nonlinear Spectra of Model Solid–Water Interfaces


Abstract

1. Introduction
2. Models and Computations
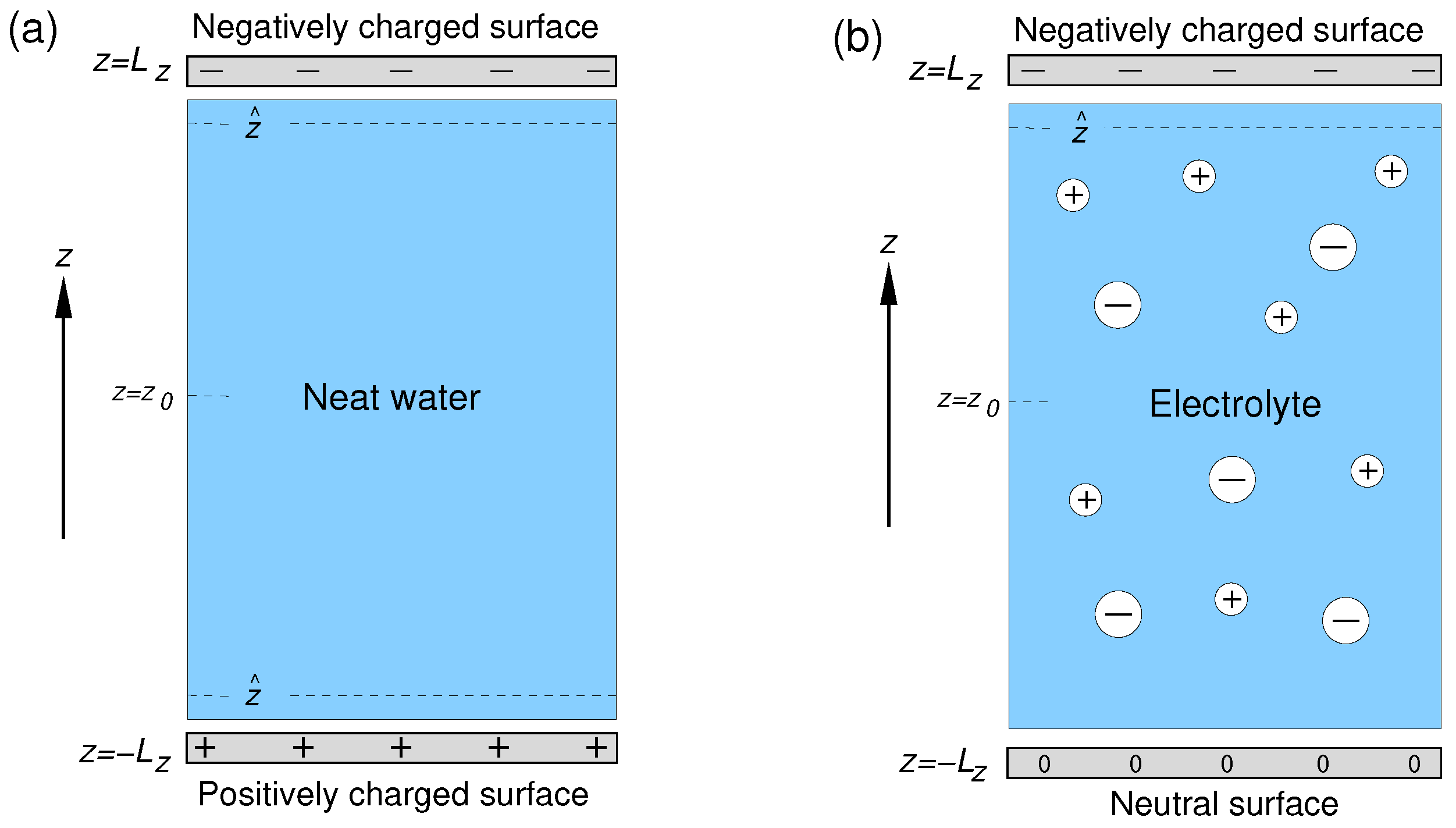
2.1. Simulation Setup
2.2. Interatomic Potentials
2.3. MD Simulations
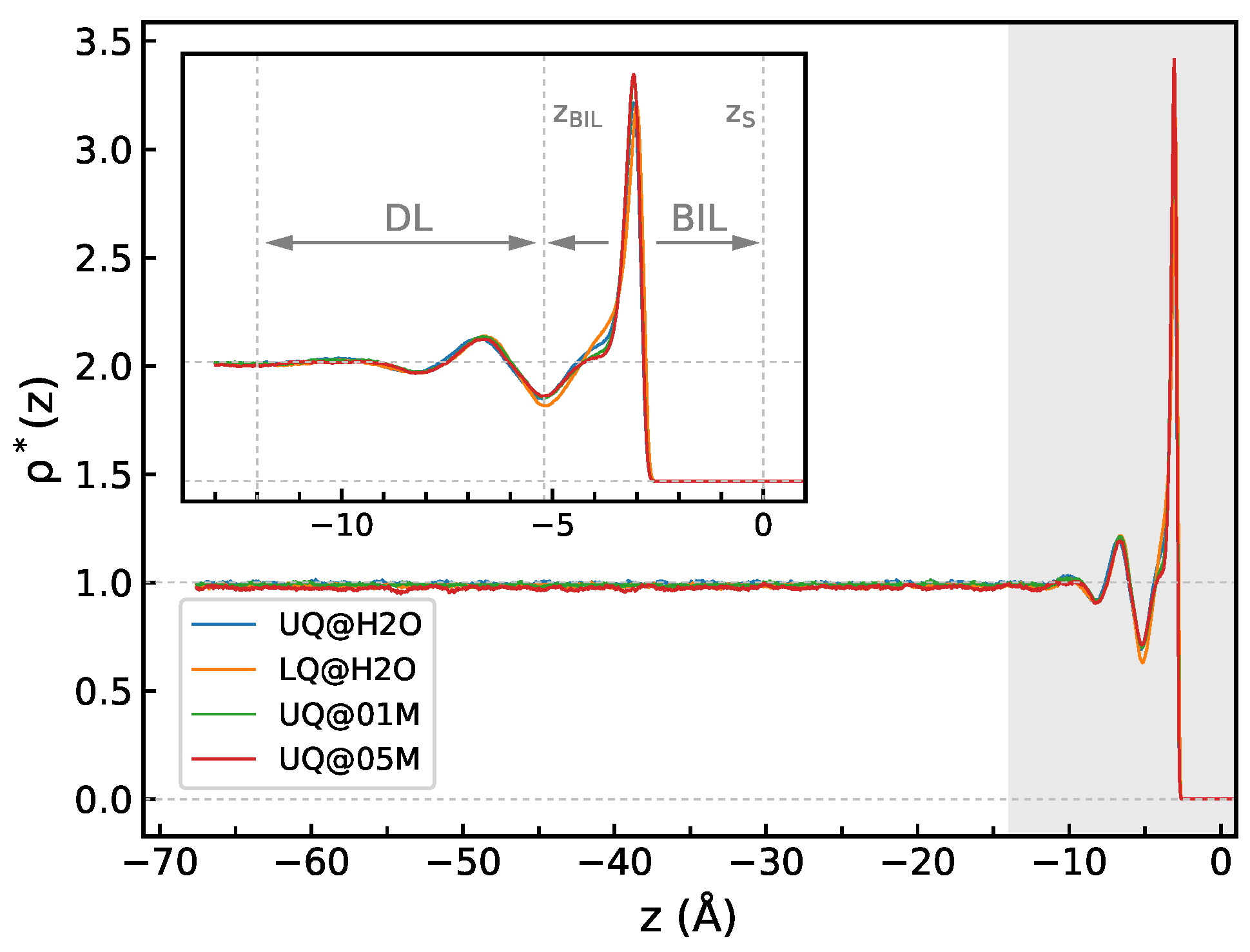
2.4. Structural Characteristics
2.5. Nonlinear Spectra Computation
2.6. Analysis of the Spectra
3. Results and Discussion
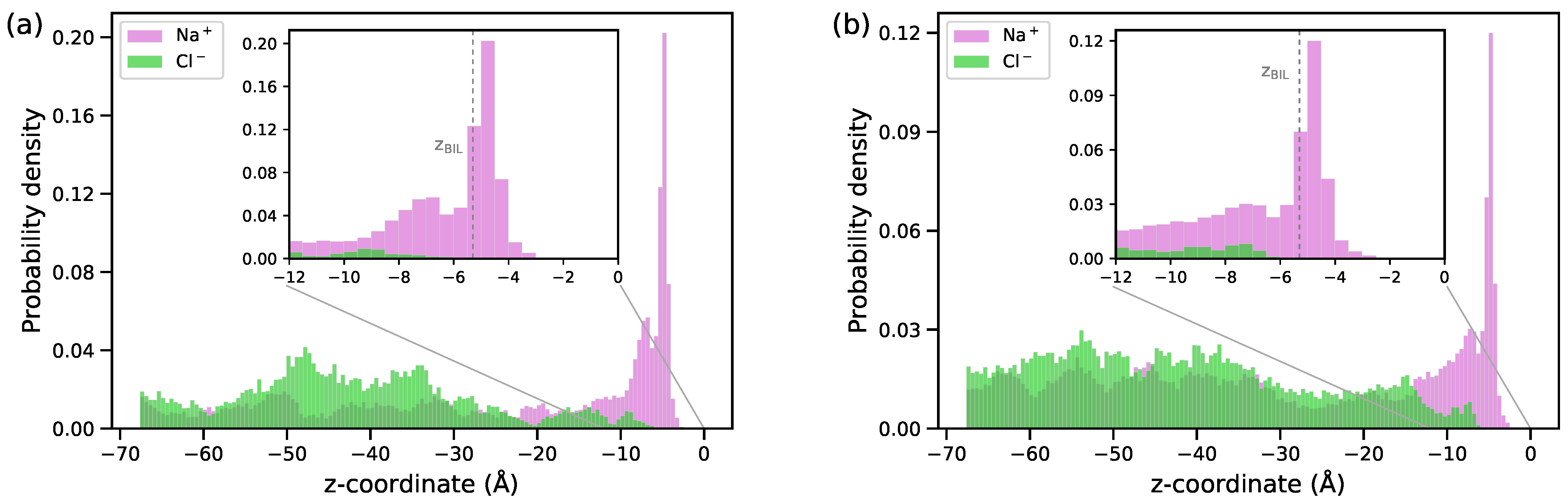
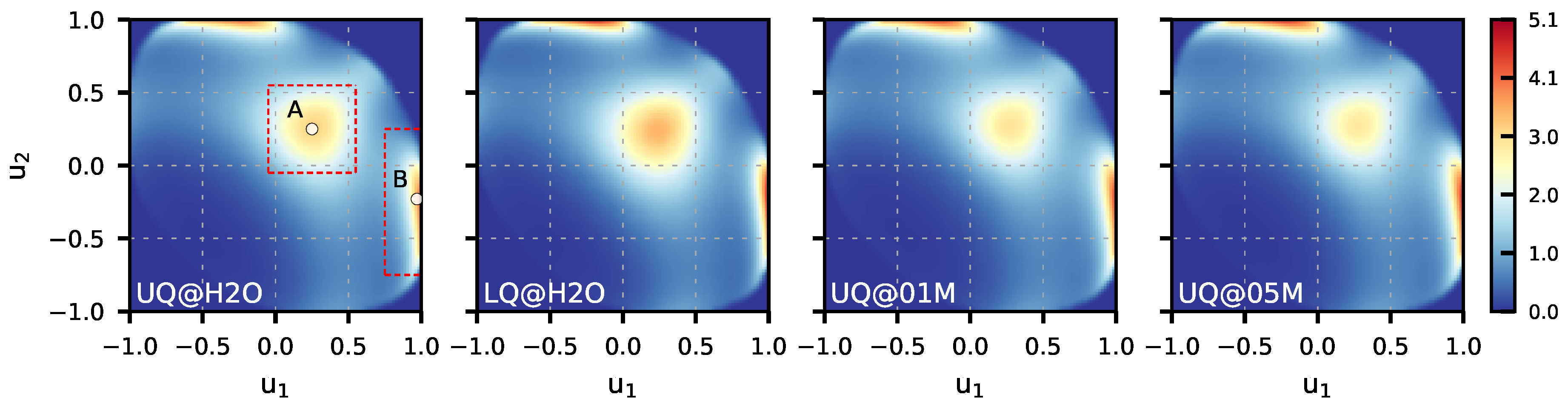
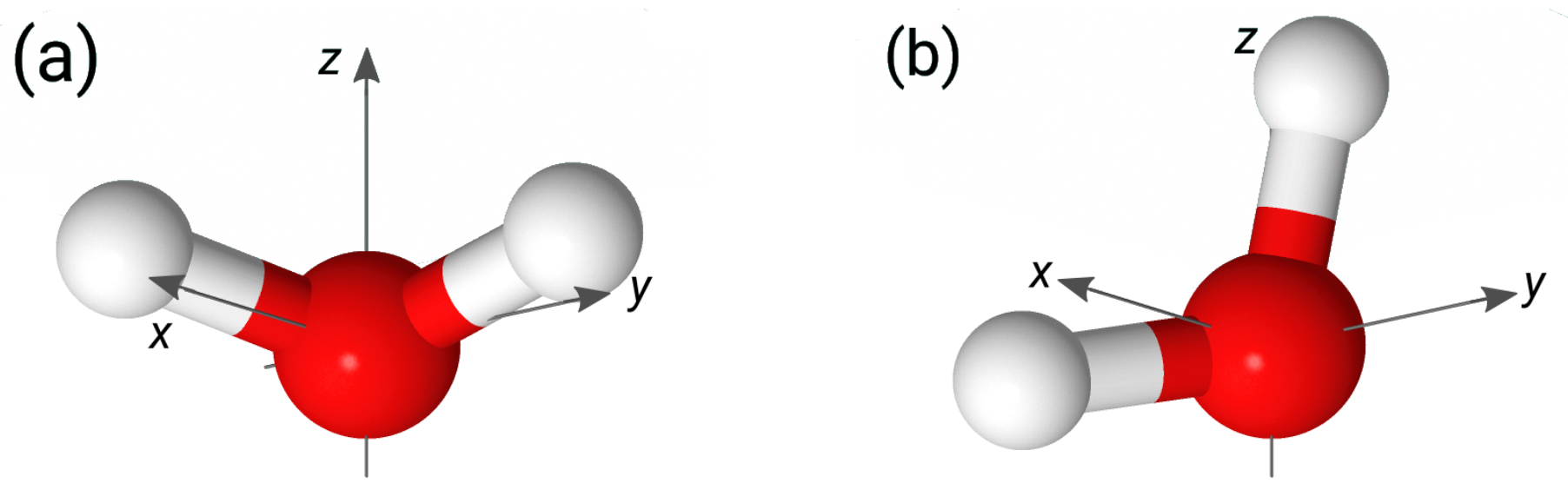
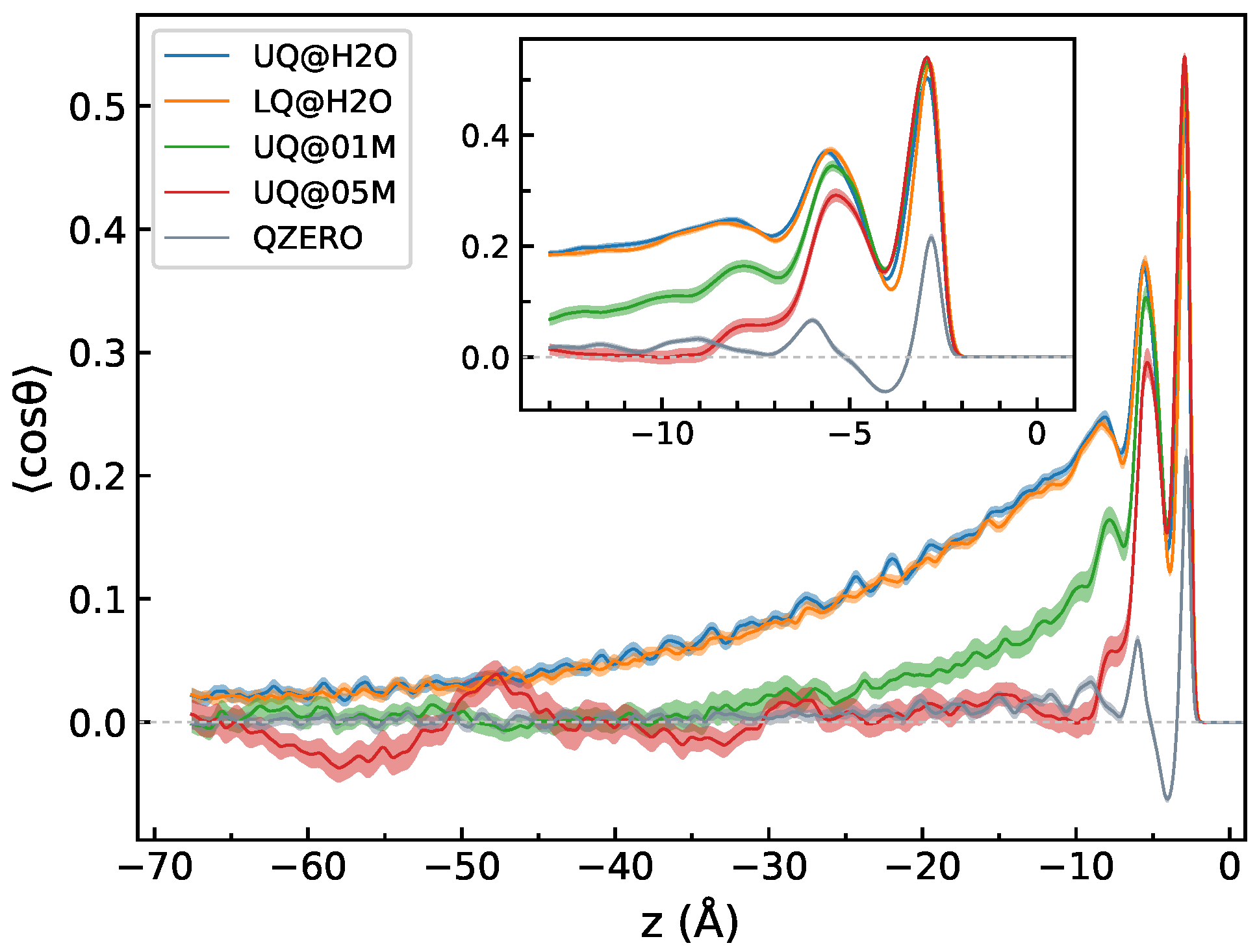
3.1. Structural Characteristics
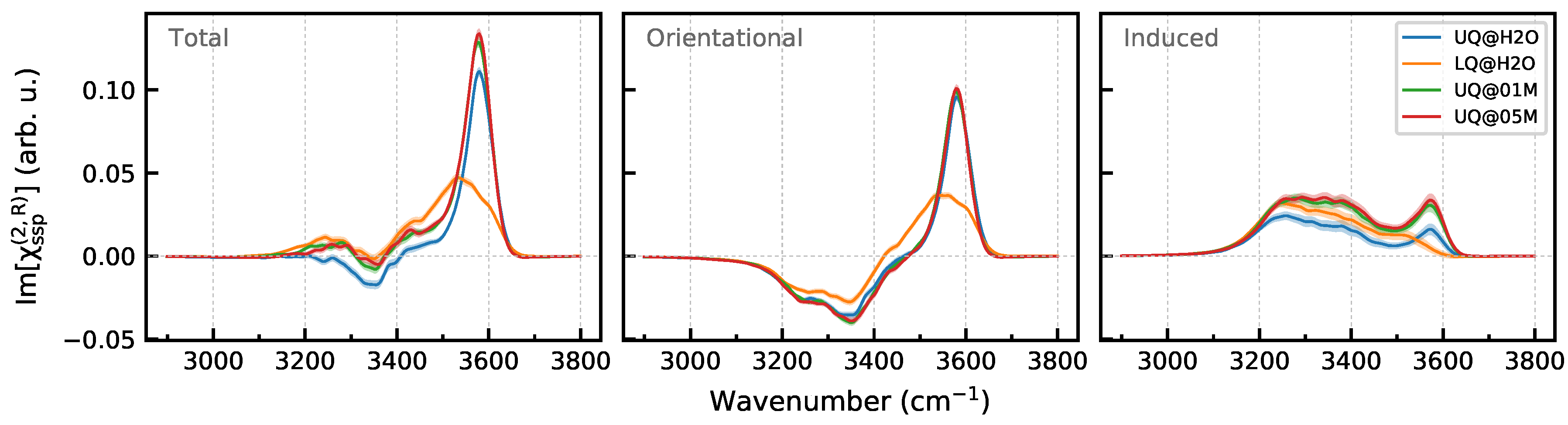
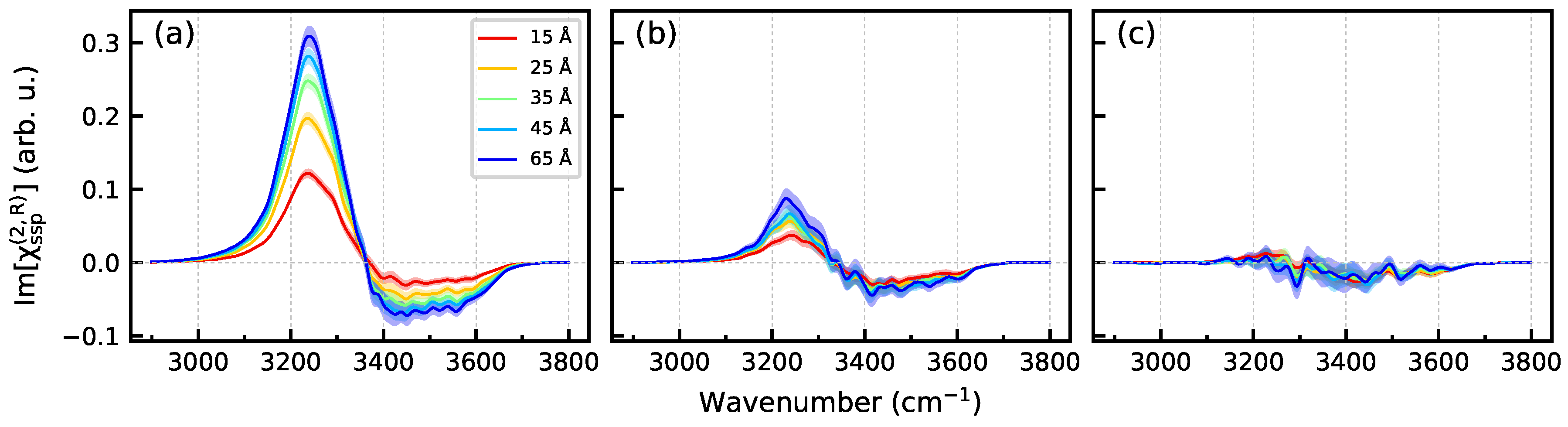
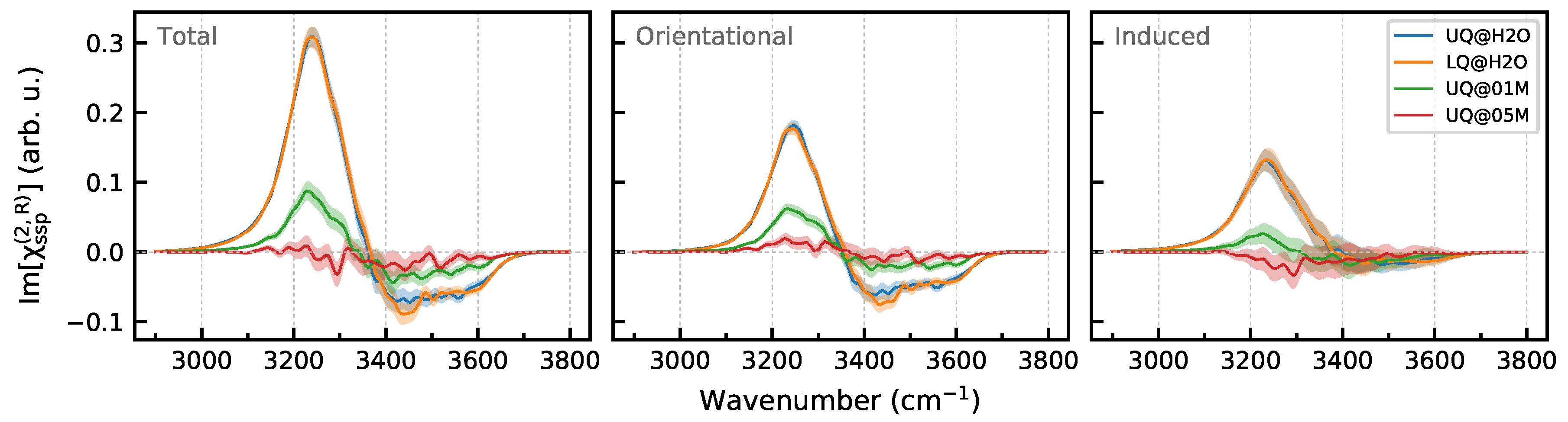
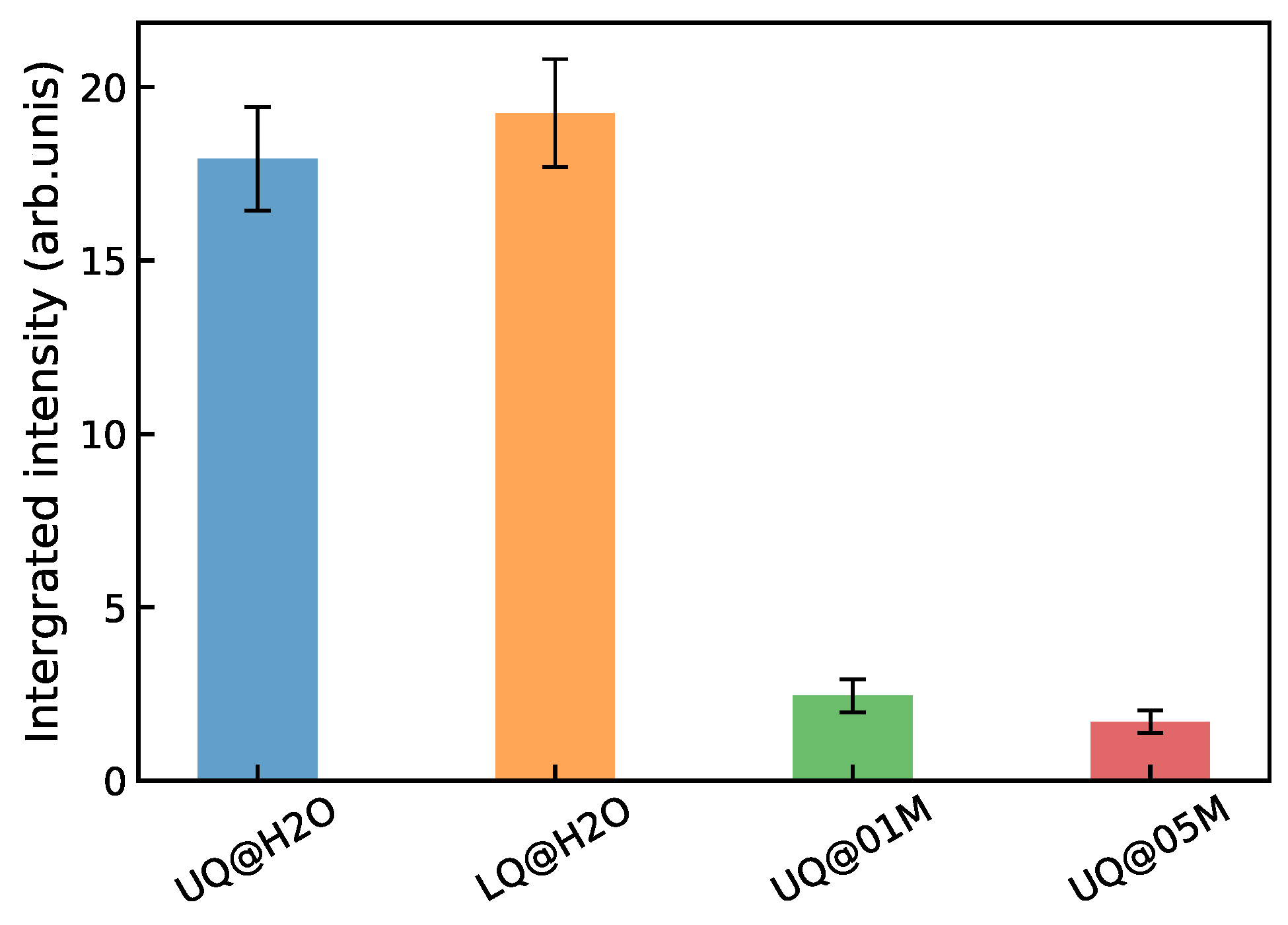
3.2. Nonlinear Spectra
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| VSFG | Vibrational sum-frequency generation spectroscopy |
| SHG | Second harmonic generation spectroscopy |
| HD-VSFG | Heterodyne-detected VSFG |
| MD | Molecular dynamics |
| EDL | Electrical double layer |
| BIL | Bonded interfacial layer |
| DL | Diffuse layer |
| SDL | Spectroscopic diffuse layer |
Appendix A. Structural Information from Im[χ(2)] Spectra
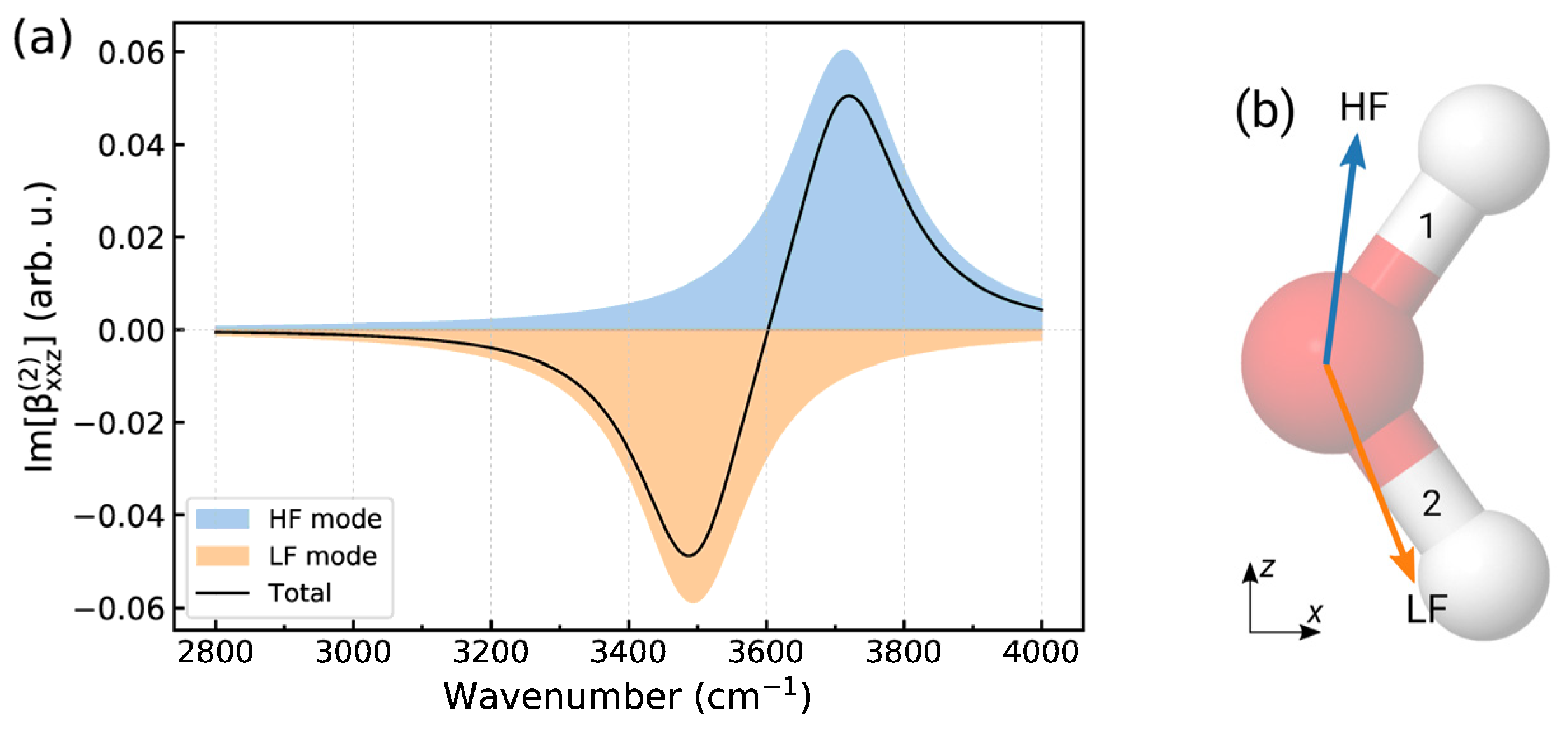
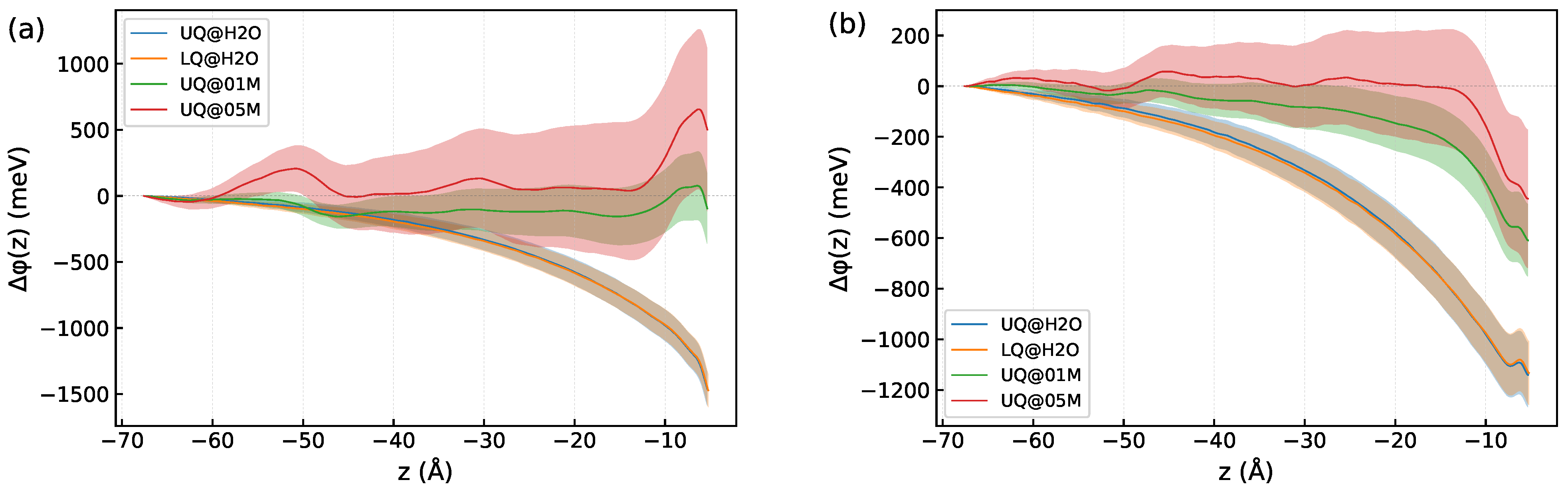
Appendix B. Computation of Electric Field
References
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Water at Interfaces. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar] [CrossRef]
- Bañuelos, J.L.; Borguet, E.; Brown, G.E., Jr.; Cygan, R.T.; DeYoreo, J.J.; Dove, P.M.; Gaigeot, M.P.; Geiger, F.M.; Gibbs, J.M.; Grassian, V.H.; et al. Oxide– and Silicate–Water Interfaces and Their Roles in Technology and the Environment. Chem. Rev. 2023, 123, 6413–6544. [Google Scholar] [CrossRef]
- Zaera, F. Probing Liquid/Solid Interfaces at the Molecular Level. Chem. Rev. 2012, 112, 2920–2986. [Google Scholar] [CrossRef]
- Shen, Y. The Principles of Nonlinear Optics; Wiley: New York, NY, USA, 1984. [Google Scholar]
- Shen, Y.R. Fundamentals of Sum-Frequency Spectroscopy; Cambridge University Press: Cambridge, UK, 2016; p. 316. [Google Scholar] [CrossRef]
- Stiopkin, I.V.; Jayathilake, H.D.; Bordenyuk, A.N.; Benderskii, A.V. Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy. J. Am. Chem. Soc. 2008, 130, 2271–2275. [Google Scholar] [CrossRef]
- Nihonyanagi, S.; Mondal, J.A.; Yamaguchi, S.; Tahara, T. Structure and Dynamics of Interfacial Water Studied by Heterodyne-Detected Vibrational Sum-Frequency Generation. Annu. Rev. Phys. Chem. 2013, 64, 579–603. [Google Scholar] [CrossRef]
- Roy, S.; Saha, S.; Mondal, J.A. Classical- and Heterodyne-Detected Vibrational Sum Frequency Generation (VSFG) Spectroscopy and Its Application to Soft Interfaces. In Modern Techniques of Spectroscopy: Basics, Instrumentation, and Applications; Singh, D.K., Pradhan, M., Materny, A., Eds.; Springer: Singapore, 2021; pp. 87–115. [Google Scholar] [CrossRef]
- Backus, E.; Schaefer, J.; Bonn, M. The mineral/water interface probed with nonlinear optical spectroscopy. Angew. Chem. Int. Ed. Engl. 2020, 60, 10482–10501. [Google Scholar] [CrossRef]
- Piontek, S.M.; Borguet, E. Vibrational Dynamics at Aqueous-Mineral Interfaces. J. Phys. Chem. C 2022, 126, 2307–2324. [Google Scholar] [CrossRef]
- Sparnaay, M. The Electrical Double Layer; Pergamon Press: Oxford, UK, 1972. [Google Scholar]
- Gonella, G.; Backus, E.H.G.; Nagata, Y.; Bonthuis, D.J.; Loche, P.; Schlaich, A.; Netz, R.R.; Kühnle, A.; McCrum, I.T.; Koper, M.T.M.; et al. Water at charged interfaces. Nat. Rev. Chem. 2021, 5, 466–485. [Google Scholar] [CrossRef]
- Wen, Y.C.; Zha, S.; Liu, X.; Yang, S.; Guo, P.; Shi, G.; Fang, H.; Shen, Y.R.; Tian, C. Unveiling Microscopic Structures of Charged Water Interfaces by Surface-Specific Vibrational Spectroscopy. Phys. Rev. Lett. 2016, 116, 016101. [Google Scholar] [CrossRef]
- Pezzotti, S.; Galimberti, D.R.; Shen, Y.R.; Gaigeot, M.P. Structural definition of the BIL and DL: A new universal methodology to rationalize non-linear χ(2) SFG signals at charged interfaces, including χ(3) contributions. Phys. Chem. Chem. Phys. 2018, 20, 5190–5199. [Google Scholar] [CrossRef]
- Smirnov, K.S. A molecular dynamics study of the nonlinear spectra and structure of charged (101) quartz/water interfaces. Phys. Chem. Chem. Phys. 2022, 24, 25118–25133. [Google Scholar] [CrossRef] [PubMed]
- Urashima, S.h.; Myalitsin, A.; Nihonyanagi, S.; Tahara, T. The Topmost Water Structure at a Charged Silica/Aqueous Interface Revealed by Heterodyne-Detected Vibrational Sum Frequency Generation Spectroscopy. J. Phys. Chem. Lett. 2018, 9, 4109–4114. [Google Scholar] [CrossRef] [PubMed]
- Rehl, B.; Gibbs, J.M. Role of Ions on the Surface-Bound Water Structure at the Silica/Water Interface: Identifying the Spectral Signature of Stability. J. Phys. Chem. Lett. 2021, 12, 2854–2864. [Google Scholar] [CrossRef] [PubMed]
- Rehl, B.; Ma, E.; Parshotam, S.; DeWalt-Kerian, E.L.; Liu, T.; Geiger, F.M.; Gibbs, J.M. Water Structure in the Electrical Double Layer and the Contributions to the Total Interfacial Potential at Different Surface Charge Densities. J. Am. Chem. Soc. 2022, 144, 16338–16349. [Google Scholar] [CrossRef] [PubMed]
- Parshotam, S.; Rehl, B.; Busse, F.; Brown, A.; Gibbs, J.M. Influence of the Hydrogen-Bonding Environment on Vibrational Coupling in the Electrical Double Layer at the Silica/Aqueous Interface. J. Phys. Chem. C 2022, 126, 21734–21744. [Google Scholar] [CrossRef]
- Wang, R.; Klein, M.L.; Carnevale, V.; Borguet, E. Investigations of water/oxide interfaces by molecular dynamics simulations. WIREs Comput. Mol. Sci. 2021, 11, e1537. [Google Scholar] [CrossRef]
- Pezzotti, S.; Galimberti, R.D.; Shen, R.Y.; Gaigeot, M.P. What the Diffuse Layer (DL) Reveals in Non-Linear SFG Spectroscopy. Minerals 2018, 8, 305. [Google Scholar] [CrossRef]
- Joutsuka, T.; Hirano, T.; Sprik, M.; Morita, A. Effects of third-order susceptibility in sum frequency generation spectra: A molecular dynamics study in liquid water. Phys. Chem. Chem. Phys. 2018, 20, 3040–3053. [Google Scholar] [CrossRef] [PubMed]
- Pezzotti, S.; Galimberti, D.R.; Gaigeot, M.P. Deconvolution of BIL-SFG and DL-SFG spectroscopic signals reveals order/disorder of water at the elusive aqueous silica interface. Phys. Chem. Chem. Phys. 2019, 21, 22188–22202. [Google Scholar] [CrossRef]
- Smirnov, K.S. Structure and sum-frequency generation spectra of water on uncharged Q4 silica surfaces: A molecular dynamics study. Phys. Chem. Chem. Phys. 2020, 22, 2033–2045. [Google Scholar] [CrossRef]
- Kroutil, O.; Pezzotti, S.; Gaigeot, M.P.; Předota, M. Phase-Sensitive Vibrational SFG Spectra from Simple Classical Force Field Molecular Dynamics Simulations. J. Phys. Chem. C 2020, 124, 15253–15263. [Google Scholar] [CrossRef]
- Smirnov, K.S. Structure and sum-frequency generation spectra of water on neutral hydroxylated silica surfaces. Phys. Chem. Chem. Phys. 2021, 23, 6929–6949. [Google Scholar] [CrossRef]
- Pezzotti, S.; Serva, A.; Sebastiani, F.; Brigiano, F.S.; Galimberti, D.R.; Potier, L.; Alfarano, S.; Schwaab, G.; Havenith, M.; Gaigeot, M.P. Molecular Fingerprints of Hydrophobicity at Aqueous Interfaces from Theory and Vibrational Spectroscopies. J. Phys. Chem. Lett. 2021, 12, 3827–3836. [Google Scholar] [CrossRef]
- Hunger, J.; Schaefer, J.; Ober, P.; Seki, T.; Wang, Y.; Praedel, L.; Nagata, Y.; Bonn, M.; Bonthuis, D.J.; Backus, E.H.G. Nature of cations critically affects water at negatively charged silica interface. J. Am. Chem. Soc. 2022, 144, 19726–19738. [Google Scholar] [CrossRef] [PubMed]
- Cyran, J.D.; Donovan, M.A.; Vollmer, D.; Siro Brigiano, F.; Pezzotti, S.; Galimberti, D.R.; Gaigeot, M.P.; Bonn, M.; Backus, E.H.G. Molecular hydrophobicity at a macroscopically hydrophilic surface. Proc. Natl. Acad. Sci. USA 2019, 116, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Milonjić, S.K. Determination of surface ionization and complexation constants at colloidal silica/electrolyte interface. Colloids Surf. 1987, 23, 301–312. [Google Scholar] [CrossRef]
- Gulicovski, J.J.; Čerović, L.S.; Milonjić, S.K. Point of Zero Charge and Isoelectric Point of Alumina. Mater. Manuf. Process. 2008, 23, 615–619. [Google Scholar] [CrossRef]
- Brown, M.A.; Goel, A.; Abbas, Z. Effect of Electrolyte Concentration on the Stern Layer Thickness at a Charged Interface. Angew. Chem. Int. Ed. Engl. 2016, 55, 3790–3794. [Google Scholar] [CrossRef]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 024503. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Keblinski, P.; Phillpot, S.R.; Eggebrecht, J. Exact method for the simulation of Coulombic systems by spherically truncated, pairwise r−1 summation. J. Chem. Phys. 1999, 110, 8254–8282. [Google Scholar] [CrossRef]
- Fennell, C.J.; Gezelter, J.D. Is the Ewald summation still necessary? Pairwise alternatives to the accepted standard for long-range electrostatics. J. Chem. Phys. 2006, 124, 234104. [Google Scholar] [CrossRef] [PubMed]
- Prada-Gracia, D.; Shevchuk, R.; Rao, F. The quest for self-consistency in hydrogen bond definitions. J. Chem. Phys. 2013, 139, 084501. [Google Scholar] [CrossRef] [PubMed]
- Willard, A.P.; Chandler, D. Instantaneous Liquid Interfaces. J. Phys. Chem. B 2010, 114, 1954–1958. [Google Scholar] [CrossRef]
- Morita, A.; Hynes, J.T. A Theoretical Analysis of the Sum Frequency Generation Spectrum of the Water Surface. II. Time-Dependent Approach. J. Phys. Chem. B 2002, 106, 673–685. [Google Scholar] [CrossRef]
- Morita, A.; Hynes, J.T. A theoretical analysis of the sum frequency generation spectrum of the water surface. Chem. Phys. 2000, 258, 371–390. [Google Scholar] [CrossRef]
- Morita, A. Theory of Sum Frequency Generation Spectroscopy; Springer: Singapore, 2018. [Google Scholar]
- Applequist, J.; Carl, J.R.; Fung, K.K. Atom dipole interaction model for molecular polarizability. Application to polyatomic molecules and determination of atom polarizabilities. J. Am. Chem. Soc. 1972, 94, 2952–2960. [Google Scholar] [CrossRef]
- Ong, S.; Zhao, X.; Eisenthal, K.B. Polarization of water molecules at a charged interface: Second harmonic studies of the silica/water interface. Chem. Phys. Lett. 1992, 191, 327–335. [Google Scholar] [CrossRef]
- Dreier, L.; Nagata, Y.; Lutz, H.; Gonella, G.; Hunger, J.; Backus, E.; Bonn, M. Saturation of charge-induced water alignment at model membrane surfaces. Sci. Adv. 2018, 4, eaap7415. [Google Scholar] [CrossRef]
- Chen, W.; Sanders, S.E.; Ãzdamar, B.; Louaas, D.; Brigiano, F.S.; Pezzotti, S.; Petersen, P.B.; Gaigeot, M.P. On the Trail of Molecular Hydrophilicity and Hydrophobicity at Aqueous Interfaces. J. Phys. Chem. Lett. 2023, 14, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Elgabarty, H.; Spura, T.; Karhan, K.; Partovi-Azar, P.; Hassanali, A.A.; Kühne, T.D. Structure and Dynamics of the Instantaneous Water/Vapor Interface Revisited by Path-Integral and Ab Initio Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 10079–10086. [Google Scholar] [CrossRef] [PubMed]
- Jena, K.C.; Covert, P.A.; Hore, D.K. The Effect of Salt on the Water Structure at a Charged Solid Surface: Differentiating Second- and Third-order Nonlinear Contributions. J. Phys. Chem. Lett. 2011, 2, 1056–1061. [Google Scholar] [CrossRef]
- Schaefer, J.; Gonella, G.; Bonn, M.; Backus, E.H.G. Surface-specific vibrational spectroscopy of the water/silica interface: Screening and interference. Phys. Chem. Chem. Phys. 2017, 19, 16875–16880. [Google Scholar] [CrossRef] [PubMed]
- Ohno, P.E.; Chang, H.; Spencer, A.P.; Liu, Y.; Boamah, M.D.; Wang, H.f.; Geiger, F.M. Beyond the Gouy-Chapman Model with Heterodyne-Detected Second Harmonic Generation. J. Phys. Chem. Lett. 2019, 10, 2328–2334. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface | Surface Charge 1 | 2 () | No. H2O 3 | No. Na+/Cl− 4 | Acronym |
|---|---|---|---|---|---|
| Solid/neat water | Uniform | −0.025 | 2058 | – | UQ@H2O |
| Solid/neat water | Localized | −0.25 | 2058 | – | LQ@H2O |
| Solid/electrolyte | Uniform | −0.025 | 2045 | 9/4 | UQ@01M |
| Solid/electrolyte | Uniform | −0.025 | 2013 | 25/20 | UQ@05M |
| System | UQ@H2O | LQ@H2O | UQ@01M | UQ@05M |
|---|---|---|---|---|
| Orientation A | 24.2 | 26.1 | 23.1 | 22.8 |
| Orientation B | 22.6 | 20.5 | 25.2 | 26.0 |
| System | Bulk | DL | BIL | |||||
|---|---|---|---|---|---|---|---|---|
| UQ@H2O | 3.50 (0.01) | 1.74 (0.03) | 3.51 (0.06) | 1.71 (0.07) | 2.73 (0.08) | 1.97 (0.11) | ||
| LQ@H2O | 3.50 (0.02) | 1.75 (0.03) | 3.49 (0.06) | 1.70 (0.07) | 2.77 (0.09) | 2.09 (0.12) | ||
| UQ@01M | 3.46 (0.01) | 1.73 (0.03) | 3.45 (0.06) | 1.68 (0.07) | 2.63 (0.08) | 1.89 (0.11) | ||
| UQ@05M | 3.35 (0.01) | 1.67 (0.03) | 3.48 (0.06) | 1.67 (0.07) | 2.63 (0.08) | 1.91 (0.11) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnov, K.S. Effects of Surface Charge Distribution and Electrolyte Ions on the Nonlinear Spectra of Model Solid–Water Interfaces. Molecules 2024, 29, 3758. https://doi.org/10.3390/molecules29163758
Smirnov KS. Effects of Surface Charge Distribution and Electrolyte Ions on the Nonlinear Spectra of Model Solid–Water Interfaces. Molecules. 2024; 29(16):3758. https://doi.org/10.3390/molecules29163758
Chicago/Turabian StyleSmirnov, Konstantin S. 2024. "Effects of Surface Charge Distribution and Electrolyte Ions on the Nonlinear Spectra of Model Solid–Water Interfaces" Molecules 29, no. 16: 3758. https://doi.org/10.3390/molecules29163758
APA StyleSmirnov, K. S. (2024). Effects of Surface Charge Distribution and Electrolyte Ions on the Nonlinear Spectra of Model Solid–Water Interfaces. Molecules, 29(16), 3758. https://doi.org/10.3390/molecules29163758