Abstract
Currently, antimicrobial resistance (AMR) is a serious health problem in the world, mainly because of the rapid spread of multidrug-resistant (MDR) bacteria. These include bacteria that produce β-lactamases, which confer resistance to β-lactams, the antibiotics with the most prescriptions in the world. Carbapenems are particularly noteworthy because they are considered the ultimate therapeutic option for MDR bacteria. However, this group of antibiotics can also be hydrolyzed by β-lactamases, including metallo-β-lactamases (MBLs), which have one or two zinc ions (Zn2+) on the active site and are resistant to common inhibitors of serine β-lactamases, such as clavulanic acid, sulbactam, tazobactam, and avibactam. Therefore, the design of inhibitors against MBLs has been directed toward various compounds, with groups such as nitrogen, thiols, and metal-binding carboxylates, or compounds such as bicyclic boronates that mimic hydrolysis intermediates. Other compounds, such as dipicolinic acid and aspergillomarasmin A, have also been shown to inhibit MBLs by chelating Zn2+. In fact, recent inhibitors are based on Zn2+ chelation, which is an important factor in the mechanism of action of most MBL inhibitors. Therefore, in this review, we analyzed the current strategies for the design and mechanism of action of metal-ion-binding inhibitors that combat MDR bacteria.
1. Introduction
Human bacterial pathogens with antimicrobial resistance (AMR) are a critical health problem and are considered a global emergency. In recent years, deaths caused by multidrug-resistant (MDR) bacteria have been on the rise, with around 1.27 million deaths reported in 2019, and these were associated with 4.95 million deaths worldwide [1]. It is estimated that AMR will be the leading cause of death worldwide by 2050 [2]. The most known cause of AMR is the inappropriate use of antibiotics for therapeutic and non-therapeutic purposes. However, it should be noted that not only has the undiscriminating use of antimicrobials promoted AMR but horizontal gene transfer (HGT) is also key in the spread of antibiotic resistance genes (ARGs) between bacteria from different environments, which encourages resistance [3], as this provides organisms with traits for greater environmental adaptability and survival owing to the presence of antibiotics in different niches [4], thus reducing therapeutic options and increasing the problem.
HGT is facilitated by mobile genetic elements (MGEs), such as insertion sequences, transposable elements, or integrons, which can be mobilized between bacteria by other MGEs, such as plasmids and integrative conjugative elements (ICEs), which generate its rapid evolution to adapt to its environment [5]. The three major mechanisms of bacterial HGT are conjugation, natural transformation, and transduction (via plasmids and ICEs, extracellular DNA uptake, and bacteriophages, respectively), of which conjugation is the main mechanism for the spread of ARGs [6] and involves contact between the donor (carrying the plasmid and ARG) and the recipient bacteria; a conjugation bridge is synthesized between the donor and the recipient through the conjugative pili, and MGEs (plasmids or ICEs) can be moved from the donor to the recipient through this channel [7].
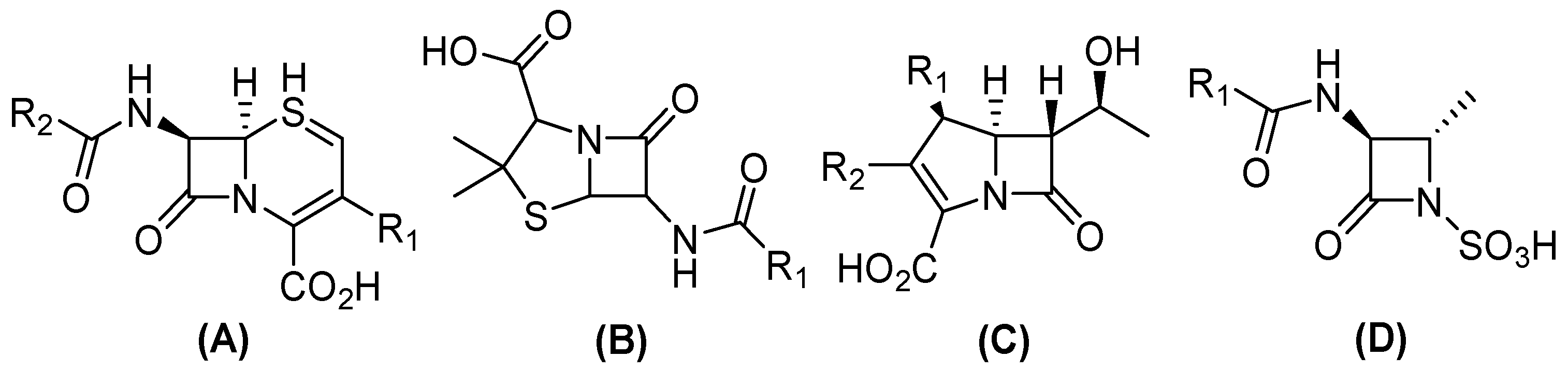
In this sense, conjugation genes encoding (β) lactamases are one of the most frequently transferred ARGs among Gram-negative bacteria, which contribute to the emergence of β-lactam resistance [8]. Currently, more than thirty-four known β-lactam antibiotics have been approved by the United States Food and Drug Administration (FDA), which can be divided into four groups (Figure 1). Three have a bicyclic structure: (i) penicillin, a β-lactam ring fused with a thiazolidine ring; (ii) cephalosporins, fused with a dihydrothiazine; and (iii) carbapenems, in which the bicyclic system is completed by a pyrroline. Conversely, (iv) monobactams have a monocyclic structure [9,10]. Nevertheless, the distribution of β-lactamase results in the development of resistance to a variety of antibiotics within this family, including carbapenems, contributes to the appearance of pan-resistant bacteria [11].
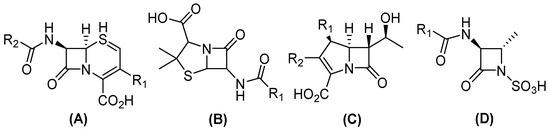
Figure 1.
General structure of clinical β-lactam antibiotics. (A) Cephalosporines, (B) penicillin, (C) carbapenems, and (D) monobactams.
Furthermore, as of 2019, only six of the thirty-two antibiotics in development are classified as innovators [12]. Owing to the increase in carbapenemase-producing bacteria and their rapid spread, it is important to develop strategies to control the spread and develop new antimicrobial molecules. Therefore, given the rise in infections that are caused by these specific antibiotic-resistant bacteria, this review will focus on metallo-beta-lactamase (MBL) inhibitors that are currently approved for clinical use or in preclinical stages.
In this review, we aimed to summarize metallo-β-lactamase inhibitors based on Zn2+ chelation that have been evaluated between 2015 and 2024. For this purpose, a literature search was performed on the PubMed and Google Scholar databases using the keywords metallo-beta-lactamases, MBL, inhibitors, metal ion binding, and Zn2+.
2. Beta (β)-Lactam and Carbapenem Antibiotics
Beta(β)-lactam antibiotics are the most used drugs around the world, and Gram-negative bacteria have developed resistance to them due to enzymatic inactivation caused by the production of a group of plasmid-encoded beta-lactamase enzymes [13,14,15]. For instance, within this class of antibiotics, carbapenems (meropenem, biapenem, ertapenem, doripenem, and imipenem) are the most important β-lactams and are incorporated in the World Health Organization (WHO) list of essential drugs because they are considered the last therapeutic choice for treating complicated MDR infections when other antibiotics, such as penicillin, have failed, especially members of Enterobacteriaceae, due to their capability to adapt to selective pressure, promoting survival [16,17,18].
Unfortunately, carbapenems are becoming less effective with the emergence of new β-lactamases that are not inactivated by available inhibitors. Due to high associated mortality, in 2017, the WHO classified carbapenem-resistant Gram-negative infections—that is, carbapenem-resistant Enterobacteriaceae (CRE), carbapenem-resistant Pseudomonas aeruginosa, and carbapenem-resistant Acinetobacter baumannii—as critically important [19] and in urgent need of new drugs [20]; carbapenem-resistant Enterobacteriaceae and third-generation cephalosporins (ceftriaxone, cefpodoxime, ceftazidime, cefdinir, ceftibuten, and others) represent the highest priority for new antibiotic development [21,22].
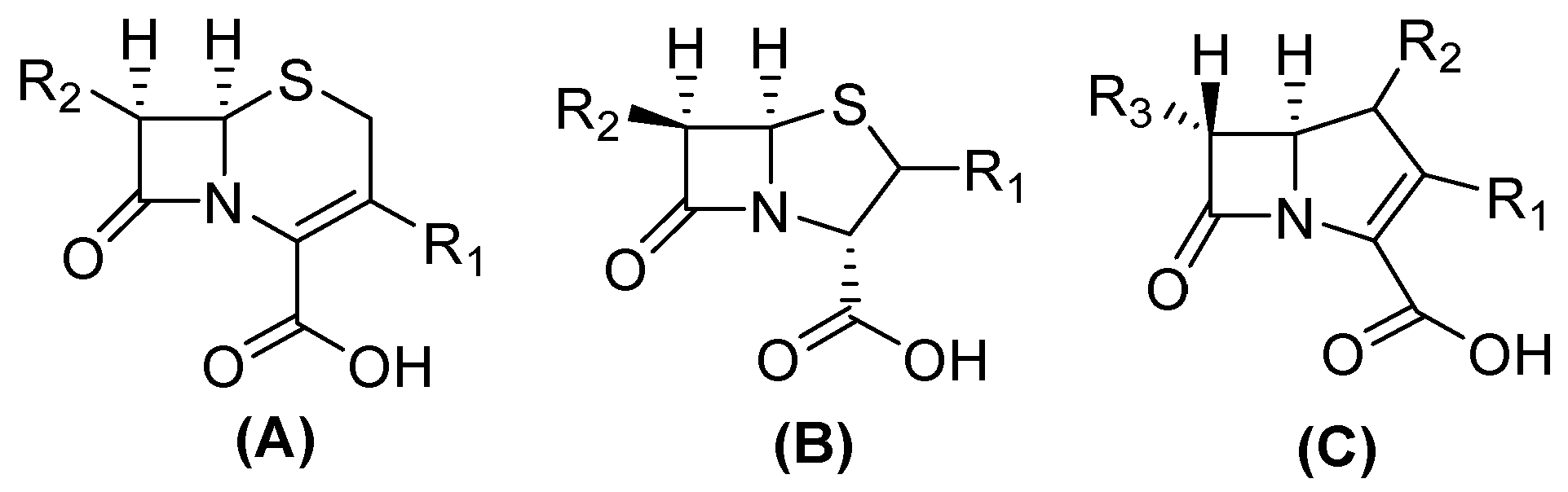
The structure of carbapenems is a five-membered ring, yet they have a carbon atom at position 1 with a sulfur atom, as in penicillin and cephalosporins, and they have a double bond between C-2 and C-3 of the pentameric ring [23]. The trans-configuration (Figure 2) at C5–C6 and the C-6 (R)-hydroxyethyl substituent provide better resistance to the action of β-lactamases versus the cis-configuration of penicillin and cephalosporin (Figure 2). Their mechanism of action is the same as that of β-lactams because they are structural analogs of peptidoglycan precursors; thus, they act by interfering with bacterial cell wall biosynthesis by covalently inactivating D, D-transpeptidases, or penicillin-binding proteins (PBPs) through the acylation of a serine active site in these crucial enzymes, leading to the inhibition of peptidoglycan cross-linking in cell walls, resulting in cell lysis and, at the same time, cell death [10,15,24,25].
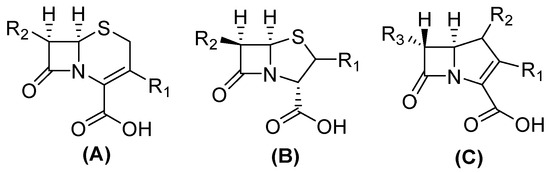
Figure 2.
Chemical structure of cephalosporin in cis-configuration (A), penicillin in cis-configuration (B), and carbapenem in trans-configuration (C).
3. Beta(β)-Lactamases
Due to their broad spectrum of activity, ß-lactams are considered one of the most effective antibiotics; however, their excessive and indiscriminate use promotes bacteria survival and has led to the rise of resistant bacteria. This resistance is principally caused by enzymatic inactivation, although mechanisms such as efflux pumps and second mutations that alter the expression and/or function of porins and PBPs are also involved [26,27].
The predominant mechanism observed in clinical isolates is the production of β-lactamases [10]. These enzymes are characterized by the hydrolysis of the β-lactam ring [25]. They are classified into four classes (A, B, C, and D) according to their amino acid sequence homology [13], based on the Ambler classification. Meanwhile, Bush–Jacoby–Medeiros functional categorization considers substrate and inhibitor response profiles and classifies enzymes into three groups (1, 2, and 3) [28,29].
Furthermore, in the Ambler classification classes (A, C, and D), serine β-lactamases use a serine residue to hydrolyze the β-lactam ring. On the other hand, class B, also known as MBLs, possesses Zn2+ in the active site, which facilitates the formation of a non-covalent reactive complex with the β-lactam [30,31,32].
4. Metallo-Beta-Lactamases (MBLs) as Drug Targets
In this work, we will focus on MBLs, which are characterized by hydrolyzing carbapenems, penicillin, and cephalosporins, but are unable to hydrolyze monobactams [33]. These types of enzymes are usually present in combination with other ESBLs in clinical isolates [28] and account for about 10% of reported β-lactamases, of which only 3% confer resistance exclusively to carbapenems [14,34]. These enzymes use Zn2+ as a fundamental cofactor to cut the β-lactam ring and deactivate the antibacterial agents; possess one or two zinc ions (Zn2+) in the active site; and are inhibited by chelator agents. In addition, MBL bacteria are resistant to common inhibitors of β-lactamases, such as clavulanic acid, sulbactam, tazobactam, and avibactam [33,34,35].
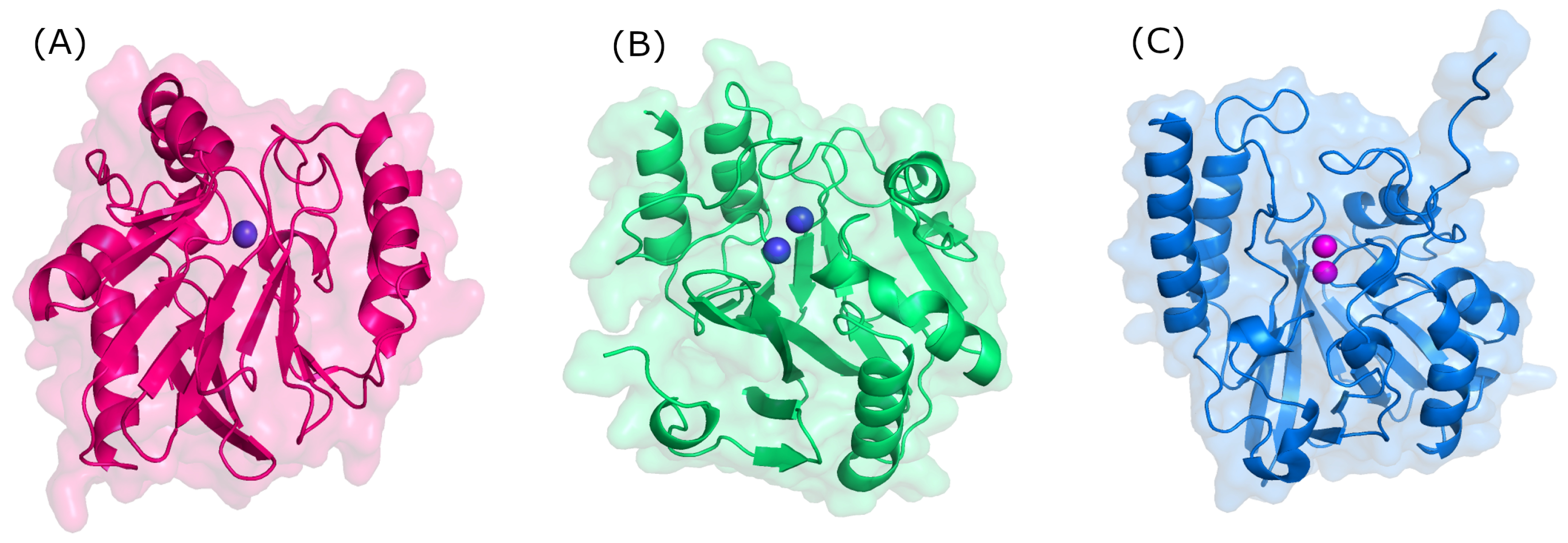
Structurally, MBLs possess a single, highly conserved globular domain in groups B1, B2, and B3 and present an αβ/βα fold. The active site is situated at the interface between αβ units that employ one or two zinc ions, critical for catalytic activity, and are also flanked by flexible loops (Figure 3) [35,36,37]. Zinc ions are relevant at several stages: substrate positioning on the active site, the activation of nucleophiles, the stabilization of the various species formed, and proton donor positioning [38]. Group B1 and B3 MBLs are characterized by two zinc-binding sites (Zn1 and Zn2), whereas group B2 has only one binding site (Zn2); thus, the only structural feature conserved in all MBLs is the Zn2 site [38,39]. It is believed that the loop region of the active site dictates differences in substrate specificity, although recent structures suggest otherwise [40].
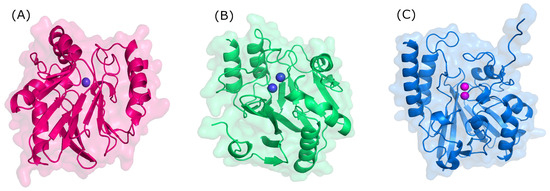
Figure 3.
Three-dimensional structure of metallo-β-lactamases. (A) B1 (NDM-1, PDB: 3SPU); (B) B2 (CphA, PDB: 3F90); and (C) B3 (L1, PDB: 1SML).
MBL genes are commonly harbored in integrons, transposons, and plasmids and are transferred horizontally, allowing for spread and persistence among bacteria, although they have also been reported to be part of the chromosome [25,33,41].
MBLs in Ambler’s classification are divided into groups B1, B2, and B3 according to the amino acid sequence on the active site, the zinc ligands, zinc stoichiometry, the loop architecture, and the substrate profiles [35] Group B1 is the most extensive and includes the most reported MBLs, such as imipenemase (IMP), integron-encoded Verona MBL (VIM), and New Dheli (NDM) [13]. It should be noted that the amino acid homology between the subclasses is <20% [35,36].
5. MBL Inhibitors
The use of β- lactamase inhibitors is one useful strategy for restoring β-lactam activity [42]. However, there are currently no FDA-approved inhibitors of MBLs [43] due to their unsuccessful design, which is a result of either the nature of the zinc ligands and catalytic mechanisms or differences in the active site architecture [44].
Inhibitors of MBLs are often grouped according to structural similarities, but they can also be grouped according to their mechanism of inhibition: (1) inhibition by metal ion binding, (2) inhibition by covalent bond formation, (3) allosteric inhibitors, and (4) inhibitors with uncharacterized mechanisms [45]. Most of the reported MBL inhibitors contain a zinc-coordinating group (Figure 4), and numerous metal-binding pharmacophores have been used. The main single or multiple groups reported are the thiol and carboxylate groups [46].
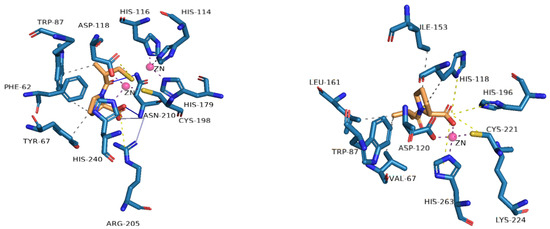
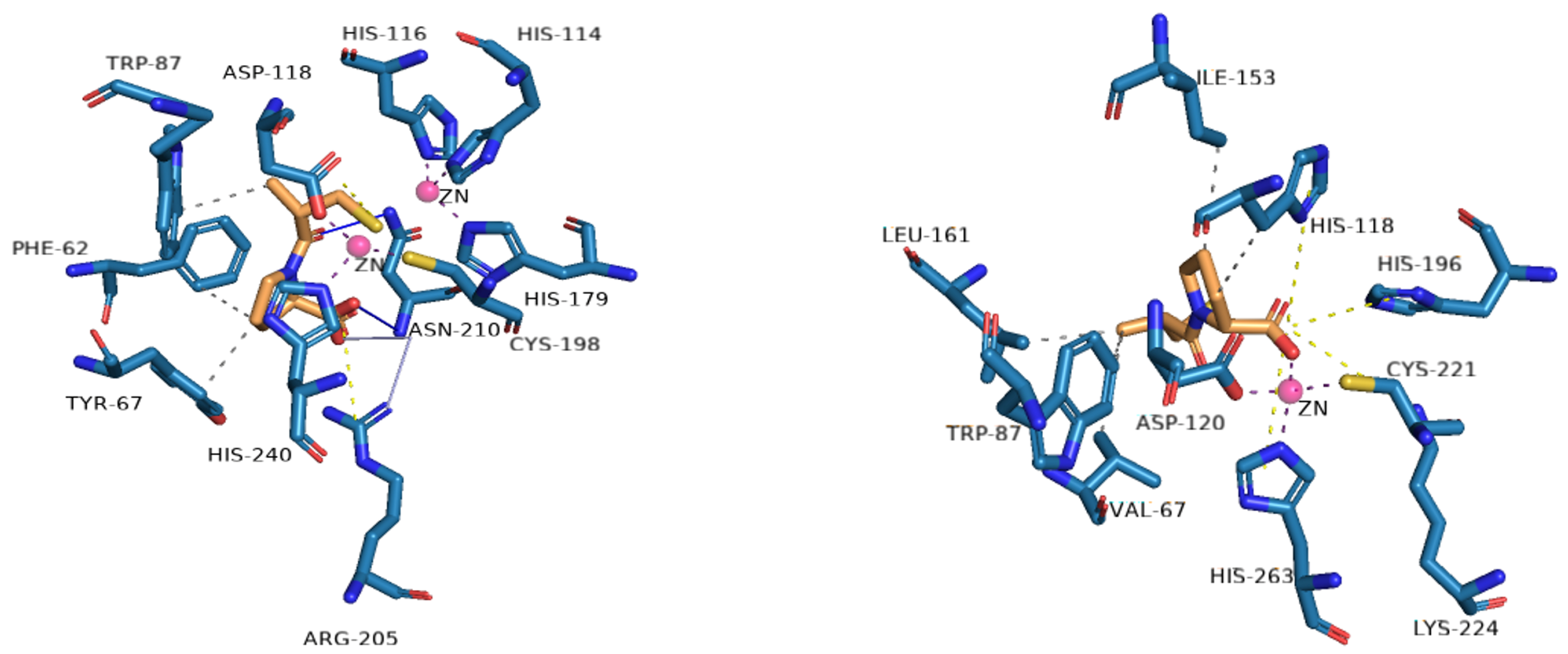
Figure 4.
Profile of interactions of zinc on the active site of VIM-2 (PDB: 4C1E) and CphA (2QDS) with D-captopril, an inhibitor.
MBL inhibitors bind with zinc, an important factor in their mechanism of action; therefore, this work focuses on metal-ion-binding-mediated inhibition, which, in turn, involves two possible modes of action: a) the removal of metal ions (Zn2+), wherein the inhibitor removes metal ions from the active site of the enzyme, or b) alternatively, the inhibitor can form a ternary complex with the metal ions (Zn2+) of the active site of the MBLs and the surrounding protein residues. This process effectively prevents antibiotics from binding to the enzyme, causing the formation of an MBL:Zn2+:inhibitor complex [45,47].
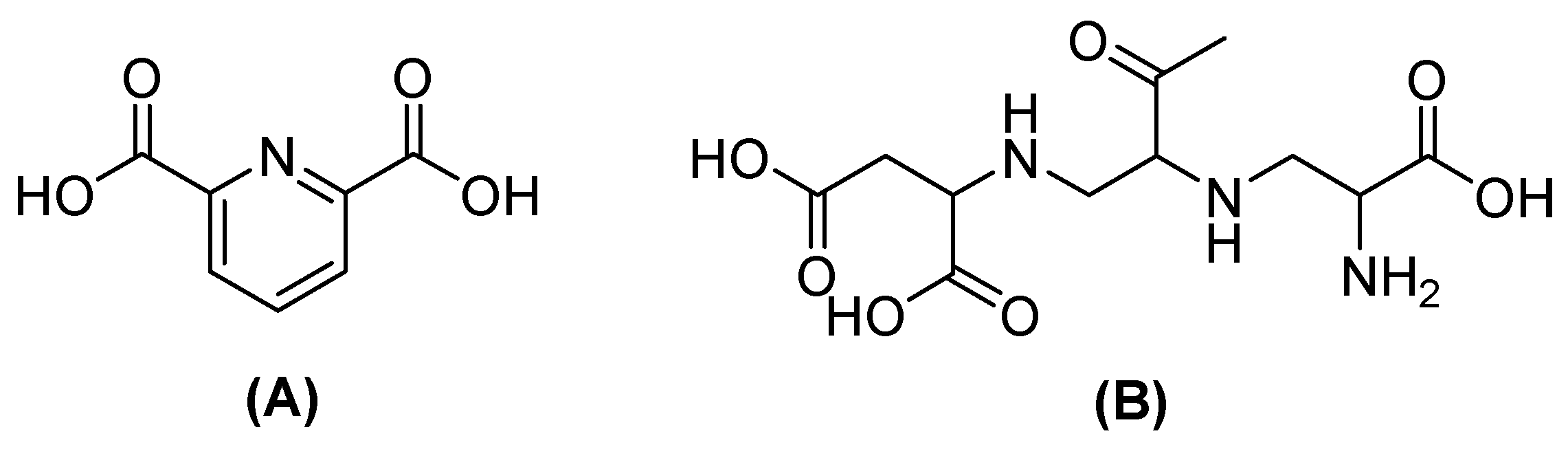
Nevertheless, the co-administration of inhibitors with β-lactam agents has shown efficacy against serine-dependent β-lactamases but not against MBLs. Therefore, the design of MBL inhibitors implicates compounds with carboxylate, nitrogen, and thiol groups that cause metal binding or that simulate hydrolysis intermediates [48]. One of the most promising is captopril, which acts by interfering with nucleophilic hydroxide production due to the thiol group, which binds with Zn2+ on the active site in NDM-1; boronic acids also inhibit NDM-1, simulating the transition state of hydrolyzed β-lactamases. Other compounds, such as dipicolinic acid (DPA) or aspergillomarasmin A (AMA) (Figure 5), have also been shown to inhibit MBLs by chelating Zn2+ [49].
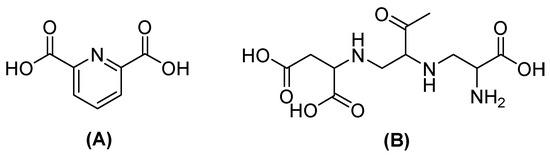
Figure 5.
Chemical structure of dipicolinic acid (A) and aspergillomarasmin (B).
5.1. Metal-Complex-Forming Inhibitors
Several compounds from different groups have been proposed as potential inhibitors against MBLs, with the B1 subclass being the focus. The results show that NDM is more susceptible to inhibition than VIM and IMP.
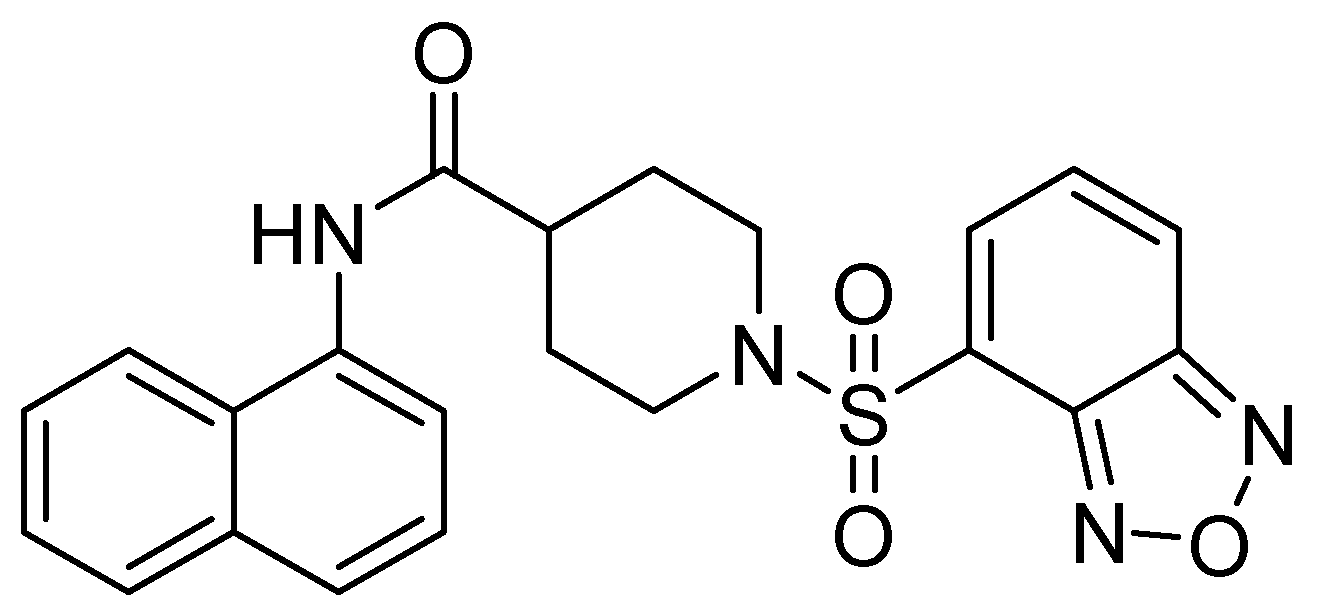
In their study, Wang and cols. (2015) identified a potent inhibitor (VNI-41) (Figure 6) that affected NDM-1 activity with an IC50 of 29.6 ± 1.3 µM through a broad interaction with the active site (His122, His120, His189, Asn220 His250, and Ile30). The sulfonamide group of compound VNI-41 directly interacts with Zn1, suggesting that compounds with the sulfonamide group could be important inhibitors of MBLs [50].
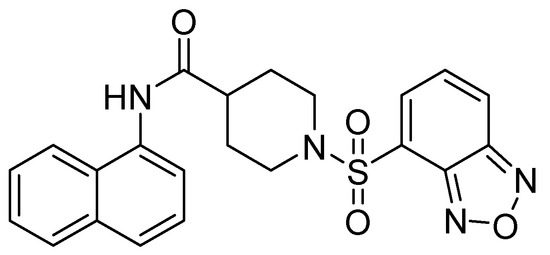
Figure 6.
Chemical structure of VNI-41, a potent inhibitor of NDM-1 activity.
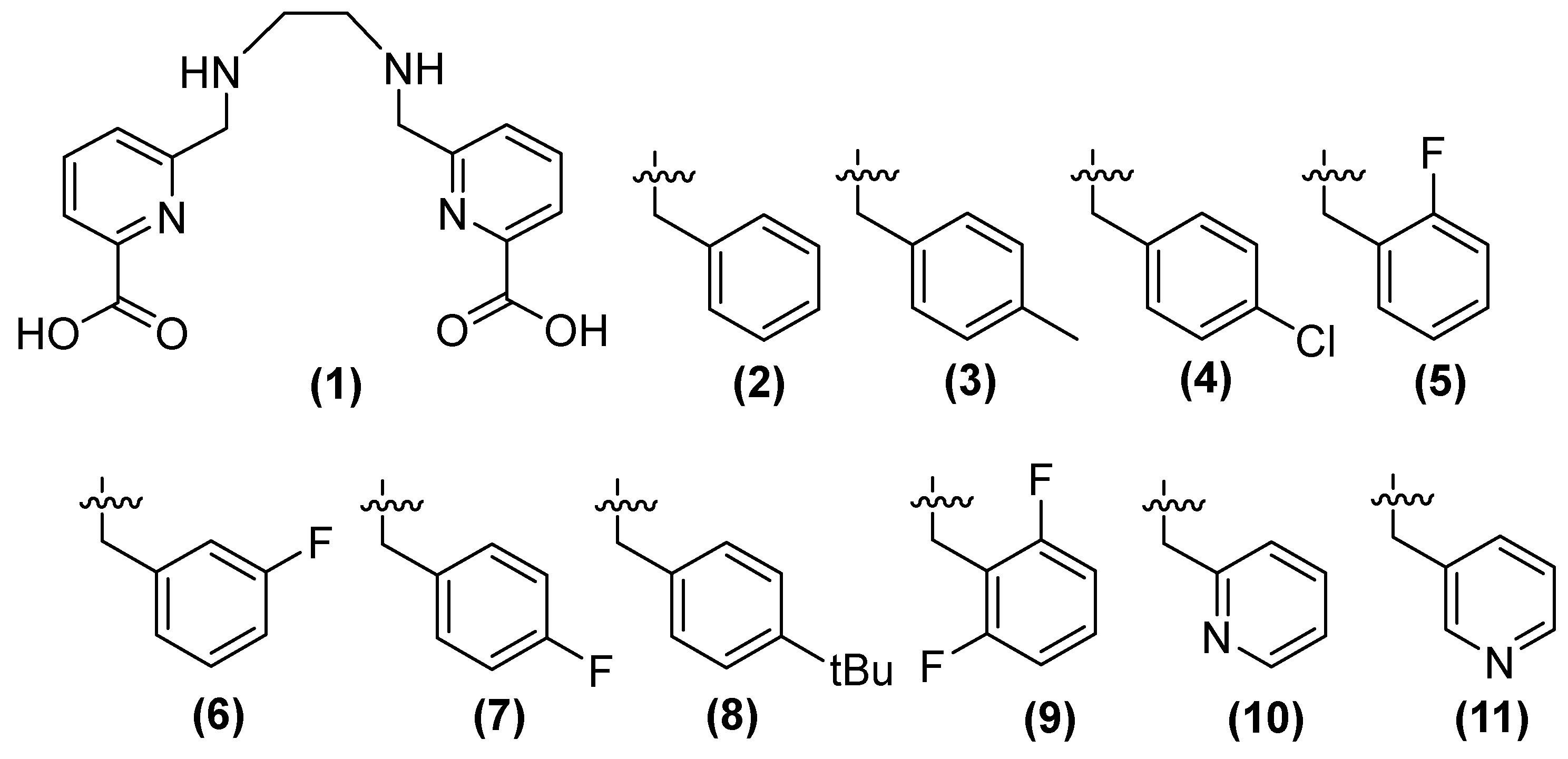
Compound 1 (h2dedpa [1,2-[[6-carboxy-pyridin-2-yl]-methylamino]ethane]) (Figure 7) is a metal chelator and inhibitor of NDM-1. Consequently, Cui and cols. synthesized derivatives (2–11) of this compound, observing that compounds 9, 7 (substituted aryl), and 11 (substituted aromatic heterocycle) showed the inhibition of NDM-1 at 0.11, 0.06, and 0.46 μM, respectively, when alkyl chain elongation was performed in 9 and 8 and pyridine derivatives were used in 11 [51].
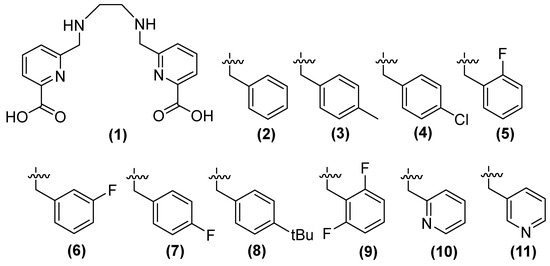
Figure 7.
Chemical structure of derivatives of h2dedpa, a metal chelator and inhibitor of NDM-1.
The antibacterial activity of meropenem (MEM) (Figure 8) can be restored by compounds 8, 9, and 11 against bacteria that produce NDM-1, such as Klebsiella pneumoniae, Klebsiella oxytoca, Enterobacter aerogenes, Escherichia coli, and Proteus. Compound 8, in combination with MEM, reduces the MIC at 1 μg/mL and 0.25 μg/mL in K. pneumoniae and Enterobacter aerogenes, respectively; 11 shows better results (0.25 μg/mL) in K. oxytoca; the same compounds (8, 9, and 11) show the same MIC (0.25 μg/mL) against E. coli, and the best MIC among bacteria has been observed in P. mirabilis, with a MIC of 0.06 μg/mL for these three compounds. Other carbapenemases, such as NDM-5, NDM-7, and IMP, have shown less inhibition than NDM-1 [51].
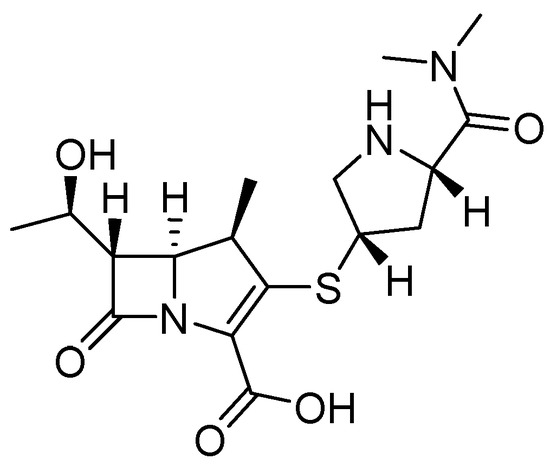
Figure 8.
Chemical structure of meropenem (MEM).
Compound 8 has been observed to exhibit cytotoxic activity. While 9 and 11 have been tested using a computational approach, both compounds have also been demonstrated to inhibit NDM-1 by binding to Zn2+. Compound 9 forms a coordinated bound with two zinc ions of NDM-1, while the carbonyl of the other carboxylic acid forms two hydrogen bonds with Lys211 and a fluorine atom in 2,6-difluorobenzene, which binds to Asn220. In compound 11, the hydroxyl group of one of the carboxylic acids forms a coordinated bond with Zn2 of NDM-1. It has been observed that these compounds increase the efficacy of MEM against E. coli, E. aerogenes, K. pneumoniae, K. oxytoca, and P. mirabilis, all of which carry the NDM-1 gene [51].
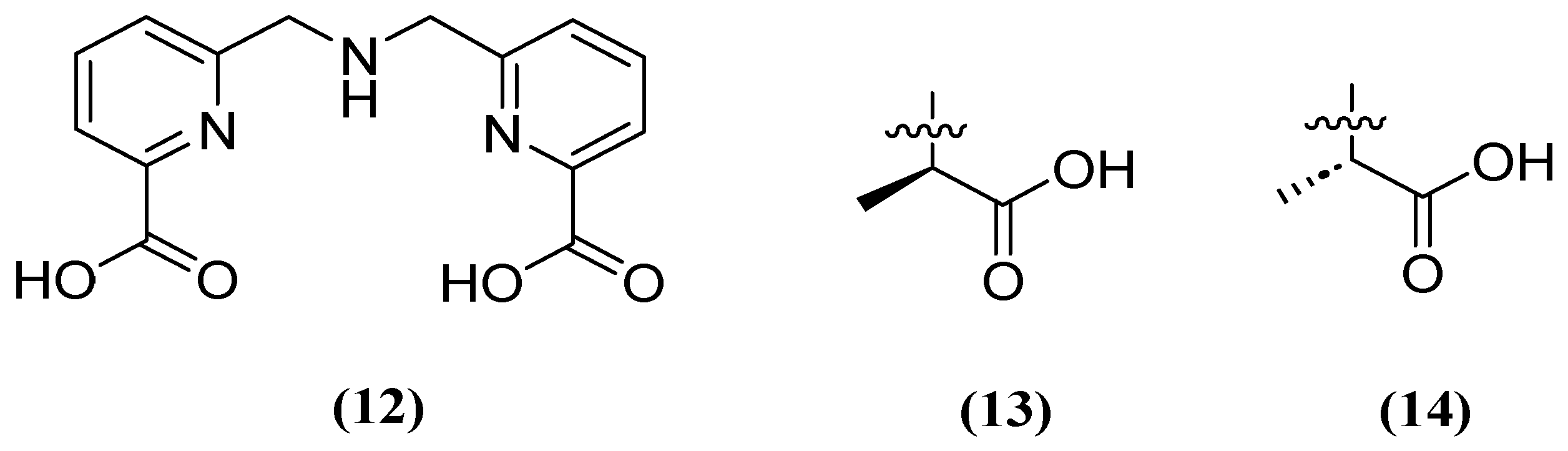
Similarly, H2dpa (12) derivatives (Figure 9) containing pentadentate-chelating ligands have shown inhibitory activity against NDM, VIM, and IMP at the molecular, cellular, and in vivo levels. Furthermore, the derivatized compounds designated as 13 and 14 (Figure 9) have been found to be non-competitive and have the ability to restore the activity of MEM against clinical isolates that produce NDM (1.59 μM and 1.57 μM, respectively) and IMP (1.70 μM and 1.54 μM, respectively) [52].
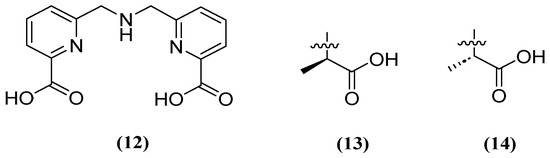
Figure 9.
Chemical structure of H2dpa (12) and derivatives 13 and 14.
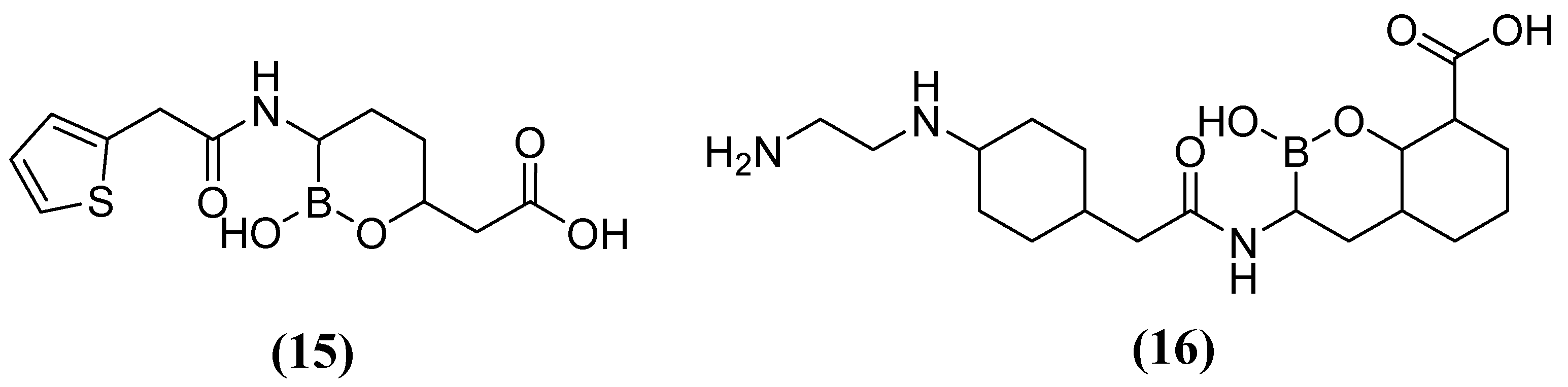
On the other hand, acyclic boronic acids are known to inhibit nucleophilic enzymes such as serine β-lactamases (SBLs), although cyclic boronates have been observed to exhibit the potent inhibition of MBLs [53]. Vaborbactam 15 (Figure 10), in combination with MEM, has demonstrated the capacity to inhibit SBLs (class A), including KPC carbapenemases. However, it has not exhibited efficacy against MBLs (MIC 32 mg/L) or certain clinically relevant SBLs (MIC 32 mg/L) [54]. Cyclic boronate derivatives such as taniborbactam (TAN) 16 (also known as VNRX-5133) are promising compounds that inhibit serine-dependent β-lactamases and MBLs such as NDM-1, VIM-1, and VIM-2 [55]. TAN has been shown to bind to the metal ion of Zn1 and the conserved amino acid Asn233, located in the VIM-2 active site. In phase I clinical trials, the TAN-CEF (cefepime) combination has been demonstrated to be effective against carbapenemase and extended-spectrum beta-lactamase Enterobacteriaceae (ESBL). Specifically, a 4 mg/L TAN dose was required to reduce the cefepime MIC90 of the Enterobacterales from 256 to 1 mg/L. Additionally, in Pseudomonas aeruginosa cefepime, the MIC90 was reduced from 128 to 16 mg/L [56,57,58].
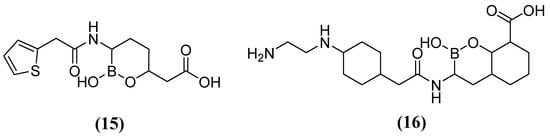
Figure 10.
Chemical structure of vaborbactam (15) and taniborbactam (16).
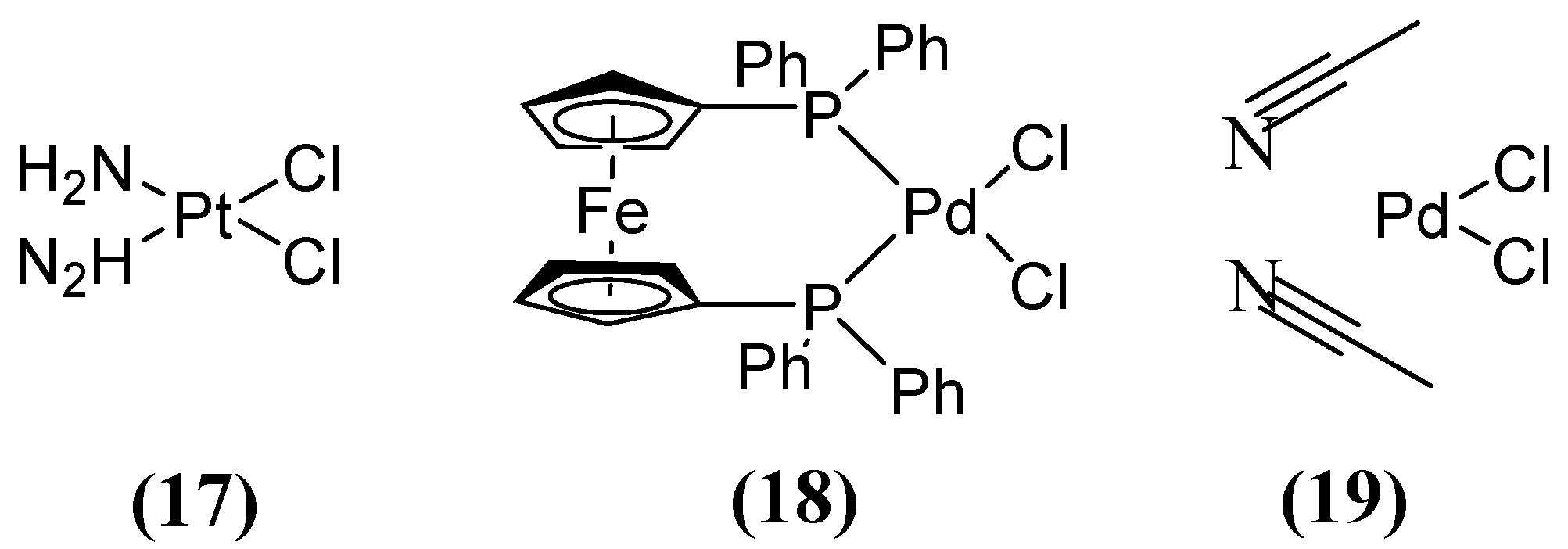
Other compounds, such as those used in chemotherapy against various cancers, like cisplatin (used in approximately 50 % of treated patients) [59], have shown inhibitory activity when combining MEM (Figure 8) with cisplatin 17 or Pd(II)s 18 and 19 (Figure 11). This inhibitory effect has been observed against a broad spectrum of B1 (MIC 8 µg/mL for NDM-1-producing E. coli) and B2 MBL subclasses, thereby restoring efficacy against MEM. This demonstrates that these metal complexes are irreversible inhibitors of NDM-1 through a novel mode of inhibition that involves binding to Cys208 and the displacement of the Zn2+ ion of the enzyme caused by a Pt(II) ion containing two NH3 groups or a Pd(II) ion [60].
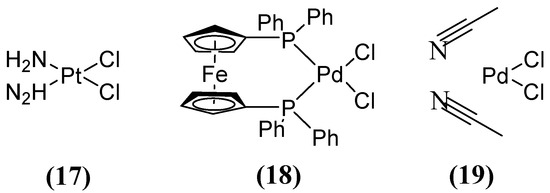
Figure 11.
Chemical structures of platinum compound and palladium complexes.
Compounds of natural origin have also shown activity as inhibitors, one of which is AMA (Figure 5), a secondary fungal metabolite derived from Aspergillus versicolor that can restore MEM antibiotic activity against E. coli [61]. This compound alone is not antimicrobial, but in combination with β-lactam antibiotics, it inhibits the catalysis of MBLs. The mechanism of inhibition is based on the sequestration of the Zn2+ ion, which spontaneously dissociates from the MBL, but the effect is different for different MBL families and alleles [47].
Previously, the AMA restored effective susceptibility to MEM in E. coli and K. pneumoniae in strains expressing blaNDM-1 and blaVIM-2 (MICs of 4 to 8 μg/mL); NDM-4, NDM-5, NDM-6, NDM-7, IMP-1, IMP-7, and IMP-27 were moderately sensitive (12 to 16 μg/mL). In contrast, subclass B2 and B3 MBLs, CphA2, and AIM-1 exhibit the least susceptibility to AMA inhibition, with MIC values exceeding 64 μg/mL [62]. For example, Sychantha and cols. (2021) evaluated the mechanism of inhibition in vivo and found that AMA leads to the loss of zinc ions (Zn2+) from a low-affinity binding site of NDM-1 (8 and 16 μg/mL). This loss contributes to the rapid degradation of NDM-1, which is a crucial factor in the efficacy of AMA as a β-lactam enhancer. However, MBLs with a higher affinity for Zn2+, such as NDM-6 or IMP-7, demonstrate increased tolerance to AMA (MICs of 16 and 32 μg/mL) [47].
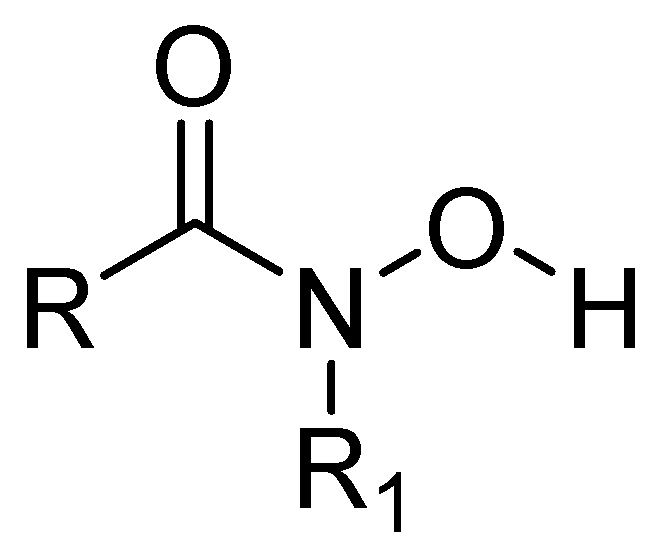
Some metalloprotease inhibitors, such as hydroxamic acid (Figure 12), have a higher capacity to chelate Zn2+ and Fe3+ and can be used as inhibitors of MBLs [63]. About this matter, the MBL from Bacillus anthracis Bla2, which is a member of the B1 subclass of MBLs, contains strongly bound Zn1 and weakly bound Zn2 in its active site. The presence or absence of Zn2 has no significant effect on the catalytic activity of this MBL [64].
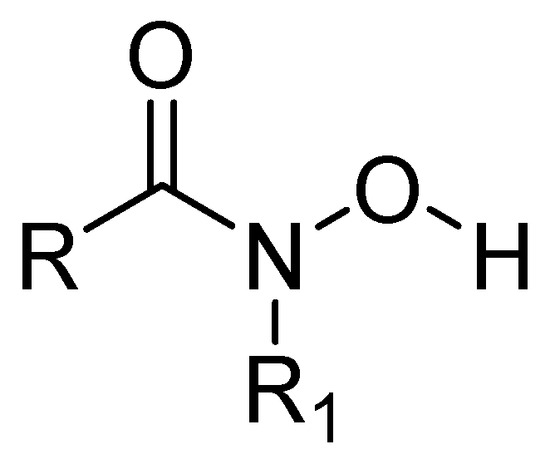
Figure 12.
Chemical structure of hydroxamic acid, a metalloprotease inhibitor.
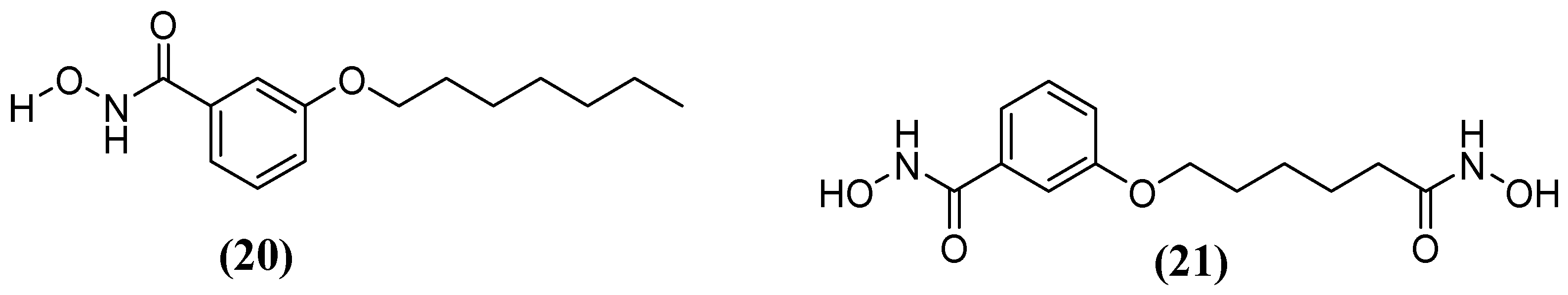
Kim and cols. (2016) synthetized hydroxamic acid derivatives, including 3-(heptyloxy)-N-hydroxybenzamide) (20) and N-hydroxy-3-((6-(hydroxyamino)-6-oxohexyl)oxy)benzamide (21) (Figure 13). They did so to evaluate these derivatives against Bla2. The compound 3-(heptyloxy)-N-hydroxybenzamide) was unable to inhibit Bla2, whereas the compound N-hydroxy-3-((6-(hydroxyamino)-6-oxohexyl)oxy) benzamide revealed a competitive reversible inhibition, obtaining a Ki value of 0.18 ± 0.06 μM. Molecular docking analysis indicated that the affinity of the inhibitor compound for the active site of Bla2 is due to coordination with Zn1, with a binding energy of −7.8 kcal/mol. A stable complex was formed between Zn1 and the amino acids His117, Cys197, and His178. The N-hydroxy-3-((6-(hydroxyamino)-6-oxohexyl)oxy) benzamide compound has dihydroxamic acids on both sides, which play a crucial role in its binding affinity. This is in contrast to monohydroxamic acid, which is unable to inhibit Bla2 [65].
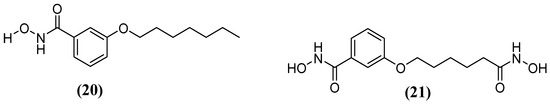
Figure 13.
Chemical structure of 3-(heptyloxy)-N-hydroxybenzamide) (20) and hydroxamic acid derivative N-hydroxy-3-((6-(hydroxyamino)-6-oxohexyl)oxy)benzamide (21).
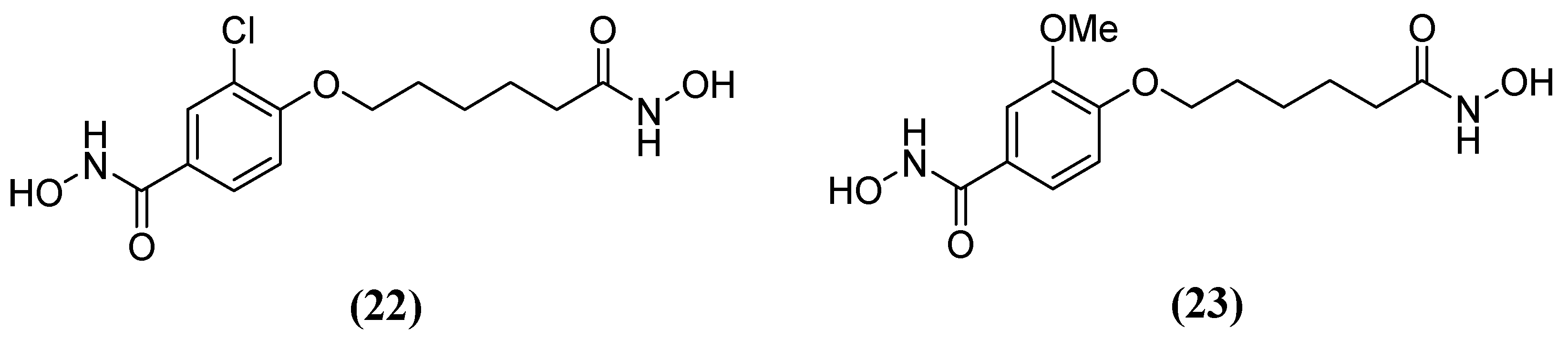
Hydroxamic acid derivatives 3-chloro-N-hydroxy-4-(7-(hydroxyamino)-7-oxoheptyl) benzamide 22 and N-hydroxy-4-(7-(hydroxyamino)-7-oxoheptyl)-3-methoxybenzamide 23 (Figure 14) have also been tested against Bla2. The results showed that the hydroxamate group is attached to the aromatic end and binds both Zn2+ ions during molecular docking. Additionally, the hydrophobic nature of the active site residues Phe63 and Val68 seems to create a more non-polar environment that favors the aromatic end of the compounds. Several potential hydrogen bonds have been identified between these compounds and active site residues. In one study, Lys200, Ser201, and His239 forming a potential hydrogen bond with a hydroxamate group attached to the aliphatic end—referred to as the aliphatic hydroxamate group—with IC50 values of 20.0 ± 5.0 µM and 14.9 ± 9.8 µM, were obtained in inhibition assays for 22 and 23, respectively [64], as well as Asn209 forming a potential hydrogen bond with the aromatic hydroxamate group.
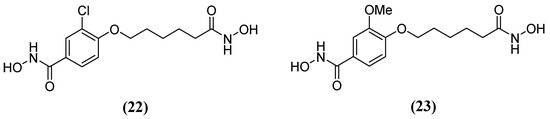
Figure 14.
Chemical structure of hydroxamic acid derivatives: (22) 3-chloro-N-hydroxy-4-(7-(hydroxyamino)-7-oxoheptyl)benzamide and (23) N-hydroxy-4-(7-(hydroxyamino)-7-oxoheptyl)-3-methoxybenzamide.
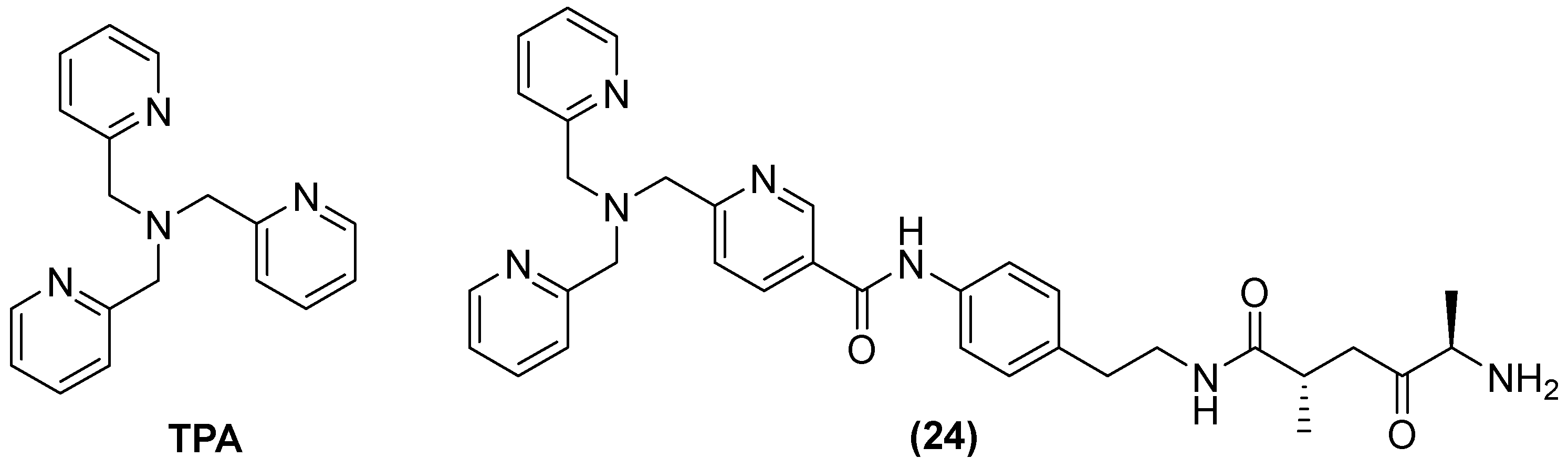
Tris-picolylamine (TPA) (Figure 15) alone has also shown re-sensitization in clinical isolates of K. pneumoniae-producing NDM-1 and P. aeruginosa-producing VIM-2, with MIC reductions from 32 to 64 mg/L and 0.125 to 1 mg/L, but toxic effects against the human liver cancer cell line HepG2 were also observed. Therefore, modifications of its structure have also been made by observing the bactericidal activity of MEM in P. aeruginosa and K. pneumoniae expressing VIM-2 and NDM-1, respectively. The compound designated as 24 reduced the MIC concentration of MEM from 32 to 256 µg/mL at 50 μM in all clinical isolates expressing MBLs [66].
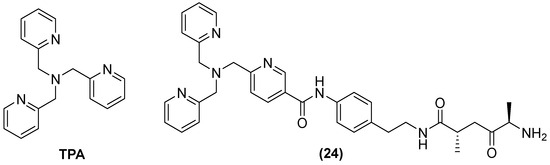
Figure 15.
Chemical structure of tris-picolylamine and its derivative, compound 24.
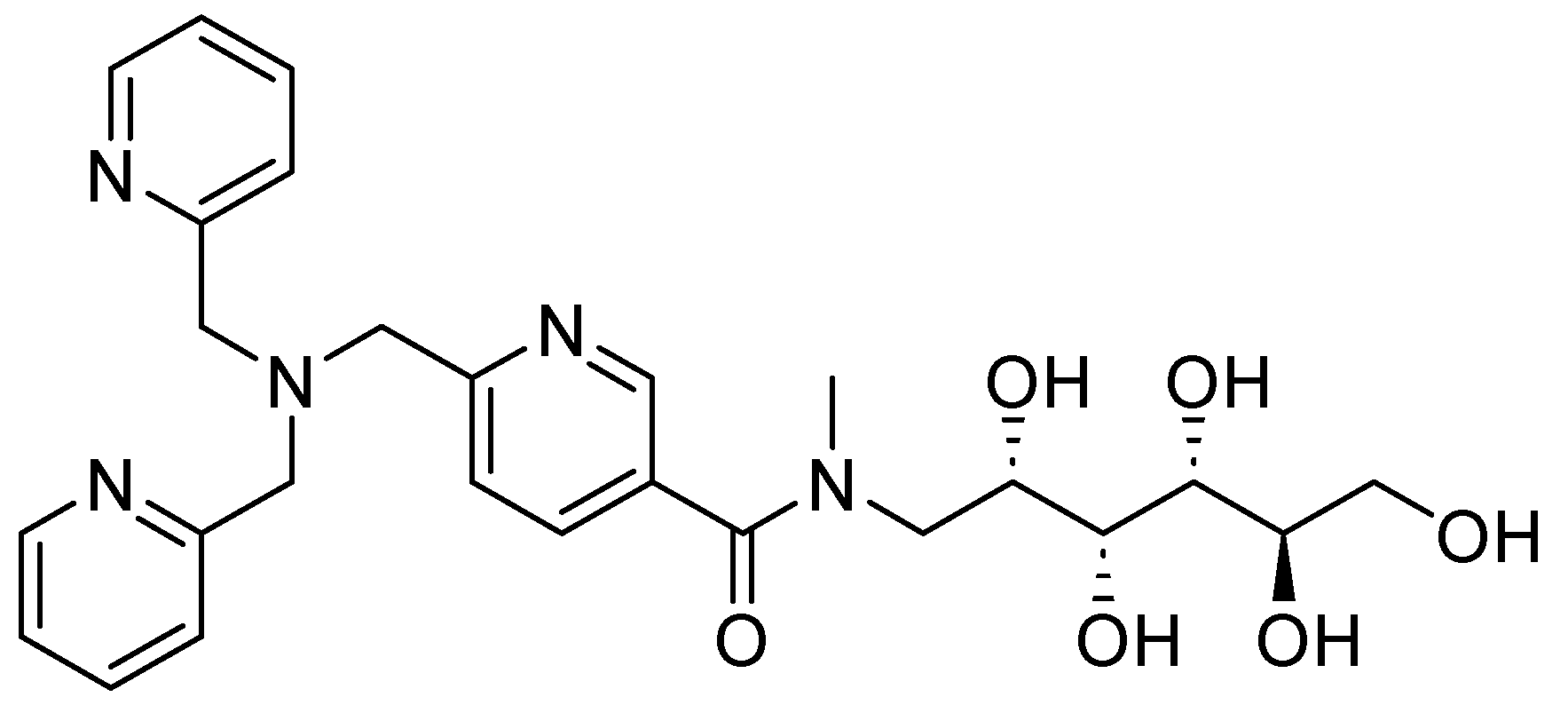
Other derivatives of TPA have also shown promising results; compound 25 (Figure 16), also a zinc chelator, can restore the bactericidal effect of MEM and in vitro clinical susceptibility to carbapenems by >98%, achieving MICs of ≤2 mg/L in Enterobacteriaceae-producing MBLs expressing the enzyme variants NDM, VIM, and IMP. When tested with other carbapenemic agents, such as doripenem and imipenem, 25 reduced the MIC to susceptible levels by >99%. However, this compound has shown favorable aromatic interactions involving Phe61, Tyr67, Tyr224, and Arg228 cation–π interactions with Arg228 between 25 and the active site of VIM-2 in NDM-1. The stacking of aromatic side chains likely results in greater hydrophobic and cation–π interactions between 25 and the VIM-2 active site compared with those involving the equivalent residues in Met61, Val67, Lys224, and Ala228 in NDM-1, indicating that NDM-1 may exhibit reduced enzyme-inhibitor interactions and, consequently, weaker binding. Furthermore, in vivo models at accumulative doses of up to 128 mg/kg, no acute toxicity has been observed [67]. Also, the activity of NDM-1 inhibitors for other metallo-β-lactamases could be influenced by assay conditions, as well as the characteristics of each protein; for this reason, it is important to perform more studies on the mechanisms of action and toxicity [68]. Likewise, it has been noted that potent inhibitors of NDM-1 and other representative MBLs have little inhibition of human zinc-binding enzymes. Further, pharmacokinetic analysis can develop potent β-lactamases using molecular docking and in silico through more studies. However, there is no information on toxicity in this regard.
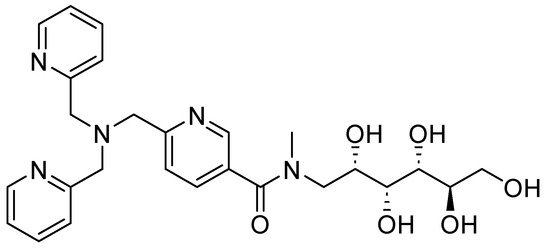
Figure 16.
Chemical structure of compound 25, a zinc chelator.
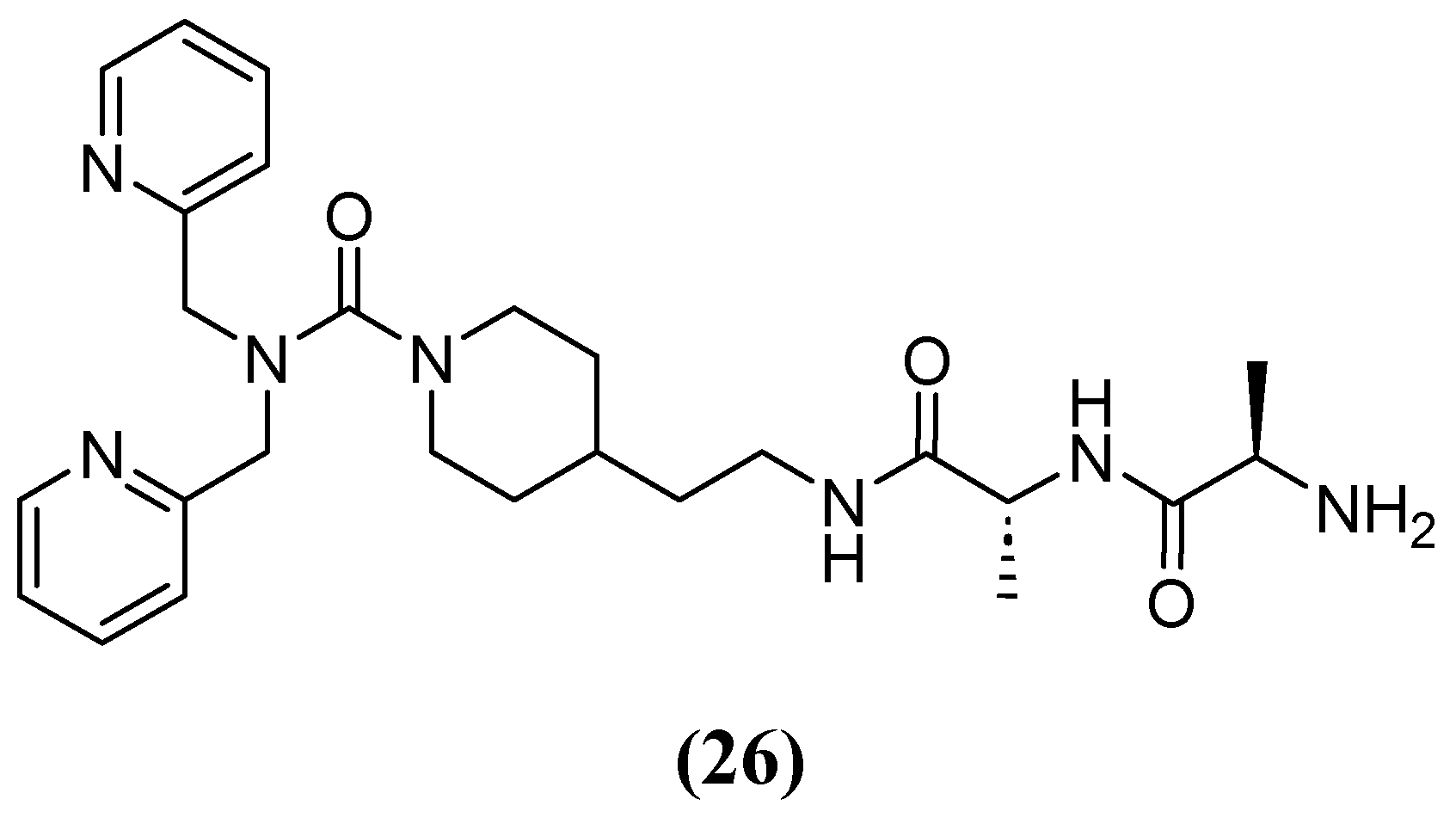
Most chelators are not zinc-specific, but dipicolylamine derivatives have a higher affinity for zinc. Consequently, a series of bivalent hybrids were constructed, consisting of a lipophilic chelator selective for zinc (dipicolylamine) attached to a connector with selective affinity for the bacterial cell structure (D-Ala-D-Ala), with the connector varying in each hybrid. Compound 26 (Figure 17), which has an ethylpiperidine moiety, had the best synergistic effect in combination with MEM, reducing the MIC of MEM from 32 and 64 mg/L to 2 and 1–2 mg/L against clinical isolates producing VIM-2 and NDM-1, respectively. The IC50 of 26 against VIM-2 was 9.8 and 2.2 µM at 5 and 20 min, respectively [69].
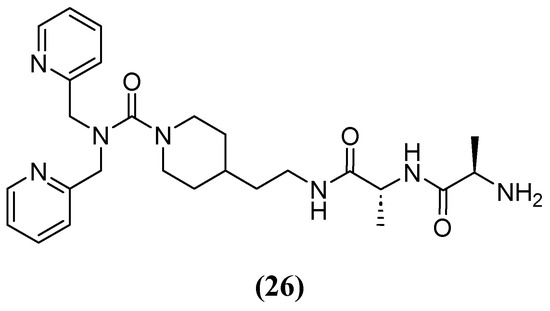
Figure 17.
Chemical structure of compound 26, a derivate of dipicolylamine.
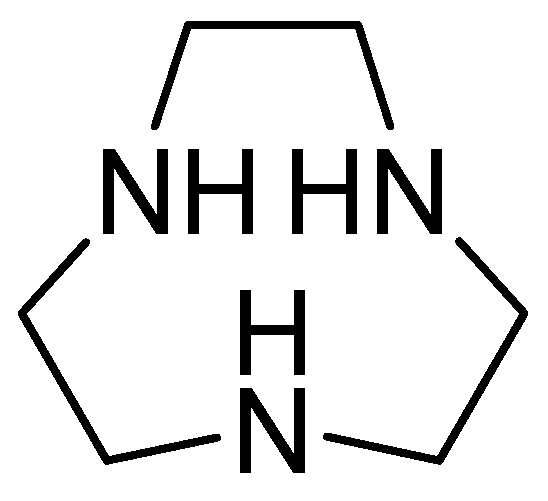
1,4,7-triazacyclononane (Figure 18) is derived from cyclononane by substituting three intermediate CH2 groups with NH groups and is characterized by a low molecular weight. Its inhibitory activity against MBLs has been observed after joint evaluation with MEM against carbapenem-resistant Enterobacteriaceae, obtaining a MIC value = 0.03 mg/L. Furthermore, the combination of MEM with 1,4,7-triazacyclononane causes a significant decrease in the colony-forming units (CFUs) per milliliter. Additionally, kill-time kinetics have shown that this combination causes a 103-fold reduction in the number of CFUs per mL at 4 h in E. coli NDM-1, E. coli VIM-2, and Enterobacter cloacae IMP-1. The inhibitor’s effect has been observed against two key enzymes, NDM-1 and VIM-2, using the new compound, with binding energies of −39.163 kcal/mol and −41.2199 kcal/mol, respectively. Furthermore, the enzyme–ligand complex remains stable. His250 and Asp124 contribute to the hydrogen-bonding capacity, while His122, Asn220, and Trp93 cover the hydrophobic pocket of the active site [70].
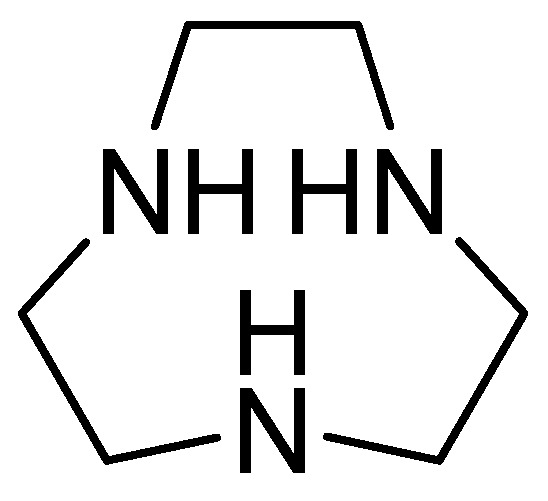
Figure 18.
Chemical structure of 1,4,7-triazacyclononane.
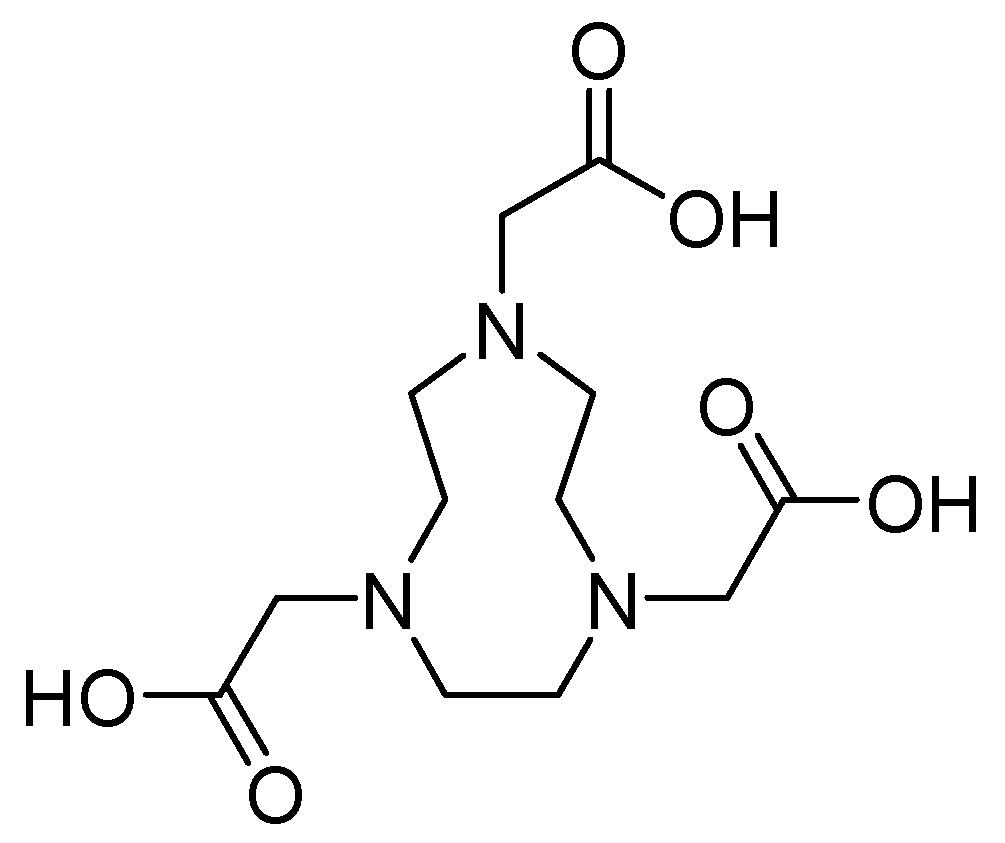
Metal chelators such as 1,4,7-triazacyclononane-1,4,7-triacetic acid 27 (Figure 19) demonstrate a high affinity for zinc. However, when used at concentrations of up to 64 mg/L, they do not inhibit this process. In contrast, the efficacy of MEM has been restored in combination with this antibiotic at a concentration of 0.06 mg/L for E. coli IMP-1 and E. cloacae NDM-1 and 0.125 mg/L for K. pneumoniae NDM-1 and E. coli NDM-1 [71].
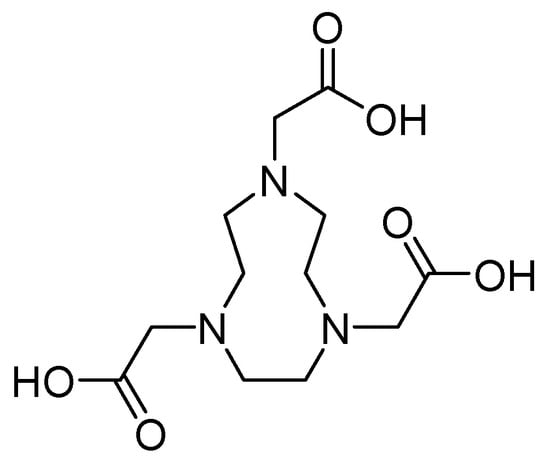
Figure 19.
Chemical structure of 1,4,7-triazacyclononane-1,4,7-triacetic acid (compound 27).
5.2. Formation of Ternary Complexes
Moreover, the inhibition of MBL activity by Zn2+ chelation is not specific. Indeed, numerous studies have documented the emergence of mutant MBLs with higher zinc affinity and augmented antibiotic resistance under conditions of Zn limitation [72]. Therefore, other compounds that directly inhibit Zn2+ have been investigated. Inhibitors that form ternary complexes include sulfur-containing compounds, such as d-captopril, which exhibit greater specificity for the active site of MBLs and, thus, are less prone to the same adverse off-target effects observed with metal-chelating inhibitors [73].
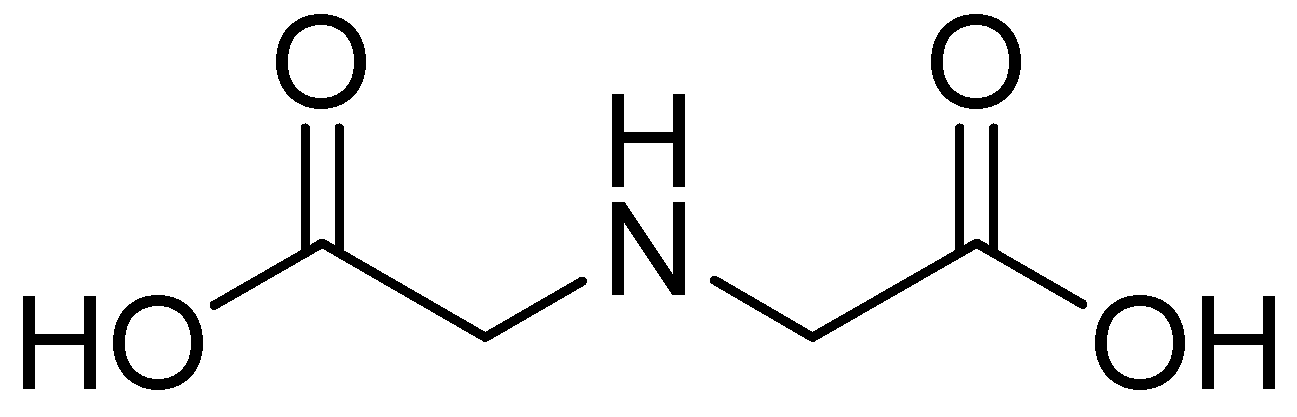
One such compound is iminodiacetic acid (IDA) (Figure 20), an aliphatic derivative of the inhibitor DPA, which is a strong tridentate metal chelator and shares structural features with the chelators AMA and EDTA, the former of which has shown activity against MBLs such as NDM-1.
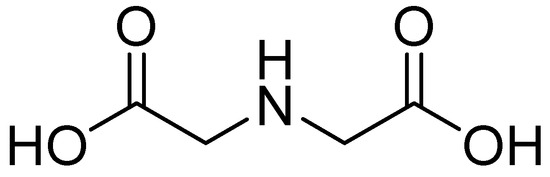
Figure 20.
Chemical structure of IDA.
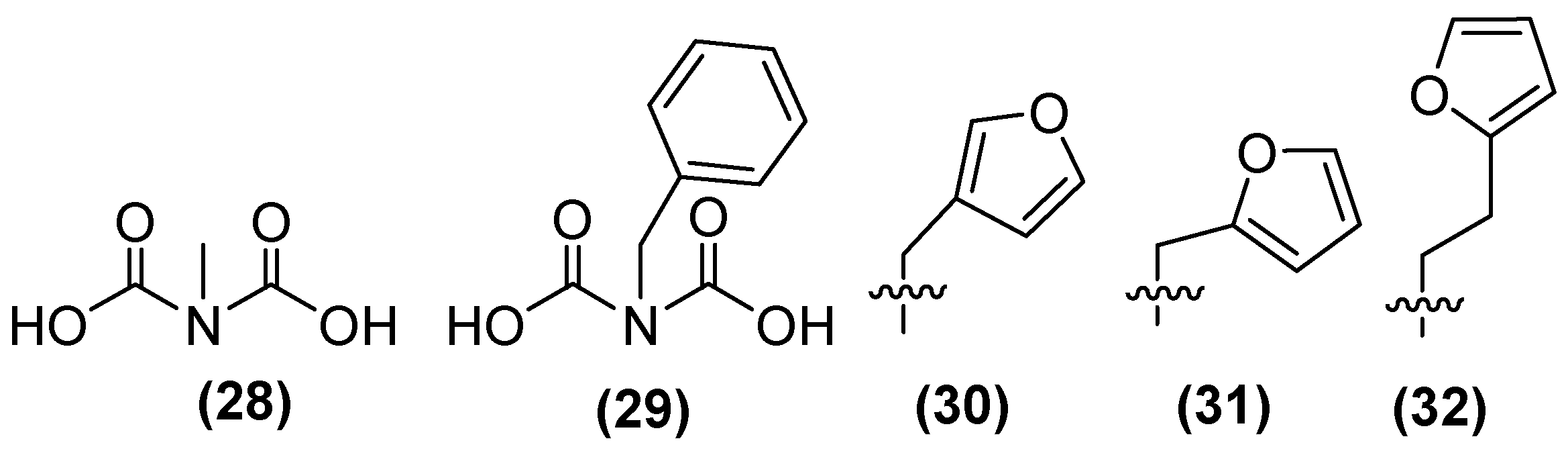
In addition, a small library of compounds has been generated using IDA as the lead structure through fragment-based drug discovery. The first methylated IDA derivative, 28 (Figure 21), exhibited the highest inhibitory activity against NDM-1, reaching 80% inhibition. In a second generation of IDA derivatives, benzylated IDA 29 showed the second highest inhibitory potential (65%). Following the selection of compounds 28 and 29 as scaffolds for inhibitor development, 30 was identified as the most potent inhibitor within this sub-library, exhibiting near complete inhibition against NDM-1 (~99%). A second library containing analogs of 30 with furan and thiophene substituents was also developed. The results showed that the extension from a methyl linker (31, IC50 = 22 μM) to an ethyl linker (32) led to a 2.5-fold enhancement in inhibitory activity, thereby producing the most potent inhibitor of this sub-library (IC50 = 8.6 μM) [74].
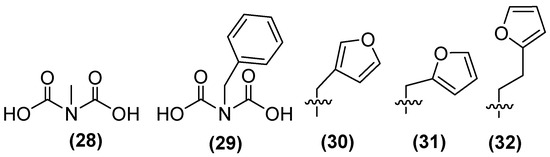
Figure 21.
Chemical structure of iminodiacetic acid derivatives as inhibitors potential against NDM-1.
Compound 32 shows the best results against NDM-1; the ethyl linker allows the furan substituent to interact more favorably with the base of the L3 β-hairpin loop of NDM-1. Ternary complex formation with NDM-1 can also be observed. This, in conjunction with the structural analogy between IDA and the β-lactam ring of the hydrolyzed antibiotic, indicates the possibility of developing analogous transition-state inhibitors of IDA and an alternative AMA scaffold for the creation of NDM-1 inhibitors.
On the other hand, taxifolin (Figure 22) is a flavonoid compound, also known as dihydroquercetin, which was originally isolated from the bark of Douglas fir trees. It is commonly found in food items such as onions and olive oil. Additionally, it is a component of various commercial preparations. Its pharmacological activities include antioxidant, anti-inflammatory, and antimicrobial effects [75].
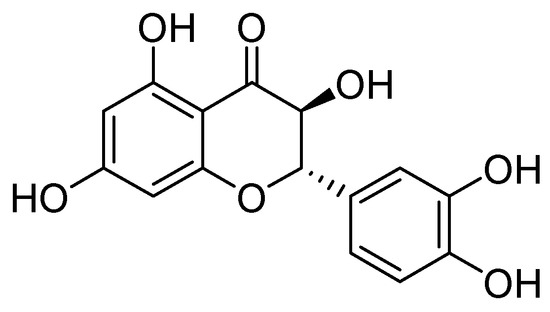
Figure 22.
Structure of taxifolin, a promising inhibitor of MBL VIM-2 in P. aeruginosa.
Taxifolin has been found to be an effective inhibitor of VIM-2 in P. aeruginosa (MIC = 3.1 µg/mL, Ki = 8 µM). Docking analysis has shown that this compound binds to the VIM-2 binding site in a similar way to quercetin. Taxifolin interacts with the amino acids Asp117 and Arg205 through hydrogen bonding, which contributes to the stabilization of the inhibitor within the active site. Flavonol-derived compounds, such as taxifolin, have the potential to serve as effective inhibitors of MBLs [72].
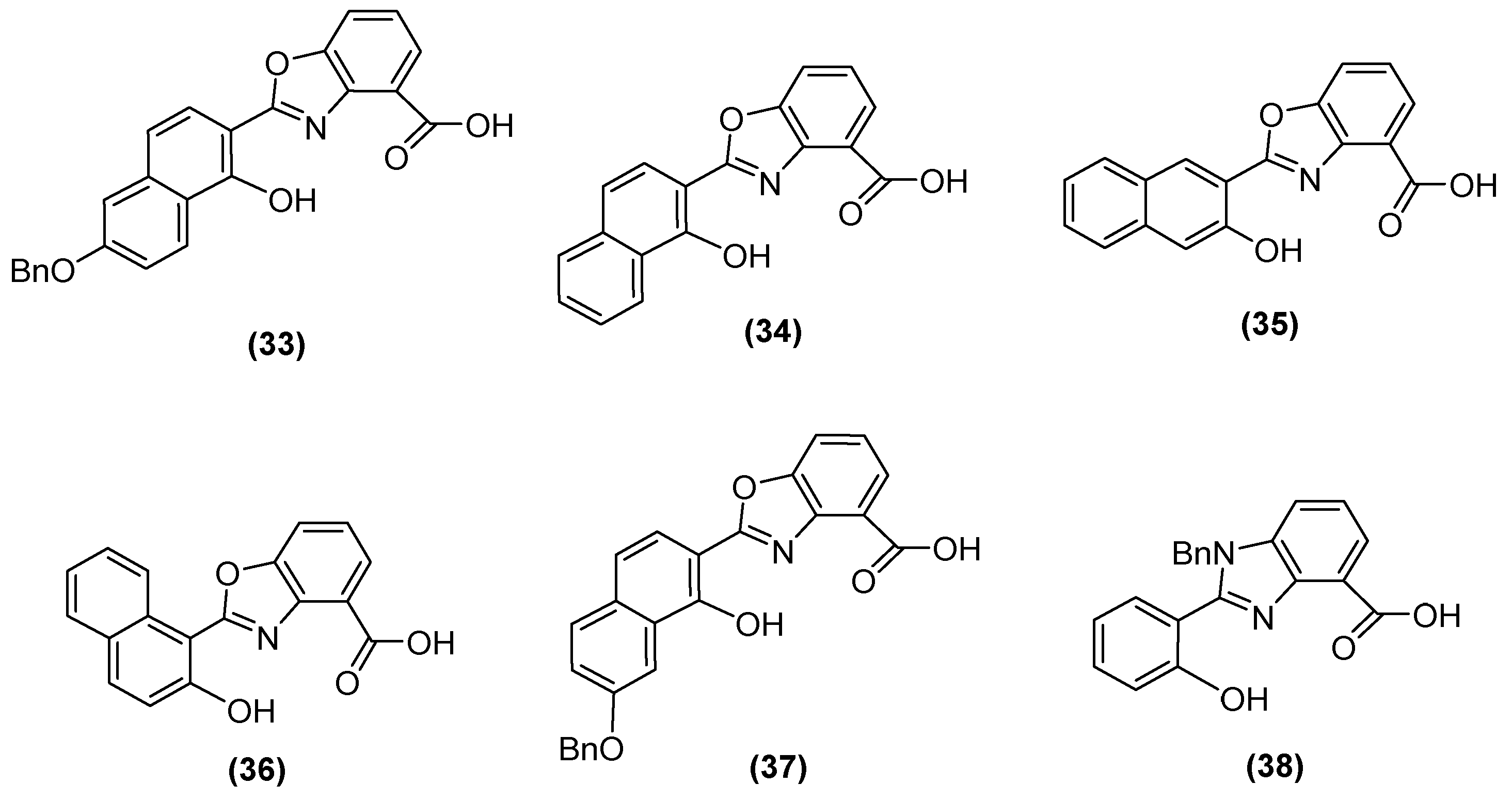
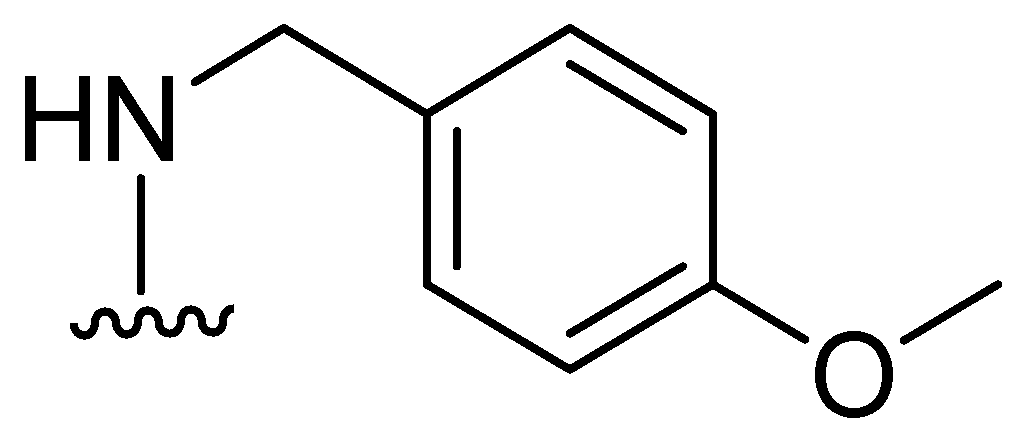
Jackson and cols. (2021) synthesized benzoxazole and benzimidazole derivatives as zinc chelators, observing that six of the compounds exhibited inhibitory activity in E. coli blaNDM-1 with an IC50 value < 10 µM, and compound 33 (Figure 23) had the best activity (IC50 = 0.38 µM). The compounds share a similar structural composition, except for the position of the benzyloxy group (compounds 33 and 37) and an extra benzyl group in compound 38. In addition, compounds 33, 34, 37, and 38 decreased the MIC of MEM (16-fold to 4 μg/mL or less), with compound 33 reaching MIC values < 0.5 μg/mL. It was observed via UV–visible spectroscopy that NDM-1 and this compound form a ternary complex. Molecular docking analysis also showed that compound 33 was coordinated at Zn1 by His120, His122, and His189, while Zn2 was coordinated by Asp124, His250, and Cys208 [73].
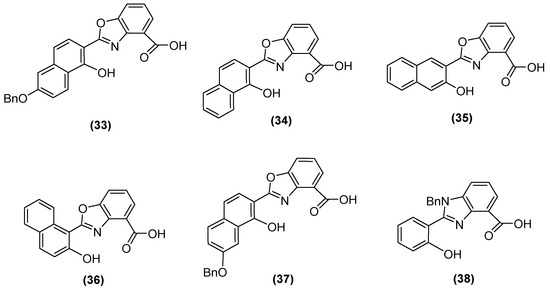
Figure 23.
Chemical structure of benzoxazole and benzimidazole derivatives as zinc chelators.
In one study, the use of DPA instead of EDTA as a chelator was tested in a disk diffusion analysis observing phenotypic inhibition patterns against MBL IMP-1 and VIM-2 in P. aeruginosa and Acinetobacter spp., respectively; the results were like the inhibition exerted by EDTA, showing the ability of DPA as a chelator [76].
Consequently, DPA-derived inhibitors—which are based on DPA and are designed to enhance binding interactions with protein residues proximal to active site Zn2+ ions (e.g., Asn220, Gln123, and Lys211)—have been investigated. One of the derivatives, 39 (Figure 24), has demonstrated notable specificity for NDM-1, with an IC50 value of 80 nM. In contrast, DPA alone demonstrates a tendency to chelate metal ions from NDM-1. However, 39 forms a ternary NDM-1 complex with Zn2+, functioning as a stable inhibitor. The inhibition of IMP-1 and VIM-2 has also been reported. The combination of compound 39 with imipenem demonstrates the restoration of antibiotic susceptibility in clinical isolates of E. coli and K. pneumoniae harboring NDM-1, as evidenced by a reduction in the MIC (0.5–1 mg/L) [77].
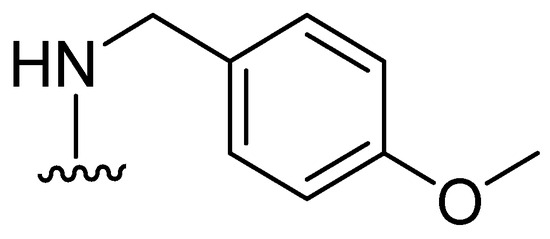
Figure 24.
Structure of DPA-derived inhibitor 39.
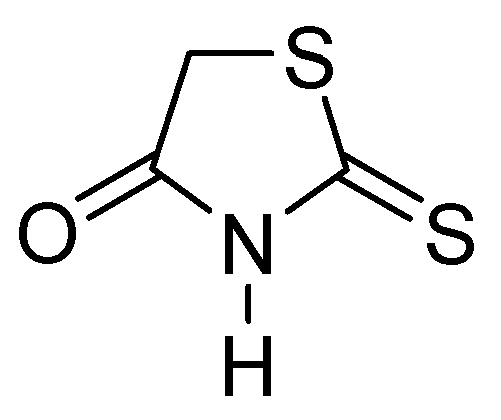
Thiazolidinones represent a significant class of heterocyclic compounds with a crucial role in human biological systems, becoming a critical element in the design of novel pharmaceutical agents [78]. Rhodanine (Figure 25) is a subtype of 4-thiazolidinone and has a broad spectrum of biological activity, demonstrating mostly antibacterial, antifungal, antiparasitic, antiviral, antineoplastic, antidiabetic, and anti-inflammatory activity [79].
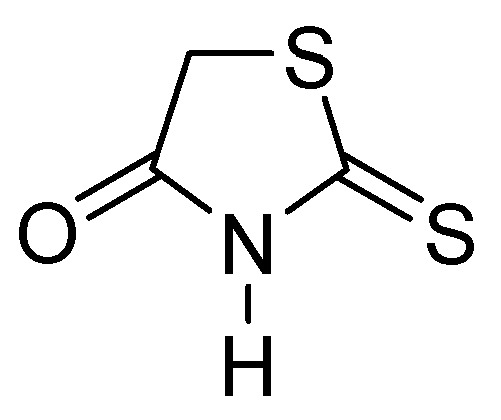
Figure 25.
Chemical structure of rhodanine monomer.
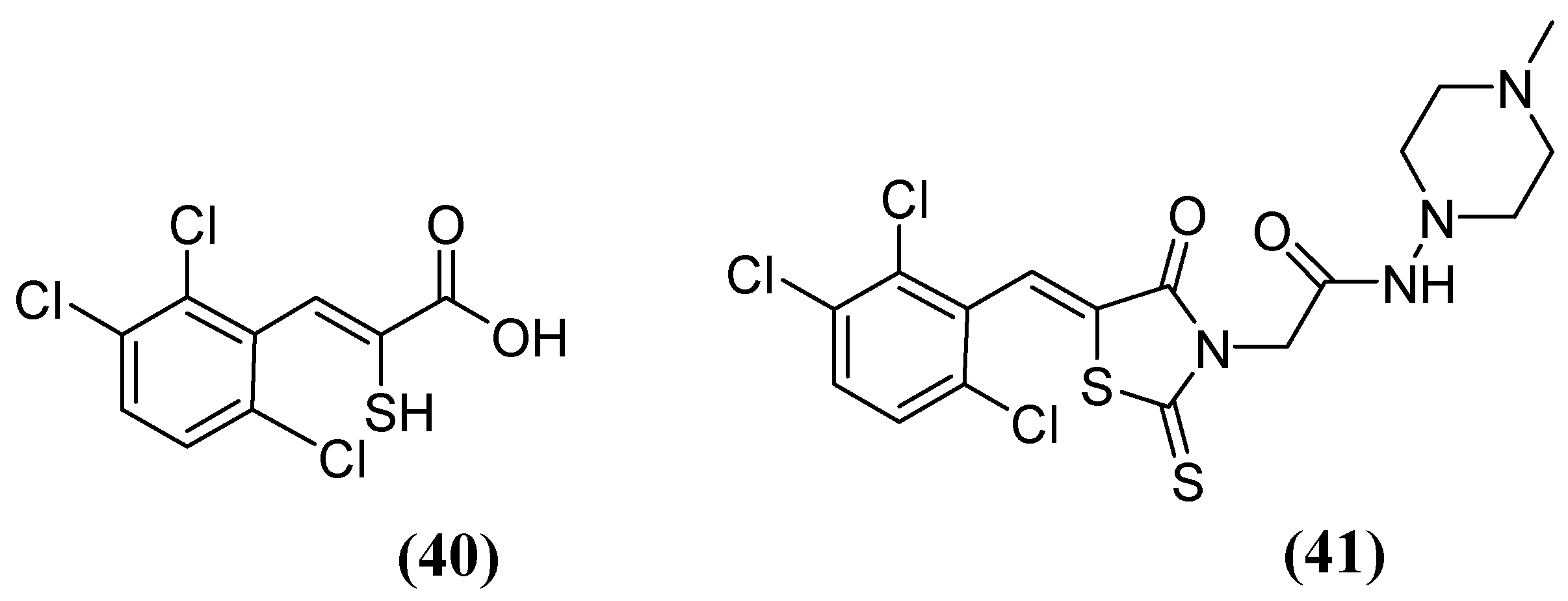
The enethiols derived from the hydrolysis of rhodanine inhibit the subclasses of MBLs. Derivative 40 (Figure 26) has been observed to inhibit MBLs by chelating the Zn2+ ion from the active site. Notably, an unusual ternary complex is formed between the enzyme and two distinct ligands, 40 and 41 (Figure 26), in the case of VIM-2, which is an unusual occurrence. The most potent inhibition has been observed for 41 against SPM-1, IMP-1, BcII, VIM-2, and NDM-1 (range, 0.02–0.08 μM), while 40 has shown inhibition against IMP-1 (0.09 μM) and VIM-2 (0.06 μM). Interaction studies have shown that thiolate interacts with both Zn2+ ions, and carboxylate interacts with Zn2 and Arg233 in VIM-2 [80].
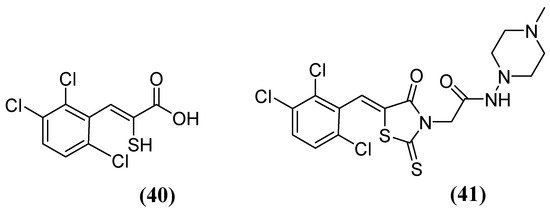
Figure 26.
Chemical structure of derivatives 40 and 41.
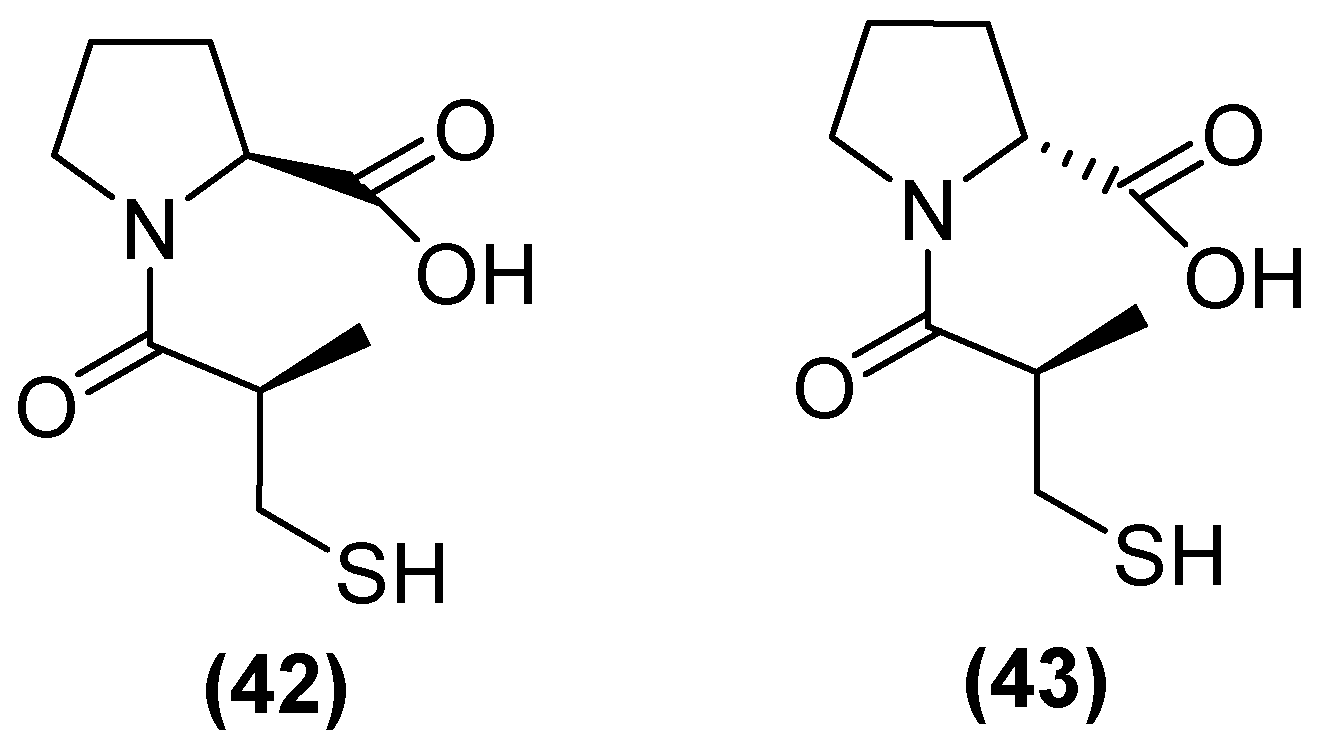
On the other hand, thiols—such as captopril, a molecule that contains a thiol and a carboxylate group—are the most widely studied inhibitors. Thiols are potent inhibitors of MBLs due to the ability of sulfur to coordinate zinc. (2 S)-1-((2 S)-2-methyl-3-sulfanylpropanoyl) pyrrolidine-2-carboxylic acid, also known as L-captopril, is a thiol-containing molecule. The D (42) and L (43) stereoisomers of captopril (Figure 27) are active against MBLs B1 and B3, with their IC50 values covering a wide range from 0.072 to >500 μM depending on the captopril stereoisomer and MBL variant [48]. However, thiol-based inhibitors tend to undergo oxidation to the corresponding disulfides, thus losing their zinc-binding and MBL-inhibitory properties [81].
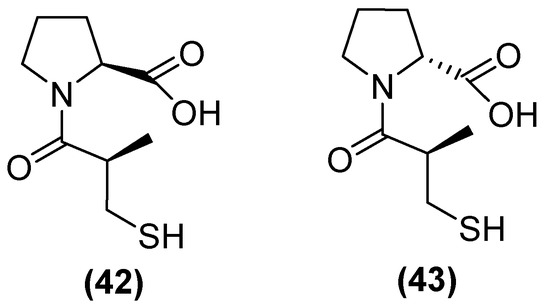
Figure 27.
Chemical structure of L-captopril (42) and D-captopril (43).
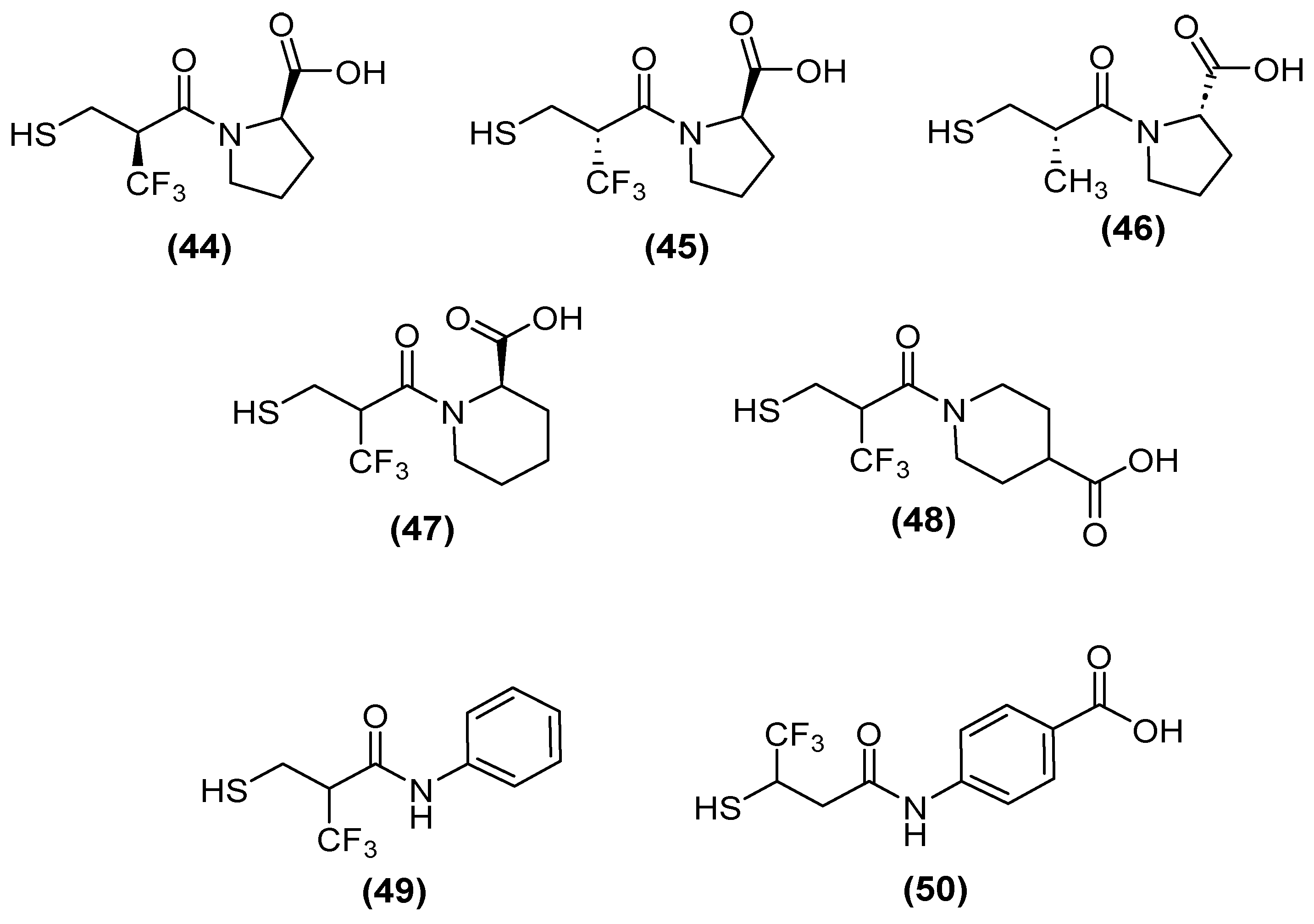
Kondratieva and cols. [81] evaluated trifluoromethylated captopril analogs, and the NDM-1 inhibition test again showed that seven of the compounds had IC50 values below 10 µM (Figure 28). However, a D-captopril derivative (45) showed an IC50 value of 0.3 µM and was considered the most active compound. The trifluoromethyl-containing analogs showed the most promising inhibitory activity [81].
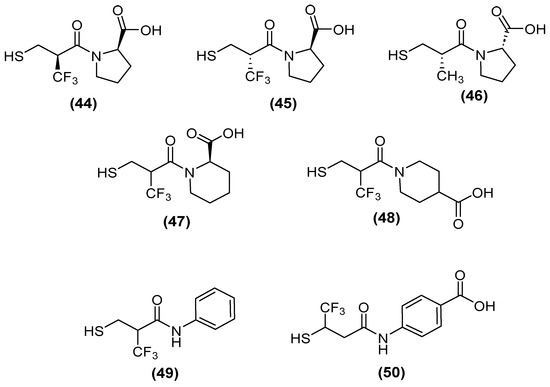
Figure 28.
Chemical trifluoromethylated derivatives, analogs of captopril, as NDM1 inhibitors.
Although, several compounds of the trifluoromethylated compounds showed synergistic effects—thus reversing the effect of MEM and reducing the MIC values in NDM-1 (up to 64-fold), VIM-2 (up to 8-fold), and IMP-26 (up to 64-fold) in E. coli (Table 1)—both native and trifluoromethyl-substituted inhibitors bind to the two zinc ions of the NDM-1 binding site through their thiol functionality (2.3 Å), and their carbonyl group hydrogen bonds to the side-chain amide of Asn220 (1.6–1.7 Å). An N-H⋯F-C hydrogen bond (2.2 Å) was predicted between the Gln123 amide proton of NDM-1 and the trifluoromethyl group of (+)-5αA [81].

Table 1.
MICs values of meropenem (MEM) with the best-trifluoromethylated compounds inhibitors against NDM-1, VIM-2, and IMP-26.
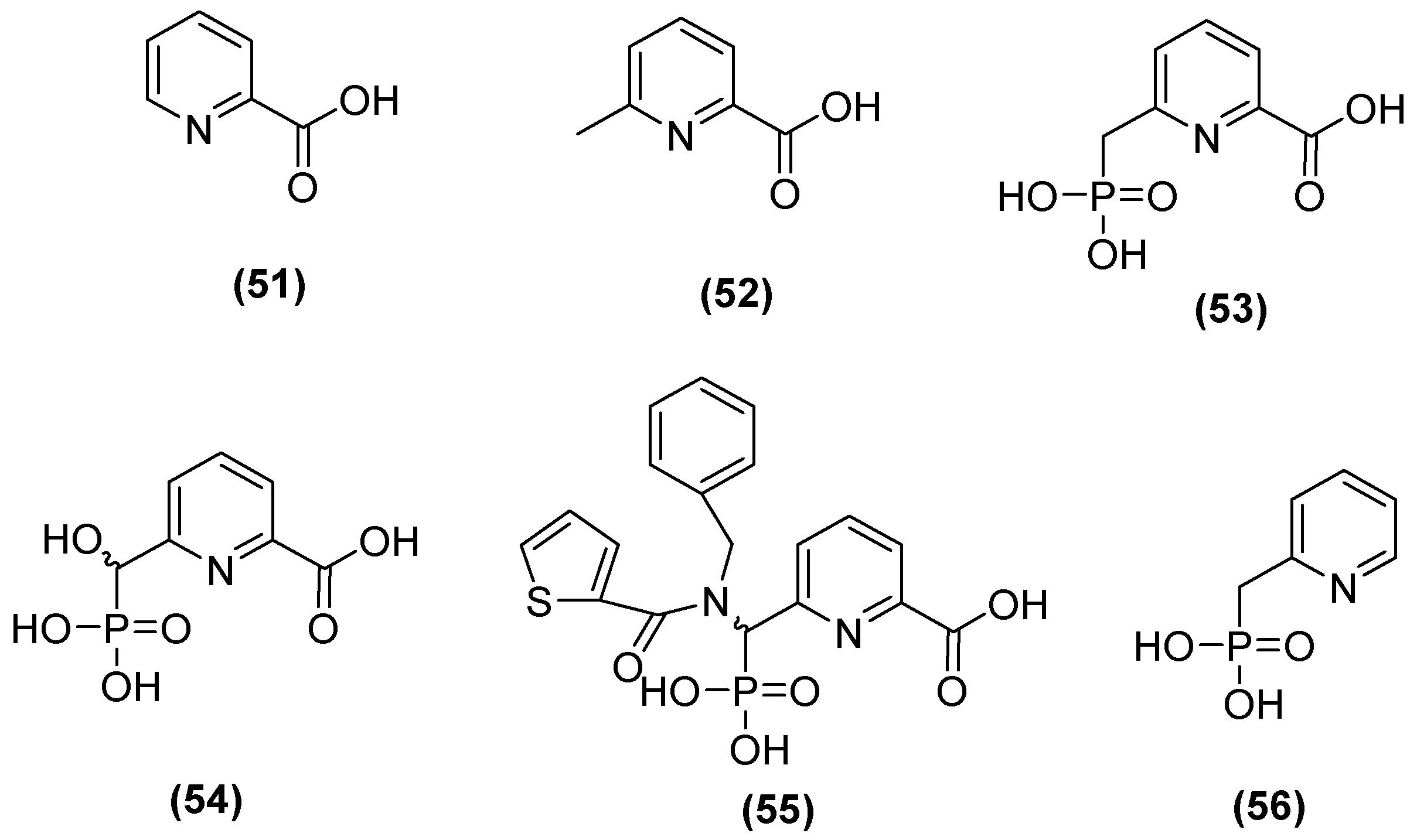
The 6-phosphonomethylpyridine-2-carboxylate (PMPC) compound has also been noted as a potent inhibitor of MBLs such as IMP-1, VIM-2, and NDM-1 in group B1 and L1 in B3. Hinchliffe and cols. [48] synthesized PMPC derivatives (Figure 29) in which the addition of a phosphonomethyl group at C6 of PMCP in 2-picolinic acid (53) resulted in significant potency against NDM-1 (IC50 = 0.374 μM). The inhibitory activity against NDM-1 (IC50 = 0.322 µM) was improved by adding a hydroxyl group to the carbon of the phosphonomethyl group (54). However, by adding an additional large hydrophobic substituent to the phosphonomethyl group, it was found that the better compound against all MBLs tested was 55, with IC50 values of 0.306, 0.464, 2.91, and 1.57 µM for NDM-1, VIM-2, IMP-1, and L1, respectively [48].
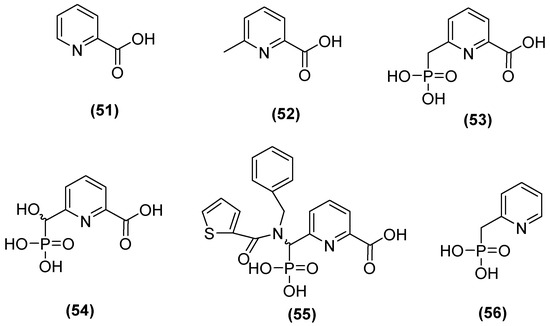
Figure 29.
Structure of 6-phosphonomethylpyridine-2-carboxylate (PMPC).
In addition, in another study, the simplest compound (53) was tested to evaluate its ability to enhance the antibacterial activity of MEM against the most clinically relevant subclass B1 MBLs. Co-administration with 53 reduced the MICs of MEM to the susceptible range against all strains tested, except E. coli MG1655, which expressed NDM-1 (MIC = 8 mg/L), although 100 mg/L of 53 was required to achieve this. In all cases, the MIC of MEM was reduced by at least 16-fold at 100 mg/mL in 53. Furthermore, it was determined that 53 binds to the zinc active site of IMP-1 but does not displace the nucleophilic hydroxide; notably, the inhibitory carboxylate group and the pyridine nitrogen atom interacted with the Zn2+ site at distances of 2.30 and 2.69 Å on the main chain of the protein (2.70 Å), and the binding was stabilized by interactions between the carboxylate and Lys224 caused by the proximity of a hydrophobic pocket containing Val61, Val67, Trp64, and Phe87 [48].
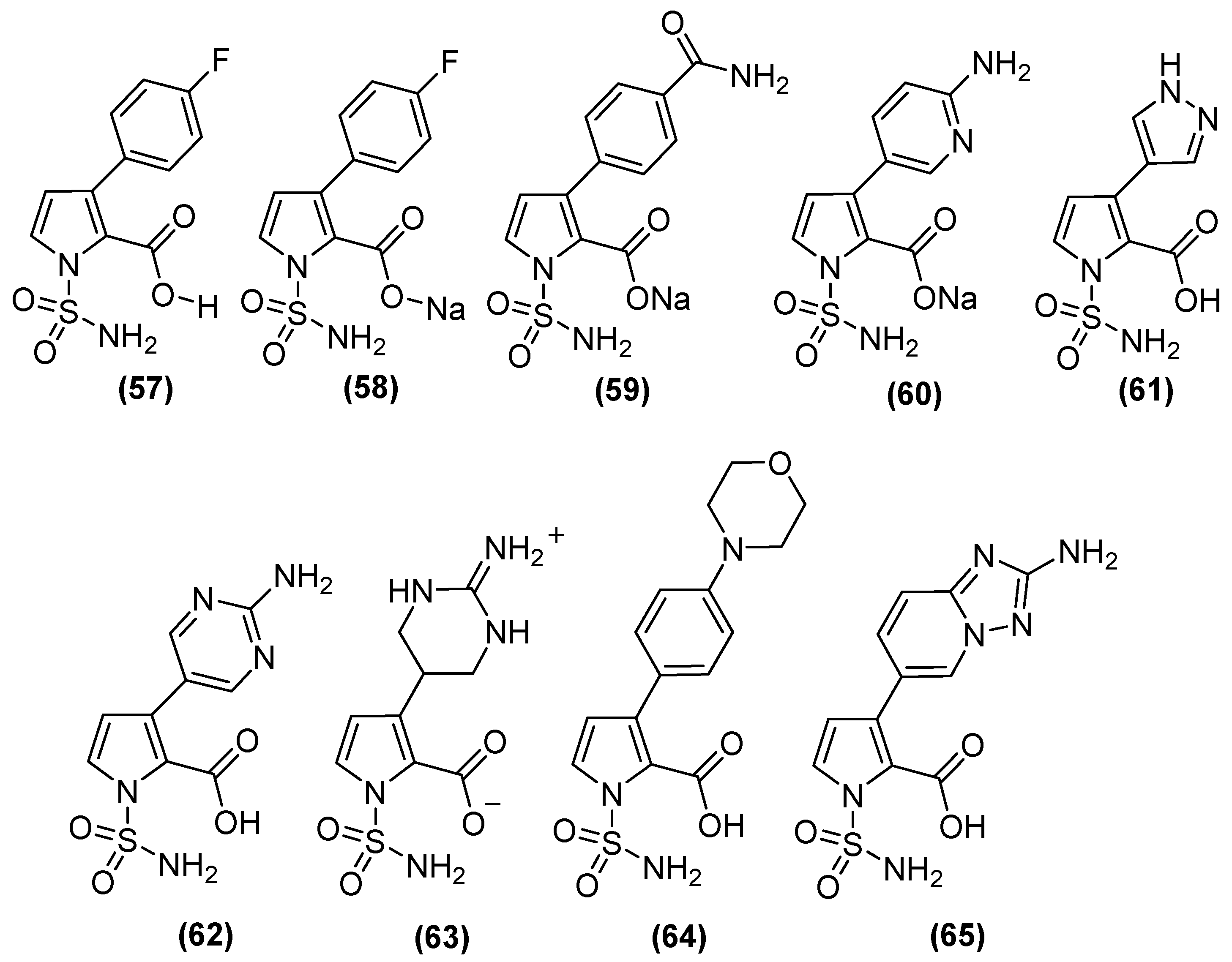
On the other hand, N-sulfamoylpyrrole-2-carboxylate (NSPC) derivatives (Figure 30) are potent inhibitors of relevant subclass B1 MBLs, including NDM-1, because they can mimic the binding mode of tetrahedral intermediates (boronates). NSPC analogs show inhibitory effects on MBLs (Table 2), of which compound 63 inhibits VIM-1 (pIC50 8.5), which is similar to the bicyclic boronate TAN but less potent against NDM-1. In contrast, analogs such as 57, 58, 61, and 64 show nanomolar potency against NDM-1. NSPC is 150 to >1500 times more potent (pIC50 of 7.3 to 9.2) than bicyclic boronates (CB2) or TAN (pIC50 values of 6.0 and 5.6, respectively) against IMP-1. Notably, none of the NSPCs can potently inhibit all four MBLs simultaneously. Antimicrobial susceptibility testing has shown that, in NDM-1-producing strains, at a concentration of 8 μg/mL, N-sulfamoylpyrroles reduces the MIC of MEM from 64 to ~0.375 μg/mL [82].
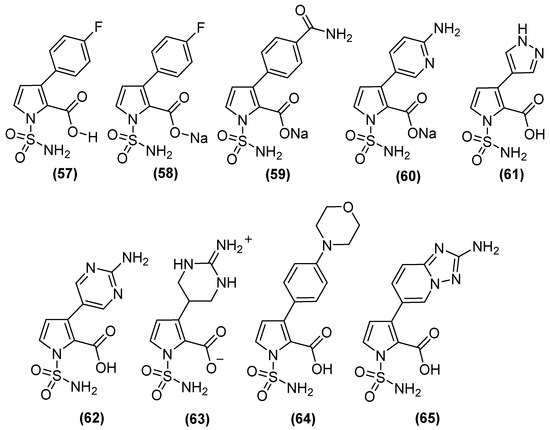
Figure 30.
N-sulfamoylpyrrole-2-carboxylate (NSPC) derivatives as potent inhibitors of clinically relevant subclass B1 MBLs.
Table 2.
Activity of NSPC derivatives against clinically relevant MBLs.
6. Conclusions
Given the growing threat of infections caused by MDR bacteria, several compounds and their derivatives have been evaluated in the search for substances capable of inhibiting the most complex group of β-lactamases, the MBLs, for which there are currently no approved inhibitors for therapeutic use, and which are capable of hydrolyzing almost all β-lactams except aztreonam. This review focuses on inhibitors that form ternary complexes with their targets or chelate zinc ions. Zn2+ chelation is an interesting strategy with promising results, as seen with AMA inhibitors. However, this mechanism presents a potential risk due to the selectivity toward human metalloenzymes; although, the information regarding this is scarce. Furthermore, the use of chelators as inhibitors may exert evolutionary pressure for the purpose of improving the Zn2+-binding affinities of MBLs. Therefore, agents that are highly selective for zinc and weakly bind to ions other than Zn2+, such as iron, calcium, manganese, potassium, sodium, and other relevant biological cations, are essential. In addition, boronic acid-based inhibitors such as TAN (VNRX-5133) are the most promising so far, as they can inhibit both serine-dependent b-lactamases and MBLs. The development of new agents against MBLs is a priority, and great efforts are being made to find inhibitors against multiple enzymes that challenge the efficacy of the latest available therapeutic options.
Author Contributions
Conceptualization, J.L.O.-B. and G.R.; methodology, formal analysis, investigation, resources, and data curation, J.L.O.-B., L.K.V.-J., G.A.-N., E.O.-P., A.D.P.-G., E.E.L.-R. and G.R.; writing—original draft preparation, J.L.O.-B.; writing—review and editing, L.K.V.-J., G.A.-N., E.O.-P., A.D.P.-G., E.E.L.-R. and G.R. All authors have read and agreed to the published version of the manuscript.
Funding
Secretaría de Investigación y Posgrado del Instituto Politécnico Nacional.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We want to express our acknowledgment to Secretaría de Investigación y Posgrado del Instituto Politécnico Nacional and the Consejo Nacional de Humanidades, Ciencia y Tecnología of Mexico.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. (The Review on Antimicrobial Resistance, London, 2016, United Kingdom). Rev. Antimicrob. Resist. 2014. [Google Scholar]
- Palm, M.; Fransson, A.; Hult, J.; Karolina, B.; Allouche, A.; Chiang, O.E.; Constandse, M.L.; Van Dijk, K.J.; Icli, S.; Klimesova, B.; et al. The Effect of Heavy Metals on Conjugation Efficiency of an F-Plasmid in Escherichia Coli. Antibiotics 2022, 11, 1123. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.Y. Antimicrobial Peptides: New Hope in the War against Multidrug Resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [PubMed]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic Resistance Genes in Bacteria: Occurrence, Spread, and Control. J. Basic Microbiol. 2021, 61, 1049–1070. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.E.; Palm, M.; Warringer, J.; Farewell, A. Inhibiting Conjugation as a Tool in the Fight against Antibiotic Resistance. Drug Dev. Res. 2019, 80, 19–23. [Google Scholar] [CrossRef]
- Li, W.; Zhang, G. Detection and Various Environmental Factors of Antibiotic Resistance Gene Horizontal Transfer. Environ. Res. 2022, 212, 113267. [Google Scholar] [CrossRef]
- Arer, V.; Kar, D. Biochemical Exploration of β-Lactamase Inhibitors. Front. Genet. 2023, 13, 1–15. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar]
- De Angelis, G.; Del Giacomo, P.; Posteraro, B.; Sanguinetti, M.; Tumbarello, M. Molecular Mechanisms, Epidemiology, and Clinical Importance of β-Lactam Resistance in Enterobacteriaceae. Int. J. Mol. Sci. 2020, 21, 5090. [Google Scholar] [CrossRef]
- Zhang, S.; Liao, X.; Ding, T.; Ahn, J. Role of β-Lactamase Inhibitors as Potentiators in Antimicrobial Chemotherapy Targeting Gram-Negative Bacteria. Antibiotics 2024, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2023 WHO Releases Report on State of Development of Antibacterials. Available online: https://www.who.int/news/item/14-06-2024-who-releases-report-on-state-of-development-of-antibacterials (accessed on 1 February 2024).
- Aurilio, C.; Sansone, P.; Barbarisi, M.; Pota, V.; Giaccari, L.G.; Coppolino, F.; Barbarisi, A.; Passavanti, M.B.; Pace, M.C. Mechanisms of Action of Carbapenem Resistance. Antibiotics 2022, 11, 421. [Google Scholar] [CrossRef] [PubMed]
- Bahr, G.; González, L.J.; Vila, A.J. Metallo-β-Lactamases in the Age of Multidrug Resistance: From Structure and Mechanism to Evolution, Dissemination, and Inhibitor Design. Chem. Rev. 2021, 121, 7957–8094. [Google Scholar]
- Hernández-Alomía, F.; Bastidas-Caldes, C.; Ballesteros, I.; Tenea, G.N.; Jarrín-V, P.; Molina, C.A.; Castillejo, P. Beta-Lactam Antibiotic Resistance Genes in the Microbiome of the Public Transport System of Quito, Ecuador. Int. J. Environ. Res. Public Health 2023, 20, 1900. [Google Scholar] [CrossRef] [PubMed]
- Iovleva, A.; Doi, Y. Carbapenem-Resistant Enterobacteriaceae. Clin. Lab. Med. 2017, 37, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, H.O.; Soliman, A.M.; Ahmed, A.M.; Shimamoto, T.; Hara, T.; Ikeda, M.; Kuroo, Y.; Kayama, S.; Sugai, M.; Shimamoto, T. High Carbapenem Resistance in Clinical Gram-Negative Pathogens Isolated in Egypt. Microb. Drug Resist. 2017, 23, 838–844. [Google Scholar] [CrossRef]
- Armstrong, T.; Fenn, S.J.; Hardie, K.R. JMM Profile: Carbapenems: A Broad-Spectrum Antibiotic. J. Med. Microbiol. 2021, 70, 001462. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-Negative Bacteria. Clin. Infect. Dis. 2019, 69, S521–S528. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Song, X.; Li, M.; Yu, Z.; Cheng, W.; Yu, Z.; Zhang, W.; Zhang, Y.; Shen, A.; Sun, H.; et al. Global Spread of Carbapenem-Resistant Enterobacteriaceae: Epidemiological Features, Resistance Mechanisms, Detection and Therapy. Microbiol. Res. 2023, 266, 127249. [Google Scholar] [CrossRef]
- Doi, Y. Treatment Options for Carbapenem-Resistant Gram-Negative Bacterial Infections. Clin. Infect. Dis. 2019, 69, S565–S575. [Google Scholar] [CrossRef]
- Noster, J.; Thelen, P.; Hamprecht, A. Detection of Multidrug-resistant Enterobacterales—From Esbls to Carbapenemases. Antibiotics 2021, 10, 1140. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lee, J.H.; Lee, J.J.; Park, K.S.; Karim, A.M.; Lee, C.R.; Jeong, B.C.; Lee, S.H. Structural Basis for Carbapenem-Hydrolyzing Mechanisms of Carbapenemases Conferring Antibiotic Resistance. Int. J. Mol. Sci. 2015, 16, 9654–9692. [Google Scholar] [CrossRef] [PubMed]
- Edoo, Z.; Arthur, M.; Hugonnet, J.E. Reversible Inactivation of a Peptidoglycan Transpeptidase by a β-Lactam Antibiotic Mediated by β-Lactam-Ring Recyclization in the Enzyme Active Site. Sci. Rep. 2017, 7, 9136. [Google Scholar] [CrossRef]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Cascella, M. Beta-Lactam Antibiotics. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [PubMed]
- Jalde, S.S.; Choi, H.K. Recent Advances in the Development of β-Lactamase Inhibitors. J. Microbiol. 2020, 58, 633–647. [Google Scholar] [PubMed]
- Feng, H.; Liu, X.; Wang, S.; Fleming, J.; Wang, D.C.; Liu, W. The Mechanism of NDM-1-Catalyzed Carbapenem Hydrolysis Is Distinct from That of Penicillin or Cephalosporin Hydrolysis. Nat. Commun. 2017, 8, 2242. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-Lactamase-Producing Pathogens. Clin. Microbiol. Rev. 2020, 33, e00047-19. [Google Scholar]
- van Haren, M.J.; Tehrani, K.H.M.E.; Kotsogianni, I.; Wade, N.; Brüchle, N.C.; Mashayekhi, V.; Martin, N.I. Cephalosporin Prodrug Inhibitors Overcome Metallo-β-Lactamase Driven Antibiotic Resistance. Chemistry 2021, 27, 3806–3811. [Google Scholar] [CrossRef]
- Chiou, J.; Leung, T.Y.C.; Chen, S. Molecular Mechanisms of Substrate Recognition and Specificity of New Delhi Metallo-β-Lactamase. Antimicrob. Agents Chemother. 2014, 58, 5372–5378. [Google Scholar] [CrossRef] [PubMed]
- Lisa, M.-N.; Palacios, A.R.; Aitha, M.; González, M.M.; Moreno, D.M.; Crowder, M.W.; Bonomo, R.A.; Spencer, J.; Tierney, D.L.; Llarrull, L.I.; et al. A General Reaction Mechanism for Carbapenem Hydrolysis by Mononuclear and Binuclear Metallo-β-Lactamases. Nat. Commun. 2017, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.E.; Livermore, D.M.; Hooper, D.C.; Hope, W.W. Metallo-β-Lactamases: Structure, Function, Epidemiology, Treatment Options, and the Development Pipeline. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Palzkill, T. Metallo-β-Lactamase Structure and Function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef]
- Yan, Y.H.; Li, G.; Li, G.B. Principles and Current Strategies Targeting Metallo-β-Lactamase Mediated Antibacterial Resistance. Med. Res. Rev. 2020, 40, 1558–1592. [Google Scholar] [PubMed]
- Palacios, A.R.; Rossi, M.-A.; Mahler, G.S.; Vila, A.J. Metallo-β-Lactamase Inhibitors Inspired on Snapshots from the Catalytic Mechanism. Biomolecules 2020, 10, 854. [Google Scholar] [CrossRef]
- Hu, Z.; Gunasekera, T.S.; Spadafora, L.; Bennett, B.; Crowder, M.W. Metal Content of Metallo-β-Lactamase L1 Is Determined by the Bioavailability of Metal Ions. Biochemistry 2008, 47, 7947–7953. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tanaka, H.; Kurisu, G.; Nakano, R.; Yano, H.; Sakai, H. Structural Insights into the Substrate Specificity of IMP-6 and IMP-1 Metallo-β-Lactamases. J. Biochem. 2023, 173, 21–30. [Google Scholar] [CrossRef]
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular Diversity of Extended-Spectrum β-Lactamases and Carbapenemases, and Antimicrobial Resistance. J. Intensive Care 2020, 8, 13. [Google Scholar]
- Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. Int. J. Mol. Sci. 2020, 21, 9308. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, K.H.M.E.; Brüchle, N.C.; Wade, N.; Mashayekhi, V.; Pesce, D.; van Haren, M.J.; Martin, N.I. Small Molecule Carboxylates Inhibit Metallo-β-Lactamases and Resensitize Carbapenem-Resistant Bacteria to Meropenem. ACS Infect. Dis. 2020, 6, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-Lactamase (NDM): A Threat to Public Health. BMC Microbiol. 2017, 17, 101. [Google Scholar]
- Ju, L.C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar]
- Gavara, L.; Verdirosa, F.; Sevaille, L.; Legru, A.; Corsica, G.; Nauton, L.; Sandra Mercuri, P.; Sannio, F.; De Luca, F.; Hadjadj, M.; et al. 1,2,4-Triazole-3-Thione Analogues with an Arylakyl Group at Position 4 as Metallo-β-Lactamase Inhibitors. Bioorg. Med. Chem. 2022, 72, 116964. [Google Scholar] [CrossRef]
- Sychantha, D.; Rotondo, C.M.; Tehrani, K.H.M.E.; Martin, N.I.; Wright, G.D. Aspergillomarasmine A Inhibits Metallo-β-Lactamases by Selectively Sequestering Zn(2). J. Biol. Chem. 2021, 297, 100918. [Google Scholar] [CrossRef]
- Hinchliffe, P.; Tanner, C.A.; Krismanich, A.P.; Labbé, G.; Goodfellow, V.J.; Marrone, L.; Desoky, A.Y.; Calvopiña, K.; Whittle, E.E.; Zeng, F.; et al. Structural and Kinetic Studies of the Potent Inhibition of Metallo-β-Lactamases by 6-Phosphonomethylpyridine-2-Carboxylates. Biochemistry 2018, 57, 1880–1892. [Google Scholar] [CrossRef]
- Thomas, C.S.; Braun, D.R.; Olmos, J.L.; Rajski, S.R.; Phillips, G.N.; Andes, D.N.; Bugni, T.S. Pyridine-2,6-Dithiocarboxylic Acid and Its Metal Complexes: New Inhibitors of New Delhi Metallo-Lactamase-1. Mar. Drugs 2020, 18, 295. [Google Scholar] [CrossRef]
- Wang, X.; Lu, M.; Shi, Y.; Ou, Y.; Cheng, X. Discovery of Novel New Delhi Metallo-β-Lactamases-1 Inhibitors by Multistep Virtual Screening. PLoS ONE 2015, 10, e0118290. [Google Scholar] [CrossRef]
- Cui, D.Y.; Yang, Y.; Bai, M.M.; Han, J.X.; Wang, C.C.; Kong, H.T.; Shen, B.Y.; Yan, D.C.; Xiao, C.L.; Liu, Y.S.; et al. Systematic Research of H2dedpa Derivatives as Potent Inhibitors of New Delhi Metallo-β-Lactamase-1. Bioorg. Chem. 2020, 101, 103965. [Google Scholar] [CrossRef]
- Chen, F.; Bai, M.; Liu, W.; Kong, H.; Zhang, T.; Yao, H.; Zhang, E.; Du, J.; Qin, S. H2dpa Derivatives Containing Pentadentate Ligands: An Acyclic Adjuvant Potentiates Meropenem Activity in Vitro and in Vivo against Metallo-β-Lactamase-Producing Enterobacterales. Eur. J. Med. Chem. 2021, 224, 113702. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.-C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.G.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase, Serine-β-Lactamase and Penicillin-Binding Protein Inhibition by Cyclic Boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.T.; Tyrrell, J.M.; Navratilova, I.H.; Calvopiña, K.; Robinson, S.W.; Lohans, C.T.; McDonough, M.A.; Cain, R.; Fishwick, C.W.G.; Avison, M.B.; et al. Studies on the Inhibition of AmpC and Other β-Lactamases by Cyclic Boronates. Biochim. Biophys. Acta-Gen. Subj. 2019, 1863, 742–748. [Google Scholar] [CrossRef]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopiña, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.J.A.G.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Interplay between β-Lactamases and New β-Lactamase Inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef]
- Behzadi, P.; García-Perdomo, H.A.; Karpiński, T.M.; Issakhanian, L. Metallo-ß-Lactamases: A Review. Mol. Biol. Rep. 2020, 47, 6281–6294. [Google Scholar]
- Kloezen, W.; Melchers, R.J.; Georgiou, P.-C.; Mouton, J.W.; Meletiadis, J. Activity of Cefepime in Combination with the Novel β-Lactamase Inhibitor Taniborbactam (VNRX-5133) against Extended-Spectrum-β-Lactamase-Producing Isolates in In Vitro Checkerboard Assays. Antimicrob. Agents Chemother. 2021, 65, e02338-20. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Chen, C.; Sun, L.Y.; Gao, H.; Kang, P.W.; Li, J.Q.; Zhen, J.B.; Yang, K.W. Identification of Cisplatin and Palladium(II) Complexes as Potent Metallo-β-Lactamase Inhibitors for Targeting Carbapenem-Resistant Enterobacteriaceae. ACS Infect. Dis. 2020, 6, 975–985. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; De Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine a Overcomes Metallo-β-Lactamase Antibiotic Resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef]
- Rotondo, C.M.; Sychantha, D.; Koteva, K.; Wright, G.D. Suppression of β-Lactam Resistance by Aspergillomarasmine A Is Influenced by Both the Metallo-β-Lactamase Target and the Antibiotic Partner. Antimicrob. Agents Chemother. 2020, 64, e01386-19. [Google Scholar] [CrossRef]
- Citarella, A.; Moi, D.; Pinzi, L.; Bonanni, D.; Rastelli, G. Hydroxamic Acid Derivatives: From Synthetic Strategies to Medicinal Chemistry Applications. ACS Omega 2021, 6, 21843–21849. [Google Scholar] [CrossRef]
- Huckleby, A.E.; Saul, J.G.; Shin, H.; Desmarais, S.; Bokka, A.; Jeon, J.; Kim, S.-K. Development of Hydroxamic Acid Compounds for Inhibition of Metallo-β-Lactamase from Bacillus Anthracis. Int. J. Mol. Sci. 2022, 23, 9163. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Demuth, M.; Schlesinger, S.R.; Kim, S.J.; Urbanczyk, J.; Shaw, R.W.; Shin, H. Inhibition of Bacillus Anthracis Metallo-β-Lactamase by Compounds with Hydroxamic Acid Functionality. J. Enzyme Inhib. Med. Chem. 2016, 31, 132–137. [Google Scholar] [CrossRef]
- Schnaars, C.; Kildahl-Andersen, G.; Prandina, A.; Popal, R.; Radix, S.; Le Borgne, M.; Gjøen, T.; Andresen, A.M.S.; Heikal, A.; Økstad, O.A.; et al. Synthesis and Preclinical Evaluation of TPA-Based Zinc Chelators as Metallo-β-Lactamase Inhibitors. ACS Infect. Dis. 2018, 4, 1407–1422. [Google Scholar] [CrossRef] [PubMed]
- Samuelsen, Ø.; Åstrand, O.A.H.; Fröhlich, C.; Heikal, A.; Skagseth, S.; Carlsen, T.J.O.; Leiros, H.-K.S.; Bayer, A.; Schnaars, C.; Kildahl-Andersen, G.; et al. ZN148 Is a Modular Synthetic Metallo-β-Lactamase Inhibitor That Reverses Carbapenem Resistance in Gram-Negative Pathogens In Vivo. Antimicrob. Agents Chemother. 2020, 64, e02415-19. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Schroeder, B.; Pfeifer, Y.; Fröhlich, C.; Deng, L.; Arkona, C.; Kuropka, B.; Sticht, J.; Ataka, K.; Bergemann, S.; et al. Kinetics, Thermodynamics, and Structural Effects of Quinoline-2-Carboxylates, Zinc-Binding Inhibitors of New Delhi Metallo-β-Lactamase-1 Re-Sensitizing Multidrug-Resistant Bacteria for Carbapenems. J. Med. Chem. 2023, 66, 11761–11791. [Google Scholar] [CrossRef] [PubMed]
- Prandina, A.; Radix, S.; Le Borgne, M.; Jordheim, L.P.; Bousfiha, Z.; Fröhlich, C.; Leiros, H.-K.S.; Samuelsen, Ø.; Frøvold, E.; Rongved, P.; et al. Synthesis and Biological Evaluation of New Dipicolylamine Zinc Chelators as Metallo-β-Lactamase Inhibitors. Tetrahedron 2019, 75, 1525–1540. [Google Scholar] [CrossRef]
- Somboro, A.M.; Amoako, D.G.; Osei Sekyere, J.; Kumalo, H.M.; Khan, R.; Bester, L.A.; Essack, S.Y. 1,4,7-Triazacyclononane Restores the Activity of β-Lactam Antibiotics against Metallo-β-Lactamase-Producing Enterobacteriaceae: Exploration of Potential Metallo-β-Lactamase Inhibitors. Appl. Environ. Microbiol. 2019, 85, e02077-18. [Google Scholar] [CrossRef]
- Omolabi, K.F.; Reddy, N.; Mdanda, S.; Ntshangase, S.; Singh, S.D.; Kruger, H.G.; Naicker, T.; Govender, T.; Bajinath, S. The in Vitro and in Vivo Potential of Metal-Chelating Agents as Metallo-Beta-Lactamase Inhibitors against Carbapenem-Resistant Enterobacterales. FEMS Microbiol. Lett. 2023, 370, fnac122. [Google Scholar] [CrossRef]
- Benin, B.M.; Hillyer, T.; Crugnale, A.S.; Fulk, A.; Thomas, C.A.; Crowder, M.W.; Smith, M.A.; Shin, W.S. Taxifolin as a Metallo-β-Lactamase Inhibitor in Combination with Augmentin against Verona Imipenemase 2 Expressing Pseudomonas Aeruginosa. Microorganisms 2023, 11, 2653. [Google Scholar] [CrossRef]
- Jackson, A.C.; Pinter, T.B.J.; Talley, D.C.; Baker-Agha, A.; Patel, D.; Smith, P.J.; Franz, K.J. Benzimidazole and Benzoxazole Zinc Chelators as Inhibitors of Metallo-β-Lactamase NDM-1. ChemMedChem 2021, 16, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Thomas, C.A.; Thomas, P.W.; Yang, K.; Cheng, Z.; Fast, W.; Crowder, M.W.; Cohen, S.M. Iminodiacetic Acid as a Novel Metal-Binding Pharmacophore for New Delhi Metallo-β-Lactamase Inhibitor Development. ChemMedChem 2020, 15, 1272–1282. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, X.; Tian, Y.; Zhai, S.; Liu, Y.; Xiong, Z.; Chu, S. An Insight into Novel Therapeutic Potentials of Taxifolin. Front. Pharmacol. 2023, 14, 1173855. [Google Scholar] [CrossRef]
- Shin, K.S.; Son, B.R.; Hong, S.B.; Kim, J. Dipicolinic Acid-Based Disk Methods for Detection of Metallo-β-Lactamase–Producing Pseudomonas spp. and Acinetobacter spp. Diagn. Microbiol. Infect. Dis. 2008, 62, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Thomas, P.W.; Stewart, A.C.; Bergstrom, A.; Cheng, Z.; Miller, C.; Bethel, C.R.; Marshall, S.H.; Credille, C.V.; Riley, C.L.; et al. Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-β-Lactamase-1. J. Med. Chem. 2017, 60, 7267–7283. [Google Scholar] [CrossRef]
- Mousavi, S.M.; Zarei, M.; Hashemi, S.A.; Babapoor, A.; Amani, A.M. A Conceptual Review of Rhodanine: Current Applications of Antiviral Drugs, Anticancer and Antimicrobial Activities. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1132–1148. [Google Scholar] [CrossRef]
- Tejchman, W.; Korona-Glowniak, I.; Kwietniewski, L.; Żesławska, E.; Nitek, W.; Suder, P.; Żylewski, M.; Malm, A. Antibacterial Properties of 5-Substituted Derivatives of Rhodanine-3-Carboxyalkyl Acids. Part II. Saudi Pharm. J. 2020, 28, 414–426. [Google Scholar] [CrossRef]
- Zhang, D.; Markoulides, M.S.; Stepanovs, D.; Rydzik, A.M.; El-Hussein, A.; Bon, C.; Kamps, J.J.A.G.; Umland, K.-D.; Collins, P.M.; Cahill, S.T.; et al. Structure Activity Relationship Studies on Rhodanines and Derived Enethiol Inhibitors of Metallo-β-Lactamases. Bioorg. Med. Chem. 2018, 26, 2928–2936. [Google Scholar] [CrossRef]
- Kondratieva, A.; Palica, K.; Frøhlich, C.; Hovd, R.R.; Leiros, H.-K.S.; Erdelyi, M.; Bayer, A. Fluorinated Captopril Analogues Inhibit Metallo-β-Lactamases and Facilitate Structure Determination of NDM-1 Binding Pose. Eur. J. Med. Chem. 2024, 266, 116140. [Google Scholar] [CrossRef] [PubMed]
- Farley, A.J.M.; Ermolovich, Y.; Calvopiña, K.; Rabe, P.; Panduwawala, T.; Brem, J.; Björkling, F.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase Inhibition by N-Sulfamoylpyrrole-2-Carboxylates. ACS Infect. Dis. 2021, 7, 1809–1817. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).