

Synthesis and Biological Activity of Homohypotaurine Obtained by the Enzyme-Based Conversion of Homocysteine Sulfinic Acid Using Recombinant Escherichia Coli Glutamate Decarboxylase

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
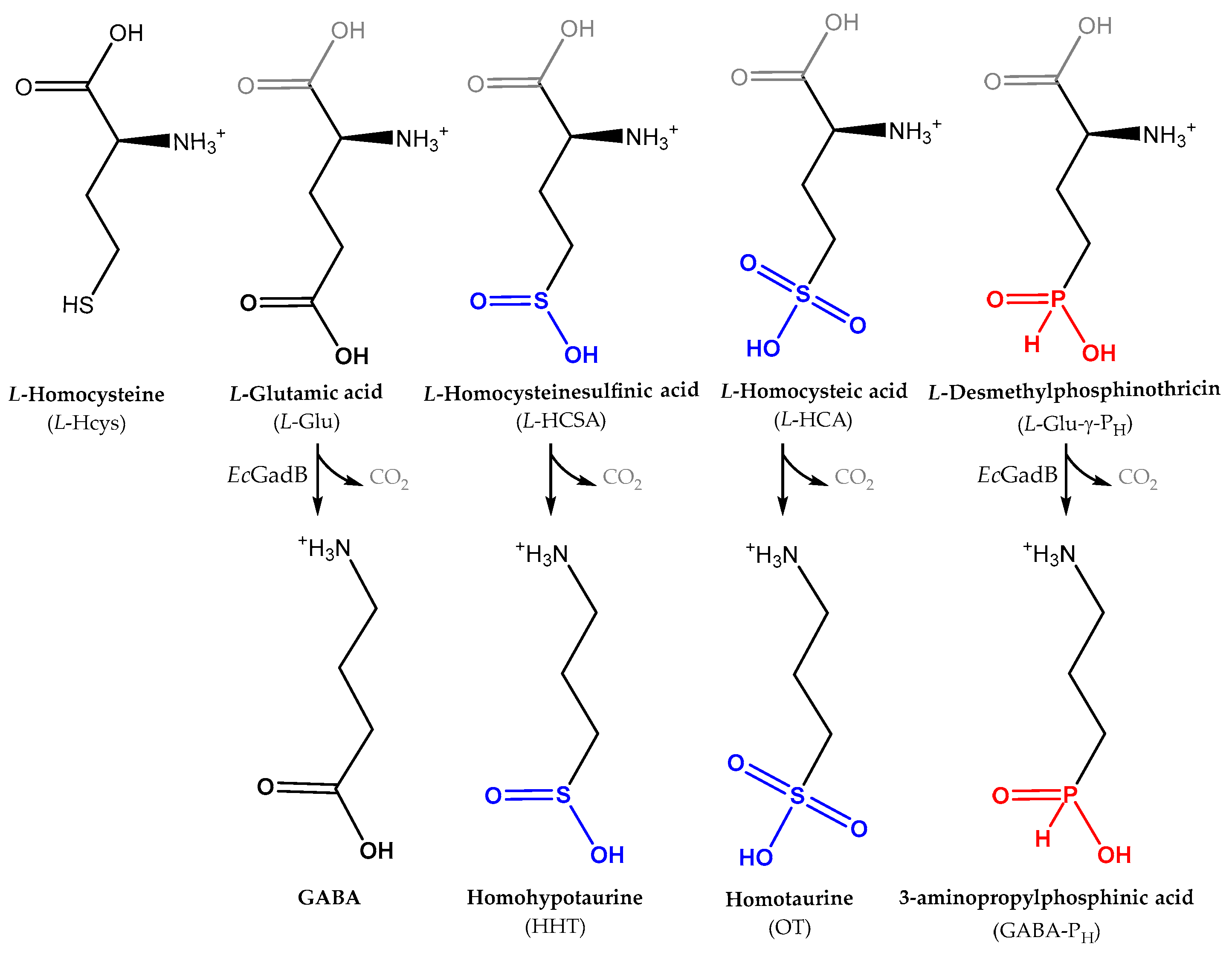
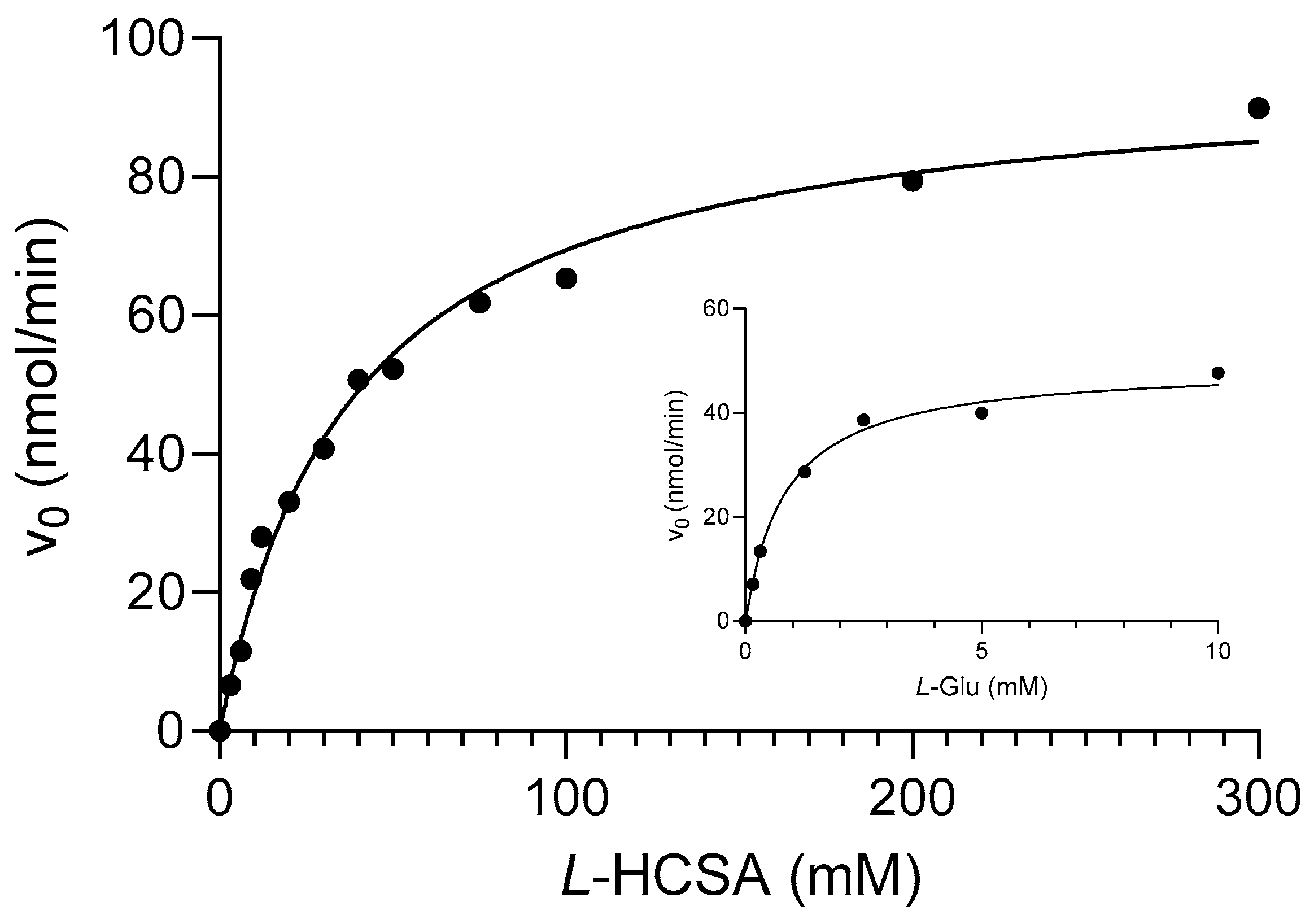
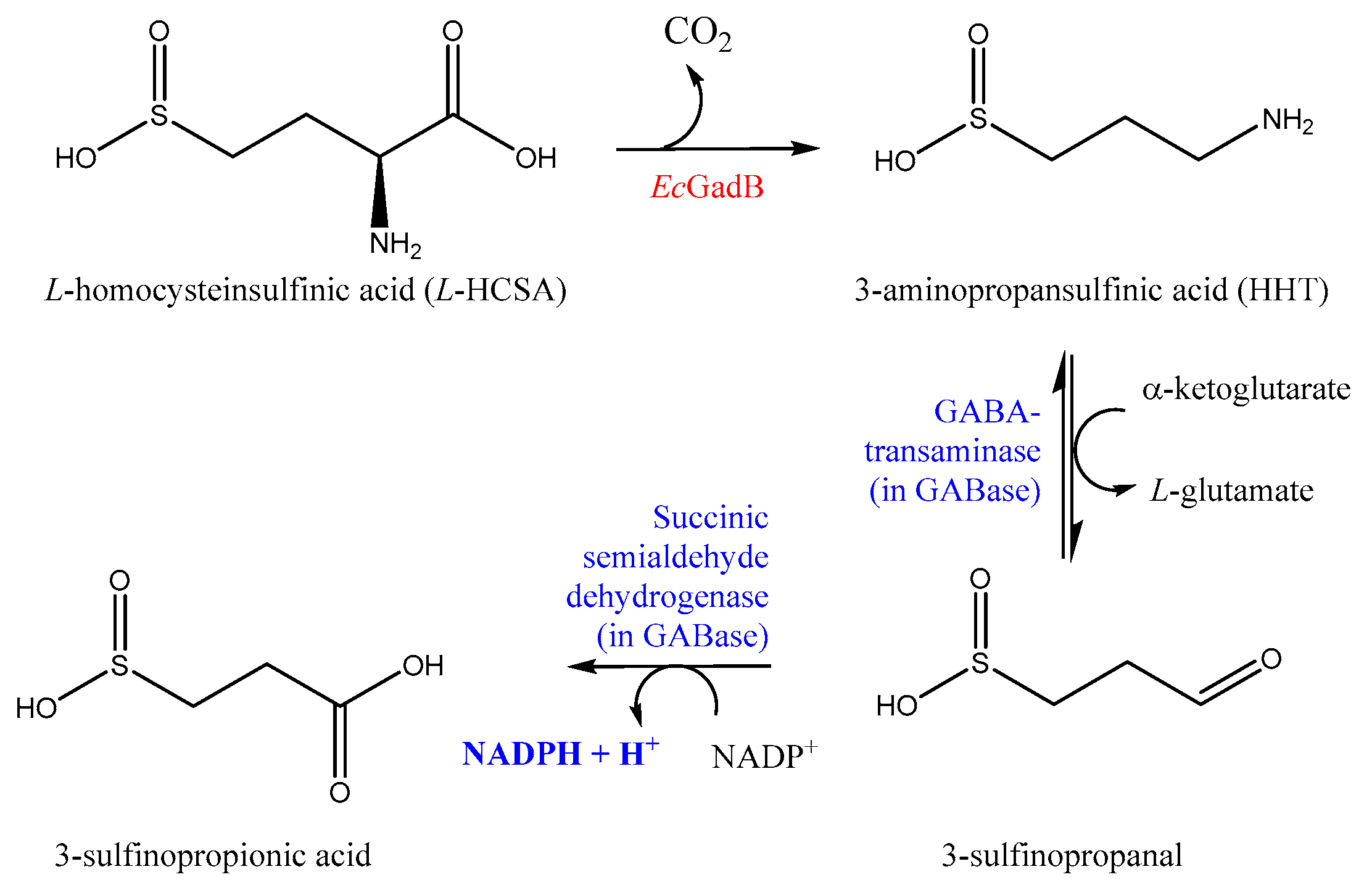
2.1. l-HCSA Is a Substrate of EcGadB and HHT Is a Substrate of GABase
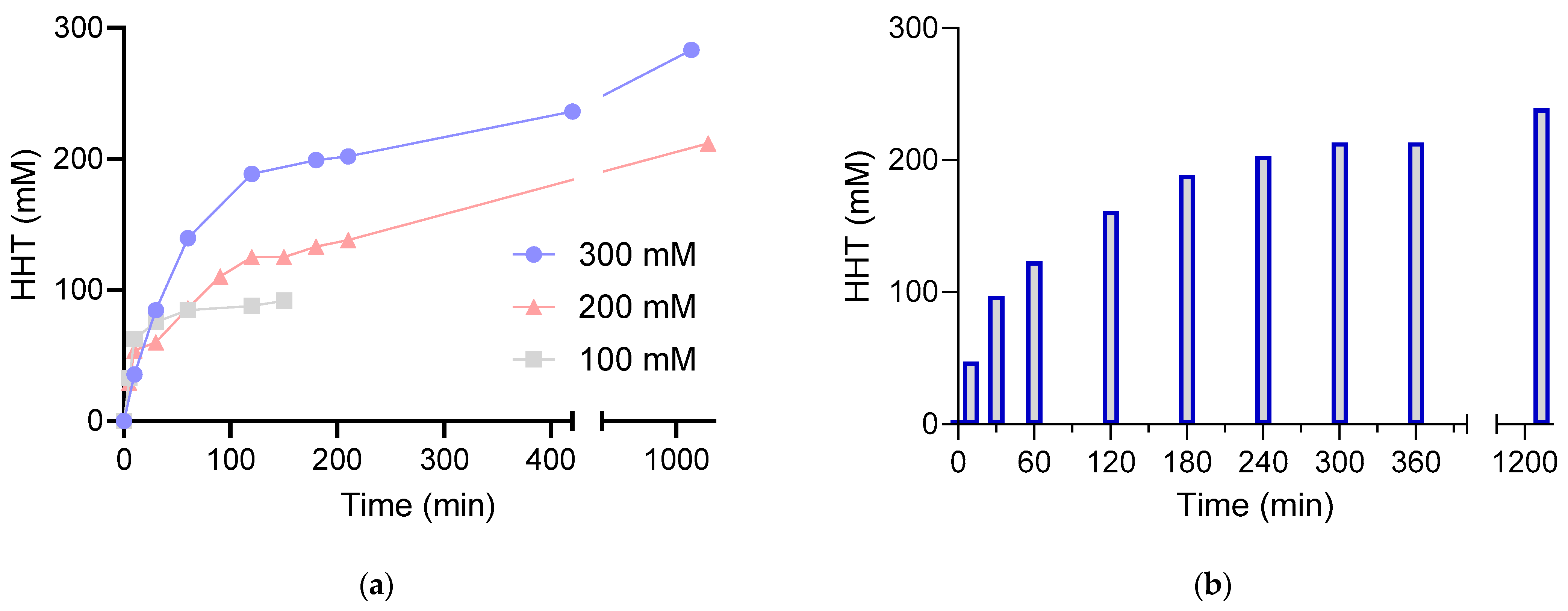
2.2. Bioconversion of l-HCSA into HHT via EcGadB and HHT Purification
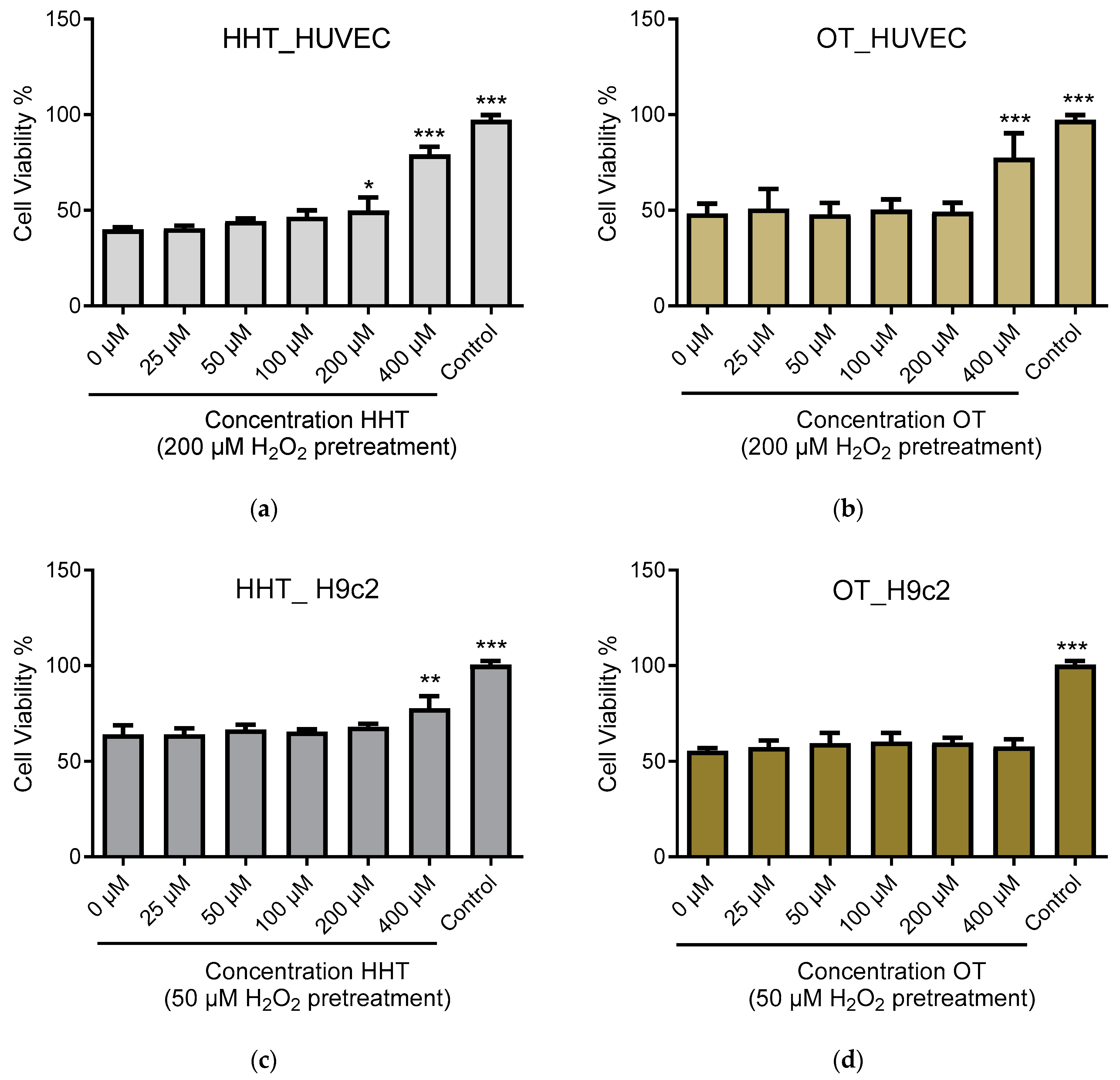
2.3. In Vitro Effect of HHT on Endothelial Cells and Cardiomyoblasts
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of l-HCSA and Purification of l-HCSA and HHT via Column Chromatography
4.3. Enzymatic Assays and Calculation of Kinetic Parameters
4.4. Bioconversion of l-HCSA into HHT by EcGadB
4.5. Biological Activity of HHT and OT
4.5.1. Cell Culture and Treatment Procedure
4.5.2. MTT Assay
4.5.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Devlin, T.M. Textbook of Biochemistry with Clinical Correlations, 7th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Skovierova, H.; Vidomanova, E.; Mahmood, S.; Sopkova, J.; Drgova, A.; Cervenova, T.; Halasova, E.; Lehotsky, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Dean, L. Methylenetetrahydrofolate Reductase Deficiency. In Medical Genetics Summaries; Pratt, V.M., Scott, S.A., Pirmohamed, M., Esquivel, B., Kattman, B.L., Malheiro, A.J., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012. [Google Scholar]
- McBean, G.J. 7 Sulfur-Containing Amino Acids. In Handbook of Neurochemistry and Molecular Neurobiology: Amino Acids and Peptides in the Nervous System; Lajtha, A., Oja, S.S., Schousboe, A., Saransaari, P., Eds.; Springer US: New York, NY, USA, 2007; pp. 133–154. [Google Scholar]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Francioso, A.; Baseggio Conrado, A.; Mosca, L.; Fontana, M. Chemistry and Biochemistry of Sulfur Natural Compounds: Key Intermediates of Metabolism and Redox Biology. Oxidative Med. Cell. Longev. 2020, 2020, 8294158. [Google Scholar] [CrossRef]
- Khan, A.; Choi, Y.; Back, J.H.; Lee, S.; Jee, S.H.; Park, Y.H. High-resolution metabolomics study revealing l-homocysteine sulfinic acid, cysteic acid, and carnitine as novel biomarkers for high acute myocardial infarction risk. Metab. Clin. Exp. 2020, 104, 154051. [Google Scholar] [CrossRef]
- Lee, Y.; Khan, A.; Hong, S.; Jee, S.H.; Park, Y.H. A metabolomic study on high-risk stroke patients determines low levels of serum lysine metabolites: A retrospective cohort study. Mol. Biosyst. 2017, 13, 1109–1120. [Google Scholar] [CrossRef]
- Sridharan, G.; Ramani, P.; Patankar, S.; Vijayaraghavan, R. Evaluation of salivary metabolomics in oral leukoplakia and oral squamous cell carcinoma. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2019, 48, 299–306. [Google Scholar] [CrossRef]
- Watkins, J.C.; Krogsgaard-Larsen, P.; Honore, T. Structure-activity relationships in the development of excitatory amino acid receptor agonists and competitive antagonists. Trends Pharmacol. Sci. 1990, 11, 25–33. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Di Lorenzo, F.; Manenti, G.; Semprini, R.; Koch, G. Homotaurine induces measurable changes of short latency afferent inhibition in a group of mild cognitive impairment individuals. Front. Aging Neurosci. 2014, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Meera, P.; Uusi-Oukari, M.; Lipshutz, G.S.; Wallner, M. GABA(A) receptors as plausible molecular targets and mediators for taurine and homotaurine actions. Front. Pharmacol. 2023, 14, 1271203. [Google Scholar] [CrossRef]
- Jollès-Bergeret, B.; de Vaucher, M.H. Decarboxylation of DL-homocysteinesulphinic acid (2-amino 4-sulphinobutyric acid) in rat brain. J. Neurochem. 1973, 20, 1797–1800. [Google Scholar] [CrossRef]
- De Marco, C.; Rinaldi, A. Synthesis of homohypotaurine (3-aminopropansulfinic acid) and homothiotaurine (3-aminopropanthiosulfonic acid). Anal. Biochem. 1973, 51, 265–273. [Google Scholar] [CrossRef]
- Francioso, A.; Dupré, S.; Fontana, M. Chemistry of Outlandish Natural Products Belonging to Sulfur Metabolism: Unrevealed Green Syntheses and Separation Strategies from the Cavallini’s Old School. Separations 2022, 9, 45. [Google Scholar] [CrossRef]
- Jollès-Bergeret, B. A new sulfinic amino acid: Homohypotaureine (3-aminopropanesulfinic acid). Isolation, determination and chromatographic behaviour compared with sulfinic and sulfonic derivatives of cysteine and homocysteine. Eur. J. Biochem. 1969, 10, 569–573. [Google Scholar] [CrossRef]
- Capitani, G.; De Biase, D.; Aurizi, C.; Gut, H.; Bossa, F.; Grutter, M.G. Crystal structure and functional analysis of Escherichia coli glutamate decarboxylase. EMBO J. 2003, 22, 4027–4037. [Google Scholar] [CrossRef] [PubMed]
- De Biase, D.; Pennacchietti, E. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: Function, distribution and biomedical implications of the gadBC operon. Mol. Microbiol. 2012, 86, 770–786. [Google Scholar] [CrossRef]
- Damiano, M.A.; Bastianelli, D.; Al Dahouk, S.; Kohler, S.; Cloeckaert, A.; De Biase, D.; Occhialini, A. Glutamate decarboxylase-dependent acid resistance in Brucella spp.: Distribution and contribution to fitness under extremely acidic conditions. Appl. Environ. Microbiol. 2015, 81, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.; Tramonti, A.; De Biase, D. Coping with low pH: Molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef]
- Lakicevic, B.Z.; Den Besten, H.M.W.; De Biase, D. Landscape of Stress Response and Virulence Genes Among Listeria monocytogenes Strains. Front. Microbiol. 2021, 12, 738470. [Google Scholar] [CrossRef]
- Gut, H.; Pennacchietti, E.; John, R.A.; Bossa, F.; Capitani, G.; De Biase, D.; Grutter, M.G. Escherichia coli acid resistance: pH-sensing, activation by chloride and autoinhibition in GadB. EMBO J. 2006, 25, 2643–2651. [Google Scholar] [CrossRef]
- Pennacchietti, E.; Lammens, T.M.; Capitani, G.; Franssen, M.C.; John, R.A.; Bossa, F.; De Biase, D. Mutation of His465 alters the pH-dependent spectroscopic properties of Escherichia coli glutamate decarboxylase and broadens the range of its activity toward more alkaline pH. J. Biol. Chem. 2009, 284, 31587–31596. [Google Scholar] [CrossRef] [PubMed]
- Lammens, T.M.; De Biase, D.; Franssen, M.C.R.; Scott, E.L.; Sanders, J.P.M. The application of glutamic acid α-decarboxylase for the valorization of glutamic acid. Green Chem. 2009, 11, 1562–1567. [Google Scholar] [CrossRef]
- De Biase, D.; Cappadocio, F.; Pennacchietti, E.; Giovannercole, F.; Coluccia, A.; Vepsäläinen, J.; Khomutov, A. Enzymatic kinetic resolution of desmethylphosphinothricin indicates that phosphinic group is a bioisostere of carboxyl group. Commun. Chem. 2020, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Fonda, M.L. Glutamate decarboxylase. Substrate specificity and inhibition by carboxylic acids. Biochemistry 1972, 11, 1304–1309. [Google Scholar] [CrossRef]
- Grassini, G.; Pennacchietti, E.; Cappadocio, F.; Occhialini, A.; De Biase, D. Biochemical and spectroscopic properties of Brucella microti glutamate decarboxylase, a key component of the glutamate-dependent acid resistance system. FEBS Open Bio 2015, 5, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Jolles-Bergeret, B.; Charton, M. [Bacterial L-glutamate decarboxylases: Their action on L homocysteinesulfinic acid and L-homocysteic acid]. Biochimie 1971, 53, 553–562. [Google Scholar] [CrossRef]
- Luchi, P.; De Marco, C. Synthesis of homocysteinesulfinic acid. Anal. Biochem. 1972, 45, 236–241. [Google Scholar] [CrossRef]
- Cuénod, M.; Do, K.Q.; Matute, C.; Streit, P. Identification of Pathways for Acidic Amino Acid Transmitters and Search for New Candidates: Sulphur-containing Amino Acids. In Excitatory Amino Acids; Roberts, P.J., Storm-Mathisen, J., Bradford, H.F., Eds.; Palgrave Macmillan UK: London, UK, 1986; pp. 117–129. [Google Scholar]
- Jollès-Bergeret, B.; Marty-Lopez, M. [Metabolism of L-homocysteinesulfinic acid in higher animals]. Comptes Rendus Hebd. Seances L’academie Sci. Ser. D Sci. Nat. 1966, 262, 930–932. [Google Scholar]
- Costa, M.; Vesci, L.; Fontana, M.; Solinas, S.P.; Dupre, S.; Cavallini, D. Displacement of [(3)H]GABA binding to bovine brain receptors by sulfur-containing analogues. Neurochem. Int. 1990, 17, 547–551. [Google Scholar] [CrossRef]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABAA and GABAB Receptors Mediate GABA-Induced Intracellular Ca2+ Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef]
- Zhu, Z.; Shi, Z.; Xie, C.; Gong, W.; Hu, Z.; Peng, Y. A novel mechanism of Gamma-aminobutyric acid (GABA) protecting human umbilical vein endothelial cells (HUVECs) against H(2)O(2)-induced oxidative injury. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2019, 217, 68–75. [Google Scholar] [CrossRef] [PubMed]
- De Biase, D.; Tramonti, A.; John, R.A.; Bossa, F. Isolation, overexpression, and biochemical characterization of the two isoforms of glutamic acid decarboxylase from Escherichia coli. Protein Expr. Purif. 1996, 8, 430–438. [Google Scholar] [CrossRef]
- Miyazaki, T. Identification of a novel enzyme and the regulation of key enzymes in mammalian taurine synthesis. J. Pharmacol. Sci. 2024, 154, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Baseggio Conrado, A.; D’Angelantonio, M.; D’Erme, M.; Pecci, L.; Fontana, M. The Interaction of Hypotaurine and Other Sulfinates with Reactive Oxygen and Nitrogen Species: A Survey of Reaction Mechanisms. Adv. Exp. Med. Biol. 2017, 975 Pt 1, 573–583. [Google Scholar] [CrossRef]
- Ramírez-Guerrero, S.; Guardo-Maya, S.; Medina-Rincón, G.J.; Orrego-González, E.E.; Cabezas-Pérez, R.; González-Reyes, R.E. Taurine and Astrocytes: A Homeostatic and Neuroprotective Relationship. Front. Mol. Neurosci. 2022, 15, 937789. [Google Scholar] [CrossRef]
- Kim, B.; Jung, J. Metabolomic Approach to Identify Potential Biomarkers in KRAS-Mutant Pancreatic Cancer Cells. Biomedicines 2024, 12, 865. [Google Scholar] [CrossRef]
- Chebib, M.; Vandenberg, R.J.; Johnston, G.A. Analogues of gamma-aminobutyric acid (GABA) and trans-4-aminocrotonic acid (TACA) substituted in the 2 position as GABAC receptor antagonists. Br. J. Pharmacol. 1997, 122, 1551–1560. [Google Scholar] [CrossRef]
- Marques, S.M.; Kouba, P.; Legrand, A.; Sedlar, J.; Disson, L.; Planas-Iglesias, J.; Sanusi, Z.; Kunka, A.; Damborsky, J.; Pajdla, T.; et al. CoVAMPnet: Comparative Markov State Analysis for Studying Effects of Drug Candidates on Disordered Biomolecules. JACS Au 2024, 4, 2228–2245. [Google Scholar] [CrossRef] [PubMed]
- Dadi, P.; Pauling, C.W.; Shrivastava, A.; Shah, D.D. Synthesis of versatile neuromodulatory molecules by a gut microbial glutamate decarboxylase. Biorxiv Prepr. Serv. Biol. 2024, preprint. [Google Scholar] [CrossRef]
- Jollés-Bergeret, B. Enzymatic and chemical synthesis of 3-sulfinopropionic acid, an analog of succinic acid. Eur. J. Biochem. 1974, 42, 349–353. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | kcat (s−1) | KM (mM) | kcat/KM (s−1 mM−1) |
|---|---|---|---|
| l-Glutamic acid (l-Glu) | 86.78 ± 2.25 | 0.84 ± 0.09 | 103.31 ± 11.38 |
| l-Homocysteinsulfinic acid (l-HCSA) | 33.92 ± 1.07 | 38.24 ± 3.45 | 0.89 ± 0.09 |
| l-Desmethylphoshinothricin (l-Glu-γ-PH) * | 13.93 ± 0.58 | 39.82 ± 5.66 | 0.35 ± 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, M.; Gunaydin Akyildiz, A.; D’Alonzo, C.; Giovannercole, F.; Zicchi, A.; Francioso, A.; Capuozzo, E.; De Biase, D. Synthesis and Biological Activity of Homohypotaurine Obtained by the Enzyme-Based Conversion of Homocysteine Sulfinic Acid Using Recombinant Escherichia Coli Glutamate Decarboxylase. Molecules 2024, 29, 3985. https://doi.org/10.3390/molecules29173985
Fontana M, Gunaydin Akyildiz A, D’Alonzo C, Giovannercole F, Zicchi A, Francioso A, Capuozzo E, De Biase D. Synthesis and Biological Activity of Homohypotaurine Obtained by the Enzyme-Based Conversion of Homocysteine Sulfinic Acid Using Recombinant Escherichia Coli Glutamate Decarboxylase. Molecules. 2024; 29(17):3985. https://doi.org/10.3390/molecules29173985
Chicago/Turabian StyleFontana, Mario, Aysenur Gunaydin Akyildiz, Chiara D’Alonzo, Fabio Giovannercole, Arianna Zicchi, Antonio Francioso, Elisabetta Capuozzo, and Daniela De Biase. 2024. "Synthesis and Biological Activity of Homohypotaurine Obtained by the Enzyme-Based Conversion of Homocysteine Sulfinic Acid Using Recombinant Escherichia Coli Glutamate Decarboxylase" Molecules 29, no. 17: 3985. https://doi.org/10.3390/molecules29173985