Current Strategy for Targeting Metallo-β-Lactamase with Metal-Ion-Binding Inhibitors

 , , , ,
, , , , 
Abstract
:1. Introduction
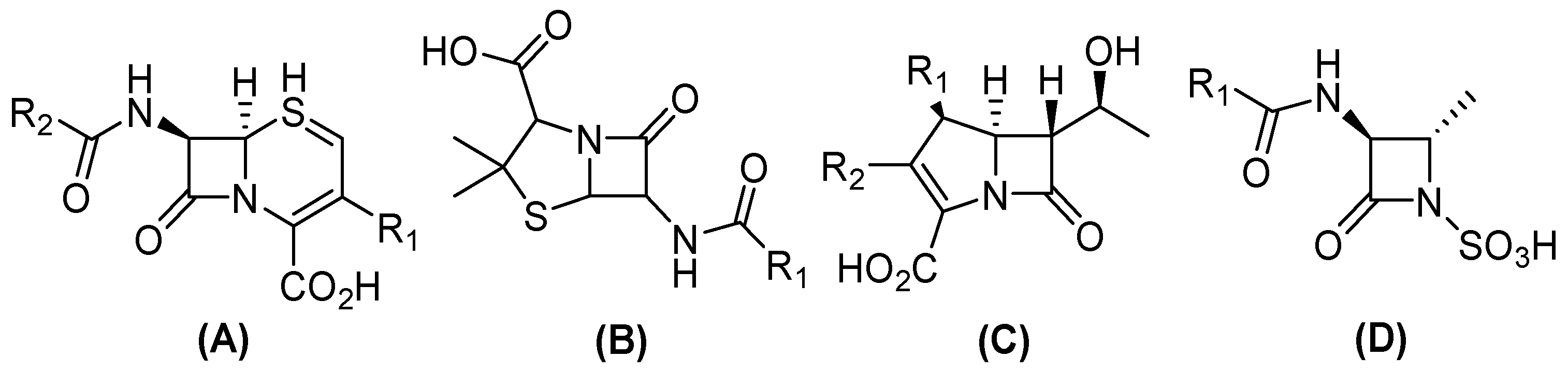
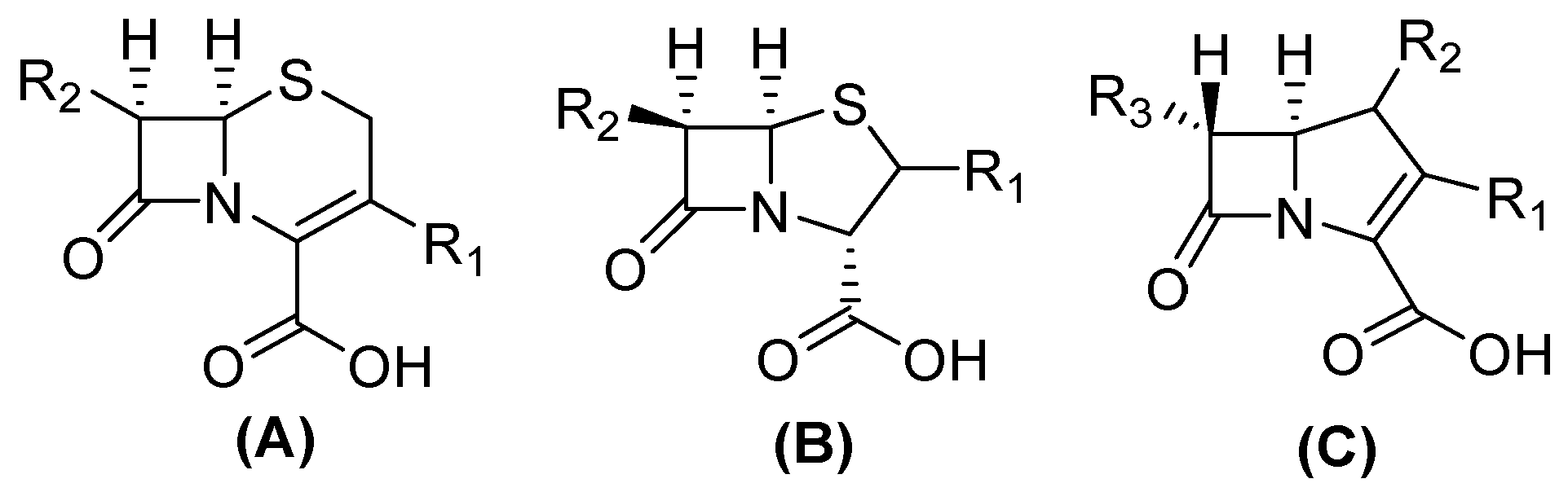
2. Beta (β)-Lactam and Carbapenem Antibiotics
3. Beta(β)-Lactamases
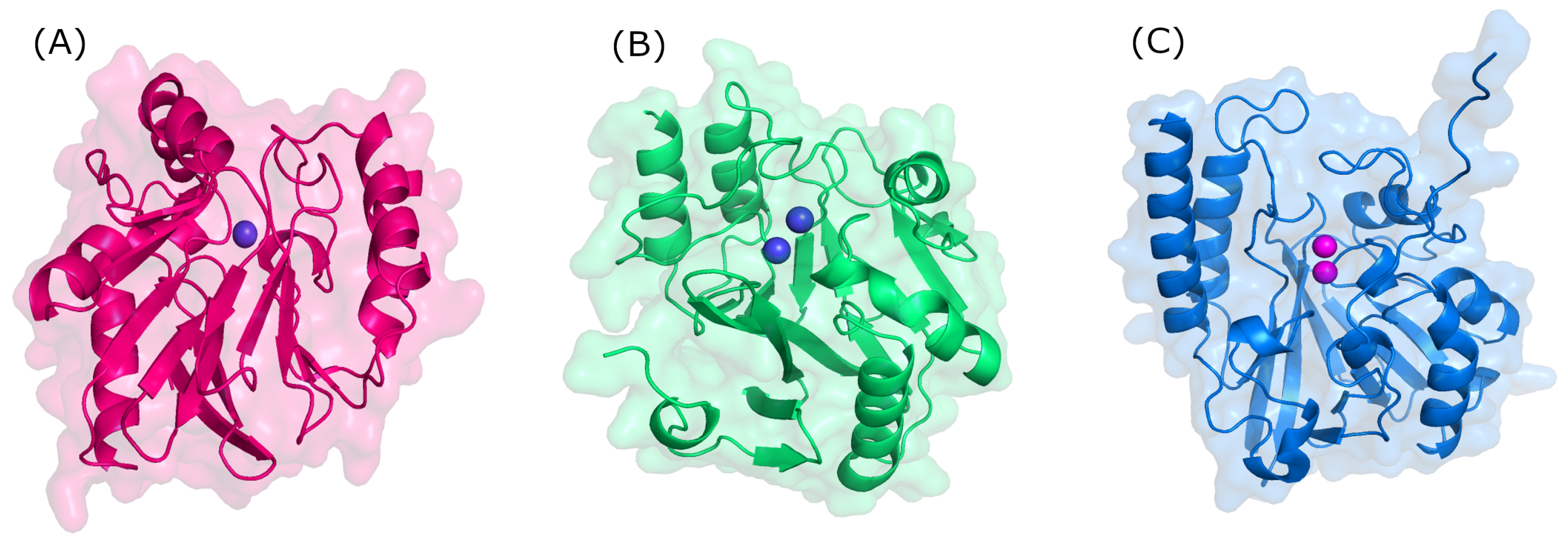
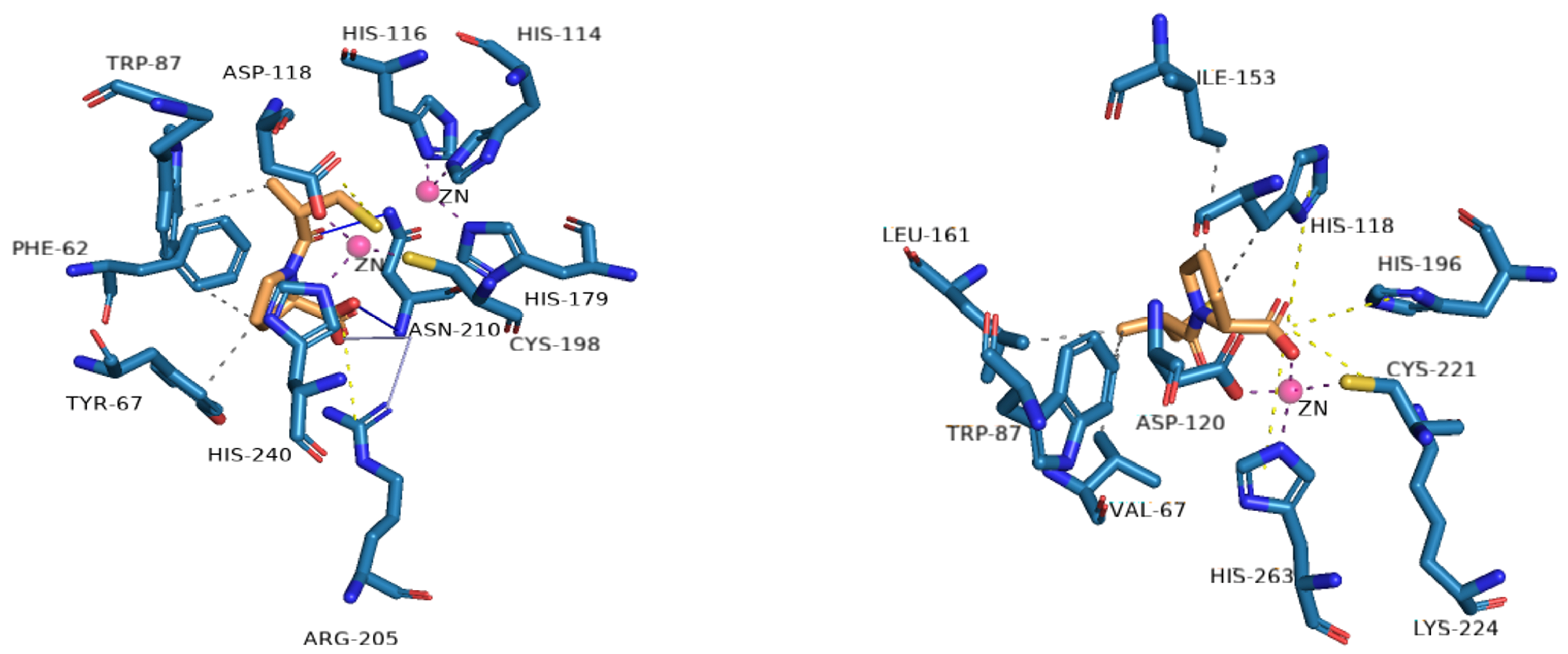
4. Metallo-Beta-Lactamases (MBLs) as Drug Targets
5. MBL Inhibitors
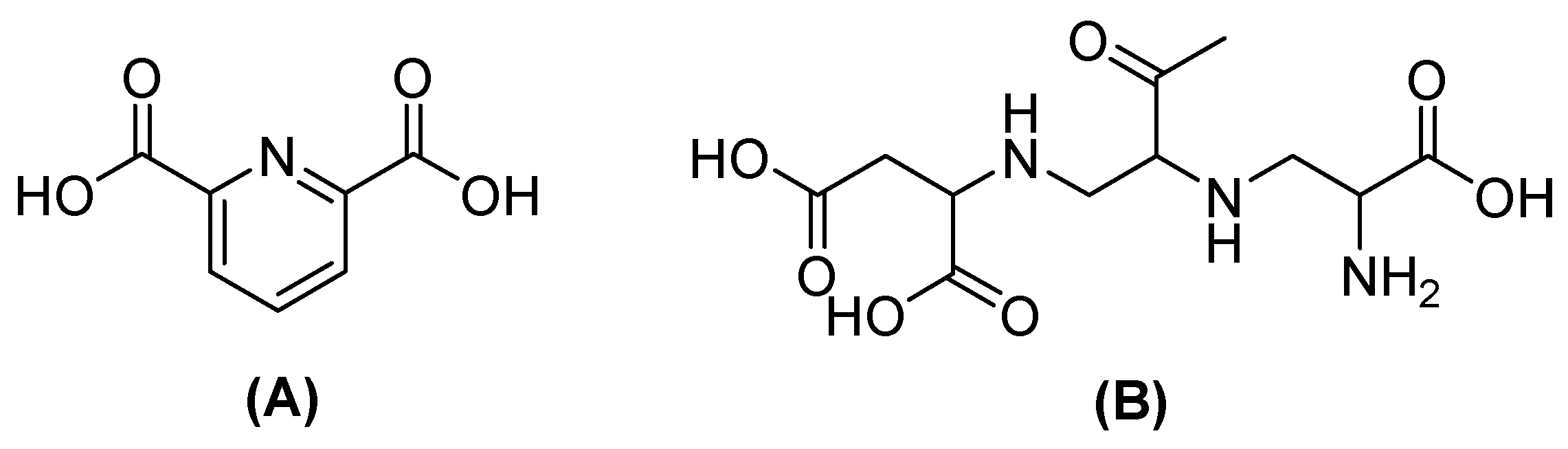
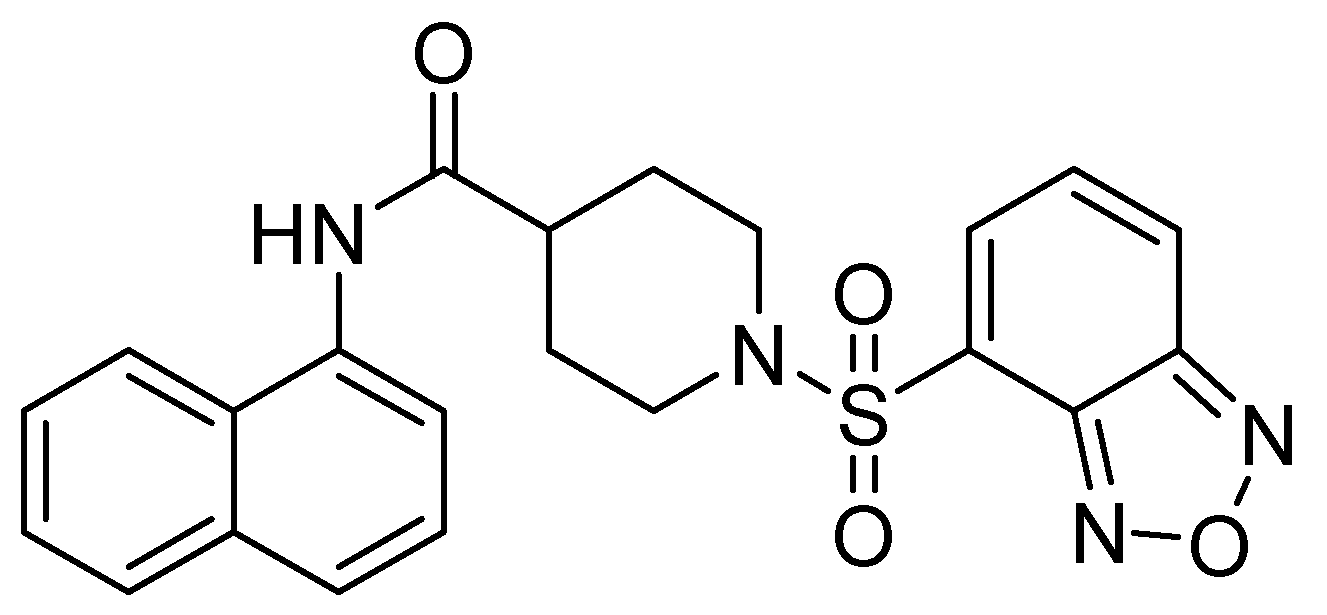
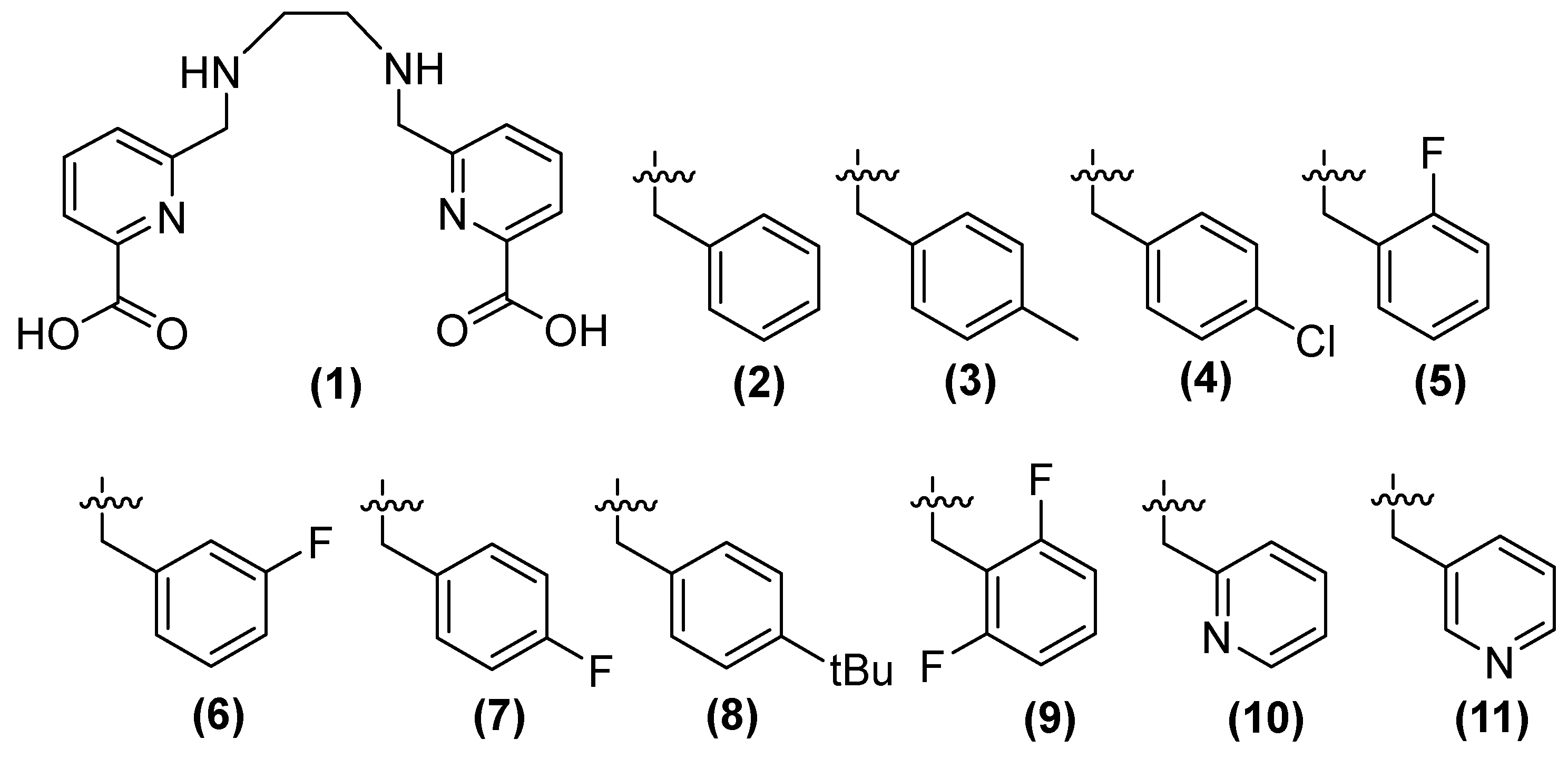
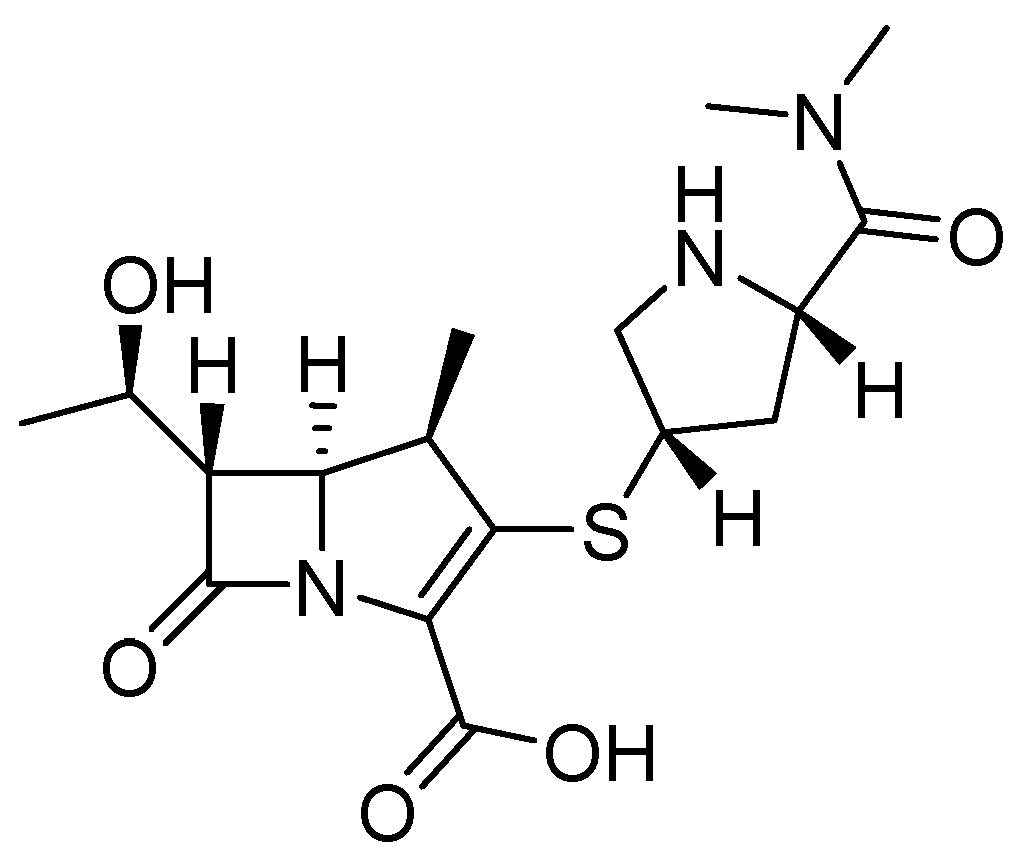
5.1. Metal-Complex-Forming Inhibitors
5.2. Formation of Ternary Complexes
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. (The Review on Antimicrobial Resistance, London, 2016, United Kingdom). Rev. Antimicrob. Resist. 2014. [Google Scholar]
- Palm, M.; Fransson, A.; Hult, J.; Karolina, B.; Allouche, A.; Chiang, O.E.; Constandse, M.L.; Van Dijk, K.J.; Icli, S.; Klimesova, B.; et al. The Effect of Heavy Metals on Conjugation Efficiency of an F-Plasmid in Escherichia Coli. Antibiotics 2022, 11, 1123. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.Y. Antimicrobial Peptides: New Hope in the War against Multidrug Resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [PubMed]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic Resistance Genes in Bacteria: Occurrence, Spread, and Control. J. Basic Microbiol. 2021, 61, 1049–1070. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.E.; Palm, M.; Warringer, J.; Farewell, A. Inhibiting Conjugation as a Tool in the Fight against Antibiotic Resistance. Drug Dev. Res. 2019, 80, 19–23. [Google Scholar] [CrossRef]
- Li, W.; Zhang, G. Detection and Various Environmental Factors of Antibiotic Resistance Gene Horizontal Transfer. Environ. Res. 2022, 212, 113267. [Google Scholar] [CrossRef]
- Arer, V.; Kar, D. Biochemical Exploration of β-Lactamase Inhibitors. Front. Genet. 2023, 13, 1–15. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar]
- De Angelis, G.; Del Giacomo, P.; Posteraro, B.; Sanguinetti, M.; Tumbarello, M. Molecular Mechanisms, Epidemiology, and Clinical Importance of β-Lactam Resistance in Enterobacteriaceae. Int. J. Mol. Sci. 2020, 21, 5090. [Google Scholar] [CrossRef]
- Zhang, S.; Liao, X.; Ding, T.; Ahn, J. Role of β-Lactamase Inhibitors as Potentiators in Antimicrobial Chemotherapy Targeting Gram-Negative Bacteria. Antibiotics 2024, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2023 WHO Releases Report on State of Development of Antibacterials. Available online: https://www.who.int/news/item/14-06-2024-who-releases-report-on-state-of-development-of-antibacterials (accessed on 1 February 2024).
- Aurilio, C.; Sansone, P.; Barbarisi, M.; Pota, V.; Giaccari, L.G.; Coppolino, F.; Barbarisi, A.; Passavanti, M.B.; Pace, M.C. Mechanisms of Action of Carbapenem Resistance. Antibiotics 2022, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Bahr, G.; González, L.J.; Vila, A.J. Metallo-β-Lactamases in the Age of Multidrug Resistance: From Structure and Mechanism to Evolution, Dissemination, and Inhibitor Design. Chem. Rev. 2021, 121, 7957–8094. [Google Scholar]
- Hernández-Alomía, F.; Bastidas-Caldes, C.; Ballesteros, I.; Tenea, G.N.; Jarrín-V, P.; Molina, C.A.; Castillejo, P. Beta-Lactam Antibiotic Resistance Genes in the Microbiome of the Public Transport System of Quito, Ecuador. Int. J. Environ. Res. Public Health 2023, 20, 1900. [Google Scholar] [CrossRef] [PubMed]
- Iovleva, A.; Doi, Y. Carbapenem-Resistant Enterobacteriaceae. Clin. Lab. Med. 2017, 37, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, H.O.; Soliman, A.M.; Ahmed, A.M.; Shimamoto, T.; Hara, T.; Ikeda, M.; Kuroo, Y.; Kayama, S.; Sugai, M.; Shimamoto, T. High Carbapenem Resistance in Clinical Gram-Negative Pathogens Isolated in Egypt. Microb. Drug Resist. 2017, 23, 838–844. [Google Scholar] [CrossRef]
- Armstrong, T.; Fenn, S.J.; Hardie, K.R. JMM Profile: Carbapenems: A Broad-Spectrum Antibiotic. J. Med. Microbiol. 2021, 70, 001462. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-Negative Bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Song, X.; Li, M.; Yu, Z.; Cheng, W.; Yu, Z.; Zhang, W.; Zhang, Y.; Shen, A.; Sun, H.; et al. Global Spread of Carbapenem-Resistant Enterobacteriaceae: Epidemiological Features, Resistance Mechanisms, Detection and Therapy. Microbiol. Res. 2023, 266, 127249. [Google Scholar] [CrossRef]
- Doi, Y. Treatment Options for Carbapenem-Resistant Gram-Negative Bacterial Infections. Clin. Infect. Dis. 2019, 69, S565–S575. [Google Scholar] [CrossRef]
- Noster, J.; Thelen, P.; Hamprecht, A. Detection of Multidrug-resistant Enterobacterales—From Esbls to Carbapenemases. Antibiotics 2021, 10, 1140. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lee, J.H.; Lee, J.J.; Park, K.S.; Karim, A.M.; Lee, C.R.; Jeong, B.C.; Lee, S.H. Structural Basis for Carbapenem-Hydrolyzing Mechanisms of Carbapenemases Conferring Antibiotic Resistance. Int. J. Mol. Sci. 2015, 16, 9654–9692. [Google Scholar] [CrossRef] [PubMed]
- Edoo, Z.; Arthur, M.; Hugonnet, J.E. Reversible Inactivation of a Peptidoglycan Transpeptidase by a β-Lactam Antibiotic Mediated by β-Lactam-Ring Recyclization in the Enzyme Active Site. Sci. Rep. 2017, 7, 9136. [Google Scholar] [CrossRef]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Cascella, M. Beta-Lactam Antibiotics. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [PubMed]
- Jalde, S.S.; Choi, H.K. Recent Advances in the Development of β-Lactamase Inhibitors. J. Microbiol. 2020, 58, 633–647. [Google Scholar] [PubMed]
- Feng, H.; Liu, X.; Wang, S.; Fleming, J.; Wang, D.C.; Liu, W. The Mechanism of NDM-1-Catalyzed Carbapenem Hydrolysis Is Distinct from That of Penicillin or Cephalosporin Hydrolysis. Nat. Commun. 2017, 8, 2242. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-Lactamase-Producing Pathogens. Clin. Microbiol. Rev. 2020, 33, e00047-19. [Google Scholar]
- van Haren, M.J.; Tehrani, K.H.M.E.; Kotsogianni, I.; Wade, N.; Brüchle, N.C.; Mashayekhi, V.; Martin, N.I. Cephalosporin Prodrug Inhibitors Overcome Metallo-β-Lactamase Driven Antibiotic Resistance. Chemistry 2021, 27, 3806–3811. [Google Scholar] [CrossRef]
- Chiou, J.; Leung, T.Y.C.; Chen, S. Molecular Mechanisms of Substrate Recognition and Specificity of New Delhi Metallo-β-Lactamase. Antimicrob. Agents Chemother. 2014, 58, 5372–5378. [Google Scholar] [CrossRef] [PubMed]
- Lisa, M.-N.; Palacios, A.R.; Aitha, M.; González, M.M.; Moreno, D.M.; Crowder, M.W.; Bonomo, R.A.; Spencer, J.; Tierney, D.L.; Llarrull, L.I.; et al. A General Reaction Mechanism for Carbapenem Hydrolysis by Mononuclear and Binuclear Metallo-β-Lactamases. Nat. Commun. 2017, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.E.; Livermore, D.M.; Hooper, D.C.; Hope, W.W. Metallo-β-Lactamases: Structure, Function, Epidemiology, Treatment Options, and the Development Pipeline. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Palzkill, T. Metallo-β-Lactamase Structure and Function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef]
- Yan, Y.H.; Li, G.; Li, G.B. Principles and Current Strategies Targeting Metallo-β-Lactamase Mediated Antibacterial Resistance. Med. Res. Rev. 2020, 40, 1558–1592. [Google Scholar] [PubMed]
- Palacios, A.R.; Rossi, M.-A.; Mahler, G.S.; Vila, A.J. Metallo-β-Lactamase Inhibitors Inspired on Snapshots from the Catalytic Mechanism. Biomolecules 2020, 10, 854. [Google Scholar] [CrossRef]
- Hu, Z.; Gunasekera, T.S.; Spadafora, L.; Bennett, B.; Crowder, M.W. Metal Content of Metallo-β-Lactamase L1 Is Determined by the Bioavailability of Metal Ions. Biochemistry 2008, 47, 7947–7953. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tanaka, H.; Kurisu, G.; Nakano, R.; Yano, H.; Sakai, H. Structural Insights into the Substrate Specificity of IMP-6 and IMP-1 Metallo-β-Lactamases. J. Biochem. 2023, 173, 21–30. [Google Scholar] [CrossRef]
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular Diversity of Extended-Spectrum β-Lactamases and Carbapenemases, and Antimicrobial Resistance. J. Intensive Care 2020, 8, 13. [Google Scholar]
- Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. Int. J. Mol. Sci. 2020, 21, 9308. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, K.H.M.E.; Brüchle, N.C.; Wade, N.; Mashayekhi, V.; Pesce, D.; van Haren, M.J.; Martin, N.I. Small Molecule Carboxylates Inhibit Metallo-β-Lactamases and Resensitize Carbapenem-Resistant Bacteria to Meropenem. ACS Infect. Dis. 2020, 6, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-Lactamase (NDM): A Threat to Public Health. BMC Microbiol. 2017, 17, 101. [Google Scholar]
- Ju, L.C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar]
- Gavara, L.; Verdirosa, F.; Sevaille, L.; Legru, A.; Corsica, G.; Nauton, L.; Sandra Mercuri, P.; Sannio, F.; De Luca, F.; Hadjadj, M.; et al. 1,2,4-Triazole-3-Thione Analogues with an Arylakyl Group at Position 4 as Metallo-β-Lactamase Inhibitors. Bioorg. Med. Chem. 2022, 72, 116964. [Google Scholar] [CrossRef]
- Sychantha, D.; Rotondo, C.M.; Tehrani, K.H.M.E.; Martin, N.I.; Wright, G.D. Aspergillomarasmine A Inhibits Metallo-β-Lactamases by Selectively Sequestering Zn(2). J. Biol. Chem. 2021, 297, 100918. [Google Scholar] [CrossRef]
- Hinchliffe, P.; Tanner, C.A.; Krismanich, A.P.; Labbé, G.; Goodfellow, V.J.; Marrone, L.; Desoky, A.Y.; Calvopiña, K.; Whittle, E.E.; Zeng, F.; et al. Structural and Kinetic Studies of the Potent Inhibition of Metallo-β-Lactamases by 6-Phosphonomethylpyridine-2-Carboxylates. Biochemistry 2018, 57, 1880–1892. [Google Scholar] [CrossRef]
- Thomas, C.S.; Braun, D.R.; Olmos, J.L.; Rajski, S.R.; Phillips, G.N.; Andes, D.N.; Bugni, T.S. Pyridine-2,6-Dithiocarboxylic Acid and Its Metal Complexes: New Inhibitors of New Delhi Metallo-Lactamase-1. Mar. Drugs 2020, 18, 295. [Google Scholar] [CrossRef]
- Wang, X.; Lu, M.; Shi, Y.; Ou, Y.; Cheng, X. Discovery of Novel New Delhi Metallo-β-Lactamases-1 Inhibitors by Multistep Virtual Screening. PLoS ONE 2015, 10, e0118290. [Google Scholar] [CrossRef]
- Cui, D.Y.; Yang, Y.; Bai, M.M.; Han, J.X.; Wang, C.C.; Kong, H.T.; Shen, B.Y.; Yan, D.C.; Xiao, C.L.; Liu, Y.S.; et al. Systematic Research of H2dedpa Derivatives as Potent Inhibitors of New Delhi Metallo-β-Lactamase-1. Bioorg. Chem. 2020, 101, 103965. [Google Scholar] [CrossRef]
- Chen, F.; Bai, M.; Liu, W.; Kong, H.; Zhang, T.; Yao, H.; Zhang, E.; Du, J.; Qin, S. H2dpa Derivatives Containing Pentadentate Ligands: An Acyclic Adjuvant Potentiates Meropenem Activity in Vitro and in Vivo against Metallo-β-Lactamase-Producing Enterobacterales. Eur. J. Med. Chem. 2021, 224, 113702. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.-C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.G.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase, Serine-β-Lactamase and Penicillin-Binding Protein Inhibition by Cyclic Boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.T.; Tyrrell, J.M.; Navratilova, I.H.; Calvopiña, K.; Robinson, S.W.; Lohans, C.T.; McDonough, M.A.; Cain, R.; Fishwick, C.W.G.; Avison, M.B.; et al. Studies on the Inhibition of AmpC and Other β-Lactamases by Cyclic Boronates. Biochim. Biophys. Acta-Gen. Subj. 2019, 1863, 742–748. [Google Scholar] [CrossRef]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopiña, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.J.A.G.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Interplay between β-Lactamases and New β-Lactamase Inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef]
- Behzadi, P.; García-Perdomo, H.A.; Karpiński, T.M.; Issakhanian, L. Metallo-ß-Lactamases: A Review. Mol. Biol. Rep. 2020, 47, 6281–6294. [Google Scholar]
- Kloezen, W.; Melchers, R.J.; Georgiou, P.-C.; Mouton, J.W.; Meletiadis, J. Activity of Cefepime in Combination with the Novel β-Lactamase Inhibitor Taniborbactam (VNRX-5133) against Extended-Spectrum-β-Lactamase-Producing Isolates in In Vitro Checkerboard Assays. Antimicrob. Agents Chemother. 2021, 65, e02338-20. [Google Scholar] [CrossRef] [PubMed]
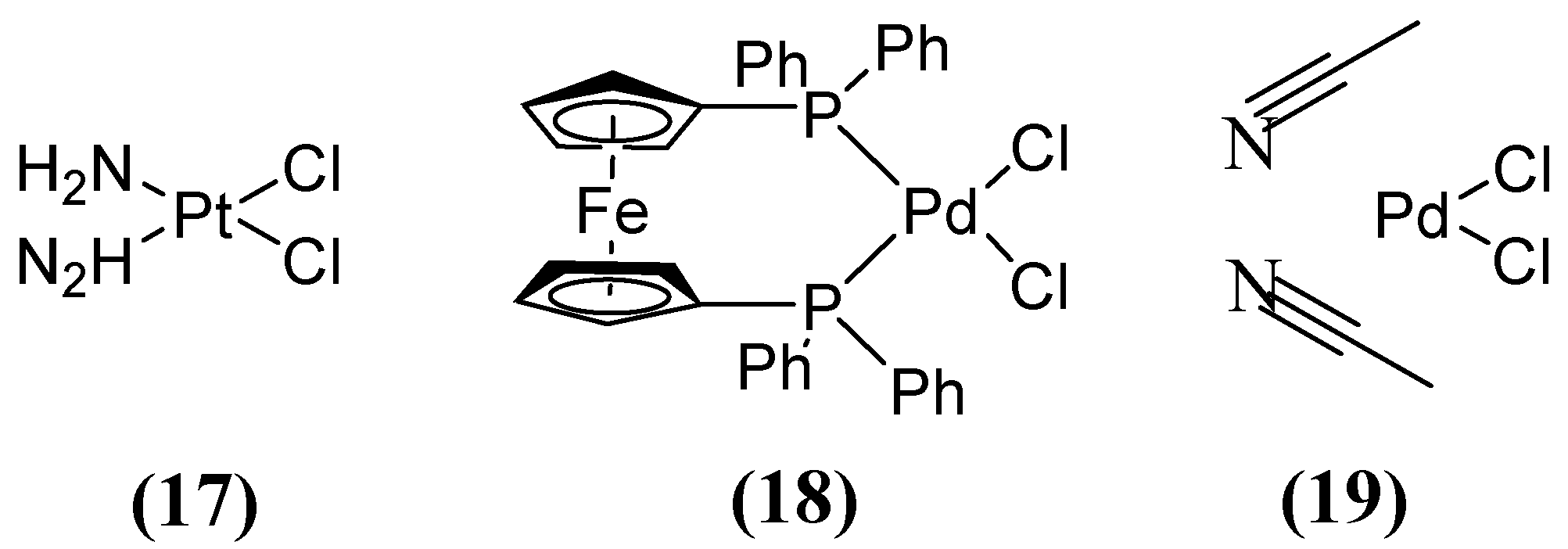
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Chen, C.; Sun, L.Y.; Gao, H.; Kang, P.W.; Li, J.Q.; Zhen, J.B.; Yang, K.W. Identification of Cisplatin and Palladium(II) Complexes as Potent Metallo-β-Lactamase Inhibitors for Targeting Carbapenem-Resistant Enterobacteriaceae. ACS Infect. Dis. 2020, 6, 975–985. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; De Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine a Overcomes Metallo-β-Lactamase Antibiotic Resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef]
- Rotondo, C.M.; Sychantha, D.; Koteva, K.; Wright, G.D. Suppression of β-Lactam Resistance by Aspergillomarasmine A Is Influenced by Both the Metallo-β-Lactamase Target and the Antibiotic Partner. Antimicrob. Agents Chemother. 2020, 64, e01386-19. [Google Scholar] [CrossRef]
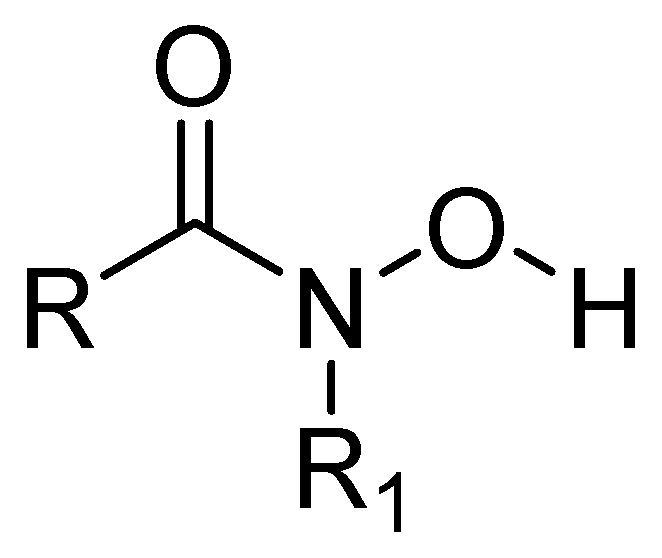
- Citarella, A.; Moi, D.; Pinzi, L.; Bonanni, D.; Rastelli, G. Hydroxamic Acid Derivatives: From Synthetic Strategies to Medicinal Chemistry Applications. ACS Omega 2021, 6, 21843–21849. [Google Scholar] [CrossRef]
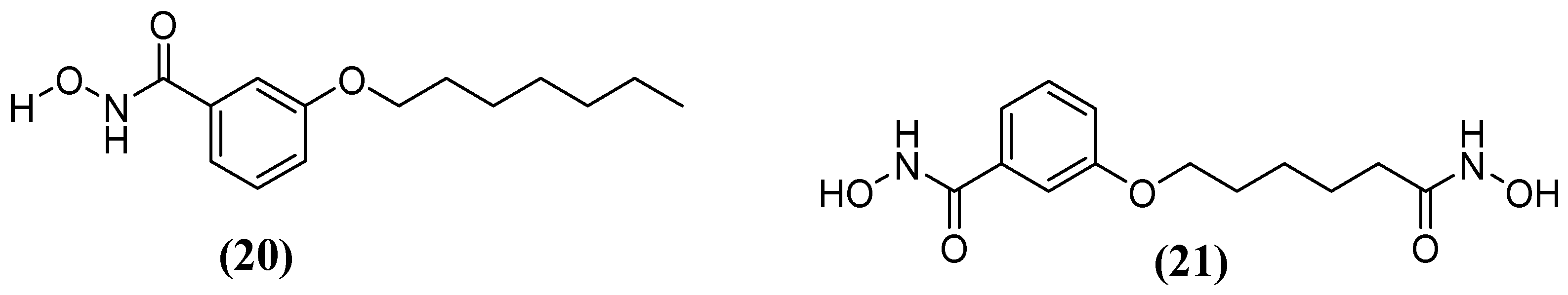
- Huckleby, A.E.; Saul, J.G.; Shin, H.; Desmarais, S.; Bokka, A.; Jeon, J.; Kim, S.-K. Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus Anthracis. Int. J. Mol. Sci. 2022, 23, 9163. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Demuth, M.; Schlesinger, S.R.; Kim, S.J.; Urbanczyk, J.; Shaw, R.W.; Shin, H. Inhibition of Bacillus Anthracis Metallo-β-Lactamase by Compounds with Hydroxamic Acid Functionality. J. Enzyme Inhib. Med. Chem. 2016, 31, 132–137. [Google Scholar] [CrossRef]
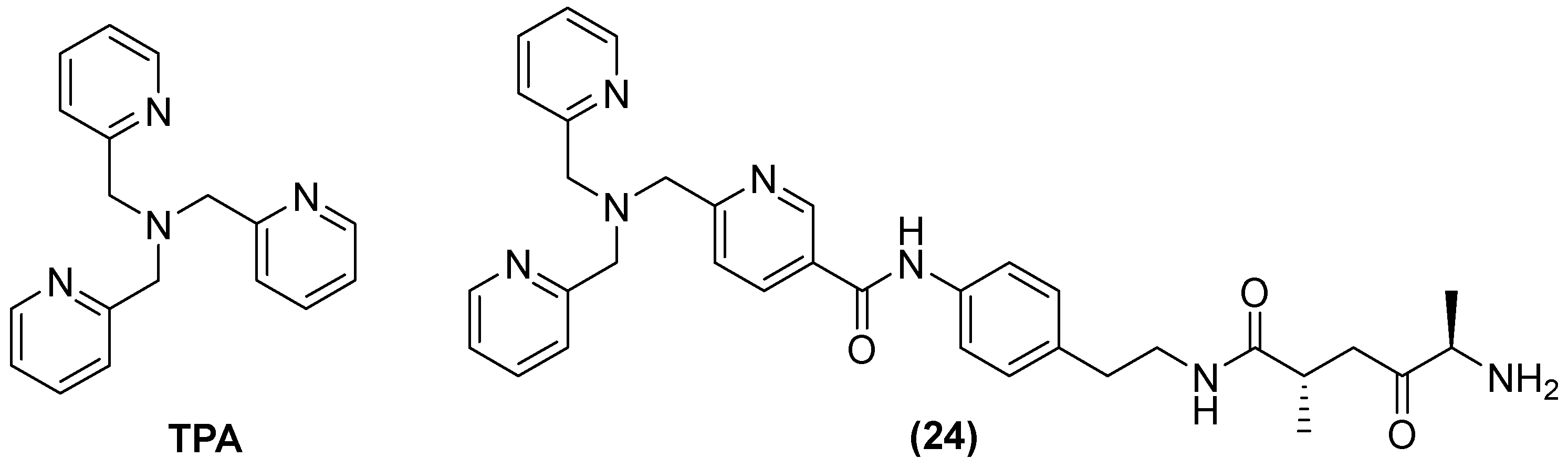
- Schnaars, C.; Kildahl-Andersen, G.; Prandina, A.; Popal, R.; Radix, S.; Le Borgne, M.; Gjøen, T.; Andresen, A.M.S.; Heikal, A.; Økstad, O.A.; et al. Synthesis and Preclinical Evaluation of TPA-Based Zinc Chelators as Metallo-β-Lactamase Inhibitors. ACS Infect. Dis. 2018, 4, 1407–1422. [Google Scholar] [CrossRef] [PubMed]
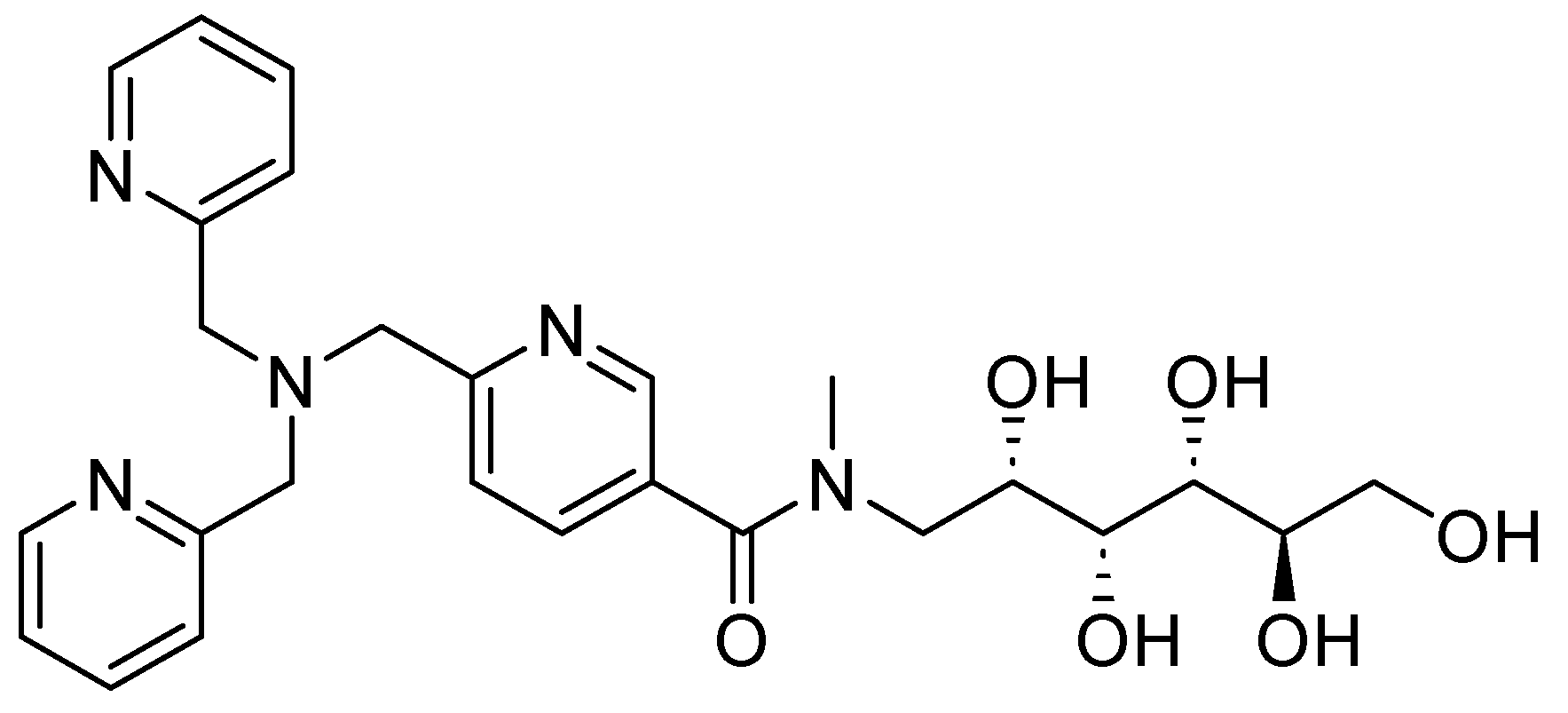
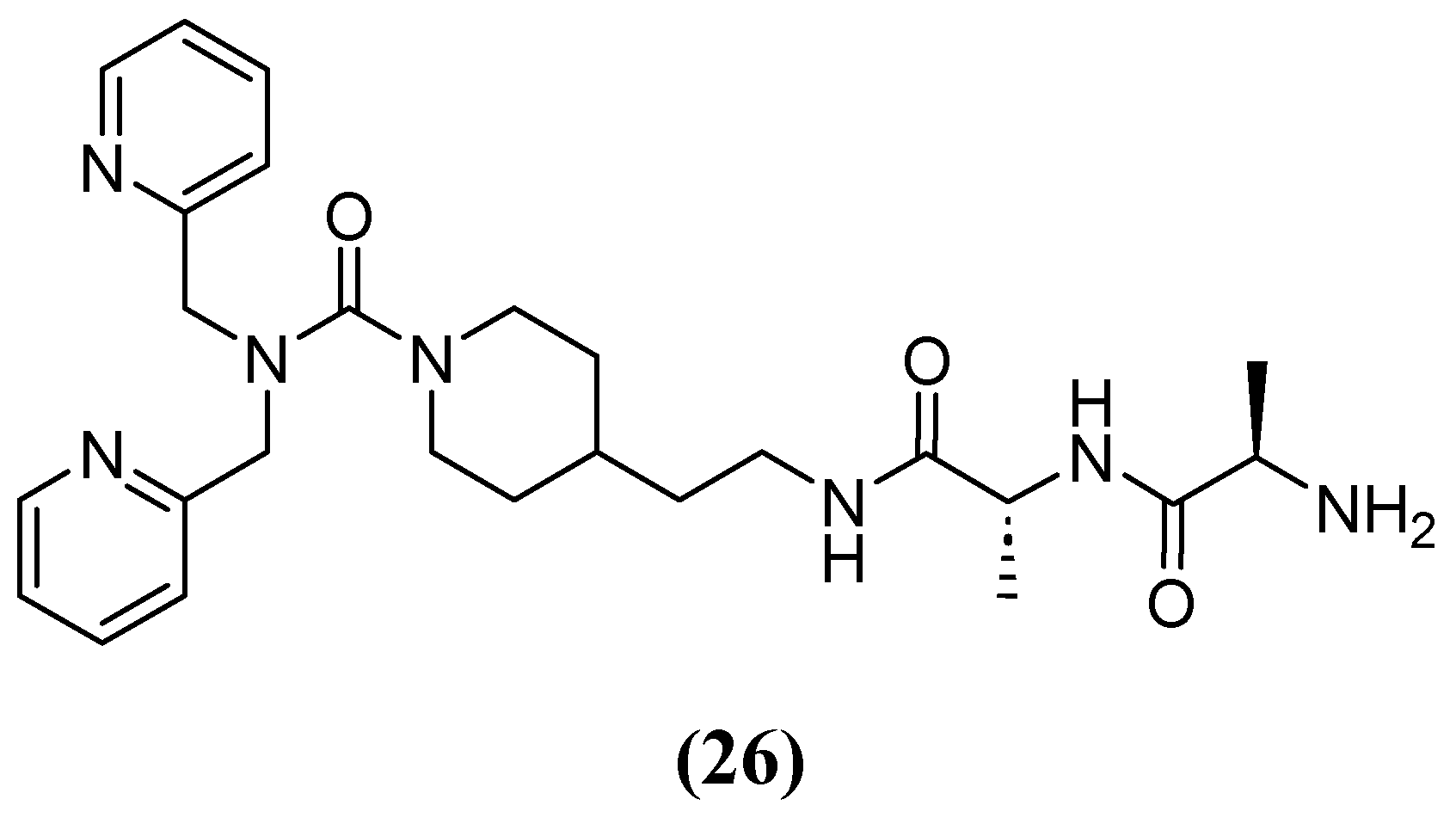
- Samuelsen, Ø.; Åstrand, O.A.H.; Fröhlich, C.; Heikal, A.; Skagseth, S.; Carlsen, T.J.O.; Leiros, H.-K.S.; Bayer, A.; Schnaars, C.; Kildahl-Andersen, G.; et al. ZN148 Is a Modular Synthetic Metallo-β-Lactamase Inhibitor That Reverses Carbapenem Resistance in Gram-Negative Pathogens In Vivo. Antimicrob. Agents Chemother. 2020, 64, e02415-19. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Schroeder, B.; Pfeifer, Y.; Fröhlich, C.; Deng, L.; Arkona, C.; Kuropka, B.; Sticht, J.; Ataka, K.; Bergemann, S.; et al. Kinetics, Thermodynamics, and Structural Effects of Quinoline-2-Carboxylates, Zinc-Binding Inhibitors of New Delhi Metallo-β-Lactamase-1 Re-Sensitizing Multidrug-Resistant Bacteria for Carbapenems. J. Med. Chem. 2023, 66, 11761–11791. [Google Scholar] [CrossRef] [PubMed]
- Prandina, A.; Radix, S.; Le Borgne, M.; Jordheim, L.P.; Bousfiha, Z.; Fröhlich, C.; Leiros, H.-K.S.; Samuelsen, Ø.; Frøvold, E.; Rongved, P.; et al. Synthesis and Biological Evaluation of New Dipicolylamine Zinc Chelators as Metallo-β-Lactamase Inhibitors. Tetrahedron 2019, 75, 1525–1540. [Google Scholar] [CrossRef]
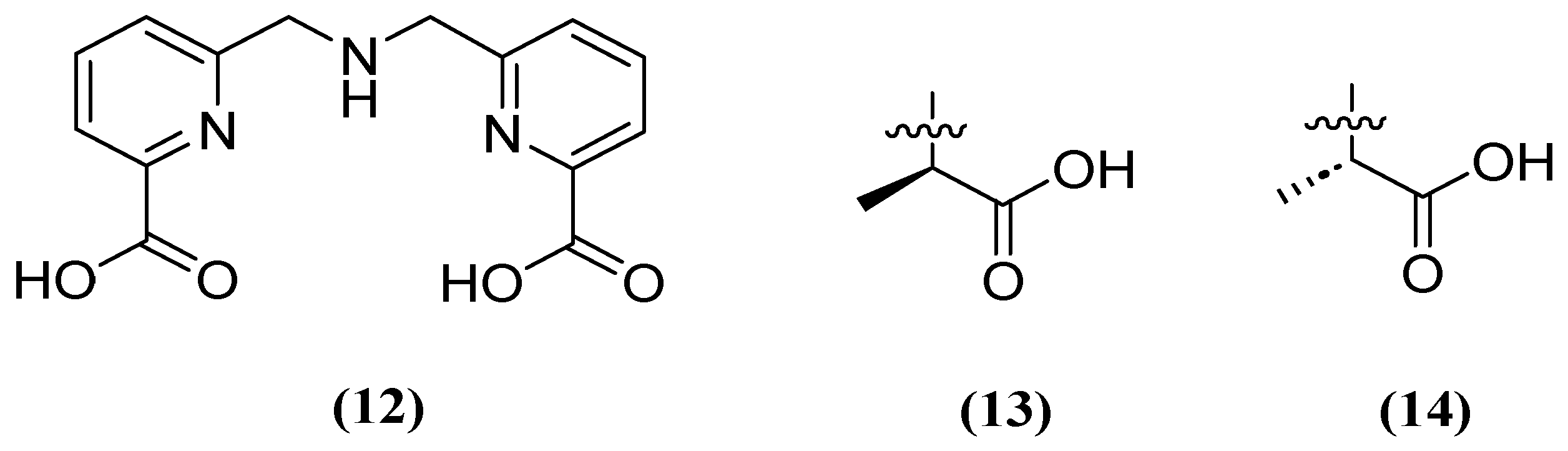
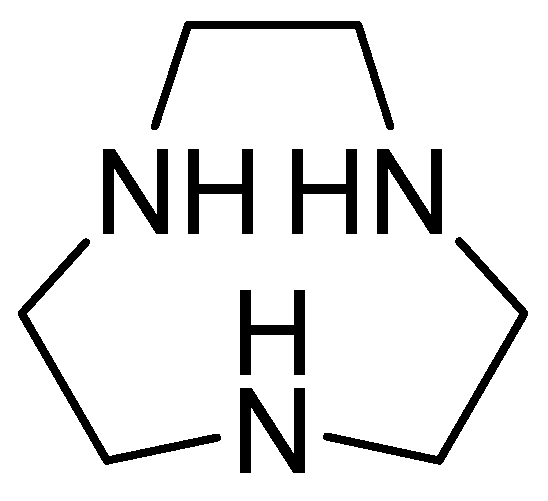
- Somboro, A.M.; Amoako, D.G.; Osei Sekyere, J.; Kumalo, H.M.; Khan, R.; Bester, L.A.; Essack, S.Y. 1,4,7-Triazacyclononane Restores the Activity of β-Lactam Antibiotics against Metallo-β-Lactamase-Producing Enterobacteriaceae: Exploration of Potential Metallo-β-Lactamase Inhibitors. Appl. Environ. Microbiol. 2019, 85, e02077-18. [Google Scholar] [CrossRef]
- Omolabi, K.F.; Reddy, N.; Mdanda, S.; Ntshangase, S.; Singh, S.D.; Kruger, H.G.; Naicker, T.; Govender, T.; Bajinath, S. The in Vitro and in Vivo Potential of Metal-Chelating Agents as Metallo-Beta-Lactamase Inhibitors against Carbapenem-Resistant Enterobacterales. FEMS Microbiol. Lett. 2023, 370, fnac122. [Google Scholar] [CrossRef]
- Benin, B.M.; Hillyer, T.; Crugnale, A.S.; Fulk, A.; Thomas, C.A.; Crowder, M.W.; Smith, M.A.; Shin, W.S. Taxifolin as a Metallo-β-Lactamase Inhibitor in Combination with Augmentin against Verona Imipenemase 2 Expressing Pseudomonas Aeruginosa. Microorganisms 2023, 11, 2653. [Google Scholar] [CrossRef]
- Jackson, A.C.; Pinter, T.B.J.; Talley, D.C.; Baker-Agha, A.; Patel, D.; Smith, P.J.; Franz, K.J. Benzimidazole and Benzoxazole Zinc Chelators as Inhibitors of Metallo-β-Lactamase NDM-1. ChemMedChem 2021, 16, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Thomas, C.A.; Thomas, P.W.; Yang, K.; Cheng, Z.; Fast, W.; Crowder, M.W.; Cohen, S.M. Iminodiacetic Acid as a Novel Metal-Binding Pharmacophore for New Delhi Metallo-β-Lactamase Inhibitor Development. ChemMedChem 2020, 15, 1272–1282. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, X.; Tian, Y.; Zhai, S.; Liu, Y.; Xiong, Z.; Chu, S. An Insight into Novel Therapeutic Potentials of Taxifolin. Front. Pharmacol. 2023, 14, 1173855. [Google Scholar] [CrossRef]
- Shin, K.S.; Son, B.R.; Hong, S.B.; Kim, J. Dipicolinic Acid-Based Disk Methods for Detection of Metallo-β-Lactamase–Producing Pseudomonas spp. and Acinetobacter spp. Diagn. Microbiol. Infect. Dis. 2008, 62, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Thomas, P.W.; Stewart, A.C.; Bergstrom, A.; Cheng, Z.; Miller, C.; Bethel, C.R.; Marshall, S.H.; Credille, C.V.; Riley, C.L.; et al. Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-β-Lactamase-1. J. Med. Chem. 2017, 60, 7267–7283. [Google Scholar] [CrossRef]
- Mousavi, S.M.; Zarei, M.; Hashemi, S.A.; Babapoor, A.; Amani, A.M. A Conceptual Review of Rhodanine: Current Applications of Antiviral Drugs, Anticancer and Antimicrobial Activities. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1132–1148. [Google Scholar] [CrossRef]
- Tejchman, W.; Korona-Glowniak, I.; Kwietniewski, L.; Żesławska, E.; Nitek, W.; Suder, P.; Żylewski, M.; Malm, A. Antibacterial Properties of 5-Substituted Derivatives of Rhodanine-3-Carboxyalkyl Acids. Part II. Saudi Pharm. J. 2020, 28, 414–426. [Google Scholar] [CrossRef]
- Zhang, D.; Markoulides, M.S.; Stepanovs, D.; Rydzik, A.M.; El-Hussein, A.; Bon, C.; Kamps, J.J.A.G.; Umland, K.-D.; Collins, P.M.; Cahill, S.T.; et al. Structure Activity Relationship Studies on Rhodanines and Derived Enethiol Inhibitors of Metallo-β-Lactamases. Bioorg. Med. Chem. 2018, 26, 2928–2936. [Google Scholar] [CrossRef]
- Kondratieva, A.; Palica, K.; Frøhlich, C.; Hovd, R.R.; Leiros, H.-K.S.; Erdelyi, M.; Bayer, A. Fluorinated Captopril Analogues Inhibit Metallo-β-Lactamases and Facilitate Structure Determination of NDM-1 Binding Pose. Eur. J. Med. Chem. 2024, 266, 116140. [Google Scholar] [CrossRef] [PubMed]
- Farley, A.J.M.; Ermolovich, Y.; Calvopiña, K.; Rabe, P.; Panduwawala, T.; Brem, J.; Björkling, F.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase Inhibition by N-Sulfamoylpyrrole-2-Carboxylates. ACS Infect. Dis. 2021, 7, 1809–1817. [Google Scholar] [CrossRef]


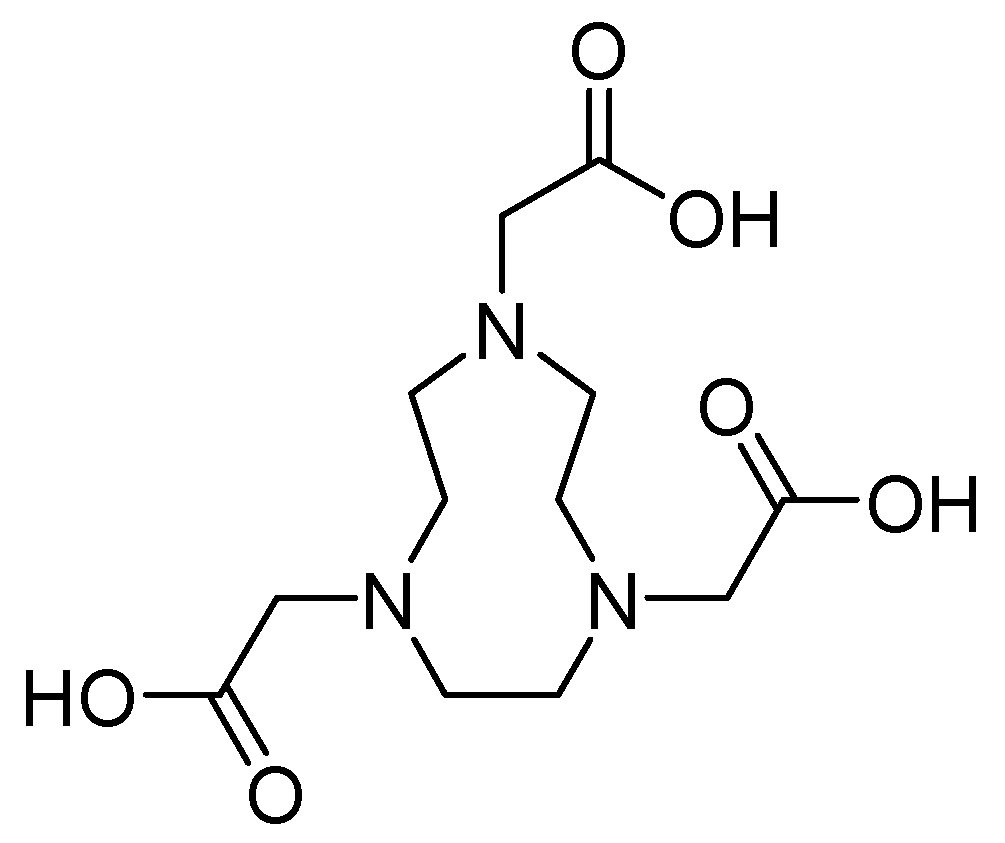
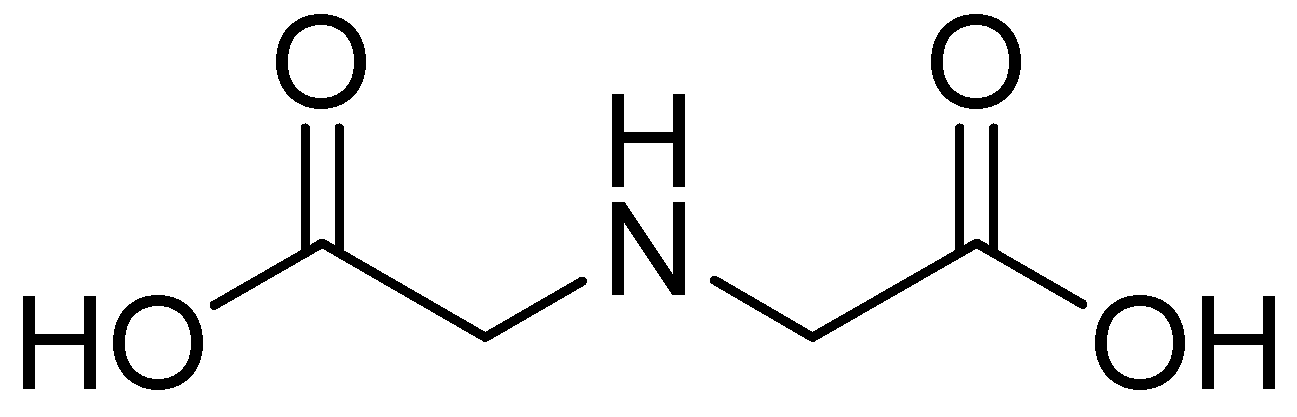
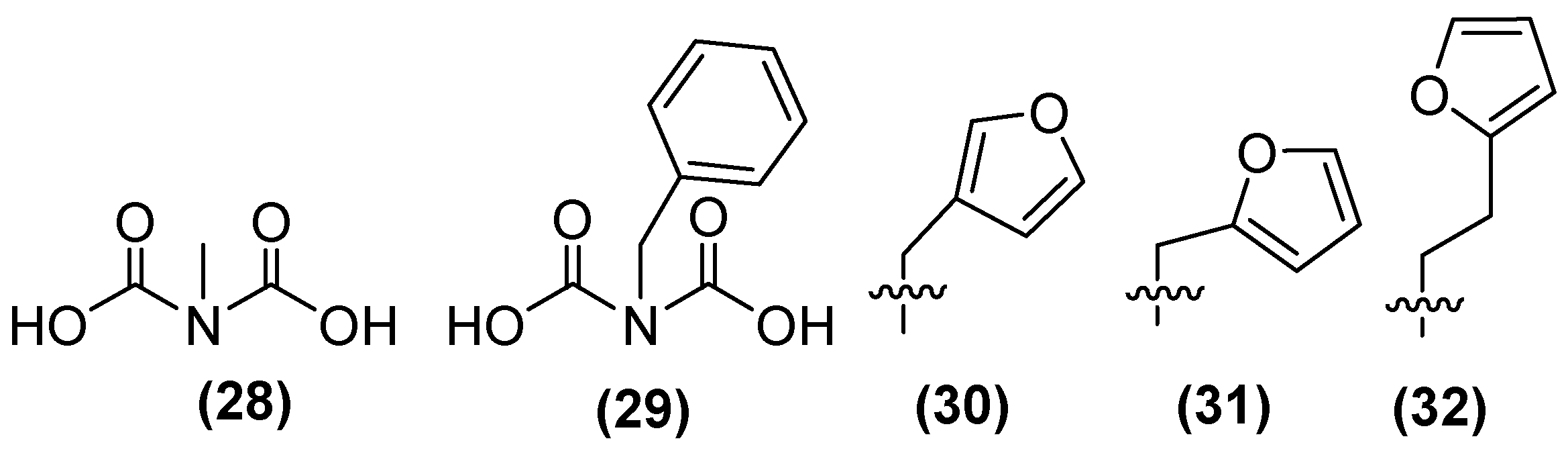
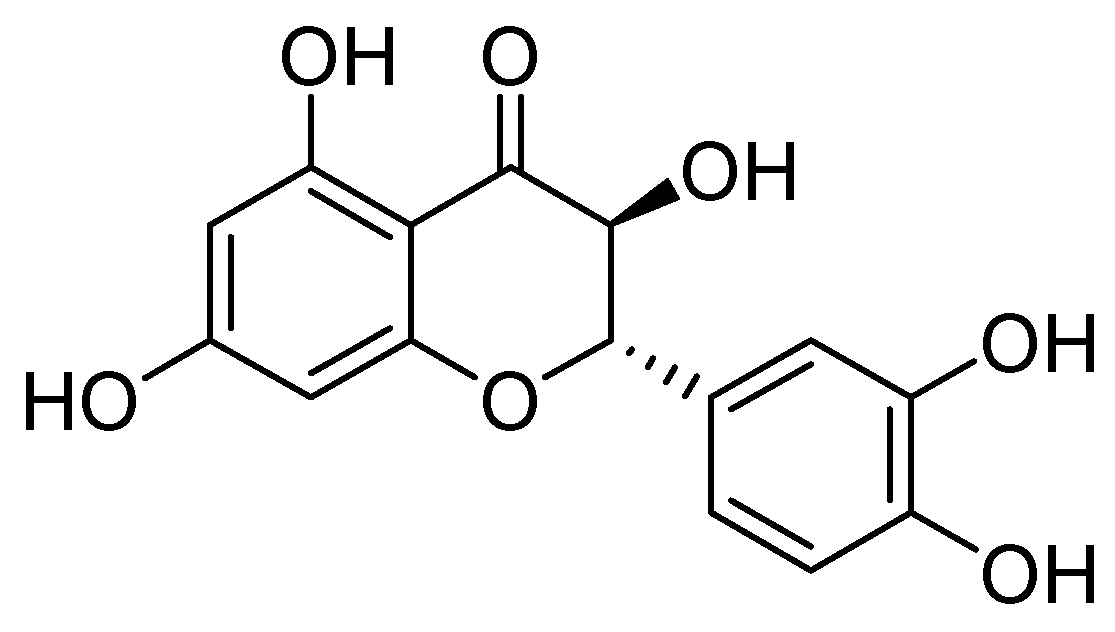

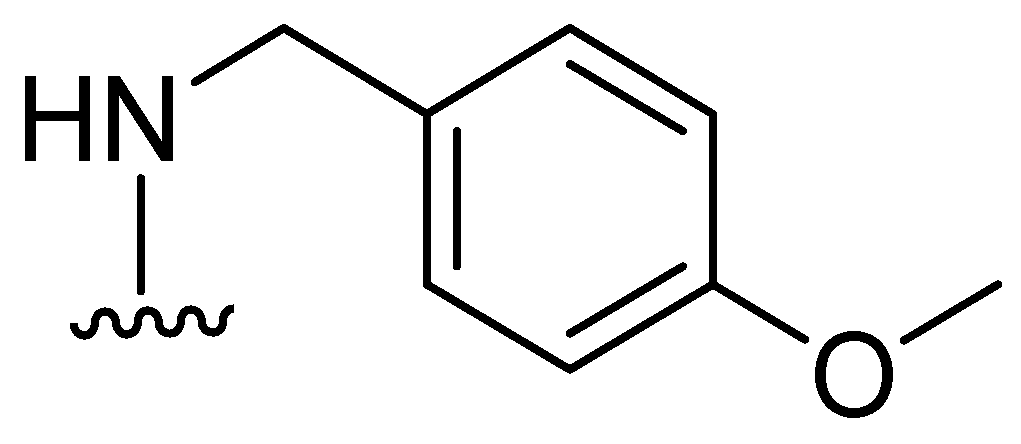
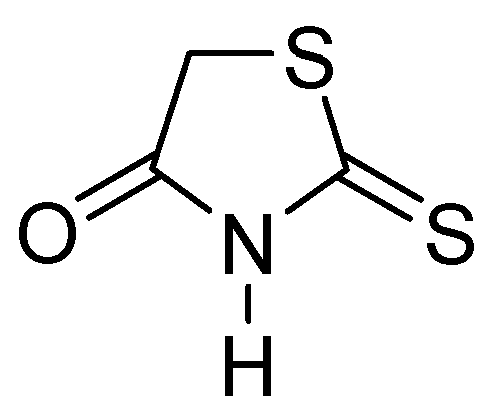
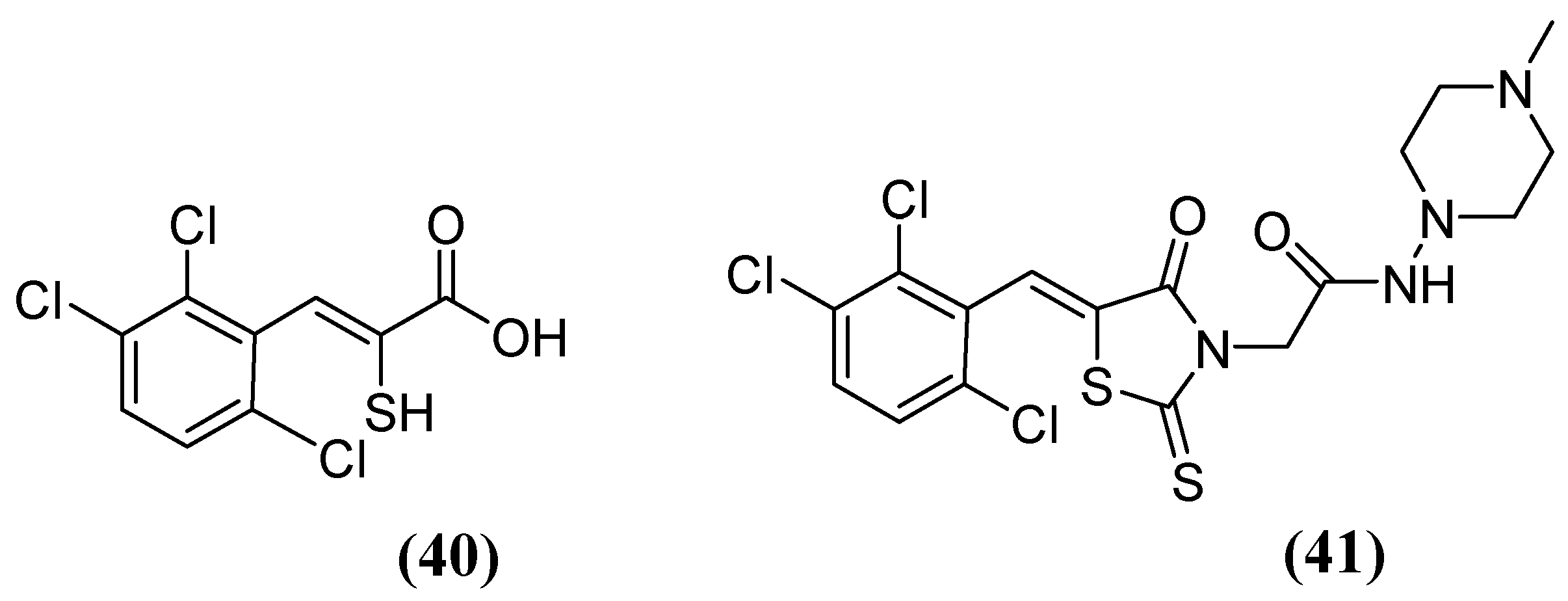
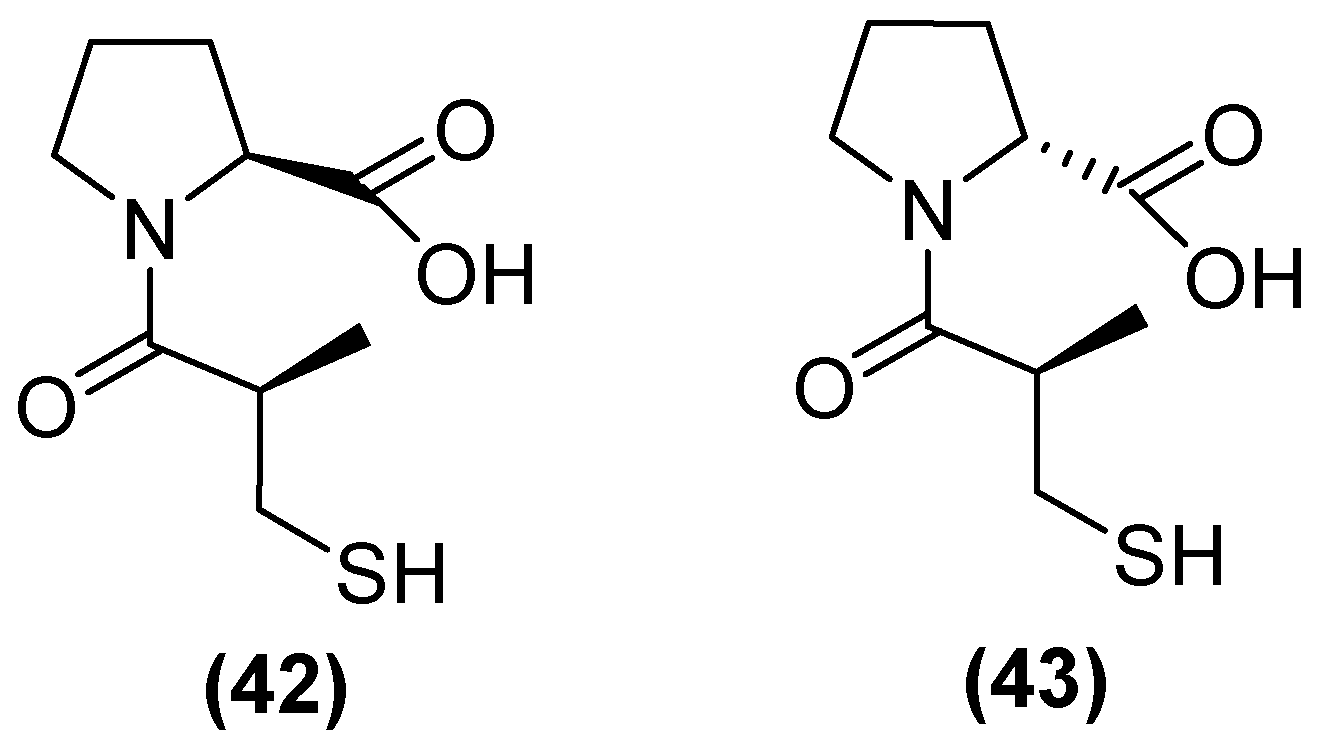

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
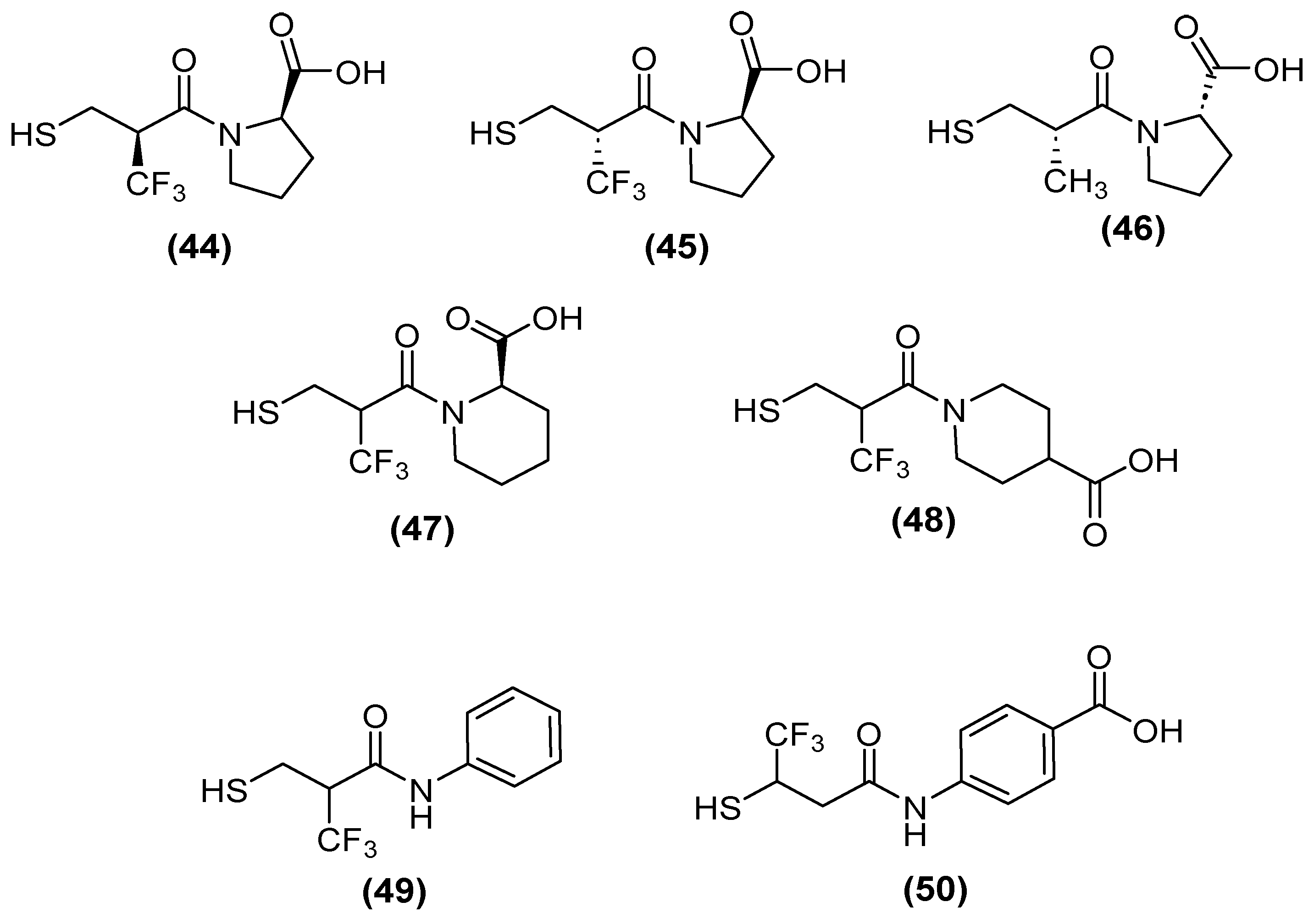
| Inhibitor | MIC Meropenem (mg/L) | ||
|---|---|---|---|
| MP30-63 blaNDM-1 | MP30-57 blaVIM-2 | MP30-58 blaIMP-26 | |
| No inhibitor control | 32 | 0.5 | 2 |
| 44 | 1 | 0.06 | 0.25 |
| 45 | 1 | 0.06 | 0.5 |
| 46 | 4 | 0.25 | 4 |
| 49 | 1 | 0.125 | 2 |
| 50 | 0.5 | 0.125 | 0.5 |
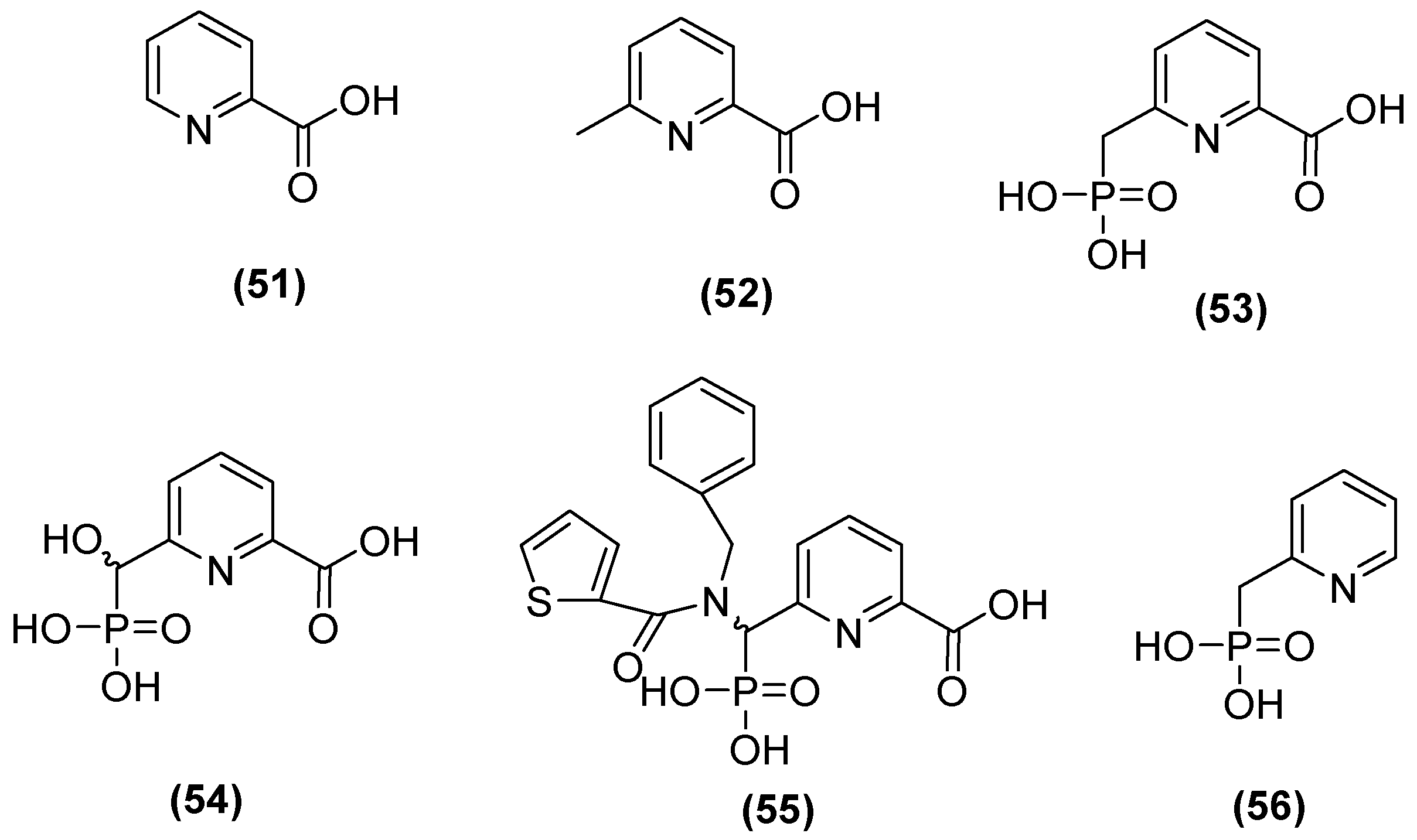
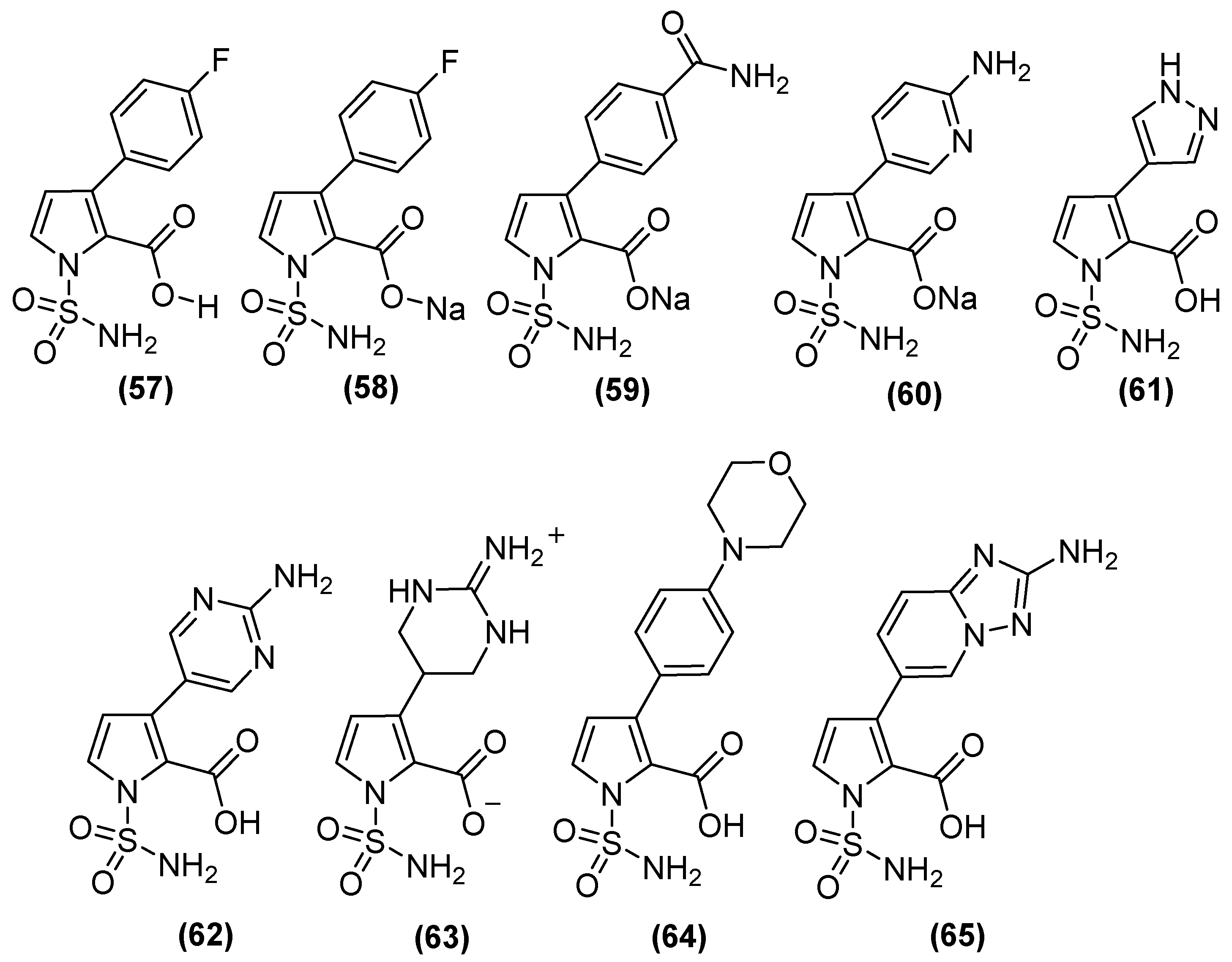
| Inhibitor | pIC50 | |||
|---|---|---|---|---|
| VIM-1 | NDM-1 | VIM-2 | IMP-1 | |
| Bicyclic boronate | 7.1 | 7.5 | 8.5 | 6.0 |
| TAN | 8.1 | 8.0 | 8.3 | 5.6 |
| 57 | 6.9 | 8.1 | 7.7 | 9.2 |
| 58 | 6.9 | 8.2 | 7.5 | 8.9 |
| 59 | 7.1 | 7.9 | 6.8 | 8.6 |
| 60 | 6.5 | 7.9 | 6.7 | 9 |
| 61 | 7.1 | 8.1 | 7.8 | 8.9 |
| 62 | 7.4 | 7.9 | 7.3 | 8.2 |
| 63 | 8.5 | 6.5 | 7.9 | 7.3 |
| 64 | 6.6 | 8.8 | 6.8 | >9.2 |
| 65 | 7.4 | 7.9 | 8.2 | 8.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega-Balleza, J.L.; Vázquez-Jiménez, L.K.; Ortiz-Pérez, E.; Avalos-Navarro, G.; Paz-González, A.D.; Lara-Ramírez, E.E.; Rivera, G. Current Strategy for Targeting Metallo-β-Lactamase with Metal-Ion-Binding Inhibitors. Molecules 2024, 29, 3944. https://doi.org/10.3390/molecules29163944
Ortega-Balleza JL, Vázquez-Jiménez LK, Ortiz-Pérez E, Avalos-Navarro G, Paz-González AD, Lara-Ramírez EE, Rivera G. Current Strategy for Targeting Metallo-β-Lactamase with Metal-Ion-Binding Inhibitors. Molecules. 2024; 29(16):3944. https://doi.org/10.3390/molecules29163944
Chicago/Turabian StyleOrtega-Balleza, Jessica L., Lenci K. Vázquez-Jiménez, Eyra Ortiz-Pérez, Guadalupe Avalos-Navarro, Alma D. Paz-González, Edgar E. Lara-Ramírez, and Gildardo Rivera. 2024. "Current Strategy for Targeting Metallo-β-Lactamase with Metal-Ion-Binding Inhibitors" Molecules 29, no. 16: 3944. https://doi.org/10.3390/molecules29163944
APA StyleOrtega-Balleza, J. L., Vázquez-Jiménez, L. K., Ortiz-Pérez, E., Avalos-Navarro, G., Paz-González, A. D., Lara-Ramírez, E. E., & Rivera, G. (2024). Current Strategy for Targeting Metallo-β-Lactamase with Metal-Ion-Binding Inhibitors. Molecules, 29(16), 3944. https://doi.org/10.3390/molecules29163944