

Molecular Docking-Based Virtual Screening of FDA-Approved Drugs Using Trypanothione Reductase Identified New Trypanocidal Agents
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
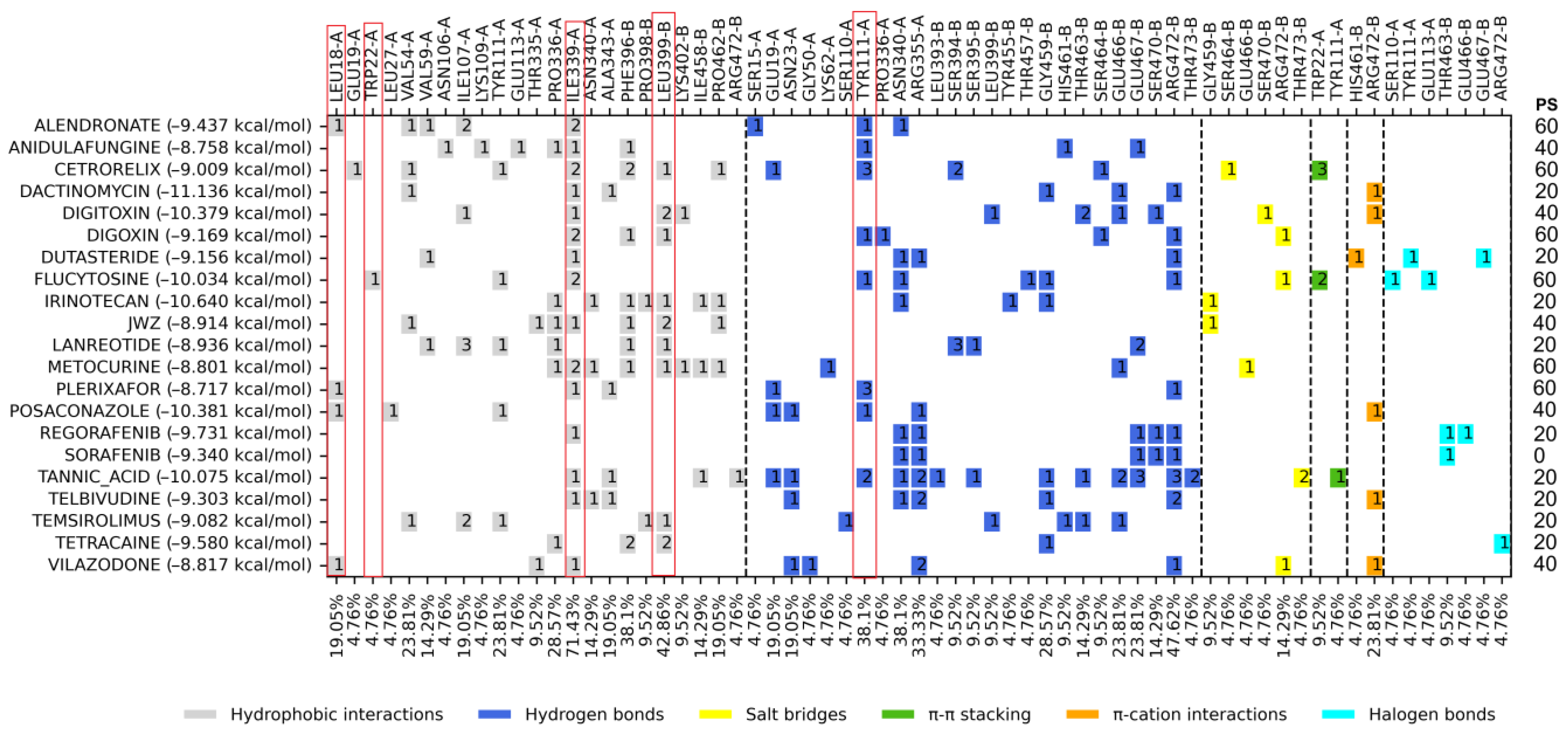
2.1. Binding Sites
2.2. Molecular Docking on Z Site
2.3. Molecular Docking on Mepacrine Site
2.4. Molecular Docking on the Catalytic Site
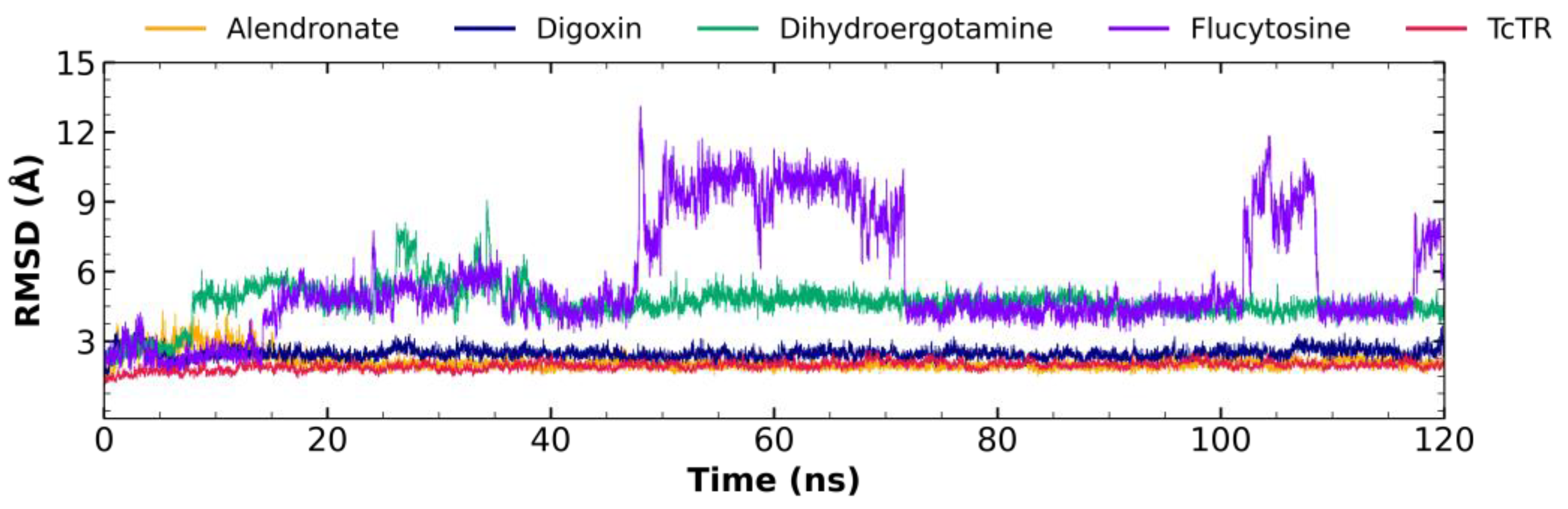
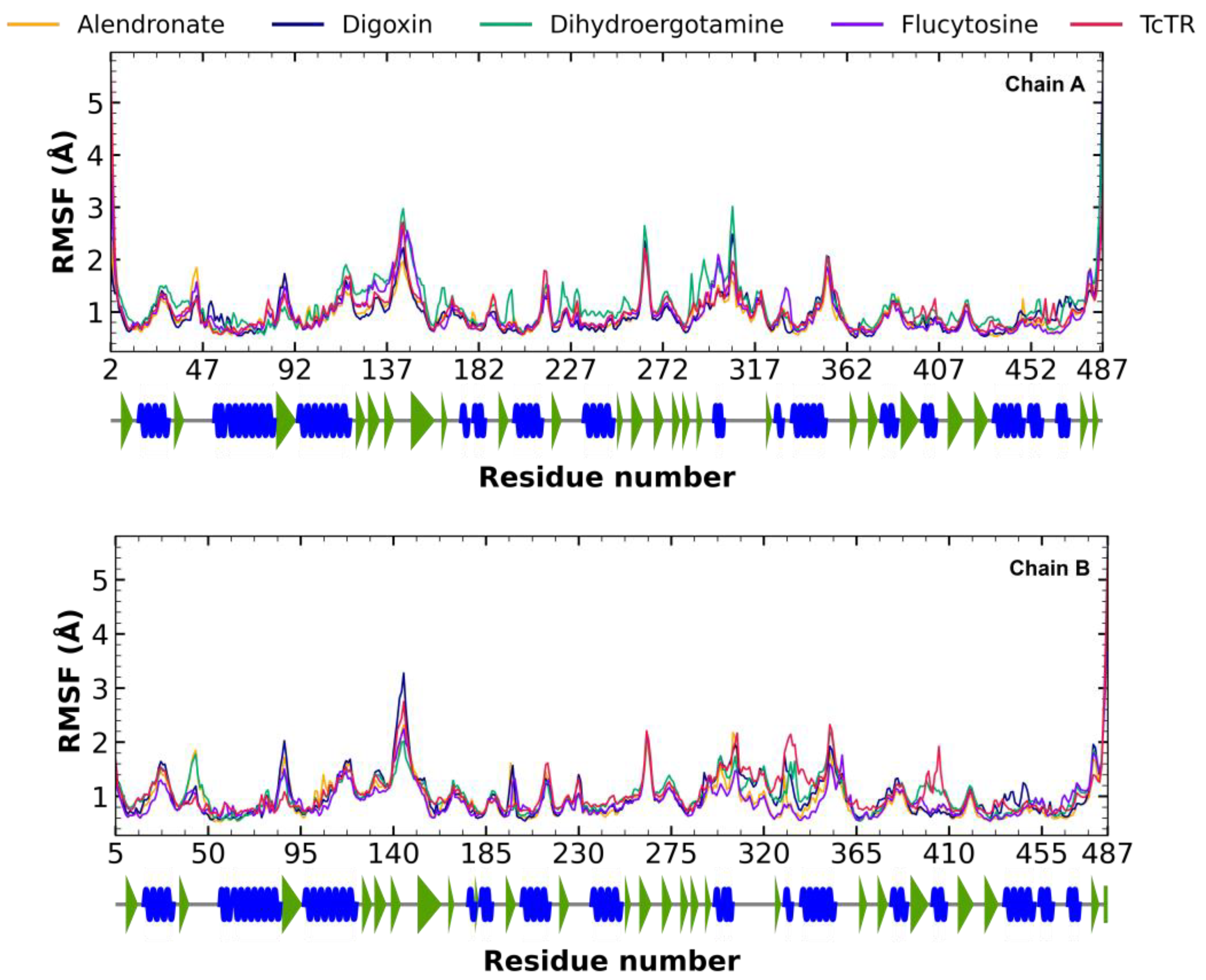
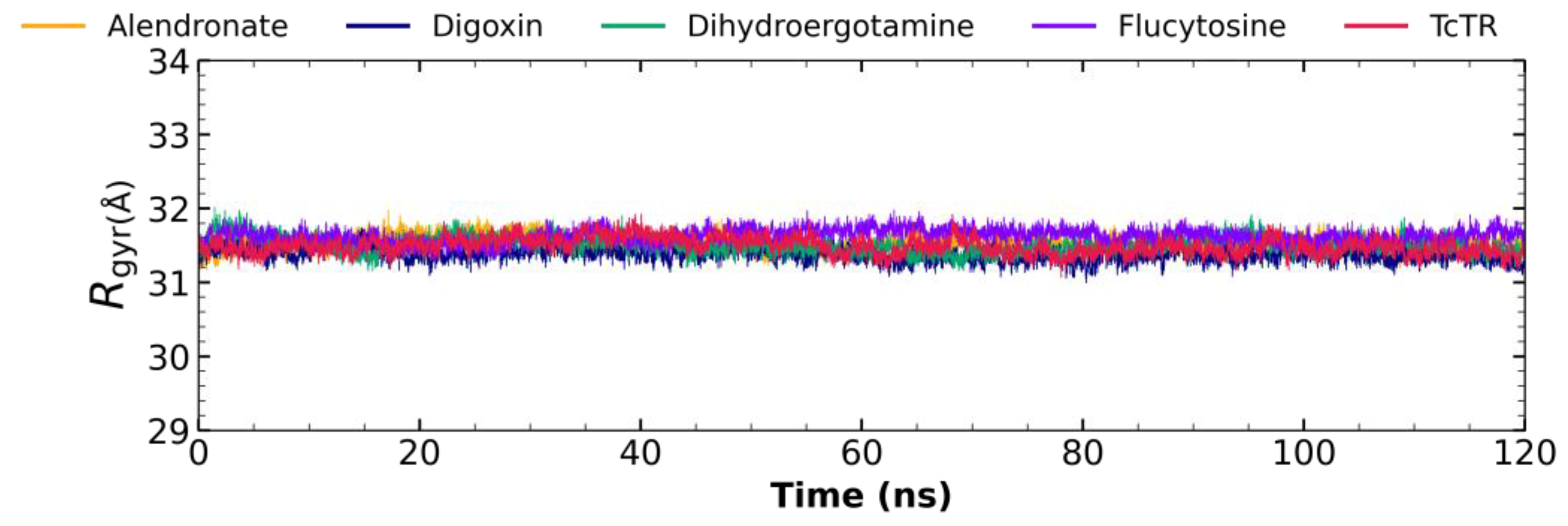
2.5. Molecular Dynamic Analysis
2.5.1. Root Mean Square Deviation (RMSD) Analysis
2.5.2. Root Mean Square Fluctuation (RMSF) Analysis
2.5.3. Radius of Gyration Analysis
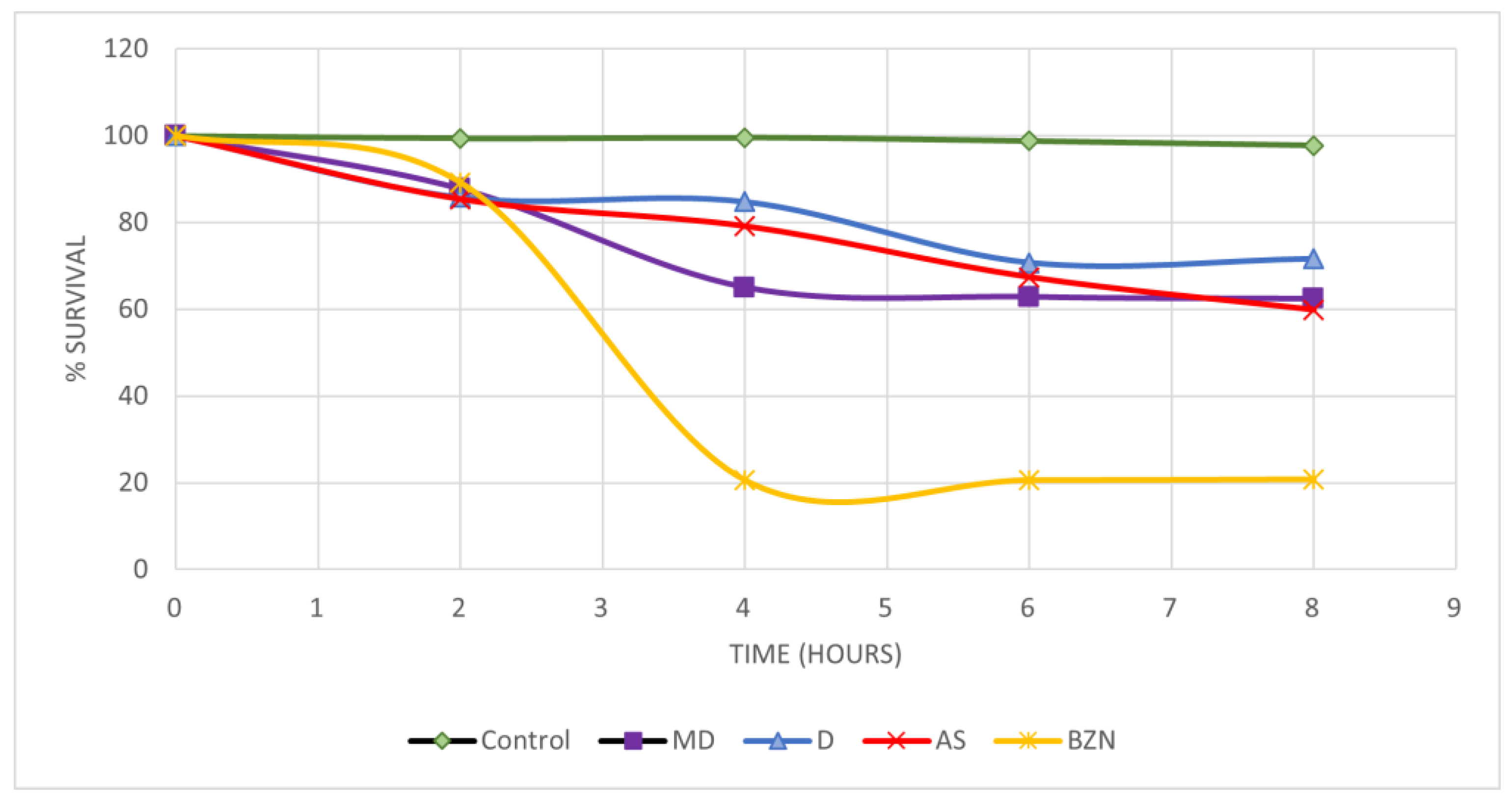
2.6. In Vitro Activity on Blood Trypomastigotes
2.7. Short-Term In Vivo Assay in a Murine Model of T. cruzi Infection
3. Discussion
3.1. Molecular Docking on Z Site
3.2. Molecular Docking on Mepacrine Site
3.3. Molecular Docking on the Catalytic Site
3.4. Molecular Dynamic Analysis
3.5. In Vitro Activity on Blood Trypomastigotes
3.6. Short-Term In Vivo Assay in a Murine Model of T. cruzi Infection
4. Materials and Methods
4.1. Protein Preparation
4.2. Ligand Library Preparation
4.3. Molecular Docking
4.4. Molecular Docking Analysis
4.5. Molecular Dynamic Analysis
4.6. In Vitro Trypanocidal Assay of Blood Trypomastigotes
4.7. Short-Term In Vivo Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Organización Panamericana de la Salud Enfermedad de Chagas. Available online: https://www.paho.org/es/temas/enfermedad-chagas#:~:text=La%20enfermedad%20de%20Chagas%20es,deficitarios%2C%20consider%C3%A1ndosela%20una%20enfermedad%20desatendida (accessed on 30 April 2024).
- World Health Organization La Enfermedad de Chagas (Tripanosomiasis Americana). Available online: https://www.who.int/es/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 30 April 2024).
- Rojo-Medina, J.; Ruiz-Matus, C.; Salazar-Schettino, P.M.; González-Roldán, J.F. Enfermedad de Chagas En México. Gac. Med. Mex. 2018, 154, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Arrúa, E.C.; Seremeta, K.P.; Bedogni, G.R.; Okulik, N.B.; Salomon, C.J. Nanocarriers for Effective Delivery of Benznidazole and Nifurtimox in the Treatment of Chagas Disease: A Review. Acta Trop. 2019, 198, 105080. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Bolz, S.N.; Adelmann, L.; Salentin, S.; Haupt, V.J.; Moreno-Rodríguez, A.; Nogueda-Torres, B.; Castillo-Campos, V.; Yepez-Mulia, L.; De Fuentes-Vicente, J.A.; et al. Repositioned Drugs for Chagas Disease Unveiled via Structure-Based Drug Repositioning. Int. J. Mol. Sci. 2020, 21, 8809. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Saldivar, A.; Schroeder, M.; Salentin, S.; Haupt, V.J.; Saavedra, E.; Vázquez, C.; Reyes-Espinosa, F.; Herrera-Mayorga, V.; Villalobos-Rocha, J.C.; García-Pérez, C.A.; et al. Computational Drug Repositioning for Chagas Disease Using Protein-Ligand Interaction Profiling. Int. J. Mol. Sci. 2020, 21, 4270. [Google Scholar] [CrossRef] [PubMed]
- Olin-Sandoval, V.; Moreno-Sanchez, R.; Saavedra, E. Targeting Trypanothione Metabolism in Trypanosomatid Human Parasites. Curr. Drug Targets 2010, 11, 1614–1630. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.O.F.; Austin, S.E.; Chan, C.; Yin, H.; Marks, D.; Vaghjiani, S.N.; Kendrick, H.; Yardley, V.; Croft, S.L.; Douglas, K.T. Use of an Additional Hydrophobic Binding Site, the Z Site, in the Rational Drug Design of a New Class of Stronger Trypanothione Reductase Inhibitor, Quaternary Alkylammonium Phenothiazines. J. Med. Chem. 2000, 43, 3148–3156. [Google Scholar] [CrossRef] [PubMed]
- Battista, T.; Colotti, G.; Ilari, A.; Fiorillo, A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules 2020, 25, 1924. [Google Scholar] [CrossRef]
- Venkatesan, S.K.; Shukla, A.K.; Dubey, V.K. Molecular Docking Studies of Selected Tricyclic and Quinone Derivatives on Trypanothione Reductase of Leishmania Infantum. J. Comput. Chem. 2010, 31, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Demoro, B.; Caruso, F.; Rossi, M.; Benítez, D.; González, M.; Cerecetto, H.; Galizzi, M.; Malayil, L.; Docampo, R.; Faccio, R.; et al. Bisphosphonate Metal Complexes as Selective Inhibitors of Trypanosoma Cruzi Farnesyl Diphosphate Synthase. Dalton Trans. 2012, 41, 6468. [Google Scholar] [CrossRef]
- Assíria Fontes Martins, T.; de Figueiredo Diniz, L.; Mazzeti, A.L.; da Silva do Nascimento, Á.F.; Caldas, S.; Caldas, I.S.; de Andrade, I.M.; Ribeiro, I.; Bahia, M.T. Benznidazole/Itraconazole Combination Treatment Enhances Anti-Trypanosoma Cruzi Activity in Experimental Chagas Disease. PLoS ONE 2015, 10, e0128707. [Google Scholar] [CrossRef]
- Bond, C.S.; Zhang, Y.; Berriman, M.; Cunningham, M.L.; Fairlamb, A.H.; Hunter, W.N. Crystal Structure of Trypanosoma Cruzi Trypanothione Reductase in Complex with Trypanothione, and the Structure-Based Discovery of New Natural Product Inhibitors. Structure 1999, 7, 81–89. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Álvarez, D.; Torres-Rojas, M.F.; Lara-Ramirez, E.E.; Marchat, L.A.; Rivera, G. Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment. Molecules 2023, 28, 4389. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein-Ligand Binding Affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- O’boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: https://cir.nii.ac.jp/crid/1570572701247244160 (accessed on 20 March 2024).
- Espinosa-Bustos, C.; Ortiz Pérez, M.; Gonzalez-Gonzalez, A.; Zarate, A.M.; Rivera, G.; Belmont-Díaz, J.A.; Saavedra, E.; Cuellar, M.A.; Vázquez, K.; Salas, C.O. New Amino Naphthoquinone Derivatives as Anti-Trypanosoma Cruzi Agents Targeting Trypanothione Reductase. Pharmaceutics 2022, 14, 1121. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, W.P.; Brylinski, M. Calculating an Optimal Box Size for Ligand Docking and Virtual Screening against Experimental and Predicted Binding Pockets. J. Cheminform. 2015, 7, 18. [Google Scholar] [CrossRef]
- Maamri, S.; Benarous, K.; Yousfi, M. Identification of 3-Methoxycarpachromene and Masticadienonic Acid as New Target Inhibitors against Trypanothione Reductase from Leishmania Infantum Using Molecular Docking and ADMET Prediction. Molecules 2021, 26, 3335. [Google Scholar] [CrossRef]
- Matadamas-Martínez, F.; Hernández-Campos, A.; Téllez-Valencia, A.; Vázquez-Raygoza, A.; Comparán-Alarcón, S.; Yépez-Mulia, L.; Castillo, R. Leishmania Mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules 2019, 24, 3216. [Google Scholar] [CrossRef]
- Saha, D.; Sharma, A. Docking-Based Screening of Natural Product Database in Quest for Dual Site Inhibitors of Trypanosoma Cruzi Trypanothione Reductase (TcTR). Med. Chem. Res. 2015, 24, 316–333. [Google Scholar] [CrossRef]
- González-González, A.; Méndez-Álvarez, D.; Vázquez-Jiménez, L.K.; Delgado-Maldonado, T.; Ortiz-Pérez, E.; Paz-González, A.D.; Bandyopadhyay, D.; Rivera, G. Molecular Docking and Dynamic Simulations of Quinoxaline 1,4-Di-N-Oxide as Inhibitors for Targets from Trypanosoma Cruzi, Trichomonas Vaginalis, and Fasciola Hepatica. J. Mol. Model. 2023, 29, 180. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Vargas, K.F.; Nogueda-Torres, B.; Sánchez-Torres, L.E.; Suarez-Contreras, E.; Villalobos-Rocha, J.C.; Torres-Martinez, Y.; Lara-Ramirez, E.E.; Fiorani, G.; Krauth-Siegel, R.L.; Bolognesi, M.L.; et al. Trypanocidal Activity of Quinoxaline 1,4 Di-N-Oxide Derivatives as Trypanothione Reductase Inhibitors. Molecules 2017, 22, 220. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Saldivar, A.; Gómez-Escobedo, R.; Corral-Ruiz, G.; Chacón-Vargas, K.F.; Horta-Montaño, V.; Sanchez-Torres, L.; Vazquez-Jimenez, L.k.; Nogueda-Torres, B.; Rivera, G. Repositioning FDA-Approved Drug Against Chagas Disease and Cutaneous Leishmaniosis by Structure-Based Virtual Screening. Arch. Med. Res. 2024, 55, 102958. [Google Scholar] [CrossRef]
- Becerra, N.A.; Espinosa-Bustos, C.; Vázquez, K.; Rivera, G.; Paulino, M.; Cantero, J.; Nogueda, B.; Chacón-Vargas, F.; Castillo-Velazquez, U.; Rodríguez, A.F.E.; et al. Expanding the Chemical Space of Aryloxy-Naphthoquinones as Potential Anti-Chagasic Agents: Synthesis and Trypanosomicidal Activity. Med. Chem. Res. 2021, 30, 2256–2265. [Google Scholar] [CrossRef]
- Romanha, A.J.; Castro, S.L.d.; Soeiro, M. de N.C.; Lannes-Vieira, J.; Ribeiro, I.; Talvani, A.; Bourdin, B.; Blum, B.; Olivieri, B.; Zani, C.; et al. In Vitro and in Vivo Experimental Models for Drug Screening and Development for Chagas Disease. Mem. Inst. Oswaldo Cruz 2010, 105, 233–238. [Google Scholar] [CrossRef]
- Wong-Baeza, C.; Nogueda-Torres, B.; Serna, M.; Meza-Toledo, S.; Baeza, I.; Wong, C. Trypanocidal Effect of the Benzyl Ester of N-Propyl Oxamate: A Bi-Potential Prodrug for the Treatment of Experimental Chagas Disease. BMC Pharmacol. Toxicol. 2015, 16, 10. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Z Site | Mepacrine Site | Catalytic Site | |||
|---|---|---|---|---|---|
| Drug | DS (kcal/mol) | Drug | DS (kcal/mol) | Drug | DS (kcal/mol) |
| Digoxin | −10.148 | Dactinomycin | −11.136 | Flucytosine | −11.725 |
| Anidulafungine | −9.873 | Irinotecan | −10.64 | Digitoxin | −11.012 |
| Tannic acid | −9.551 | Posaconazole | −10.381 | Dactinomycin | −10.973 |
| Metocurine | −9.238 | Digitoxin | −10.379 | Irinotecan | −10.489 |
| Nilotinib | −9.203 | Tannic acid | −10.075 | Anidulafungin | −10.44 |
| Palperidone | −9.186 | Flucytosine | −10.034 | Telmisartan | −10.334 |
| Crizotinib | −9.169 | Regorafenib | −9.731 | Nilotinib | −10.264 |
| Paclitaxel | −9.158 | Tetracaine | −9.58 | Regorafenib | −9.899 |
| Dibucaine | −9.085 | Alendronate | −9.437 | Vilazodone | −9.849 |
| Nafarelin | −9.051 | Sorafenib | −9.34 | Dutasteride | −9.832 |
| Conivaptan | −9.049 | Telbivudine | −9.303 | Lanreotide | −9.802 |
| Ponatinib | −9.047 | Digoxin | −9.169 | Zafirlukast | −9.777 |
| Flucytosine | −9.031 | Dutasteride | −9.156 | Lapatinib | −9.77 |
| Lomitapide | −8.979 | Temsirolimus | −9.082 | Sorafenib | −9.748 |
| Edrophonium | −8.969 | Cetrorelix | −9.009 | Tetracaine | −9.718 |
| Itraconazole | −8.945 | Lanreotide | −8.936 | Alendronate | −9.714 |
| Vilazodone | −8.881 | Vilazodone | −8.817 | Ponatinib | −9.627 |
| Regorafenib | −8.878 | Metocurine | −8.801 | Telbivudine | −9.569 |
| Tolvaptan | −8.866 | Anidulafungine | −8.758 | Digoxin | −9.55 |
| Ganirelix | −8.864 | Plerixafor | −8.717 | Dihidroergotamine | −9.536 |
| ZINC12151998 | −10.3 | JWZ | −8.914 | ZINC12151998 | −10.799 |
| 7i | −9.838 | ||||
| 7e | −9.428 | ||||
| Drug | % Lysis at 12.5 µg/mL | LC50 µmol | ||
|---|---|---|---|---|
| NINOA | INC-5 | NINOA | INC-5 | |
| Flucytosine | 13.4 ± 2.9 | 21.9 ± 1.5 | 613 ± 22 | 1272 ± 59 |
| Alendronate | 28.2 ± 6 | 17.3 ± 15.3 | 174 ± 11 | 277 ± 13.3 |
| Digoxin | 36.2 ± 2 | 33.6 ± 2.5 | 45 ± 2.8 | 76 ± 10 |
| Dihydroergotamine | 29.4 ± 1.5 | 25 ± 7 | 28.1 ± 3.1 | 57 ± 2.6 |
| Nifurtimox | 28.6 ± 4.5 | 25 ± 3.6 | 161 ± 33 | 255 ± 39 |
| Benznidazole | 32.2 ± 6 | 30.3 ± 5.5 | 220 ± 40 | 337 ± 34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Escobedo, R.; Méndez-Álvarez, D.; Vázquez, C.; Saavedra, E.; Vázquez, K.; Alcántara-Farfán, V.; Cordero-Martínez, J.; Gonzalez-Gonzalez, A.; Rivera, G.; Nogueda-Torres, B. Molecular Docking-Based Virtual Screening of FDA-Approved Drugs Using Trypanothione Reductase Identified New Trypanocidal Agents. Molecules 2024, 29, 3796. https://doi.org/10.3390/molecules29163796
Gómez-Escobedo R, Méndez-Álvarez D, Vázquez C, Saavedra E, Vázquez K, Alcántara-Farfán V, Cordero-Martínez J, Gonzalez-Gonzalez A, Rivera G, Nogueda-Torres B. Molecular Docking-Based Virtual Screening of FDA-Approved Drugs Using Trypanothione Reductase Identified New Trypanocidal Agents. Molecules. 2024; 29(16):3796. https://doi.org/10.3390/molecules29163796
Chicago/Turabian StyleGómez-Escobedo, Rogelio, Domingo Méndez-Álvarez, Citlali Vázquez, Emma Saavedra, Karina Vázquez, Verónica Alcántara-Farfán, Joaquín Cordero-Martínez, Alonzo Gonzalez-Gonzalez, Gildardo Rivera, and Benjamín Nogueda-Torres. 2024. "Molecular Docking-Based Virtual Screening of FDA-Approved Drugs Using Trypanothione Reductase Identified New Trypanocidal Agents" Molecules 29, no. 16: 3796. https://doi.org/10.3390/molecules29163796