Aqueous Phase Hydrogenation of 4-(2-Furyl)-3-buten-2-one over Different Re Phases

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
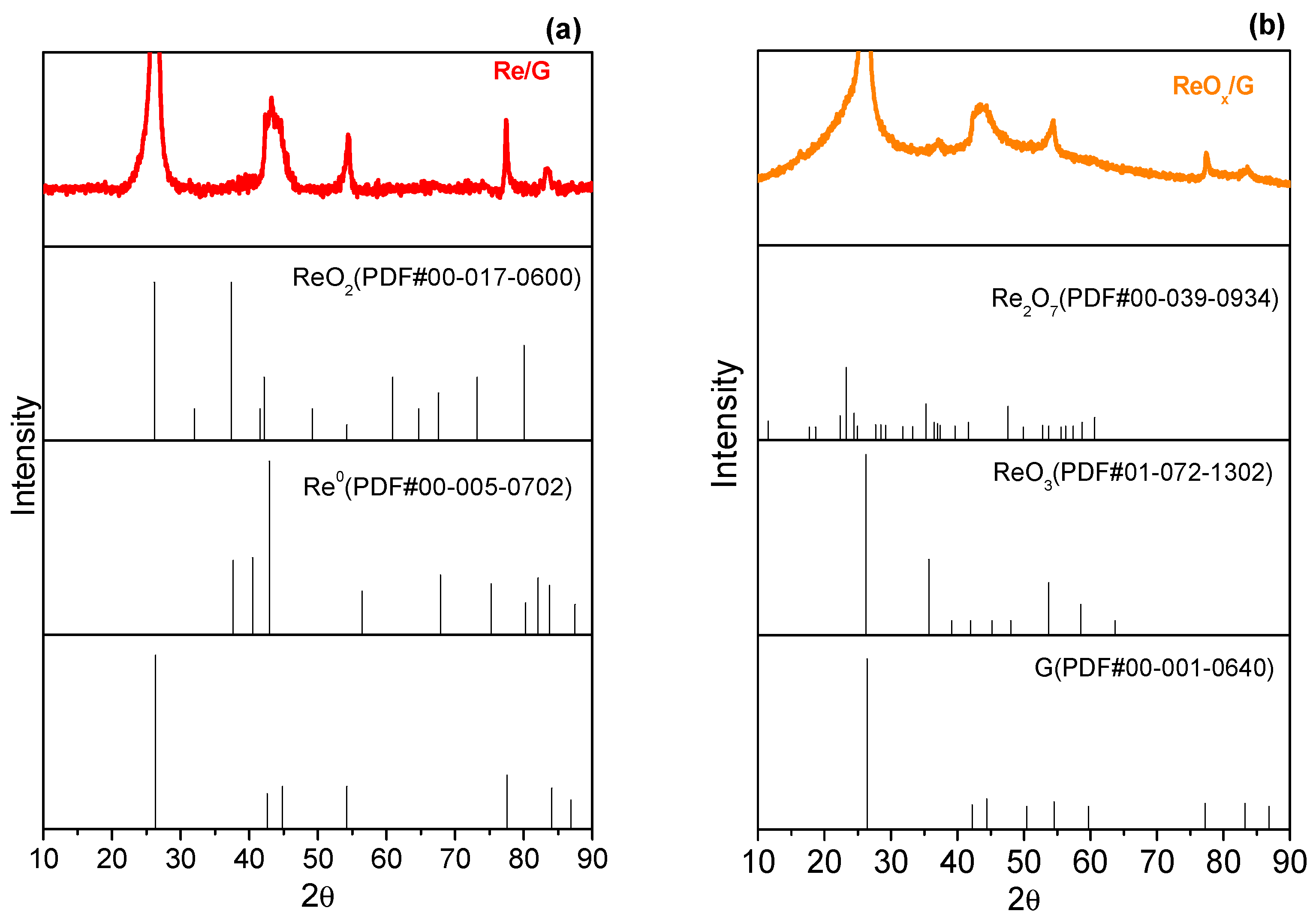
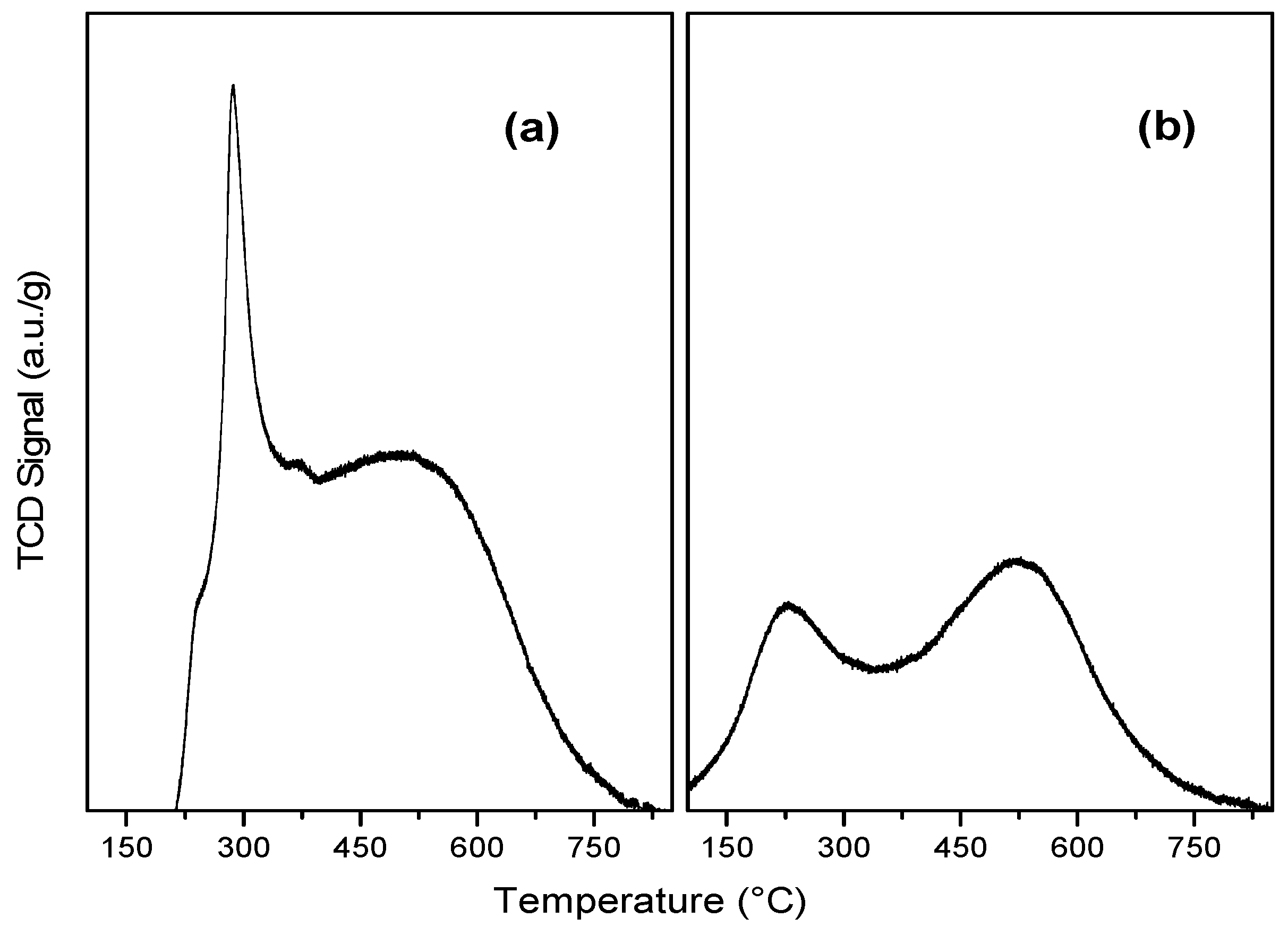
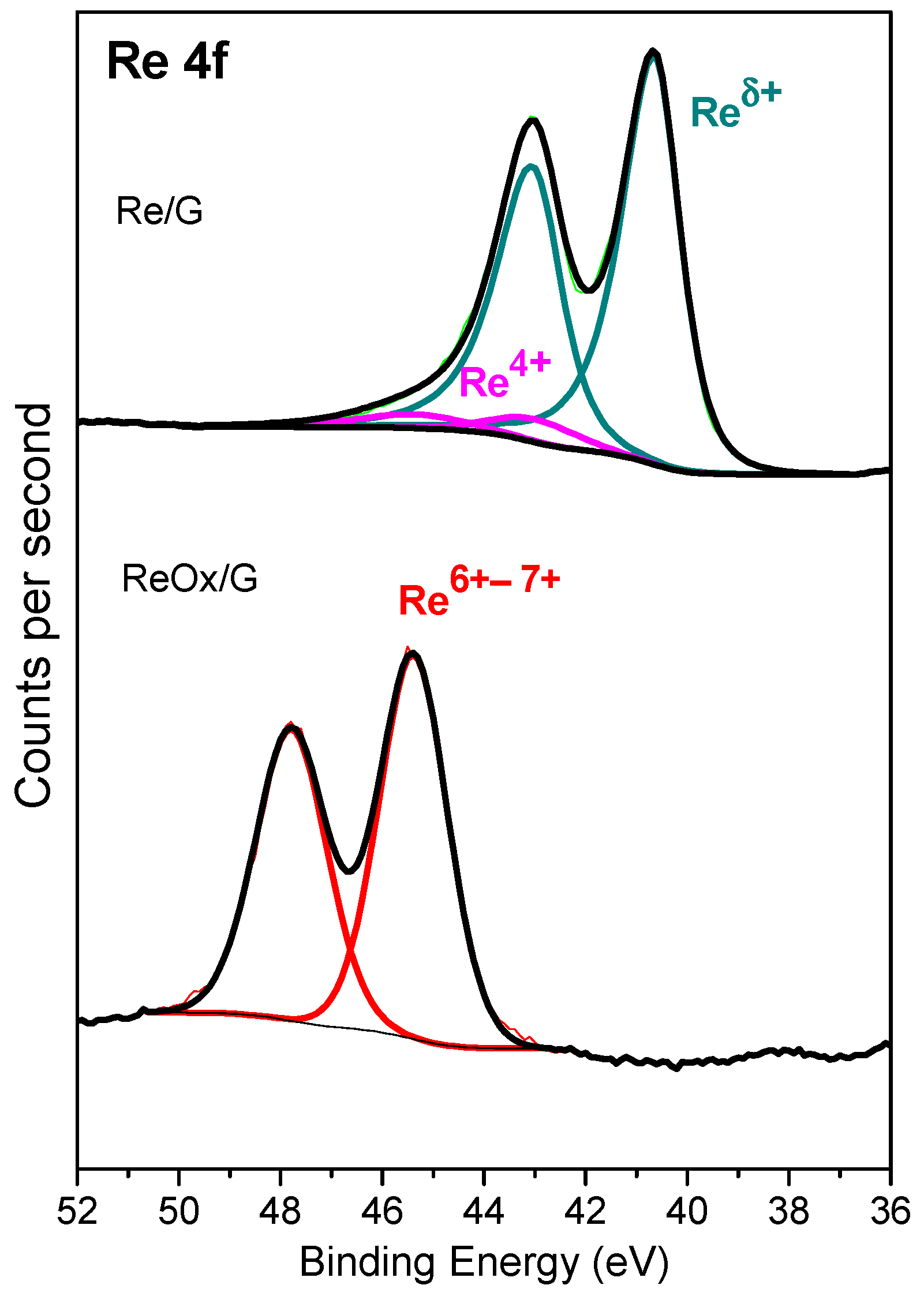
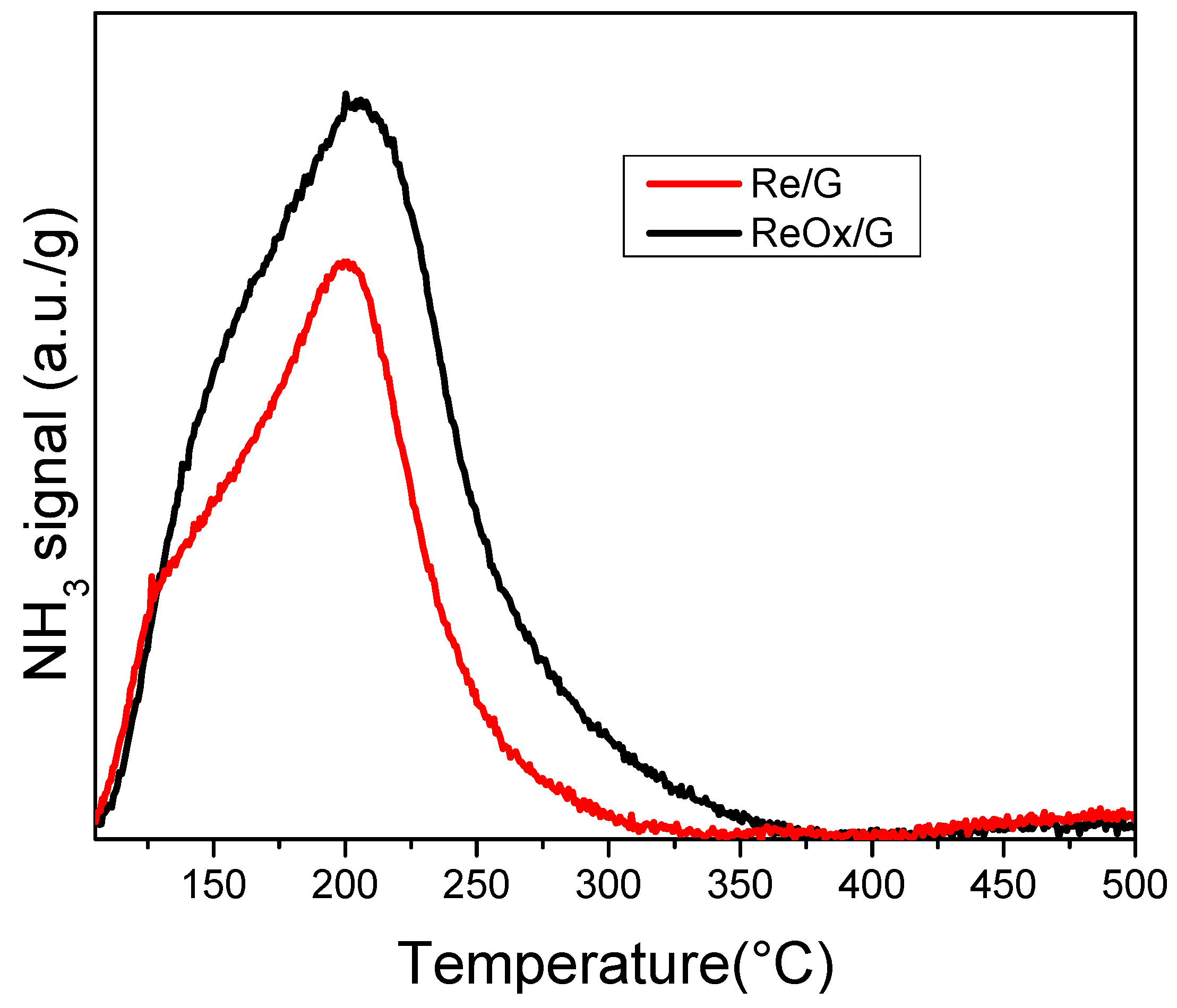
2.1. Characterization of Catalysts
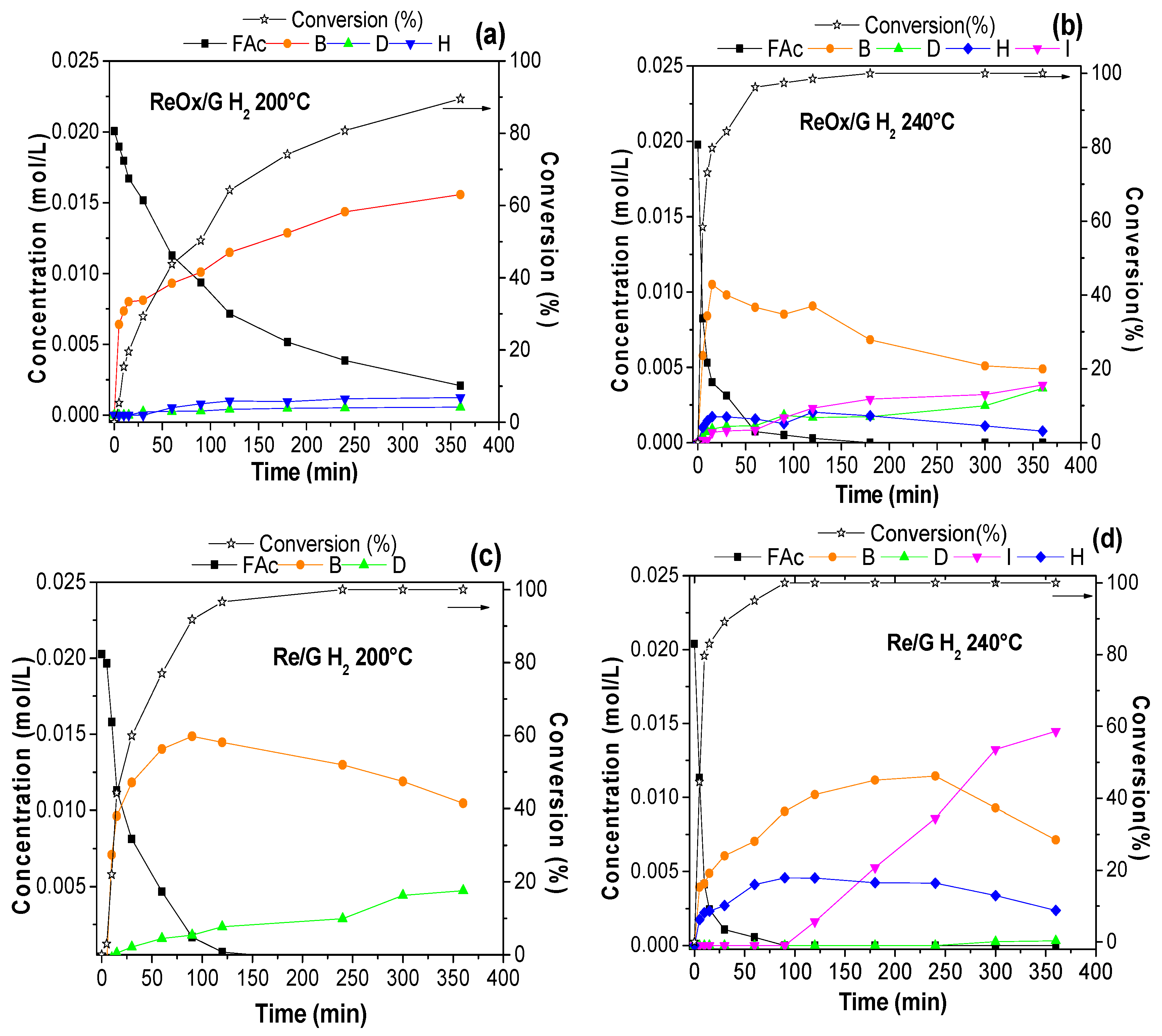
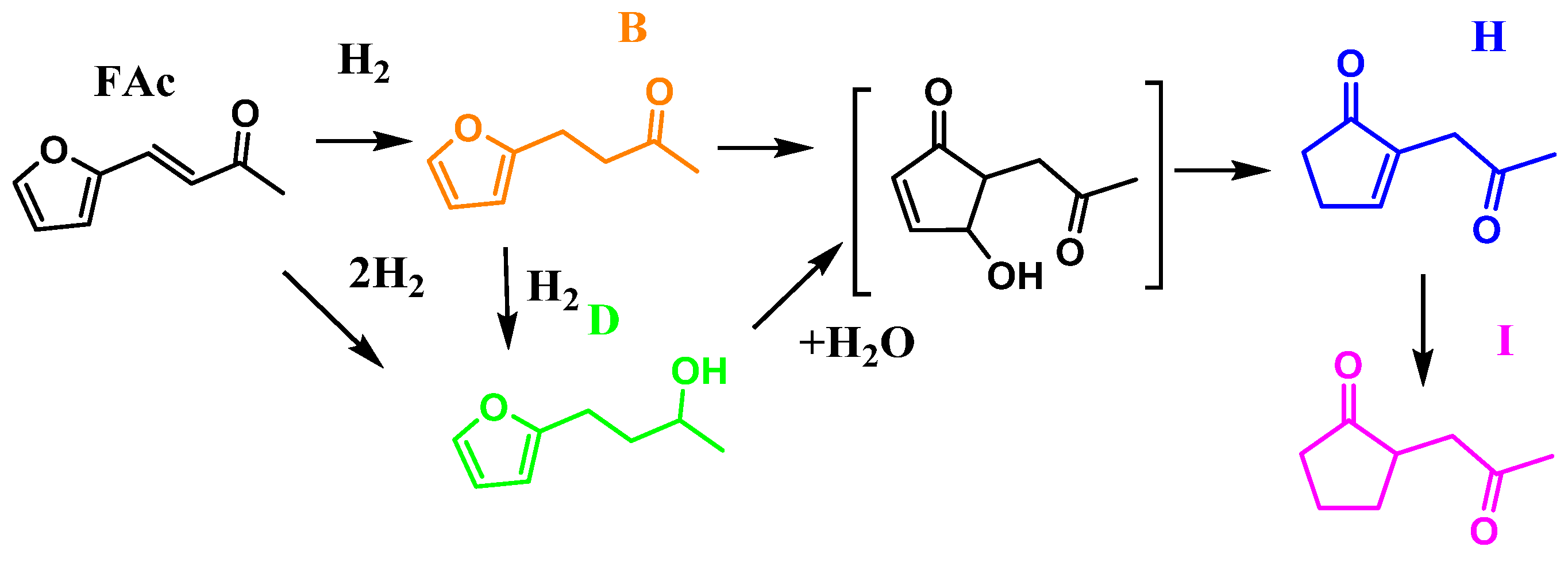
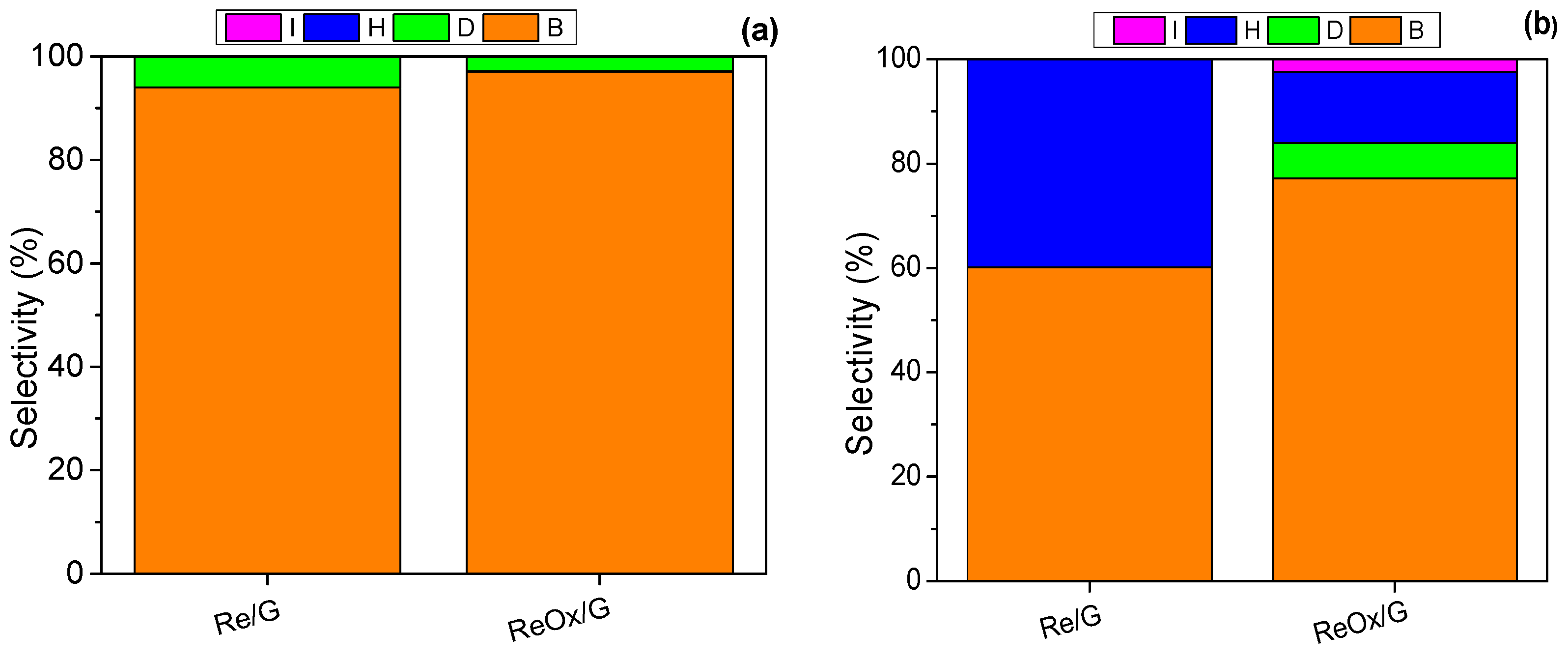
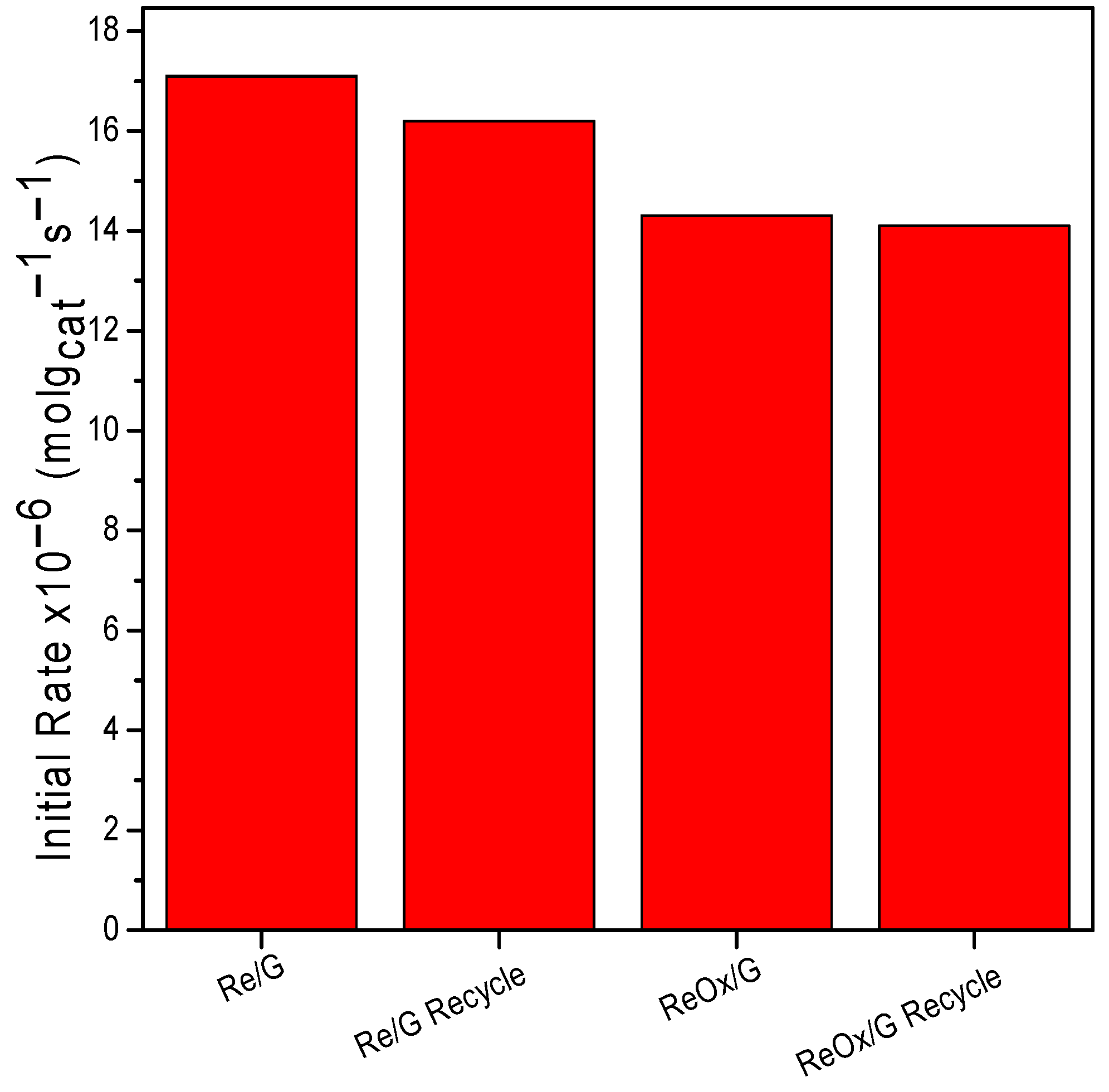
2.2. Catalytic Activity
3. Materials and Methods
3.1. Catalysts Preparation
3.2. Characterization of the Catalysts
3.3. Catalytic Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dawood, F.; Anda, M.; Shafiullah, G.M. Hydrogen production for energy: An overview. Int. J. Hydrog. Energy 2020, 45, 3847–3869. [Google Scholar] [CrossRef]
- Feng, J.; Hse, C.Y.; Yang, Z.; Wang, K.; Jiang, J.; Xu, J. Liquid phase in situ hydrodeoxygenation of biomass-derived phenolic compounds to hydrocarbons over bifunctional catalysts. Appl. Catal. A Gen. 2017, 542, 163–173. [Google Scholar] [CrossRef]
- Chandel, A.K.; Antunes, F.A.; Terán-Hilares, R.; Cota, J.; Ellilä, S.; Silveira, M.H.; dos Santos, J.C.; da Silva, S.S. Bioconversion of Hemicellulose Into Ethanol and Value-Added Products: Commercialization, Trends, and Future Opportunities. In Advances in Sugarcane Biorefinery: Technologies, Commercialization, Policy Issues and Paradigm Shift for Bioethanol and By-Products; Elsevier: Amsterdam, The Netherlands, 2018; pp. 97–134. [Google Scholar] [CrossRef]
- Iram, A.; Berenjian, A.; Demirci, A. A review on the utilization of lignin as a fermentation substrate to produce lignin-modifying enzymes and other value-added products. Molecules 2021, 26, 2960. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Pérez, M.A.; Serrano-Ruiz, J.C. Catalytic production of jet fuels from biomass. Molecules 2020, 25, 802. [Google Scholar] [CrossRef] [PubMed]
- Demirbas, A. Competitive liquid biofuels from biomass. Appl. Energy 2011, 88, 17–28. [Google Scholar] [CrossRef]
- Hongkailers, S.; Pattiya, A.; Hinchiranan, N. Hydrodeoxygenation of Oxygenates Derived from Biomass Pyrolysis Using Titanium Dioxide-Supported Cobalt Catalysts. Molecules 2023, 28, 7468. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.F.; Costa, D.; Ferraria, A.M.; Botelho do Rego, A.M.; Ribeiro, F.; Martins, Â.; Fernandes, A. Heterogenization of Heteropolyacid with Metal-Based Alumina Supports for the Guaiacol Gas-Phase Hydrodeoxygenation. Molecules 2023, 28, 2245. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wei, L.; Alsowij, M.R.; Corbin, F.; Julson, J.; Boakye, E.; Raynie, D. In situ hydrodeoxygenation upgrading of pine sawdust bio-oil to hydrocarbon biofuel using Pd/C catalyst. J. Energy Inst. 2018, 91, 163–171. [Google Scholar] [CrossRef]
- Vandevyvere, T.; Sabbe, M.K.; Smirnov, A.; Kikhtyanin, O.; Kubička, D.; Thybaut, J.W.; Lauwaert, J. Potential of (La-)NiCu catalysts for the hydrodeoxygenation of 4-(2-furanyl)-3-buten-2-one into fuels: Investigation of support and La addition. Appl. Catal. A Gen. 2024, 667, 119678. [Google Scholar] [CrossRef]
- Li, S.; Ma, Q.; Zhong, W.; Zhao, X.; Wei, X.; Zhang, X.; Liu, Q.; Wang, C.; Ma, L.; Zhang, Q. One-pot hydrodeoxygenation of bioderived furans into octane at low temperatures via an octanediol route. Green Chem. 2021, 23, 4741–4752. [Google Scholar] [CrossRef]
- Goepel, M.; Ramos, R.; Gläser, R.; Kubička, D. Novel polymer-silica composite-based bifunctional catalysts for hydrodeoxygenation of 4-(2-Furyl)-3-Buten-2-One as model substance for furfural-acetone aldol condensation products. Appl. Sci. 2019, 9, 2438. [Google Scholar] [CrossRef]
- Castillejos, E.; Serp, P. Carbon Nanotubes for Catalytic Applications. In Carbon Nanotubes and Related Structures; Wiley: Hoboken, NJ, USA, 2010; pp. 321–347. [Google Scholar] [CrossRef]
- Fernández-Morales, J.; Martínez-Quintana, L.; Dongil, A.; Asedegbega, E.; Castillejos, E.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Reactivity of n-butene using carbon supported Mo carbide and Ni, Mo, Co phosphides catalysts. Catal. Today 2023, 423, 114254–114262. [Google Scholar] [CrossRef]
- Faba, L.; Díaz, E.; Vega, A.; Ordóñez, S. Hydrodeoxygenation of furfural-acetone condensation adducts to tridecane over platinum catalysts. Catal. Today 2016, 269, 132–139. [Google Scholar] [CrossRef]
- Guerrero-Ruiz, A.; Badenes, P.; Rodrı, I. Study of some factors affecting the Ru and Pt dispersions over high surface area graphite-supported catalysts. Appl. Catal. A Gen. 1998, 173, 313–321. [Google Scholar] [CrossRef]
- Eslava, J.L.; Sun, X.; Gascon, J.; Kapteijn, F.; Rodríguez-Ramos, I. Ruthenium particle size and cesium promotion effects in Fischer–Tropsch synthesis over high-surface-area graphite supported catalysts. Catal. Sci. Technol. 2017, 7, 1235–1244. [Google Scholar] [CrossRef]
- Luo, J.; Liang, C. Rhenium in Heterogeneous Catalysis: A Rising Star for Hydrogenation Reactions. ACS Catal. 2024, 14, 7032–7049. [Google Scholar] [CrossRef]
- Toledo, F.; Ghampson, I.; Sepúlveda, C.; García, R.; Fierro, J.; Videla, A.; Serpell, R.; Escalona, N. Effect of Re content and support in the liquid phase conversion of furfural to furfuryl alcohol and 2-methyl furan over ReOx catalysts. Fuel 2019, 242, 532–544. [Google Scholar] [CrossRef]
- Leiva, K.; Leiva, K.; Garcia, R.; Garcia, R.; Sepulveda, C.; Sepulveda, C.; Laurenti, D.; Laurenti, D.; Geantet, C.; Geantet, C.; et al. Conversion of guaiacol over supported ReOx catalysts: Support. and metal loading effect. Catal. Today 2017, 296, 228–238. [Google Scholar] [CrossRef]
- Blanco, E.; Sepulveda, C.; Cruces, K.; García-Fierro, J.L.; Ghampson, I.T.; Escalona, N. Conversion of guaiacol over metal carbides supported on activated carbon catalysts. Catal. Today 2020, 356, 376–383. [Google Scholar] [CrossRef]
- Leiva, K.; Martinez, N.; Sepulveda, C.; García, R.; Jiménez, C.; Laurenti, D.; Vrinat, M.; Geantet, C.; Fierro, J.; Ghampson, I.; et al. Hydrodeoxygenation of 2-methoxyphenol over different Re active phases supported on SiO2 catalysts. Appl. Catal. A Gen. 2015, 490, 71–79. [Google Scholar] [CrossRef]
- Li, X.; Tong, Z.; Zhu, S.; Deng, Q.; Chen, S.; Wang, J.; Zeng, Z.; Zhang, Y.; Zou, J.-J.; Deng, S. Water-mediated hydrogen spillover accelerates hydrogenative ring-rearrangement of furfurals to cyclic compounds. J. Catal. 2022, 405, 363–372. [Google Scholar] [CrossRef]
- Ramos, R.; Tišler, Z.; Kikhtyanin, O.; Kubička, D. Solvent effects in hydrodeoxygenation of furfural-acetone aldol condensation products over Pt/TiO2 catalyst. Appl. Catal. A Gen. 2017, 530, 174–183. [Google Scholar] [CrossRef]
- Ramos, R.; Tišler, Z.; Kikhtyanin, O.; Kubička, D. Towards understanding the hydrodeoxygenation pathways of furfural–acetone aldol condensation products over supported Pt catalysts. Catal. Sci. Technol. 2016, 6, 1829–1841. [Google Scholar] [CrossRef]
- Faba, L.; Díaz, E.; Ordóñez, S. Performance of bifunctional Pd/MxNyO (M = Mg, Ca; N = Zr, Al) catalysts for aldolization–hydrogenation of furfural–acetone mixtures. Catal. Today 2011, 164, 451–456. [Google Scholar] [CrossRef]
- Donohue, M.D.; Aranovich, G.L. Classification of Gibbs adsorption isotherms. Adv. Colloid. Interface Sci. 1998, 76, 137–152. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Blanco, E.; Dongil, A.B.; Ghampson, I.T.; Escalona, N. Effect of the Support on Rhenium Carbide in the Hydrodeoxygenation of Guaiacol as Lignin-Derived Model Compound. Catalysts 2022, 12, 1229. [Google Scholar] [CrossRef]
- Kundu, S.; Wang, Y.; Xia, W.; Muhler, M. Thermal Stability and Reducibility of Oxygen-Containing Functional Groups on Multiwalled Carbon Nanotube Surfaces: A Quantitative High-Resolution XPS and TPD/TPR Study. J. Phys. Chem. C 2008, 112, 16869–16878. [Google Scholar] [CrossRef]
- Tamargo-Martínez, K.; Villar-Rodil, S.; Martínez-Alonso, A.; Tascón, J.M.D. Surface modification of high-surface area graphites by oxygen plasma treatments. Appl. Surf. Sci. 2022, 575, 151675. [Google Scholar] [CrossRef]
- Ting, K.W.; Mine, S.; El Fakir, A.A.; Du, P.; Li, L.; Siddiki, S.M.A.H.; Toyao, T.; Shimizu, K.-I. The reducibility and oxidation states of oxide-supported rhenium: Experimental and theoretical investigations. Phys. Chem. Chem. Phys. 2022, 24, 28621–28631. [Google Scholar] [CrossRef]
- Arnoldy, P.; van Oers, E.M.; Bruinsma, O.S.L.; de Beer, V.H.J.; Moulijn, J.A. Temperature-Programmed Reduction of Al2O3-, SiO2-, and carbon-supported Re2O7 catalysts. J. Catal. 1985, 93, 231–245. [Google Scholar] [CrossRef]
- Soto, G.; Tiznado, H.; Díaz, J.A.; Samano, E.C.; Reyes-Serrato, A. Evaluation of rhenium carbide as a prospective material for hard coating. Thin Solid. Films 2011, 519, 3236–3241. [Google Scholar] [CrossRef]
- Greiner, M.T.; Rocha, T.C.R.; Johnson, B.; Klyushin, A.; Knop-Gericke, A.; Schlögl, R. The Oxidation of Rhenium and Identification of Rhenium Oxides During Catalytic Partial Oxidation of Ethylene: An In-Situ XPS Study. Z. Für Phys. Chem. 2014, 228, 521–541. [Google Scholar] [CrossRef]
- Okal, J.; Tylus, W.; Kȩpiński, L. XPS study of oxidation of rhenium metal on γ-Al2O3 support. J. Catal. 2004, 225, 498–509. [Google Scholar] [CrossRef]
- Vorakitkanvasin, S.; Phongsawat, W.; Suriye, K.; Praserthdam, P.; Panpranot, J. In situ-DRIFTS study: Influence of surface acidity of rhenium-based catalysts in the metathesis of various olefins for propylene production. RSC Adv. 2017, 7, 38659–38665. [Google Scholar] [CrossRef]
- Tong, Z.; Li, X.; Dong, J.; Gao, R.; Deng, Q.; Wang, J.; Zeng, Z.; Zou, J.J.; Deng, S. Adsorption Configuration-Determined Selective Hydrogenative Ring Opening and Ring Rearrangement of Furfural over Metal. Phosphate. ACS Catal. 2021, 11, 6406–6415. [Google Scholar] [CrossRef]
- Ohyama, J.; Kanao, R.; Esaki, A.; Satsuma, A. Conversion of 5-hydroxymethylfurfural to a cyclopentanone derivative by ring rearrangement over supported Au nanoparticles. Chem. Commun. 2014, 50, 5633–5636. [Google Scholar] [CrossRef] [PubMed]
- Ghampson, I.T.; Sepúlveda, C.; García, R.; Fierro, J.L.G.; Escalona, N. Carbon nanofiber-supported ReOx catalysts for the hydrodeoxygenation of lignin-derived compounds. Catal. Sci. Technol. 2016, 6, 4356–4369. [Google Scholar] [CrossRef]
- Li, X.; Zhang, L.; Zhou, R.; Chen, S.; Wang, J.; Zeng, Z.; Zou, J.J.; Deng, S.; Deng, Q. Bifunctional Role of Hydrogen in Aqueous Hydrogenative Ring Rearrangement of Furfurals over Co@Co-NC. ACS Sustain. Chem. Eng. 2022, 10, 7321–7329. [Google Scholar] [CrossRef]
- Harth, F.M.; Likozar, B.; Grilc, M. Stability of solid rhenium catalysts for liquid-phase biomass valorization–various facets of catalyst deactivation and rhenium leaching. Mater. Today Chem. 2022, 26, 101191. [Google Scholar] [CrossRef]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the bet equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 2007, 160, 49–56. [Google Scholar] [CrossRef]
- Singleton, J.H.; Halsey, G.D., Jr. The Growth Of Crystalline Layers On Foreign Surfaces. Can. J. Chem. 1955, 33, 184–192. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Orfao, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2/g) | Vp (cm3/g) | Vm (cm3/g) | Vµ (cm3/g) | Dp (nm) |
|---|---|---|---|---|---|
| G | 430 | 0.87 | 0.841 | 0.040 | 5.9 |
| ReOx/G | 331 | 0.54 | 0.513 | 0.027 | 6.5 |
| Re/G | 283 | 0.51 | 0.488 | 0.022 | 7.3 |
| Phases | Re/G | ReOx/G | ||
|---|---|---|---|---|
| BE, eV (%) | Re 4f7/2 | Reδ+ | 40.6 (93) | - |
| Re4+ | 43.0(7) | - | ||
| Re6–7+ | - | 45.4 (100) | ||
| Re/C Ratio | - | 0.0346 | 0.0067 | |
| O/Re Ratio | 0.92 | 6.8 | ||
| Samples | Total Acid Sites (10−5 mol NH3·g−1) |
|---|---|
| Re/G | 1.1 |
| ReOx/G | 1.9 |
| Samples | Reaction Temperature | r0 (×10−6 mol·gcat−1·s−1) |
|---|---|---|
| Re/G | 200 | 4.41 |
| ReOx/G | 1.88 | |
| Re/G | 240 | 17.3 |
| ReOx/G | 14.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, C.I.C.; Araya-López, C.; Dongil, A.B.; Escalona, N. Aqueous Phase Hydrogenation of 4-(2-Furyl)-3-buten-2-one over Different Re Phases. Molecules 2024, 29, 3853. https://doi.org/10.3390/molecules29163853
Díaz CIC, Araya-López C, Dongil AB, Escalona N. Aqueous Phase Hydrogenation of 4-(2-Furyl)-3-buten-2-one over Different Re Phases. Molecules. 2024; 29(16):3853. https://doi.org/10.3390/molecules29163853
Chicago/Turabian StyleDíaz, Claudio Ignacio C., Claudio Araya-López, A. B. Dongil, and Nestor Escalona. 2024. "Aqueous Phase Hydrogenation of 4-(2-Furyl)-3-buten-2-one over Different Re Phases" Molecules 29, no. 16: 3853. https://doi.org/10.3390/molecules29163853