

An Overview on the Synthesis of Lamellarins and Related Compounds with Biological Interest


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthetic Strategies for the Preparation of Lamellarins and Related Compounds
2.1. Synthesis of Lamellarins
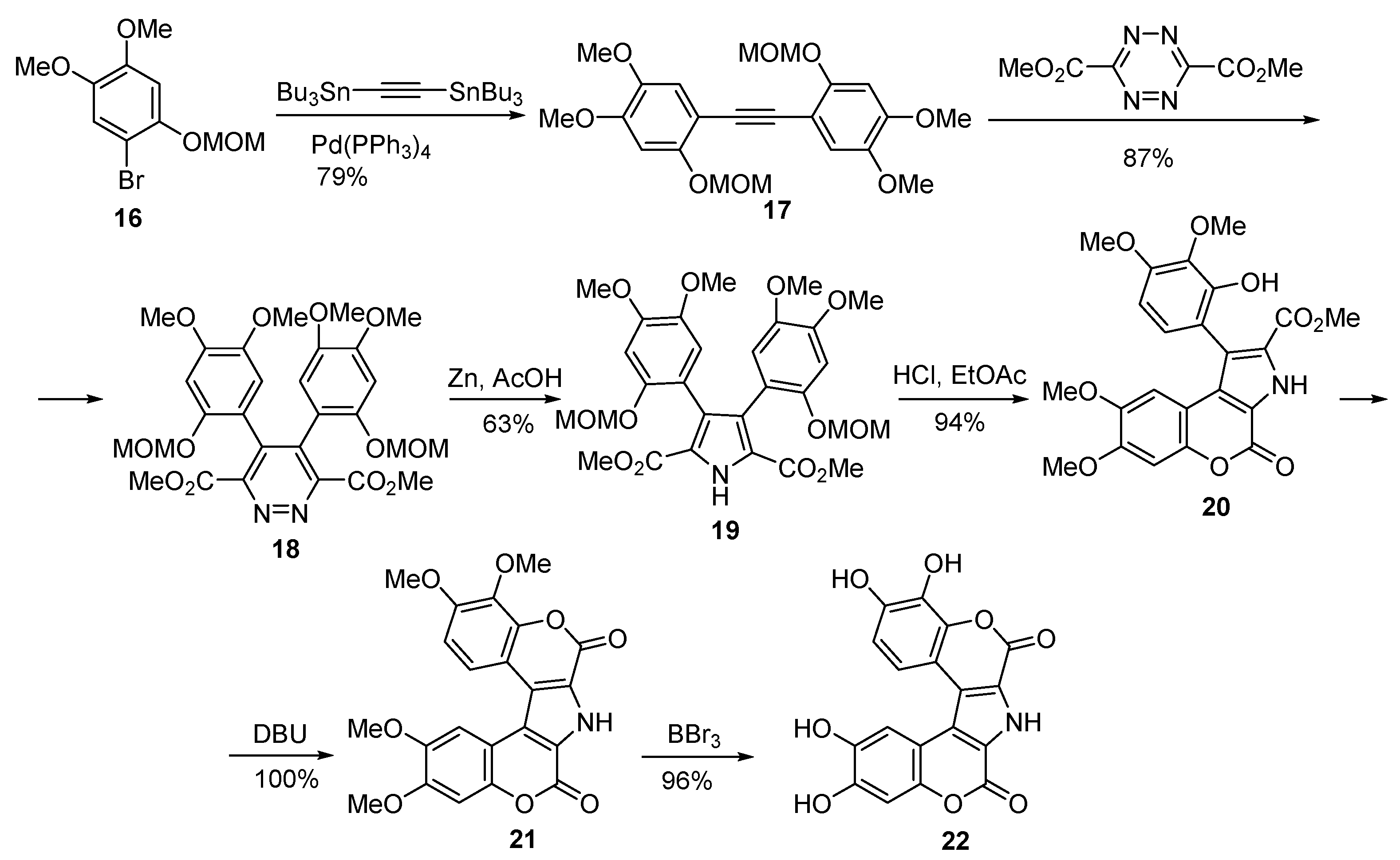
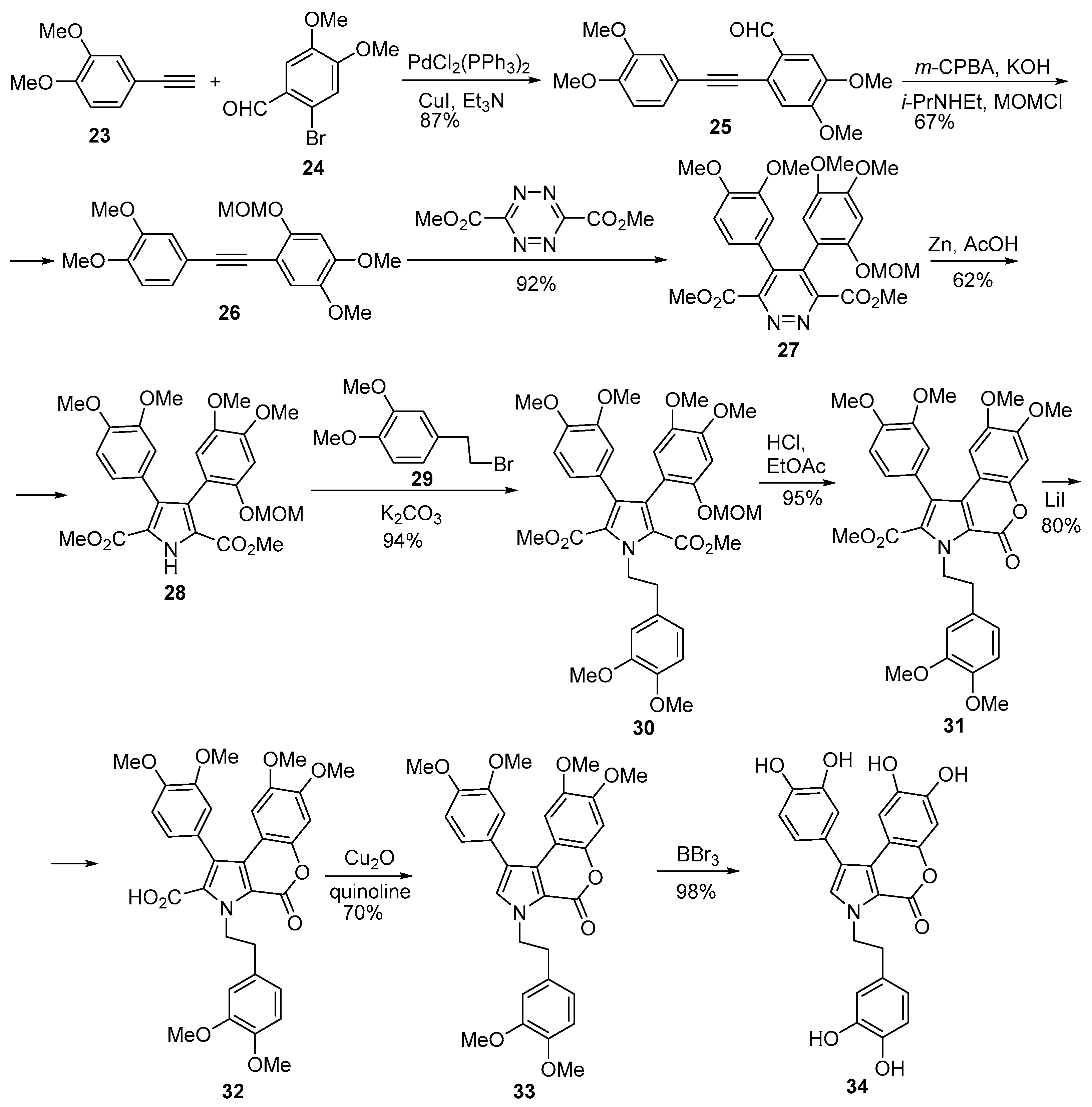
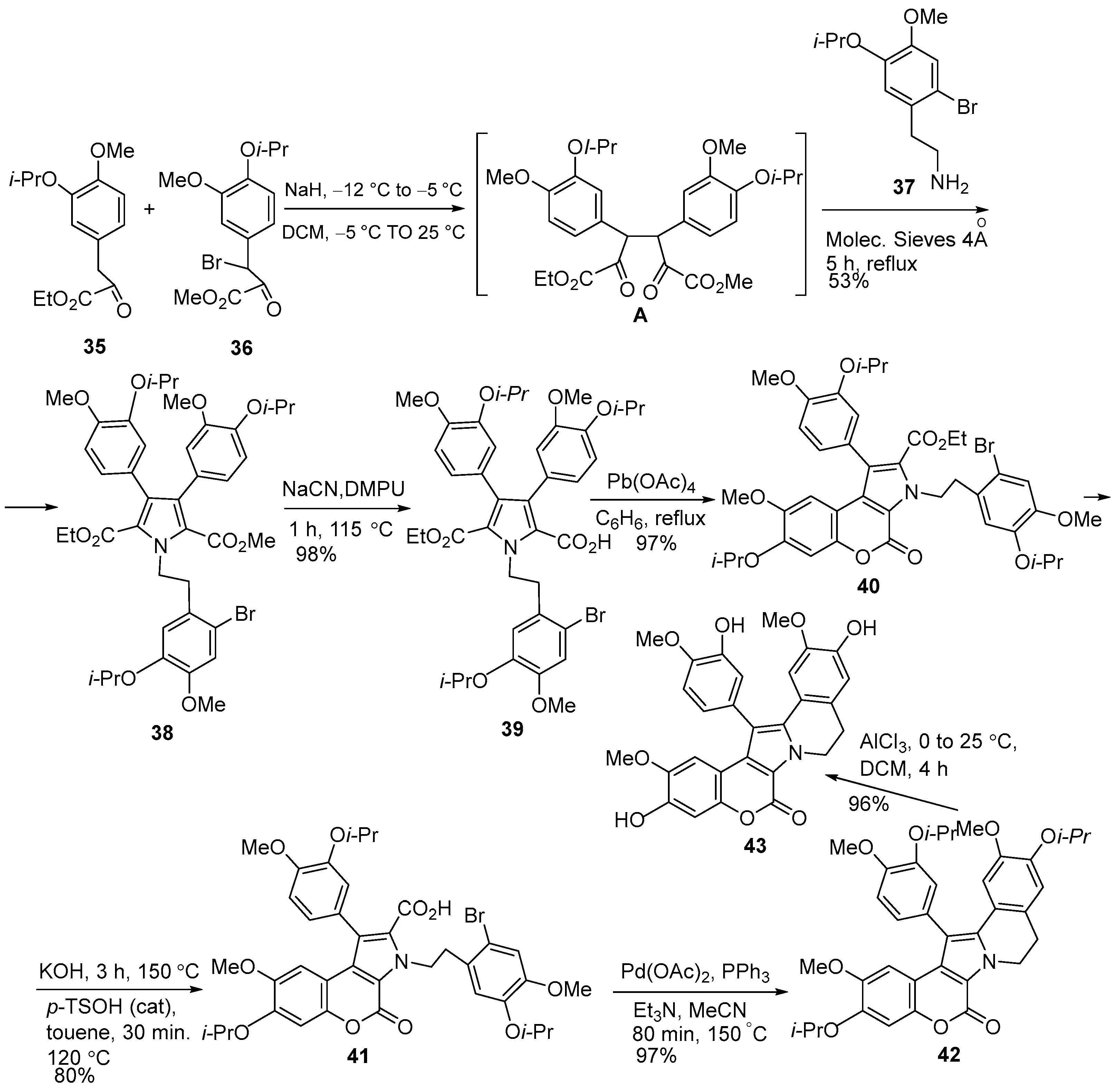
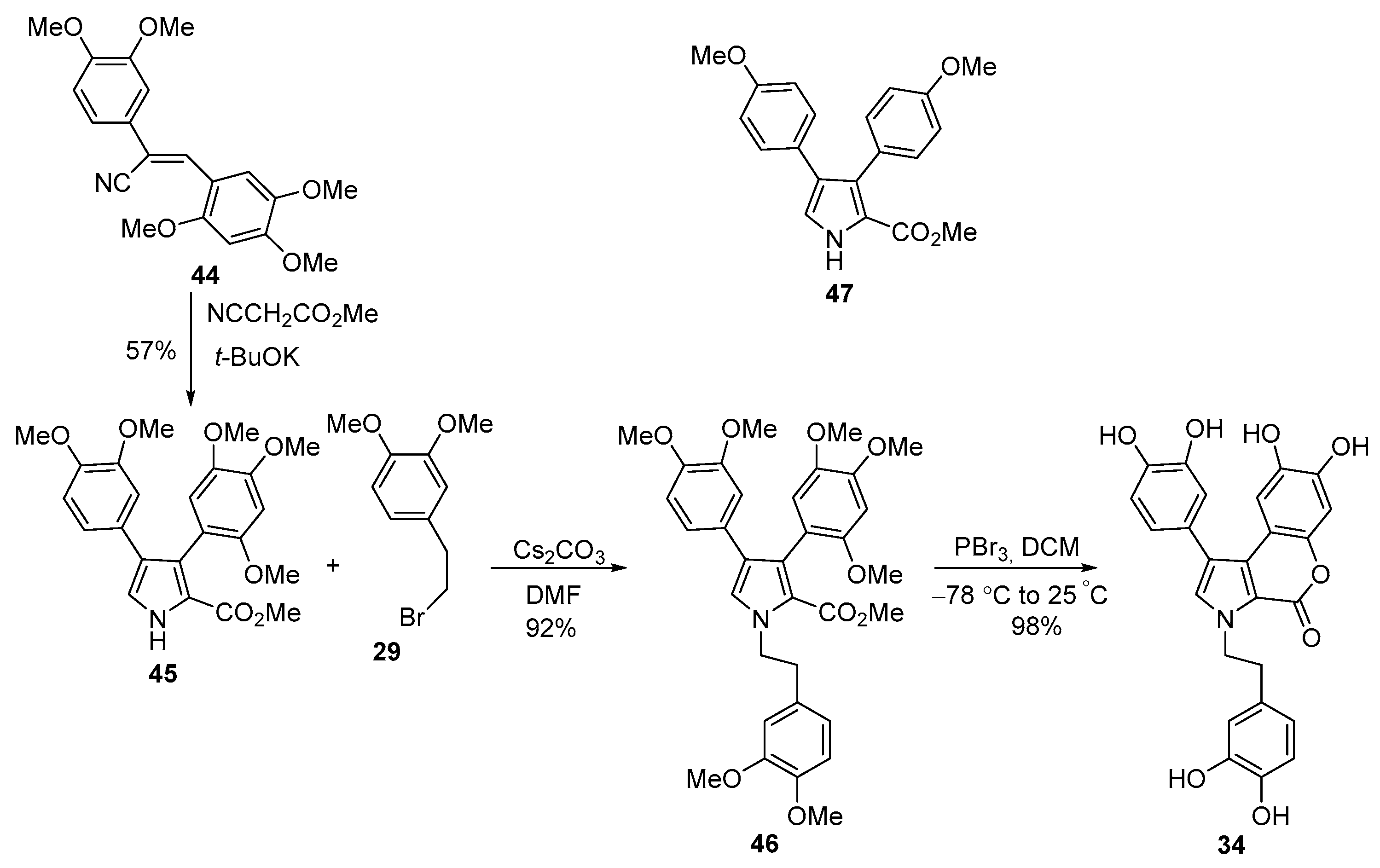
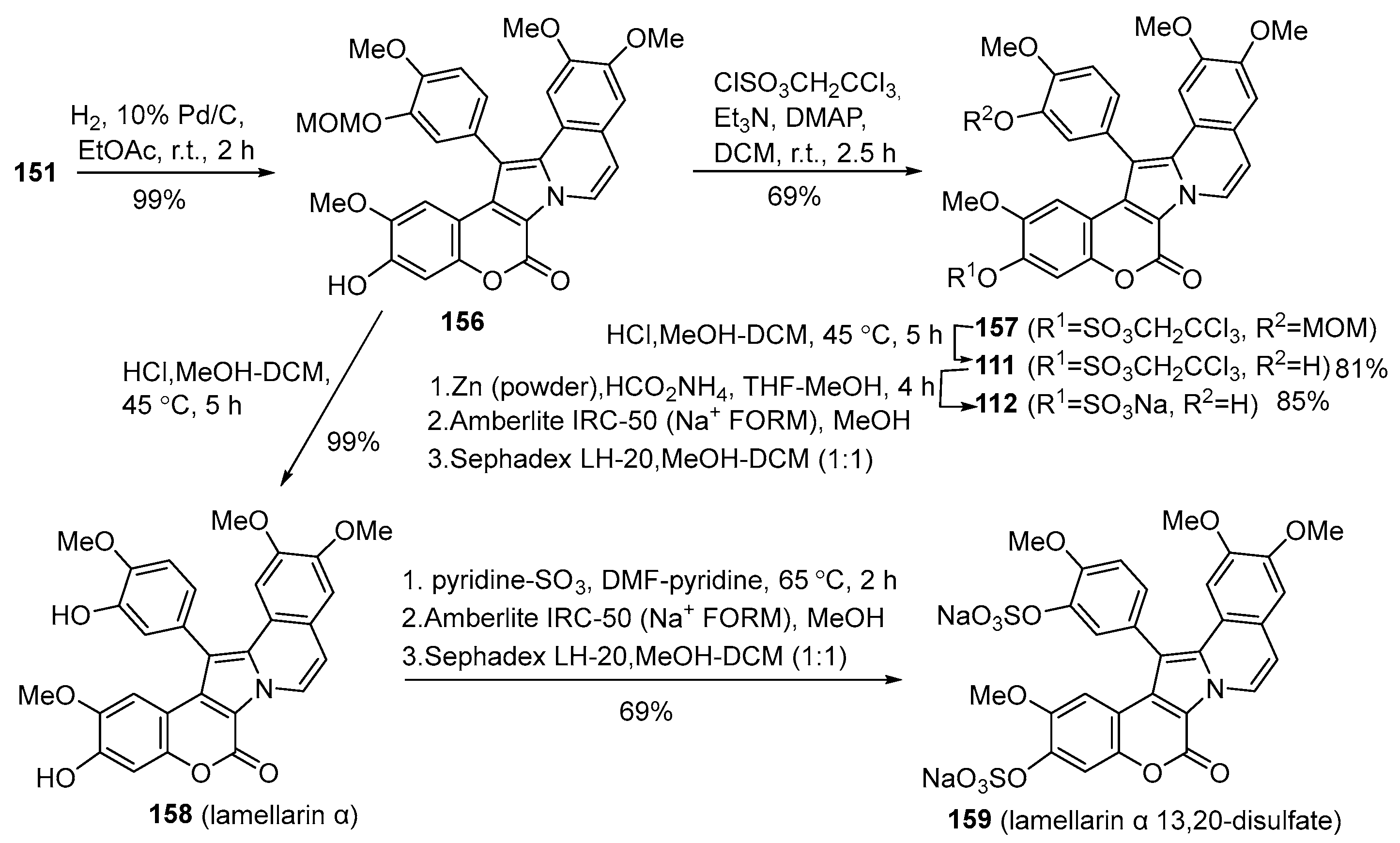
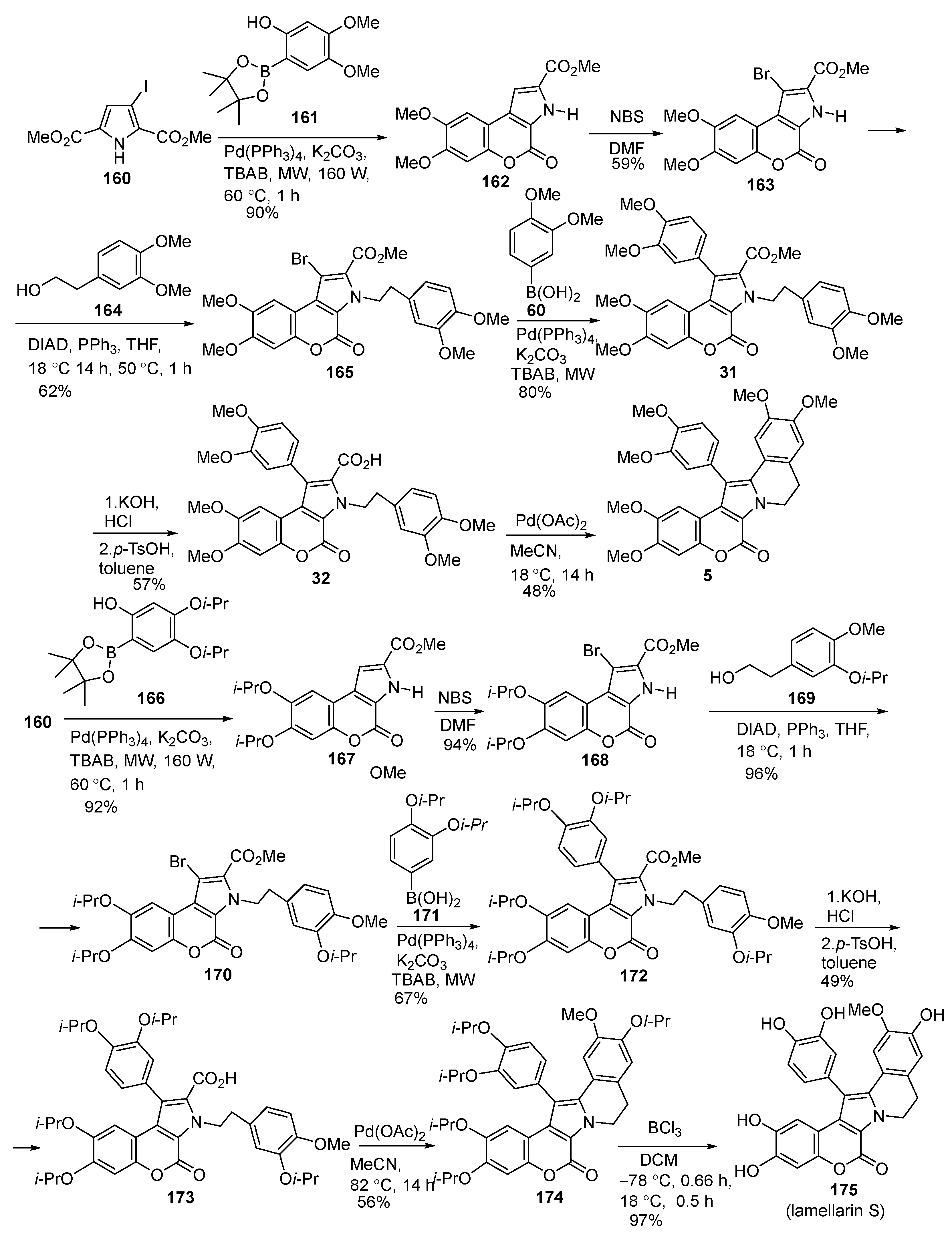
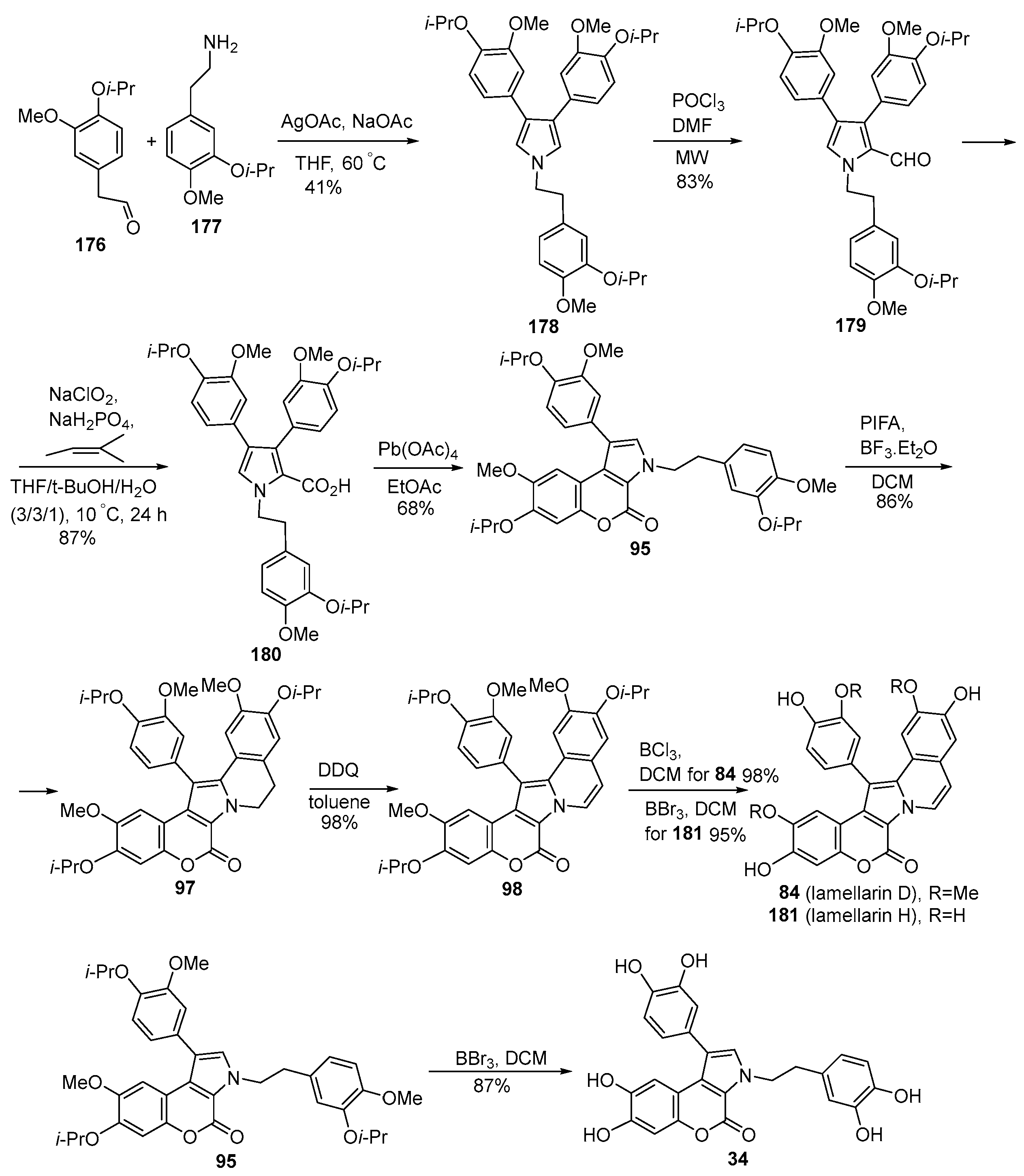
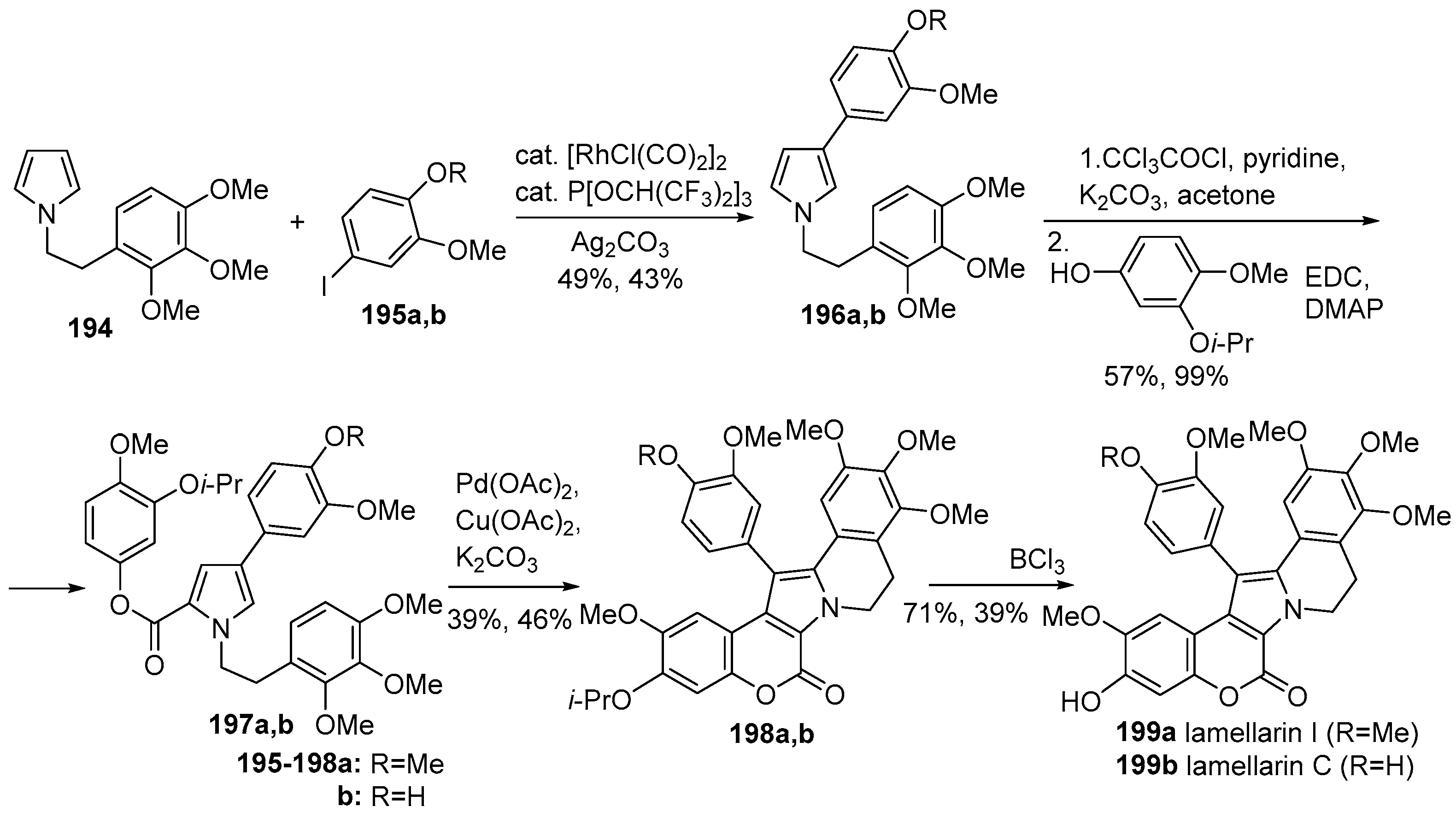
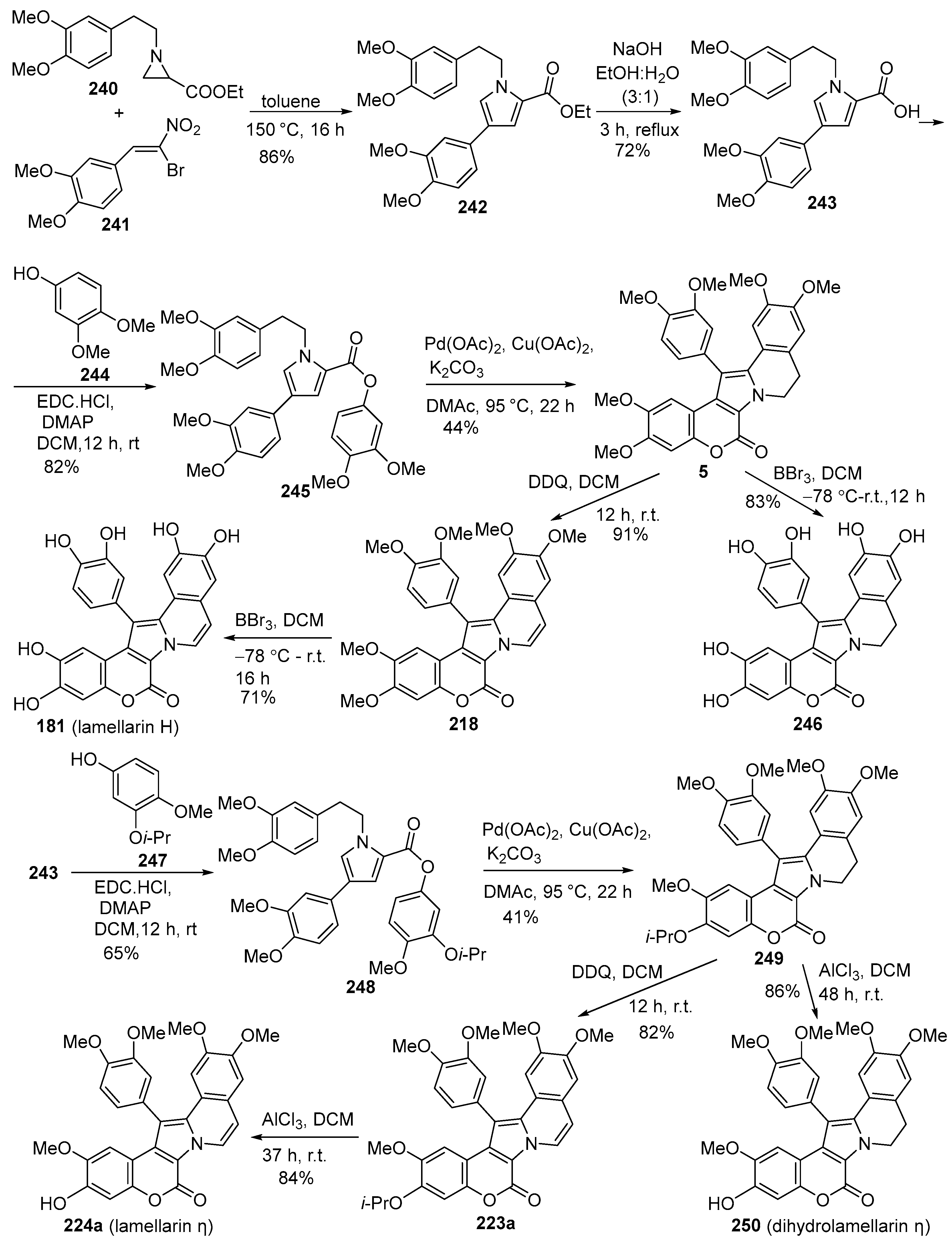
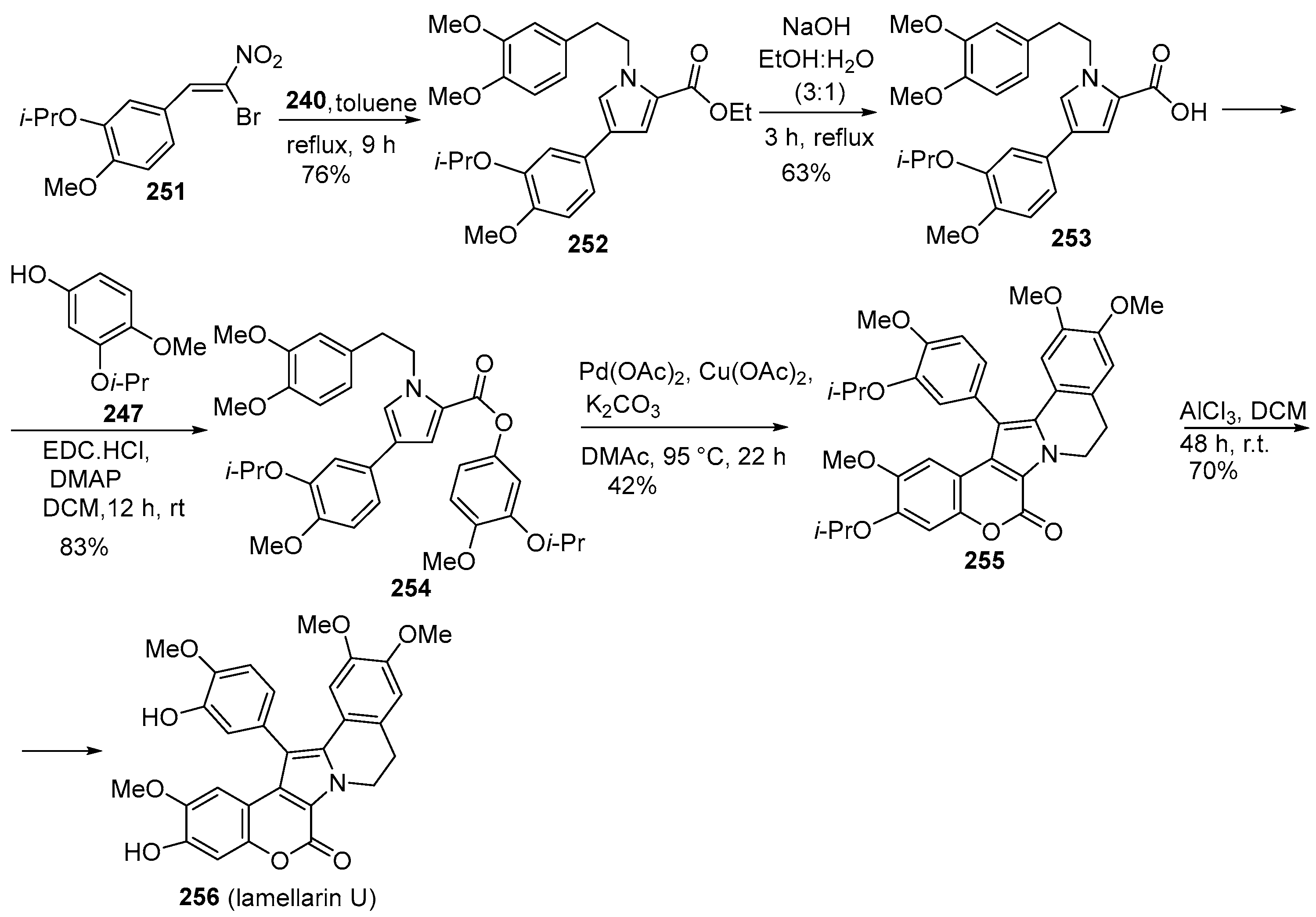
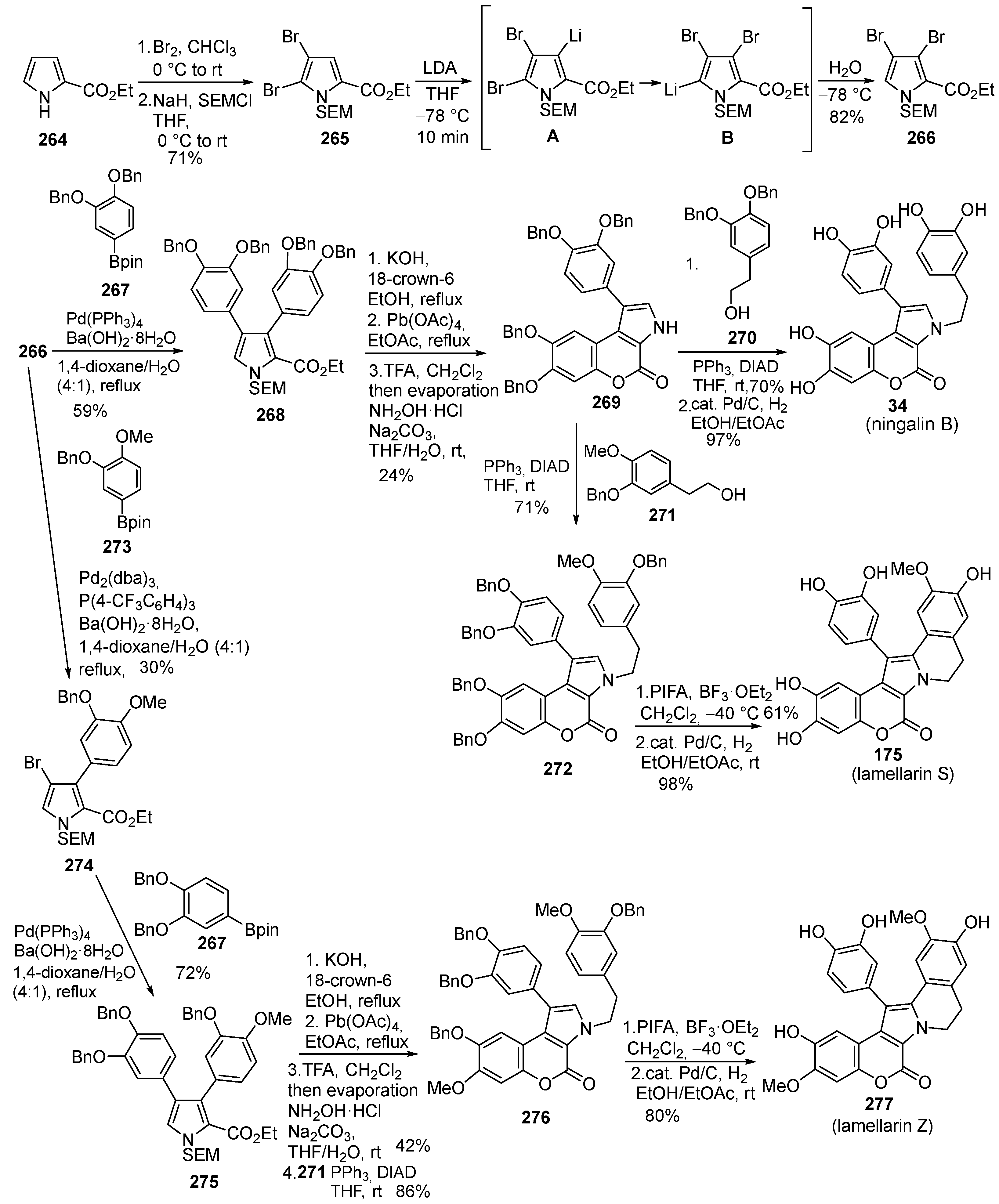
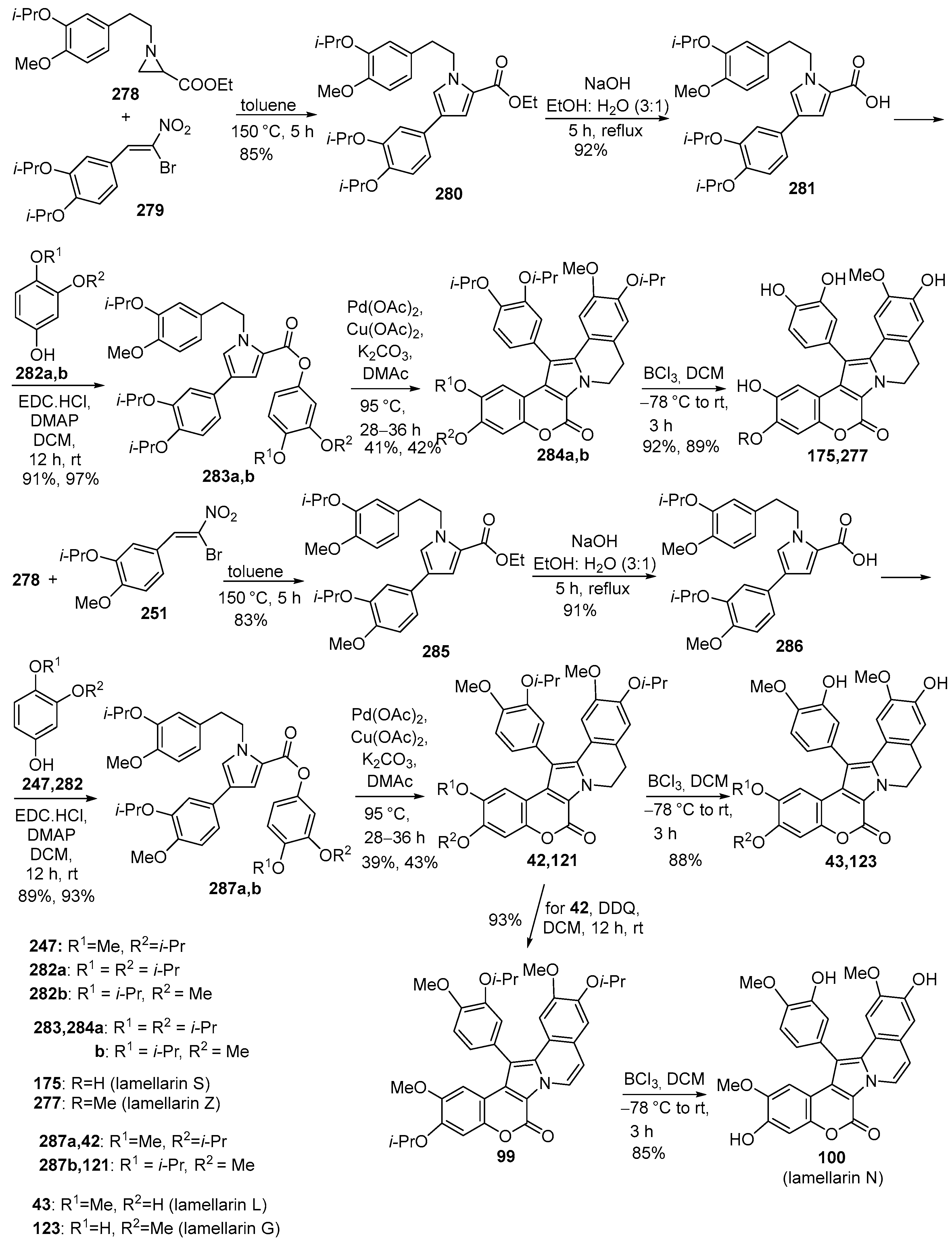
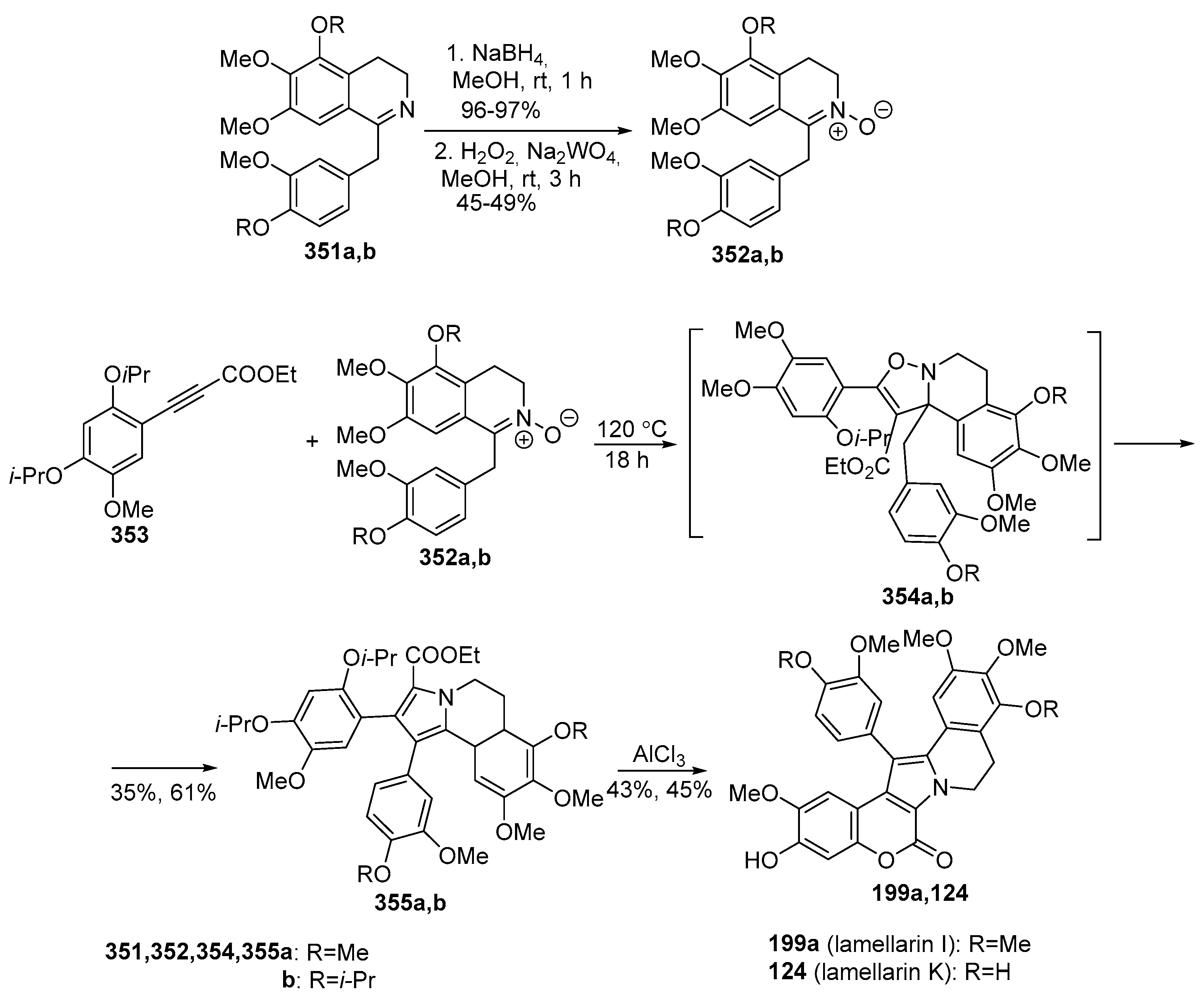
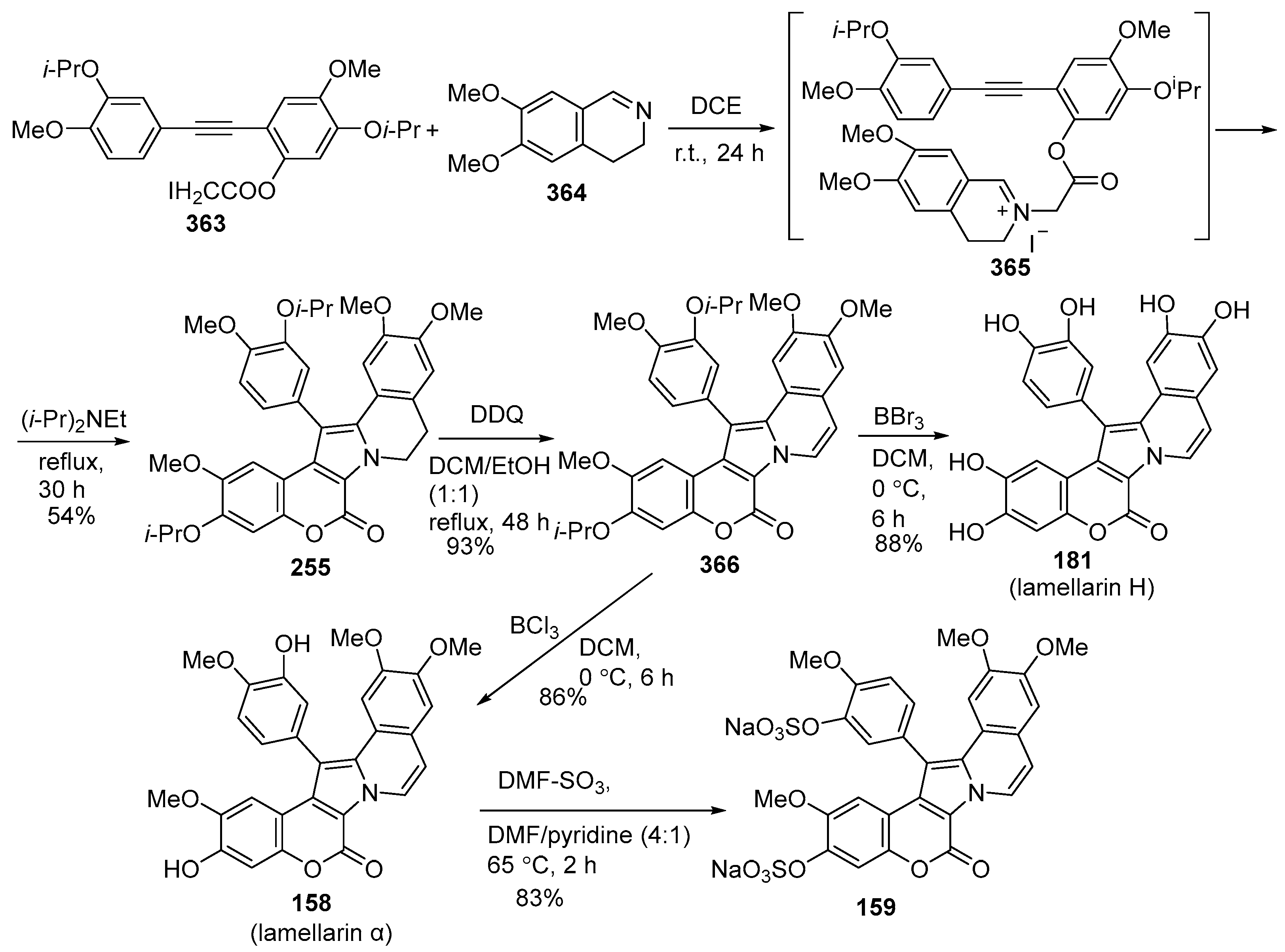
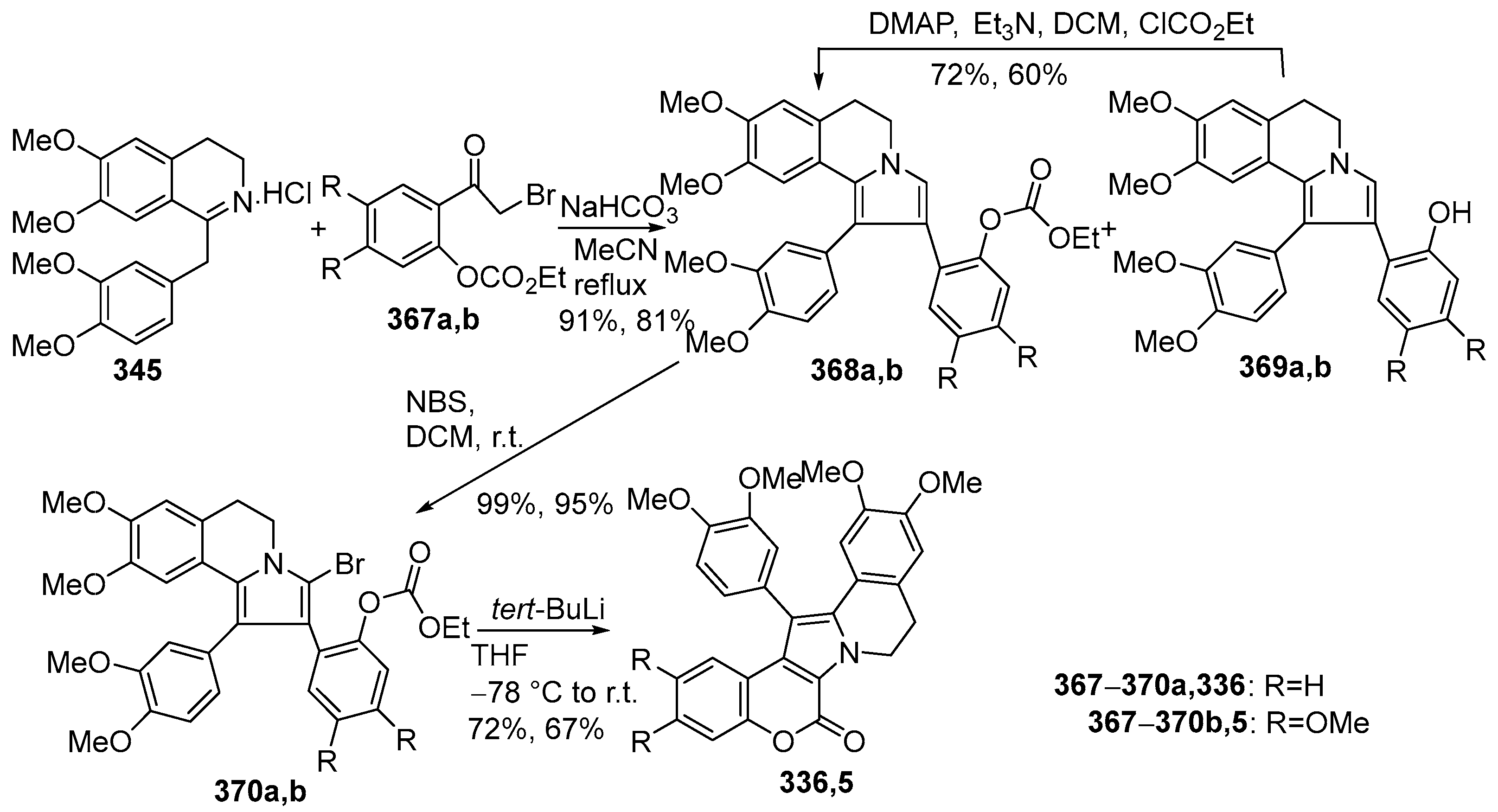
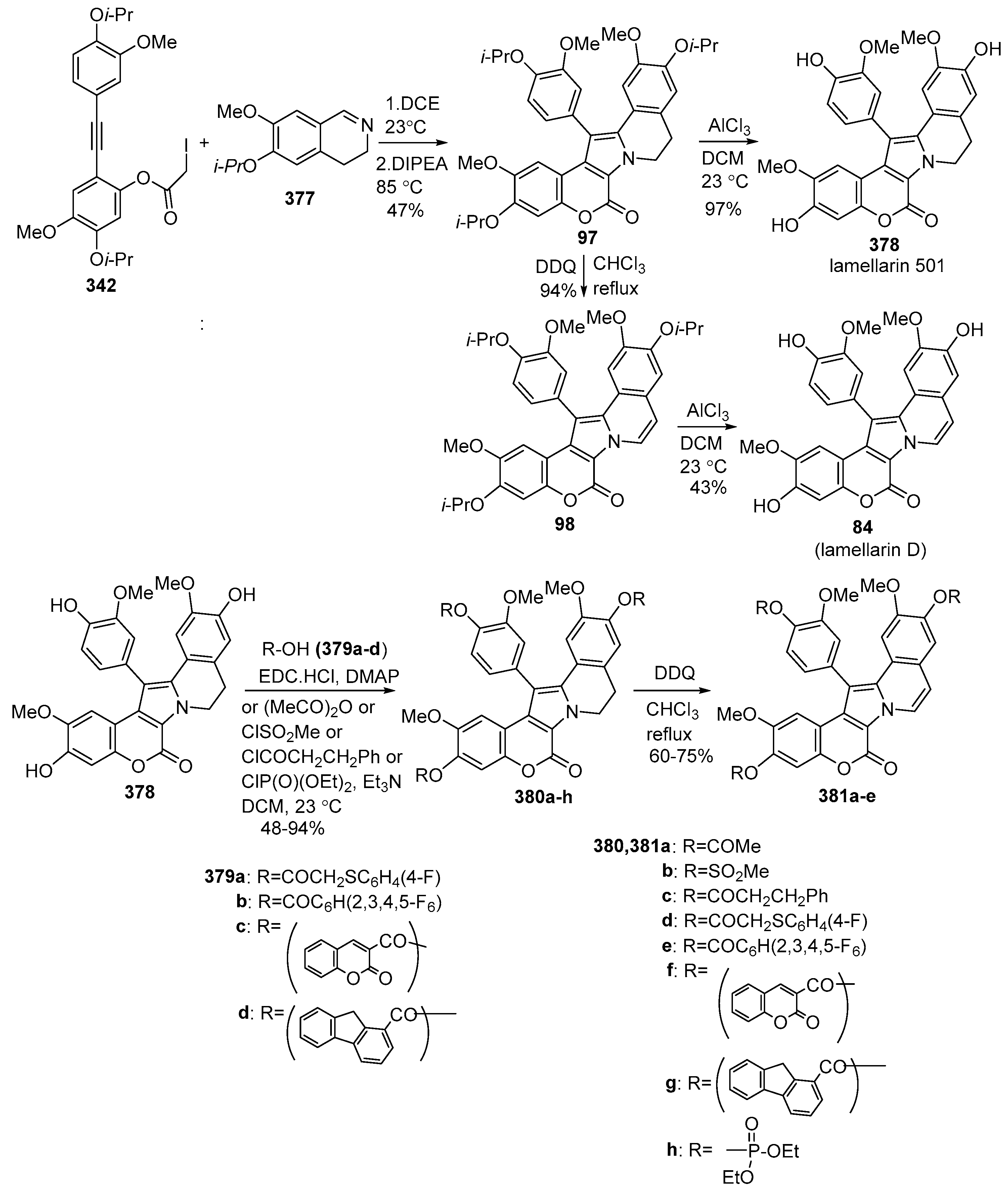
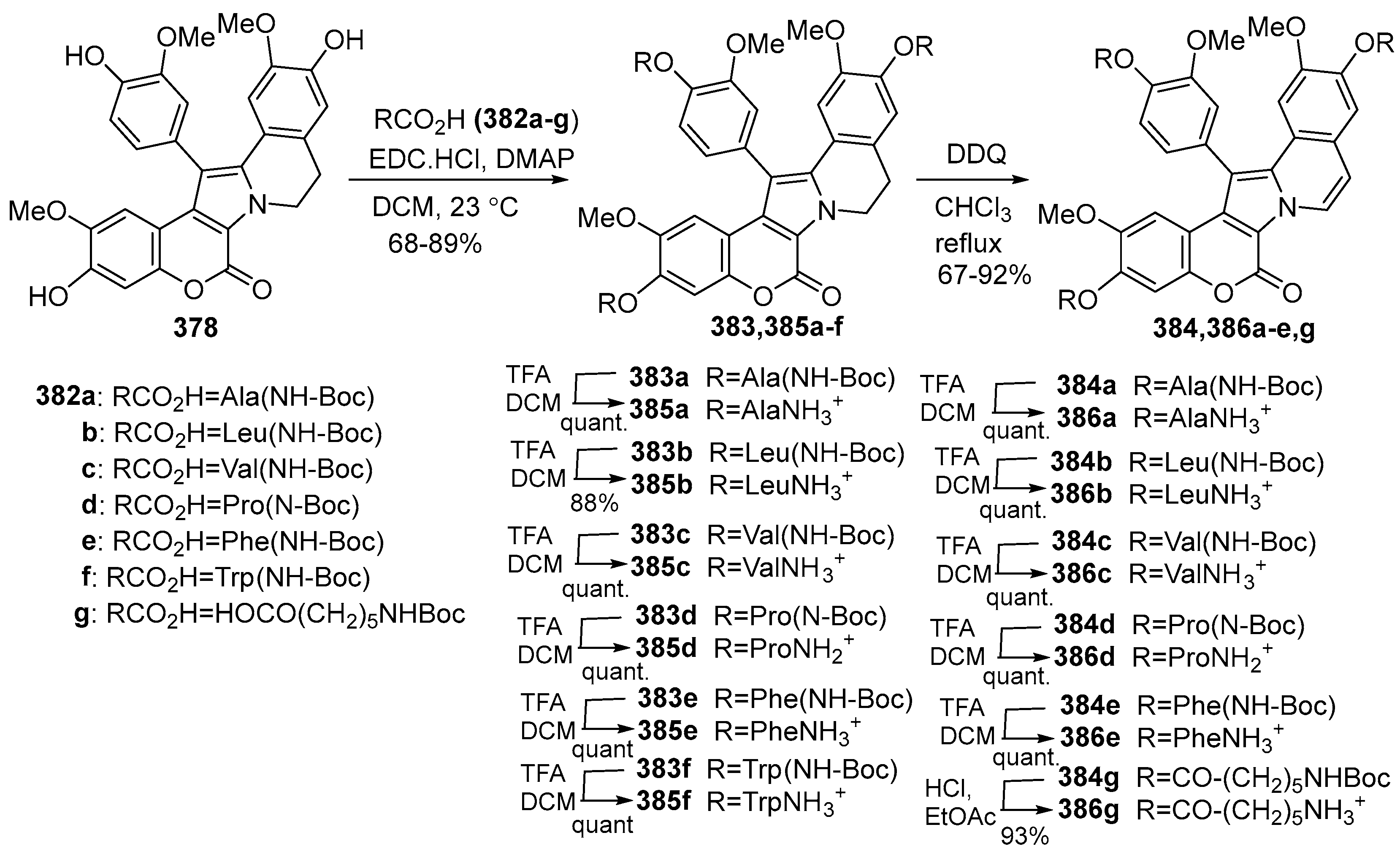
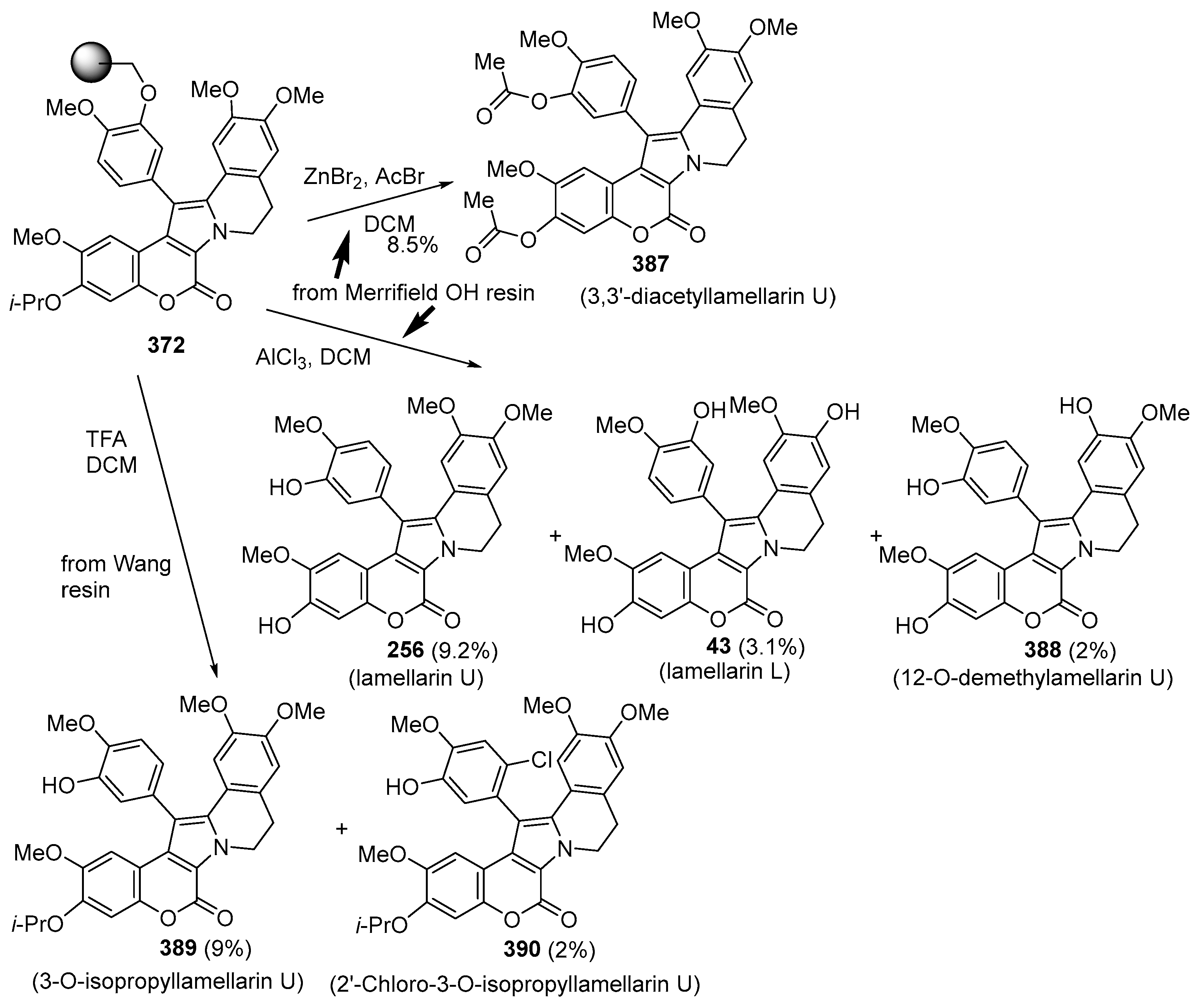
2.1.1. Synthesis Using Pyrrole Derivatives
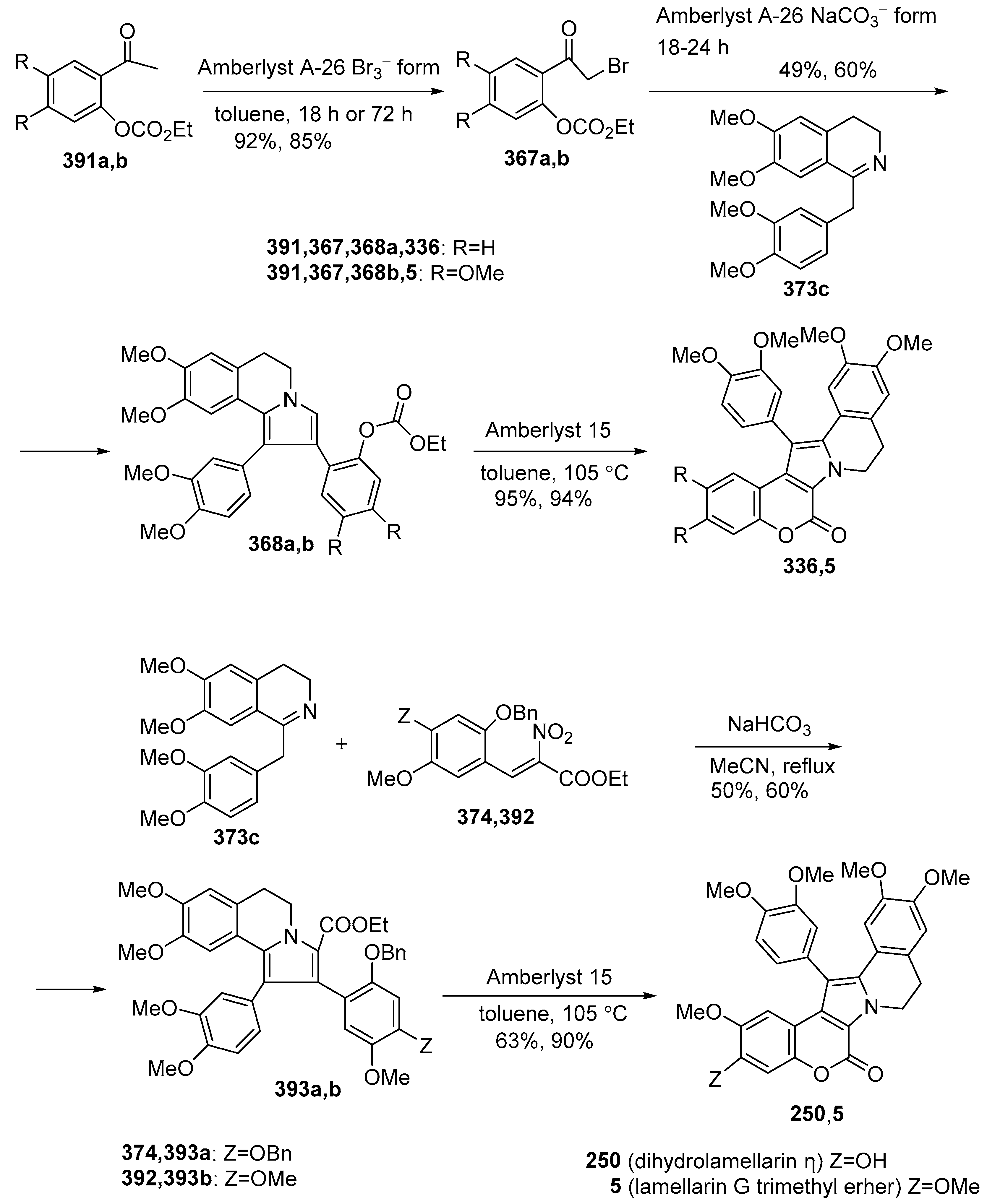
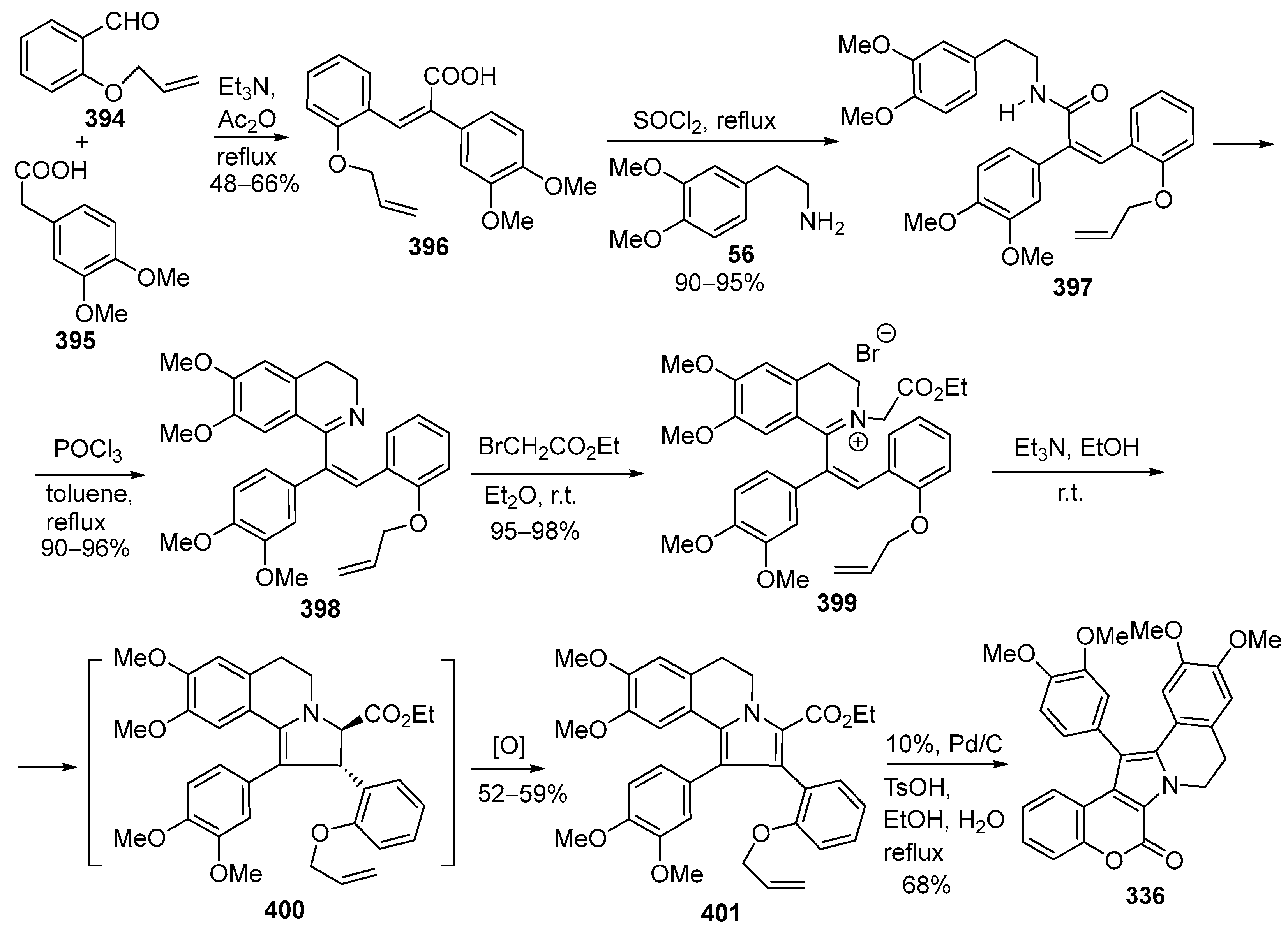
2.1.2. Synthesis Using Isoquinoline Derivatives
2.1.3. Synthesis Starting from Coumarin Derivatives
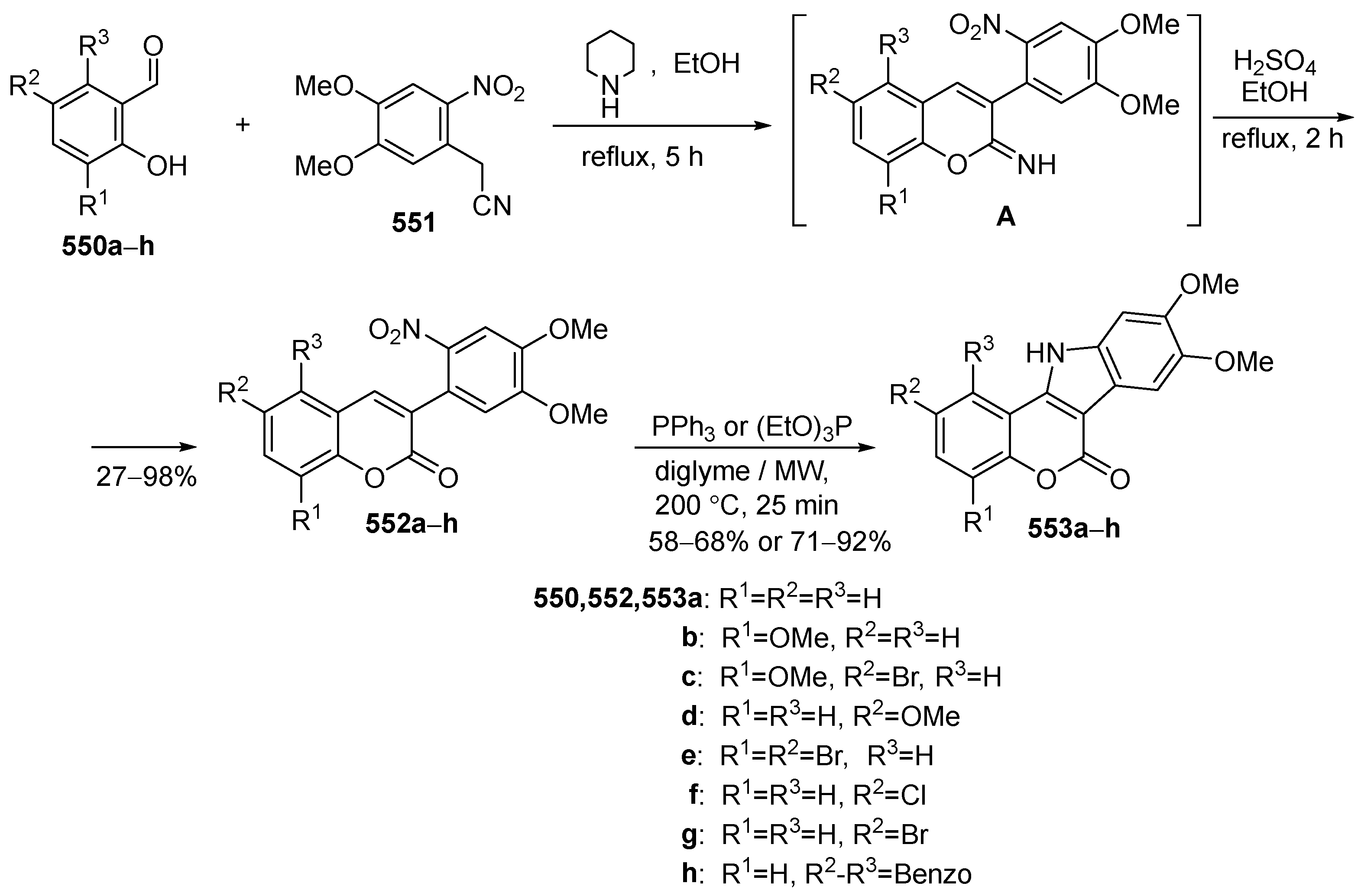
2.2. Synthesis of Isolamellarins and Analogues
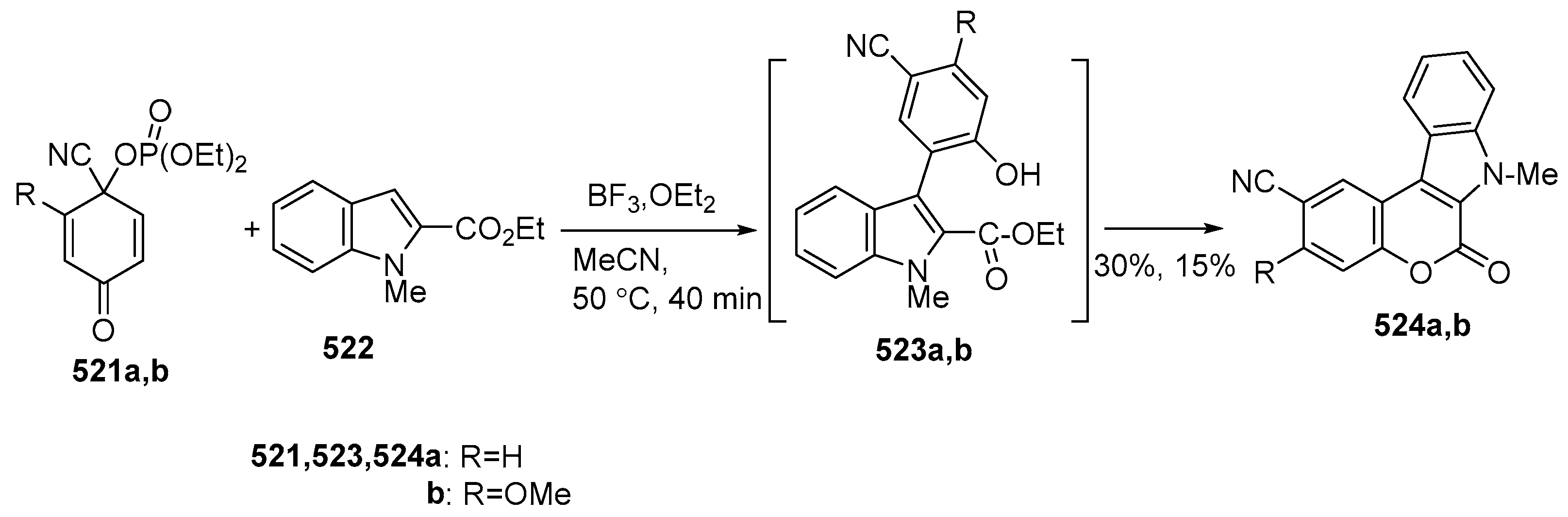
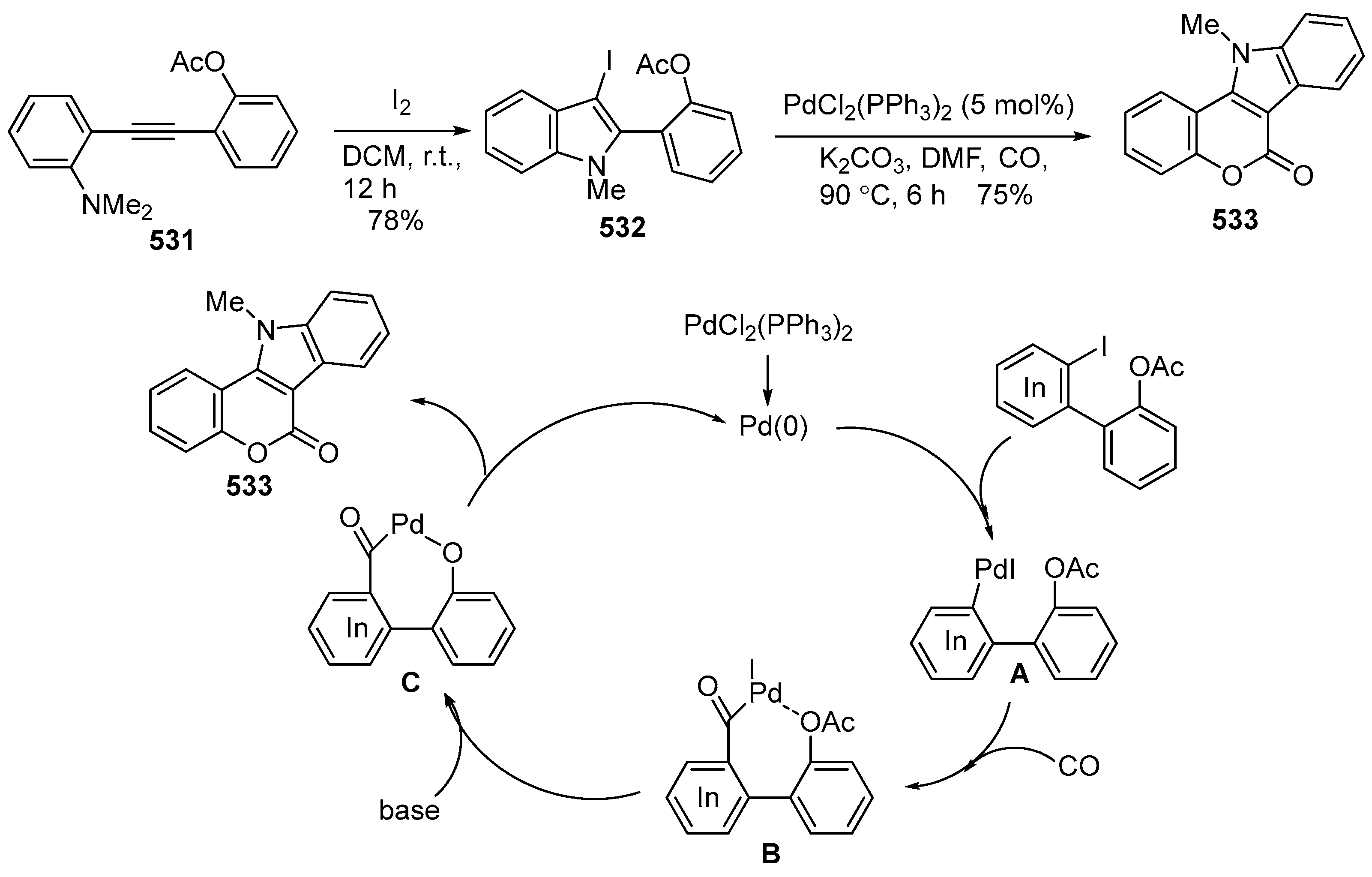
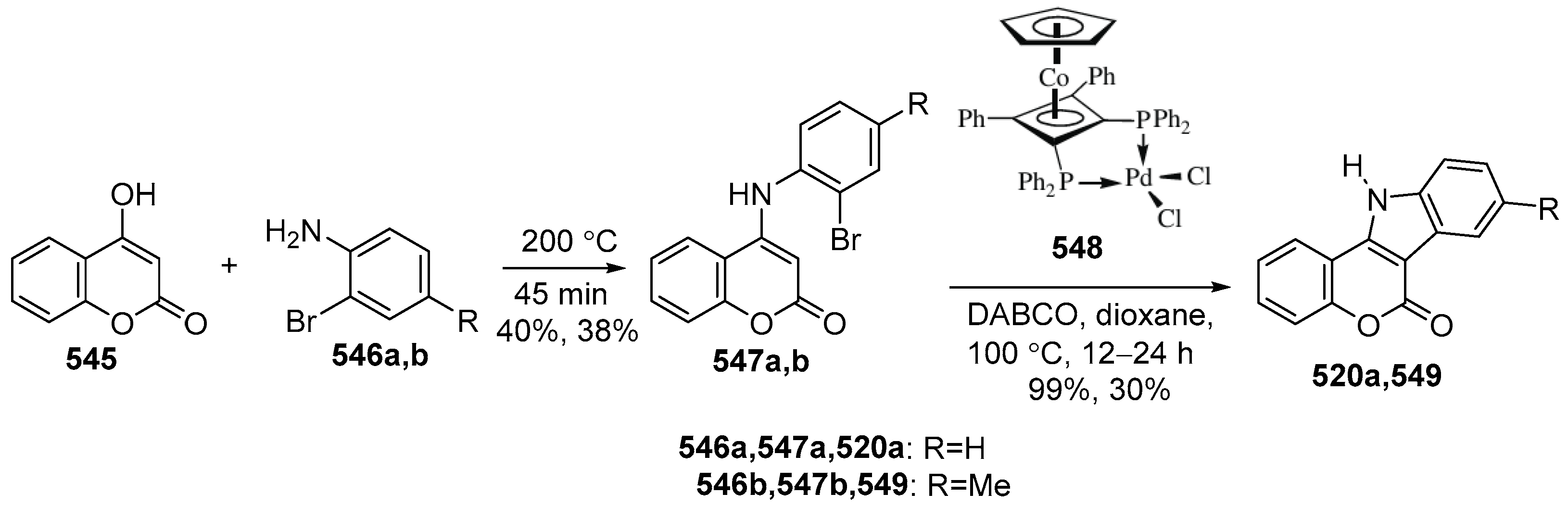
2.3. Synthesis of Azacoumestans, Isoazacoumestans, and Analogues
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernández-Peña, L.; Matos, M.J.; López, E. Recent Advances in Biologically Active Coumarins from Marine Sources: Synthesis and Evaluation. Mar. Drugs 2023, 21, 37. [Google Scholar] [CrossRef]
- Flores-Morales, V.; Villasana-Ruíz, A.P.; Garza-Veloz, I.; González-Delgado, S.; Martinez-Fierro, M.L. Therapeutic Effects of Coumarins with Different Substitution Patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, U.R.; Albohy, A.; Abdulrazik, B.S.; Bayoumi, S.A.L.; Malak, L.G.; Khallaf, I.S.A.; Bringmann, G.; Farag, S.F. Natural coumarins as potential anti-SARS-CoV-2 agents supported by docking analysis. RSC Adv. 2021, 11, 16970–16979. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.; Matos, M.J.; Uriarte, E.; Santana, L. Trending Topics on Coumarin and Its Derivatives in 2020. Molecules 2021, 26, 501. [Google Scholar] [CrossRef] [PubMed]
- Lončarić, M.; Gašo-Sokač, D.; Jokić, S.; Molnar, M. Recent Advances in the Synthesis of Coumarin Derivatives from Different Starting Materials. Biomolecules 2020, 10, 151. [Google Scholar] [CrossRef]
- Stringlis, I.A.; De Jonge, R.; Pieterse, C.M.J. The Age of Coumarins in Plant–Microbe Interactions. Plant Cell Physiol. 2019, 60, 1405–1419. [Google Scholar] [CrossRef]
- Hussain, M.I.; Syed, Q.A.; Khattak, M.N.K.; Hafez, B.; Reigosa, M.J.; El-Keblawy, A. Natural product coumarins: Biological and pharmacological perspectives. Biologia 2019, 74, 863–888. [Google Scholar] [CrossRef]
- Voges, M.J.E.E.E.; Bai, Y.; Schulze-Lefert, P.; Sattely, E.S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 12558–12565. [Google Scholar] [CrossRef]
- Matos, M.J.; Santana, L.; Uriarte, E.; Abreu, O.A.; Molina, E.; Yordi, E.M.A.E.G. Coumarins—An Important Class of Phytochemicals. In Phytochemicals—Isolation, Characterisation and Role in Human Health; Rao, A.V., Rao, L.G., Eds.; IntechOpen: London, UK, 2015; pp. 113–140. [Google Scholar] [CrossRef]
- O’Kennedy, R.; Thornes, R.D. Coumarins: Biology, Applications and Mode of Action; John Wiley & Sons: Chichester, UK, 1997. [Google Scholar]
- Murray, D.H.; Mendez, J.; Brown, S.A. The Natural Coumarins: Occurrence, Chemistry and Biochemistry; John Wiley & Sons: New York, NY, USA, 1982. [Google Scholar]
- Pereira, T.M.; Franco, D.P.; Vitorio, F.; Kummerle, A.E. Coumarin Compounds in Medicinal Chemistry: Some Important Examples from the Last Years. Curr. Top. Med. Chem. 2018, 18, 124–148. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef]
- Rubab, L.; Afroz, S.; Ahmad, S.; Hussain, S.; Nawaz, I.; Irfan, A.; Batool, F.; Kotwica-Mojzych, K.; Mojzych, M. An Update on Synthesis of Coumarin Sulfonamides as Enzyme Inhibitors and Anticancer Agents. Molecules 2022, 27, 1604. [Google Scholar] [CrossRef]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef]
- Alshibl, H.M.; Al-Abdullah, E.S.; Haiba, M.E.; Alkahtani, H.M.; Awad, G.E.A.; Mahmoud, A.H.; Ibrahim, B.M.M.; Bari, A.; Villinger, A. Synthesis and Evaluation of New Coumarin Derivatives as Antioxidant, Antimicrobial, and Anti-Inflammatory Agents. Molecules 2020, 25, 3251. [Google Scholar] [CrossRef]
- Fylaktakidou, K.C.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Nicolaides, D.N. Natural and Synthetic Coumarin Derivatives with Anti-Inflammatory/Antioxidant Activities. Curr. Pharm. Des. 2004, 10, 3813–3833. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, K.; Ponczek, M.B.; Owczarek, J.; Guga, P.; Budzisz, E. Antagonists of Vitamin K—Popular Coumarin Drugs and New Synthetic and Natural Coumarin Derivatives. Molecules 2020, 25, 1465. [Google Scholar] [CrossRef] [PubMed]
- Lowenthal, J.; Birnbaum, H. Vitamin K and Coumarin Anticoagulants: Dependence of Anticoagulant Effect on Inhibition of Vitamin K Transport. Science 1969, 164, 181–183. [Google Scholar] [CrossRef]
- Sharapov, A.D.; Fatykhov, R.F.; Khalymbadzha, I.A.; Zyryanov, G.V.; Chupakhin, O.N.; Tsurkan, M.V. Plant Coumarins with Anti-HIV Activity: Isolation and Mechanisms of Action. Int. J. Mol. Sci. 2023, 24, 2839. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Q.; Zhang, Y.; Liang, C. Coumarin-based derivatives with potential anti-HIV activity. Fitoterapia 2021, 150, 104863. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, T.; Wang, X.; Sun, J. Research progress of coumarins and their derivatives in the treatment of diabetes. J. Enzym. Inhib. Med. Chem. 2022, 37, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Y.; Li, L. Coumarins as potential antidiabetic agents. J. Pharm. Pharmacol. 2017, 69, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.A.R.; De Menezes, I.R.A.; Sousa, A.K.; Farias, P.A.M.; dos Santos, F.A.V.; Freitas, T.S.; Figueredo, F.G.; Ribeiro-Filho, J.; Carvalho, D.T.; Coutinho, H.D.M.; et al. In vitro and in silico antibacterial evaluation of coumarin derivatives against MDR strains of Staphylococcus aureus and Escherichia coli. Microb. Pathog. 2023, 177, 106058. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Liu, Y.; Guo, R.; Tang, Y.; Shi, Z.; Zhang, M.; Wu, W.; Chen, Y.; Hou, K. An In Vitro Coumarin-Antibiotic Combination Treatment of Pseudomonas aeruginosa Biofilms. Nat. Prod. Commun. 2021, 16, 1–7. [Google Scholar] [CrossRef]
- Reddy, D.S.; Kongot, M.; Kumar, A. Coumarin hybrid derivatives as promising leads to treat tuberculosis: Recent developments and critical aspects of structural design to exhibit anti-tubercular activity. Tuberculosis 2021, 127, 102050. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.-W.; Lv, Z.-S.; Feng, L.-S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, L.M.G.; Carreira, R.B.; de Oliveira, J.V.R.; Nascimento, R.P.D.; Souza, C.d.S.; Trias, E.; da Silva, V.D.A.; Costa, S.L. Impact of Plant-Derived Compounds on Amyotrophic Lateral Sclerosis. Neurotox. Res. 2023, 41, 288–309. [Google Scholar] [CrossRef]
- Lin, T.-H.; Chang, K.-H.; Chiu, Y.-J.; Weng, Z.-K.; Sun, Y.-C.; Lin, W.; Lee-Chen, G.-J.; Chen, C.-M. Neuroprotective Action of Coumarin Derivatives through Activation of TRKB-CREB-BDNF Pathway and Reduction of Caspase Activity in Neuronal Cells Expressing Pro-Aggregated Tau Protein. Int. J. Mol. Sci. 2022, 23, 12734. [Google Scholar] [CrossRef]
- El-Sawy, E.R.; Abdelwahab, A.B.; Kirsch, G. Synthetic Routes to Coumarin(Benzopyrone)-Fused Five-Membered Aromatic Heterocycles Built on the α-Pyrone Moiety. Part 1: Five-Membered Aromatic Rings with One Heteroatom. Molecules 2021, 26, 483. [Google Scholar] [CrossRef]
- Woźniak, Ł.; Połaska, M.; Marszałek, K.; Skąpska, S. Photosensitizing Furocoumarins: Content in Plant Matrices and Kinetics of Supercritical Carbon Dioxide Extraction. Molecules 2020, 25, 3805. [Google Scholar] [CrossRef]
- Vlachou, E.-E.N.; Litinas, K.E. An Overview on Pyranocoumarins: Synthesis and Biological Activities. Curr. Org. Chem. 2019, 23, 2679–2721. [Google Scholar] [CrossRef]
- Douka, M.D.; Litinas, K.E. An Overview on the Synthesis of Fused Pyridocoumarins with Biological Interest. Molecules 2022, 27, 7256. [Google Scholar] [CrossRef]
- Patra, P.; Patra, S. 4-Aminocoumarin Derivatives as Multifaceted Building Blocks for the Development of Various Bioactive Fused Coumarin Heterocycles: A Brief Review. Curr. Org. Chem. 2022, 26, 1585–1614. [Google Scholar] [CrossRef]
- Patra, P.; Manna, S.; Patra, S.; Samanta, K.; Roy, D. A Brief Review on the Synthesis of Pyrrolo [2,3-c]coumarins, including Lamellarin and Ningalin Scaffolds. Org. Prep. Proced. Int. 2023, 55, 63–83. [Google Scholar] [CrossRef]
- Samanta, K.; Patra, P.; Kar, G.K.; Dinda, S.K.; Mahanty, D.S. Diverse synthesis of pyrrolo/indolo[3,2-c]coumarins as isolamellarin-A scaffolds: A brief update. New J. Chem. 2021, 45, 7450–7485. [Google Scholar] [CrossRef]
- Salehian, F.; Nadri, H.; Jalili-Baleh, L.; Youseftabar-Miri, L.; Bukhari, S.N.A.; Foroumadi, A.; Küçükkilinç, T.T.; Sharifzadeh, M.; Khoobi, M. A review: Biologically active 3,4-heterocycle-fused coumarins. Eur. J. Med. Chem. 2020, 212, 113034. [Google Scholar] [CrossRef]
- Soman, S.S.; Thaker, T.H.; Rajput, R.A. Novel synthesis and cytotoxic activity of some chromeno[3,4-b]pyrrol-4(3H)-ones. Chem. Heterocycl. Compd. 2011, 46, 1514–1519. [Google Scholar] [CrossRef]
- Litinas, K.E.; Balalas, T.D.; Kanelli, M.G.; Gabriel, C.; Pontiki, E.; Hadjipavlou-Litina, D.J. Pd-Catalyzed N–H or C–H Functionalization/Oxidative Cyclization for the Efficient Synthesis of N-Aryl-Substituted [3,4]-Fused Pyrrolocoumarins. Synthesis 2022, 54, 2894–2906. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Litinas, K.E.; Makri, A.; Nicolaides, D.N.; Vronteli, A.; Hadjipavlou-Litina, D.J.; Pontiki, E.; Siohou, A. Synthesis and biological evaluation of novel angular fused Pyrrolocoumarins. J. Enzym. Inhib. Med. Chem. 2008, 23, 43–49. [Google Scholar] [CrossRef]
- Thakur, A.; Thakur, M.; Khadikar, P. Topological modeling of benzodiazepine receptor binding. Bioorganic Med. Chem. 2003, 11, 5203–5207. [Google Scholar] [CrossRef] [PubMed]
- Colotta, V.; Cecchi, L.; Melani, F.; Filacchioni, G.; Martini, C.; Giannaccini, G.; Lucacchini, A. Tricyclic heteroaromatic systems. [1]benzopyranopyrrol-4-ones and [1]benzopyrano-1,2,3-triazol-4-ones as benzodiazepine receptor ligands. Synthesis and structure-activity relationships. J. Med. Chem. 1990, 33, 2646–2651. [Google Scholar] [CrossRef]
- Dakshanamurthy, S.; Kim, M.; Brown, M.L.; Byers, S.W. In-silico fragment-based identification of novel angiogenesis inhibitors. Bioorganic Med. Chem. Lett. 2007, 17, 4551–4556. [Google Scholar] [CrossRef]
- Neagoie, C.; Vedrenne, E.; Buron, F.; Mérour, J.-Y.; Rosca, S.; Bourg, S.; Lozach, O.; Meijer, L.; Baldeyrou, B.; Lansiaux, A.; et al. Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: A novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012, 49, 379–396. [Google Scholar] [CrossRef]
- Friese, A.; Kapoor, S.; Schneidewind, T.; Vidadala, S.R.; Sardana, J.; Brause, A.; Förster, T.; Bischoff, M.; Wagner, J.; Janning, P.; et al. Chemical Genetics Reveals a Role of dCTP Pyrophosphatase 1 in Wnt Signaling. Angew. Chem. Int. Ed. 2019, 58, 13009–13013. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hazra, S.; Chowdhury, S.; Sarkar, S.; Chattopadhyay, K.; Pramanik, A. A novel pyrrole fused coumarin based highly sensitive and selective fluorescence chemosensor for detection of Cu2+ ions and applications towards live cell imaging. J. Photochem. Photobiol. A Chem. 2018, 364, 635–644. [Google Scholar] [CrossRef]
- Vazquez-Rodriguez, S.; Matos, M.J.; Borges, F.; Uriarte, E.; Santana, L. Bioactive coumarins from marine sources: Origin, structural features and pharmacological properties. Curr. Top. Med. Chem. 2015, 15, 1755–1766. [Google Scholar] [CrossRef]
- Duc, D.X.; Van Quoc, N. Isolation, Bioactivities, and Synthesis of Lamellarin Alkaloids: A Review. Curr. Org. Chem. 2022, 26, 961–990. [Google Scholar] [CrossRef]
- Seipp, K.; Geske, L.; Opatz, T. Marine Pyrrole Alkaloids. Mar. Drugs 2021, 19, 514. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Ishibashi, F.; Iwao, M. Chapter One—Lamellarin Alkaloids: Isolation, Synthesis, and Biological Activity. In The Alkaloids: Chemistry and Biology, 1st ed.; Elsevier: Cambridge, MA, USA; San Diego, CA, USA; London, UK; Oxford, UK, 2020; Volume 83, pp. 1–112. [Google Scholar]
- Fan, H.; Peng, J.; Hamann, M.T.; Hu, J.-F. Lamellarins and Related Pyrrole-Derived Alkaloids from Marine Organisms. Chem. Rev. 2007, 108, 264–287. [Google Scholar] [CrossRef]
- Andersen, R.J.; Faulkner, D.J.; He, C.H.; Van Duyne, G.D.; Clardy, J. Metabolites of the marine prosobranch mollusk Lamellaria sp. J. Am. Chem. Soc. 1985, 107, 5492–5495. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; Van Duyne, G.D.; Clardy, J. New alkaloids of the lamellarin class from the marine ascidian Didemnum chartaceum (Sluiter, 1909). J. Org. Chem. 1988, 53, 4570–4574. [Google Scholar] [CrossRef]
- Carroll, A.R.; Bowden, B.F.; Coll, J.C. Studies of Australian Ascidians. I. Six New Lamellarin-Class Alkaloids From a Colonial Ascidian, Didemnum sp. Aust. J. Chem. 1993, 46, 489–501. [Google Scholar] [CrossRef]
- Urban, S.; Hobbs, L.; Hooper, J.N.A.; Capon, R.J. Lamellarins Q and R: New Aromatic Metabolites From an Australian Marine Sponge, Dendrilla cactos. Aust. J. Chem. 1995, 48, 1491–1494. [Google Scholar] [CrossRef]
- Urban, S.; Capon, R.J. Lamellarin-S: A New Aromatic Metabolite from an Australian Tunicate, Didemnum sp. Aust. J. Chem. 1996, 49, 711–713. [Google Scholar] [CrossRef]
- Reddy, M.V.R.; Faulkner, D.J.; Venkateswarlu, Y.; Rao, M.R. New lamellarin alkaloids from an unidentified ascidian from the Arabian Sea. Tetrahedron 1997, 53, 3457–3466. [Google Scholar] [CrossRef]
- Reddy, M.V.R.; Rao, M.R.; Rhodes, D.; Hansen, M.S.T.; Rubins, K.; Bushman, F.D.; Venkateswarlu, Y.; Faulkner, D.J. Lamellarin α 20-Sulfate, an Inhibitor of HIV-1 Integrase Active against HIV-1 Virus in Cell Culture. J. Med. Chem. 1999, 42, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Carroll, A.R.; Pierens, G.K.; Quinn, R.J. New Lamellarin Alkaloids from the Australian Ascidian, Didemnum chartaceum. J. Nat. Prod. 1999, 62, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.; Kang, H. A Novel Cytotoxic Alkaloid of Lamellarin Class from a Marine Ascidian Didemnum sp. Bull. Korean Chem. Soc. 2002, 23, 163–166. [Google Scholar] [CrossRef]
- Krishnaiah, P.; Reddy, V.L.N.; Venkataramana, G.; Ravinder, K.; Srinivasulu, M.; Raju, T.V.; Ravikumar, K.; Chandrasekar, D.; Ramakrishna, S.; Venkateswarlu, Y. New Lamellarin Alkaloids from the Indian Ascidian Didemnum obscurum and Their Antioxidant Properties. J. Nat. Prod. 2004, 67, 1168–1171. [Google Scholar] [CrossRef]
- Reddy, S.M.; Srinivasulu, M.; Satyanarayana, N.; Kondapi, A.K.; Venkateswarlu, Y. New potent cytotoxic lamellarin alkaloids from Indian ascidian Didemnum obscurum. Tetrahedron 2005, 61, 9242–9247. [Google Scholar] [CrossRef]
- Plisson, F.; Huang, X.; Zhang, H.; Khalil, Z.; Capon, R.J. Lamellarins as Inhibitors of P-Glycoprotein-Mediated Multidrug Resistance in a Human Colon Cancer Cell Line. Chem. Asian J. 2012, 7, 1616–1623. [Google Scholar] [CrossRef]
- Bracegirdle, J.; Robertson, L.P.; Hume, P.A.; Page, M.J.; Sharrock, A.V.; Ackerley, D.F.; Carroll, A.R.; Keyzers, R.A. Lamellarin Sulfates from the Pacific Tunicate Didemnum ternerratum. J. Nat. Prod. 2019, 82, 2000–2008. [Google Scholar] [CrossRef]
- Kang, H.; Fenical, W. Ningalins A−D: Novel Aromatic Alkaloids from a Western Australian Ascidian of the Genus Didemnum. J. Org. Chem. 1997, 62, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Conte, M.; Khalil, Z.; Huang, X.; Piggott, A.M.; Capon, R.J. Kinase Inhibitor Scaffolds against Neurodegenerative Diseases from a Southern Australian Ascidian, Didemnum sp. ChemMedChem 2012, 7, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Li, Z.; Shen, S.; Zeng, Y.; Yang, Y.; Xu, M.; Bruhn, T.; Bruhn, H.; Morschhäuser, J.; Bringmann, G.; et al. Baculiferins A–O, O-sulfated pyrrole alkaloids with anti-HIV-1 activity, from the Chinese marine sponge Iotrochota baculifera. Bioorganic Med. Chem. 2010, 18, 5466–5474. [Google Scholar] [CrossRef]
- Kappe, T.; Stadlbauer, W. Simple and Effective Approaches to Coumestans and Azacoumestans. Heterocycles 1993, 35, 1425–1440. [Google Scholar] [CrossRef]
- Patra, P. A short review on the synthesis of pyrrolo[3,4-c]coumarins an isolamellarin-B scaffolds. Synth. Commun. 2022, 52, 1999–2018. [Google Scholar] [CrossRef]
- Pla, D.; Albericio, F.; Alvarez, M. Recent Advances in Lamellarin Alkaloids: Isolation, Synthesis and Activity. Anti-Cancer Agents Med. Chem. 2008, 8, 746–760. [Google Scholar] [CrossRef]
- Iwao, M.; Fukuda, T.; Ishibashi, F. Synthesis and Biological Activity of Lamellarin Alkaloids: An Overview. Heterocycles 2011, 83, 491–529. [Google Scholar] [CrossRef]
- Imbri, D.; Tauber, J.; Opatz, T. Synthetic Approaches to the Lamellarins—A Comprehensive Review. Mar. Drugs 2014, 12, 6142–6177. [Google Scholar] [CrossRef]
- Bailly, C. Anticancer Properties of Lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef]
- Wei, M.; Chen, J.; Song, Y.; Monserrat, J.-P.; Zhang, Y.; Shen, L. Progress on synthesis and structure-activity relationships of lamellarins over the past decade. Eur. J. Med. Chem. 2024, 269, 116294. [Google Scholar] [CrossRef]
- Cui, H.-L. Recent progress in the synthesis of pyrrolo[2,1-a]isoquinolines. Org. Biomol. Chem. 2022, 20, 2779–2801. [Google Scholar] [CrossRef] [PubMed]
- Heim, A.; Terpin, A.; Steglich, W. Biomimetic Synthesis of Lamellarin G. Trimethyl Ether. Angew. Chem. Int. Ed. Engl. 1997, 36, 155–156. [Google Scholar] [CrossRef]
- Banwell, M.G.; Hockless, D.C.R.; Flynn, B.L.; Longmore, R.W.; Rae, D. Assessment of Double-Barrelled Heck Cyclizations as a Means for Construction of the 14-Phenyl-8,9-dihydro- 6H-[1]benzopyrano[4′,3′:4,5]pyrrolo[2,1-a]isoquinolin- 6-one Core Associated with Certain Members of the Lamellarin Class of Marine Natural Product. Aust. J. Chem. 1998, 52, 755–766. [Google Scholar] [CrossRef]
- Boger, D.L.; Boyce, C.W.; Labroli, M.A.; Sehon, C.A.; Jin, Q. Total Syntheses of Ningalin A, Lamellarin O, Lukianol A, and Permethyl Storniamide A Utilizing Heterocyclic Azadiene Diels−Alder Reactions. J. Am. Chem. Soc. 1999, 121, 54–62. [Google Scholar] [CrossRef]
- Boger, D.L.; Soenen, D.R.; Boyce, C.W.; Hedrick, M.P.; Jin, Q. Total Synthesis of Ningalin B Utilizing a Heterocyclic Azadiene Diels−Alder Reaction and Discovery of a New Class of Potent Multidrug Resistant (MDR) Reversal Agents. J. Org. Chem. 2000, 65, 2479–2483. [Google Scholar] [CrossRef]
- Peschko, C.; Winklhofer, C.; Steglich, W. Biomimetic total synthesis of lamellarin L by coupling of two different arylpuru-vic acid units. Chem. Eur. J. 2000, 6, 1147–1152. [Google Scholar] [CrossRef]
- Bullington, J.L.; Wolff, R.R.; Jackson, P.F. Regioselective Preparation of 2-Substituted 3,4-Diaryl Pyrroles: A Concise Total Synthesis of Ningalin B. J. Org. Chem. 2002, 67, 9439–9442. [Google Scholar] [CrossRef] [PubMed]
- Furstner, A.; Weintritt, H.; Hupperts, A. A New, Titanium-Mediated Approach to Pyrroles: First Synthesis of Lukianol A and Lamellarin O Dimethyl Ether. J. Org. Chem. 1995, 60, 6637–6641. [Google Scholar] [CrossRef]
- Gupton, J.T.; Clough, S.C.; Miller, R.B.; Lukens, J.R.; Henry, C.A.; Kanters, R.P.; Sikorski, J.A. The application of vinylogous iminium salt derivatives to the synthesis of Ningalin B hexamethyl ether. Tetrahedron 2002, 59, 207–215. [Google Scholar] [CrossRef]
- Iwao, M.; Takeuchi, T.; Fujikawa, N.; Fukuda, T.; Ishibashi, F. Short and flexible route to 3,4-diarylpyrrole marine alkaloids: Syntheses of permethyl storniamide A, ningalin B, and lamellarin G trimethyl ether. Tetrahedron Lett. 2003, 44, 4443–4446. [Google Scholar] [CrossRef]
- Handy, S.T.; Zhang, Y.; Bregman, H. A Modular Synthesis of the Lamellarins: Total Synthesis of Lamellarin G Trimethyl Ether. J. Org. Chem. 2004, 69, 2362–2366. [Google Scholar] [CrossRef]
- Pla, D.; Marchal, A.; Olsen, C.A.; Albericio, F.; Álvarez, M. Modular Total Synthesis of Lamellarin D. J. Org. Chem. 2005, 70, 8231–8234. [Google Scholar] [CrossRef] [PubMed]
- Facompré, M.; Tardy, C.; Bal-Mahieu, C.; Colson, P.; Perez, C.; Manzanares, I.; Cuevas, C.; Bailly, C. Lamellarin D: A novel potent inhibitor of topoisomerase I. Cancer Res. 2003, 63, 7392–7399. [Google Scholar]
- Fujikawa, N.; Ohta, T.; Yamaguchi, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. Total synthesis of Lamellarins D, L, and N. Tetrahedron 2006, 62, 594–604. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. The first total synthesis of lamellarin α 20-sulfate, a selective inhibitor of HIV-1 integrase. Tetrahedron Lett. 2006, 47, 3755–3757. [Google Scholar] [CrossRef]
- Steglich, W.; Peschko, C.; Winklhofer, C.; Terpin, A. Biomimetic Syntheses of Lamellarin and Lukianol-Type Alkaloids. Synthesis 2006, 3048–3057. [Google Scholar] [CrossRef]
- Gupton, J.T.; Giglio, B.C.; Eaton, J.E.; Rieck, E.A.; Smith, K.L.; Keough, M.J.; Barelli, P.J.; Firich, L.T.; Hempel, J.E.; Smith, T.M.; et al. The application of vinylogous iminium salt derivatives to efficient formal syntheses of the marine alkaloids lamellarin G trimethyl ether and ningalin B. Tetrahedron 2009, 65, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. Design and Synthesis of Lamellarin D Analogues Targeting Topoisomerase I. J. Org. Chem. 2009, 74, 8143–8153. [Google Scholar] [CrossRef]
- Fukuda, T.; Nanjo, Y.; Fujimoto, M.; Yoshida, K.; Natsui, Y.; Ishibashi, F.; Okazaki, F.; To, H.; Iwao, M. Lamellarin-inspired potent topoisomerase I inhibitors with the unprecedented benzo[g][1]benzopyrano[4,3-b]indol-6(13H)-one scaffold. Bioorganic Med. Chem. 2019, 27, 265–277. [Google Scholar] [CrossRef]
- Iwao, M.; Fukuda, T.; Saeki, S.; Ohta, T. Divergent Synthesis of Lamellarin α 13-Sulfate, 20-Sulfate, and 13,20-Disulfate. Heterocycles 2010, 80, 841–846. [Google Scholar] [CrossRef]
- Hasse, K.; Willis, A.C.; Banwell, M.G. Modular Total Syntheses of Lamellarin G Trimethyl Ether and Lamellarin S. Eur. J. Org. Chem. 2011, 88–99. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total Synthesis of Lamellarins D, H, and R and Ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Komatsubara, M.; Umeki, T.; Fukuda, T.; Iwao, M. Modular Synthesis of Lamellarins via Regioselective Assembly of 3,4,5-Differentially Arylated Pyrrole-2-carboxylates. J. Org. Chem. 2014, 79, 529–537. [Google Scholar] [CrossRef]
- Ueda, K.; Amaike, K.; Maceiczyk, R.M.; Itami, K.; Yamaguchi, J. β-Selective C–H Arylation of Pyrroles Leading to Concise Syntheses of Lamellarins C and I. J. Am. Chem. Soc. 2014, 136, 13226–13232. [Google Scholar] [CrossRef] [PubMed]
- Gupton, J.T.; Telang, N.; Patteson, J.; Lescalleet, K.; Yeudall, S.; Sobieski, J.; Harrison, A.; Curry, W. The application of formyl group activation of bromopyrrole esters to formal syntheses of lycogarubin C, permethyl storniamide A and lamellarin G trimethyl ether. Tetrahedron 2014, 70, 9759–9767. [Google Scholar] [CrossRef]
- Iwao, M.; Fukuda, T.; Sato, D. A Synthesis of Lamellarins via Regioselective Assembly of 1,2,3-Differentially Substituted 5,6-Dihydropyrrolo[2,1-a]Isoquinoline Core. Heterocycles 2015, 91, 782–794. [Google Scholar] [CrossRef]
- Dialer, C.; Imbri, D.; Hansen, S.P.; Opatz, T. Synthesis of Lamellarin D Trimethyl Ether and Lamellarin H via 6π-Electrocyclization. J. Org. Chem. 2015, 80, 11605–11610. [Google Scholar] [CrossRef]
- Fukuda, T.; Katae, T.; Harada, I.; Iwao, M. Synthesis of Lamellarins via Regioselective Assembly of 1,2-Diarylated [1]Benzopyrano[3,4-b]pyrrol-4(3H)-one Core. Heterocycles 2017, 95, 950–971. [Google Scholar] [CrossRef]
- Mei, R.; Zhang, S.-K.; Ackermann, L. Concise Synthesis of Lamellarin Alkaloids by C–H/N–H Activation: Evaluation of Metal Catalysts in Oxidative Alkyne Annulation. Synlett 2017, 28, 1715–1718. [Google Scholar] [CrossRef]
- Kumar, V.; Awasthi, A.; Salam, A.; Khan, T. Scalable Total Syntheses of Some Natural and Unnatural Lamellarins: Application of a One-Pot Domino Process for Regioselective Access to the Central 1,2,4-Trisubstituted Pyrrole Core. J. Org. Chem. 2019, 84, 11596–11603. [Google Scholar] [CrossRef]
- Shirley, H.J.; Koyioni, M.; Muncan, F.; Donohoe, T.J. Synthesis of lamellarin alkaloids using orthoester-masked α-keto acids. Chem. Sci. 2019, 10, 4334–4338. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, D.; Morii, K.; Yasuda, Y.; Mori, A.; Okano, K. Convergent Total Synthesis of Lamellarins and Their Congeners. J. Org. Chem. 2020, 85, 8603–8617. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Salam, A.; Kumar, D.; Khan, T. Concise and Scalable Total Syntheses of Lamellarin Z and other Natural Lamellarins. ChemistrySelect 2020, 5, 14510–14514. [Google Scholar] [CrossRef]
- Morii, K.; Yasuda, Y.; Morikawa, D.; Mori, A.; Okano, K. Total Synthesis of Lamellarins G, J, L, and Z Using One-Pot Halogen Dance/Negishi Coupling. J. Org. Chem. 2021, 86, 13388–13401. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Okui, Y.; Yasuda, Y.; Mori, A. Total Synthesis of Lamellarins U and A3 by Interrupting Halogen Dance. Synthesis 2022, 54, 2647–2660. [Google Scholar] [CrossRef]
- Ishibashi, F.; Miyazaki, Y.; Iwao, M. Total Syntheses of Lamellarin D and H. The First Synthesis of Lamellarin-Class Marine Alkaloids. Tetrahedron 1997, 53, 5951–5962. [Google Scholar] [CrossRef]
- Banwell, M.; Flynn, B.; Hockless, D. Convergent total synthesis of lamellarin K. Chem. Commun. 1997, 23, 2259–2260. [Google Scholar] [CrossRef]
- Ruchirawat, S.; Mutarapat, T. An efficient synthesis of lamellarin alkaloids: Synthesis of lamellarin G trimethyl ether. Tetrahedron Lett. 2001, 42, 1205–1208. [Google Scholar] [CrossRef]
- Díaz, M.; Guitián, E.; Castedo, L. Syntheses of Lamellarins I and K by [3 + 2] Cycloaddition of a Nitrone to an Alkyne. Synlett 2001, 2001, 1164–1166. [Google Scholar] [CrossRef]
- Ishibashi, F.; Tanabe, S.; Oda, T.; Iwao, M. Synthesis and Structure−Activity Relationship Study of Lamellarin Derivatives. J. Nat. Prod. 2002, 65, 500–504. [Google Scholar] [CrossRef]
- Ridley, C.P.; Reddy, M.V.R.; Rocha, G.; Bushman, F.D.; Faulkner, D.J. Total synthesis and evaluation of lamellarin α 20-Sulfate analogues. Bioorganic Med. Chem. 2002, 10, 3285–3290. [Google Scholar] [CrossRef]
- Ploypradith, P.; Jinaglueng, W.; Pavaro, C.; Ruchirawat, S. Further developments in the synthesis of lamellarin alkaloids via direct metal–halogen exchange. Tetrahedron Lett. 2003, 44, 1363–1366. [Google Scholar] [CrossRef]
- Cironi, P.; Manzanares, I.; Albericio, F.; Álvarez, M. Solid-Phase Total Synthesis of the Pentacyclic System Lamellarins U and L. Org. Lett. 2003, 5, 2959–2962. [Google Scholar] [CrossRef] [PubMed]
- Ploypradith, P.; Mahidol, C.; Sahakitpichan, P.; Wongbundit, S.; Ruchirawat, S. A Highly Efficient Synthesis of Lamellarins K and L by the Michael Addition/Ring-Closure Reaction of Benzyldihydroisoquinoline Derivatives with Ethoxycarbonyl-β-nitrostyrenes. Angew. Chem. Int. Ed. 2004, 43, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Facompré, M.; Laine, W.; Baldeyrou, B.; García-Gravalos, D.; Francesch, A.; Mateo, C.; Pastor, A.; Jiménez, J.A.; Manzanares, I.; et al. Topoisomerase I-mediated DNA cleavage as a guide to the development of antitumor agents derived from the marine alkaloid lamellarin D: Triester derivatives incorporating amino acid residues. Bioorganic Med. Chem. 2004, 12, 1697–1712. [Google Scholar] [CrossRef] [PubMed]
- Cironi, P.; Cuevas, C.; Albericio, F.; Álvarez, M. Gaining diversity in solid-phase synthesis by modulation of cleavage conditions from hydroxymethyl-based supports. Application to lamellarin synthesis. Tetrahedron 2004, 60, 8669–8675. [Google Scholar] [CrossRef]
- Ploypradith, P.; Kagan, R.K.; Ruchirawat, S. Utility of Polymer-Supported Reagents in the Total Synthesis of Lamellarins. J. Org. Chem. 2005, 70, 5119–5125. [Google Scholar] [CrossRef]
- Nyerges, M.; Tőke, L. 1,5-Electrocyclisation of azomethine ylides leading to pyrrolo[2,1-a]isoquinolines—Concise construction of the lamellarin skeleton. Tetrahedron Lett. 2005, 46, 7531–7534. [Google Scholar] [CrossRef]
- Ploypradith, P.; Petchmanee, T.; Sahakitpichan, P.; Litvinas, N.D.; Ruchirawat, S. Total Synthesis of Natural and Unnatural Lamellarins with Saturated and Unsaturated D-Rings. J. Org. Chem. 2006, 71, 9440–9448. [Google Scholar] [CrossRef]
- Chittchang, M.; Batsomboon, P.; Ruchirawat, S.; Ploypradith, P. Cytotoxicities and Structure–Activity Relationships of Natural and Unnatural Lamellarins toward Cancer Cell Lines. ChemMedChem 2009, 4, 457–465. [Google Scholar] [CrossRef]
- Liermann, J.C.; Opatz, T. Synthesis of Lamellarin U and Lamellarin G Trimethyl Ether by Alkylation of a Deprotonated α-Aminonitrile. J. Org. Chem. 2008, 73, 4526–4531. [Google Scholar] [CrossRef]
- Imbri, D.; Tauber, J.; Opatz, T. A High-Yielding Modular Access to the Lamellarins: Synthesis of Lamellarin G Trimethyl Ether, Lamellarin η and Dihydrolamellarin η. Chem. Eur. J. 2013, 19, 15080–15083. [Google Scholar] [CrossRef] [PubMed]
- Klintworth, R.; de Koning, C.B.; Opatz, T.; Michael, J.P. A Xylochemically Inspired Synthesis of Lamellarin G Trimethyl Ether via an Enaminone Intermediate. J. Org. Chem. 2019, 84, 11025–11031. [Google Scholar] [CrossRef] [PubMed]
- Klintworth, R.; de Koning, C.B.; Michael, J.P. Demethylative Lactonization Provides a Shortcut to High-Yielding Syntheses of Lamellarins. J. Org. Chem. 2020, 85, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Manjappa, K.B.; Jhang, J.; Lakshmi, K.C.S.; Yang, D. Four-Component Construction of Coumarin-Fused Pyrrolo[2,1-a]isoquinoline: Expedient Synthesis of Lamellarins and Their Regioselective Demethylation. Asian J. Org. Chem. 2022, 11, e202100659. [Google Scholar] [CrossRef]
- Manjappa, K.B.; Lin, J.-M.; Yang, D.-Y. Construction of Pentacyclic Lamellarin Skeleton via Grob Reaction: Application to Total Synthesis of Lamellarins H and D. J. Org. Chem. 2017, 82, 7648–7656. [Google Scholar] [CrossRef]
- Silyanova, E.A.; Samet, A.V.; Semenov, V.V. A Two-Step Approach to a Hexacyclic Lamellarin Core via 1,3-Dipolar Cycloaddition of Isoquinolinium Ylides to Nitrostilbenes. J. Org. Chem. 2022, 87, 6444–6453. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Gayathri, K.U.; Reddy, B.V.S.; Prasad, A.R. Modular Total Synthesis of Lamellarin G Trimethyl Ether. Synlett 2009, 2009, 43–46. [Google Scholar] [CrossRef]
- Chen, L.; Xu, M. A New Approach to Pyrrolocoumarin Derivatives by Palladium-Catalyzed Reactions: Expedient Construction of Polycyclic Lamellarin Scaffold. Adv. Synth. Catal. 2009, 351, 2005–2012. [Google Scholar] [CrossRef]
- Manjappa, K.B.; Syu, J.-R.; Yang, D.-Y. Visible-Light-Promoted and Yb(OTf)3-Catalyzed Constructions of Coumarin-Pyrrole-(Iso)quinoline-Fused Pentacycles: Synthesis of Lamellarin Core, Lamellarin D Trimethyl Ether, and Lamellarin H. Org. Lett. 2016, 18, 332–335. [Google Scholar] [CrossRef]
- Wu, C.-K.; Weng, Z.; Yang, D.-Y. One-Pot Construction of 1-Phenylchromeno[3,4-b]pyrrol-4(3H)-one: Application to Total Synthesis of Ningalin B and a Pyrrolocoumarin-Based Electrochromic Switch. Org. Lett. 2019, 21, 5225–5228. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Immunosuppressive Ningalin Compounds. WO2006026496A2, 2006. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006026496 (accessed on 9 March 2006).
- Thasana, N.; Worayuthakarn, R.; Kradanrat, P.; Hohn, E.; Young, L.; Ruchirawat, S. Copper(I)-Mediated and Microwave-Assisted Caryl−Ocarboxylic Coupling: Synthesis of Benzopyranones and Isolamellarin Alkaloids. J. Org. Chem. 2007, 72, 9379–9382. [Google Scholar] [CrossRef] [PubMed]
- Padilha, G.; Iglesias, B.A.; Back, D.F.; Kaufman, T.S.; Silveira, C.C. Synthesis of Chromeno[4,3-b]pyrrol-4(1H)-ones, from β-Nitroalkenes and 4-Phenylaminocoumarins, under Solvent–free Conditions. ChemistrySelect 2017, 2, 1297–1304. [Google Scholar] [CrossRef]
- Vyasamudri, S.; Yang, D.-Y. Base-Dependent Divergent Annulation of 4-Chloro-3-formylcoumarin and Tetrahydroisoquinoline: Application to the Synthesis of Isolamellarins and Hydroxypyrrolones. J. Org. Chem. 2019, 84, 3662–3670. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Samanta, R. Weakly Coordinating tert-Amide-Assisted Ru(II)-Catalyzed Synthesis of Azacoumestans via Migratory Insertion of Quinoid Carbene: Application in the Total Synthesis of Isolamellarins. Org. Lett. 2022, 24, 4536–4541. [Google Scholar] [CrossRef]
- King, H.I.; Holland, R.H.; Reed, F.P.; Robertson, A. Analogues of rotenone and related compounds. Part II. Coumarono-(2?:3?:3:4)-coumarins. J. Chem. Soc. 1948, 3, 1672–1674. [Google Scholar] [CrossRef]
- Stadlbauer, W.; Kappe, T. Synthese von Indolen und Isochinolonen aus Phenylmalonylheterocyclen. Monatshefte Fur Chemie/Chem. Mon. 1984, 115, 467–475. [Google Scholar] [CrossRef]
- Stadlbauer, W.; Laschober, R.; Kappe, T. Potential non-steroidal estrogens and antiestrogens, IV Organic azides in heterocyclic synthesis, part 13: Synthesis of aza- and diazacoumestrols via azido derivatives. Monatshefte Fur Chemie/Chem. Mon. 1991, 122, 853–861. [Google Scholar] [CrossRef]
- Kurihara, T.; Harusawa, S.; Hirai, J.-I.; Yoneda, R. Regiospecific arylation of 1,4-benzoquinone cyanohydrin phosphate: Synthesis of 3-aryl-4-hydroxybenzonitriles. J. Chem. Soc. Perkin Trans. 1 1987, 1771–1776. [Google Scholar] [CrossRef]
- Yao, T.; Yue, D.; Larock, R.C. An Efficient Synthesis of Coumestrol and Coumestans by Iodocyclization and Pd-Catalyzed Intramolecular Lactonization. J. Org. Chem. 2005, 70, 9985–9989. [Google Scholar] [CrossRef]
- James, C.A.; Coelho, A.L.; Gevaert, M.; Forgione, P.; Snieckus, V. Combined Directedortho and Remote Metalation−Suzuki Cross-Coupling Strategies. Efficient Synthesis of Heteroaryl-Fused Benzopyranones from BiarylO-Carbamates. J. Org. Chem. 2009, 74, 4094–4103. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-P.; Pradiuldi, S.V.; Hong, F.-E. Synthesis of coumarin derivatives by palladium complex catalyzed intramolecular Heck reaction: Preparation of a 1,2-cyclobutadiene-substituted CpCoCb diphosphine chelated palladium complex. Inorg. Chem. Commun. 2009, 12, 596–598. [Google Scholar] [CrossRef]
- Irgashev, R.A.; Karmatsky, A.A.; Slepukhin, P.A.; Rusinov, G.L.; Charushin, V.N. A convenient approach to the design and synthesis of indolo[3,2-c]coumarins via the microwave-assisted Cadogan reaction. Tetrahedron Lett. 2013, 54, 5734–5738. [Google Scholar] [CrossRef]
- Wu, J.; Lan, J.; Guo, S.; You, J. Pd-Catalyzed C–H Carbonylation of (Hetero)arenes with Formates and Intramolecular Dehydrogenative Coupling: A Shortcut to Indolo[3,2-c]coumarins. Org. Lett. 2014, 16, 5862–5865. [Google Scholar] [CrossRef]
- Cheng, C.; Chen, W.-W.; Xu, B.; Xu, M.-H. Intramolecular cross dehydrogenative coupling of 4-substituted coumarins: Rapid and efficient access to coumestans and indole[3,2-c]coumarins. Org. Chem. Front. 2016, 3, 1111–1115. [Google Scholar] [CrossRef]
- Cheng, C.; Chen, W.-W.; Xu, B.; Xu, M.-H. Access to Indole-Fused Polyheterocycles via Pd-Catalyzed Base-Free Intramolecular Cross Dehydrogenative Coupling. J. Org. Chem. 2016, 81, 11501–11507. [Google Scholar] [CrossRef] [PubMed]
- Balalas, T.; Abdul-Sada, A.; Hadjipavlou-Litina, D.J.; Litinas, K.E. Pd-Catalyzed Efficient Synthesis of Azacoumestans Via Intramolecular Cross Coupling of 4-(Arylamino)coumarins in the Presence of Copper Acetate under Microwaves. Synthesis 2017, 49, 2575–2583. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, G.; Jiang, X. Ligand-Controlled Palladium(II)-Catalyzed Regiodivergent Carbonylation of Alkynes: Syntheses of Indolo[3,2-c]coumarins and Benzofuro[3,2-c]quinolinones. Angew. Chem. Int. Ed. 2018, 57, 9028–9032. [Google Scholar] [CrossRef]
- Gu, C.-X.; Chen, W.-W.; Xu, B.; Xu, M.-H. Synthesis of indolo[2,3-c]coumarins and indolo[2,3-c]quinolinones via microwave-assisted base-free intramolecular cross dehydrogenative coupling. Tetrahedron 2019, 75, 1605–1611. [Google Scholar] [CrossRef]
- Rusanov, D.A.; Alfadul, S.M.; Portnyagina, E.Y.; Silyanova, E.A.; Kuznetsov, N.A.; Podpovetny, K.E.; Samet, A.V.; Semenov, V.V.; Babak, M.V. Toward “E-Ring-Free” Lamellarin Analogues: Synthesis and Preliminary Biological Evaluation. ChemBioChem 2023, 24, e202300161. [Google Scholar] [CrossRef]
- Das, B.; Dahiya, A.; Chakraborty, N.; Patel, B.K. Synthesis of Chromenopyrroles (Azacoumestans) from Functionalized Enones and Alkyl Isocyanoacetates. Org. Lett. 2023, 25, 5209–5213. [Google Scholar] [CrossRef] [PubMed]









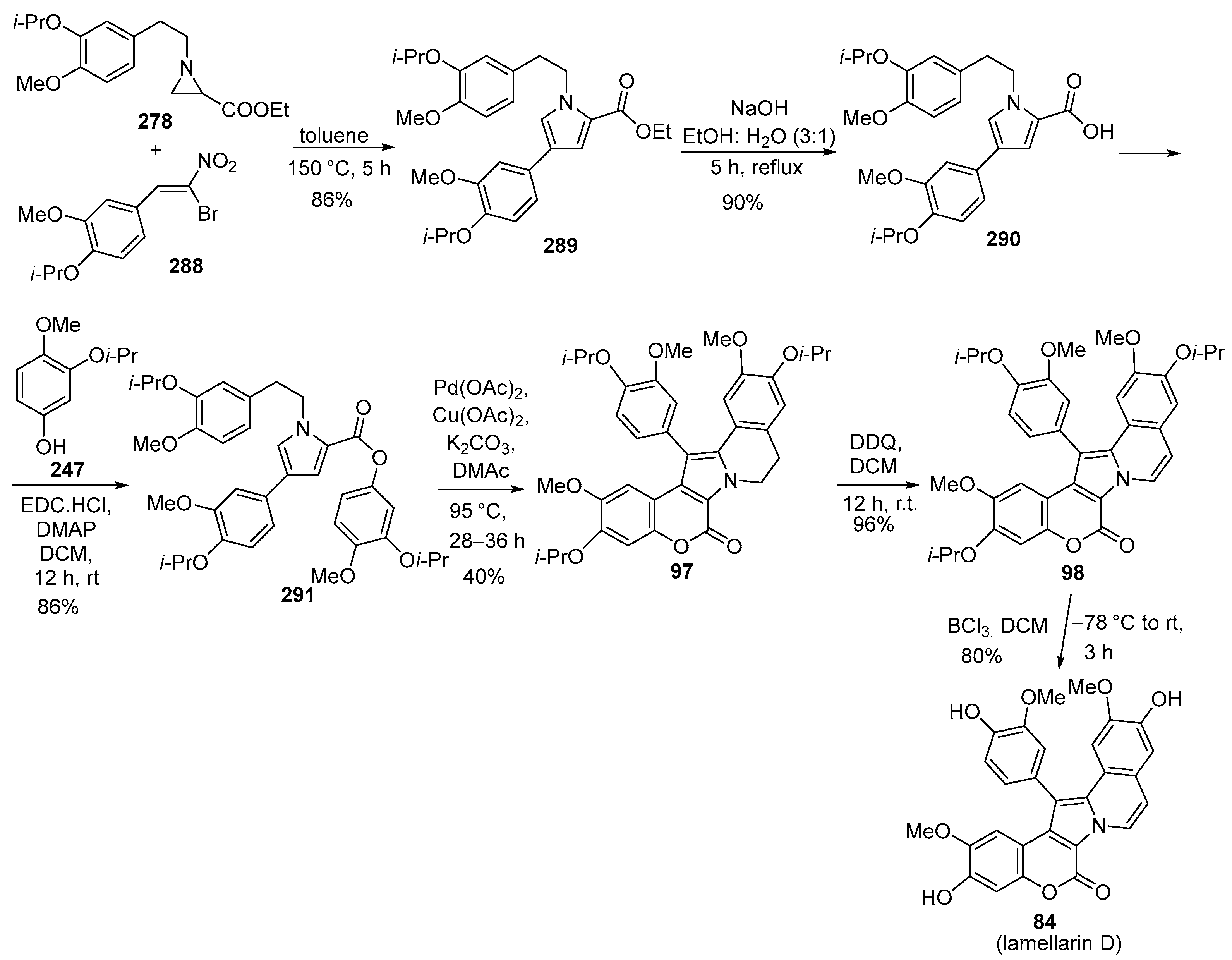
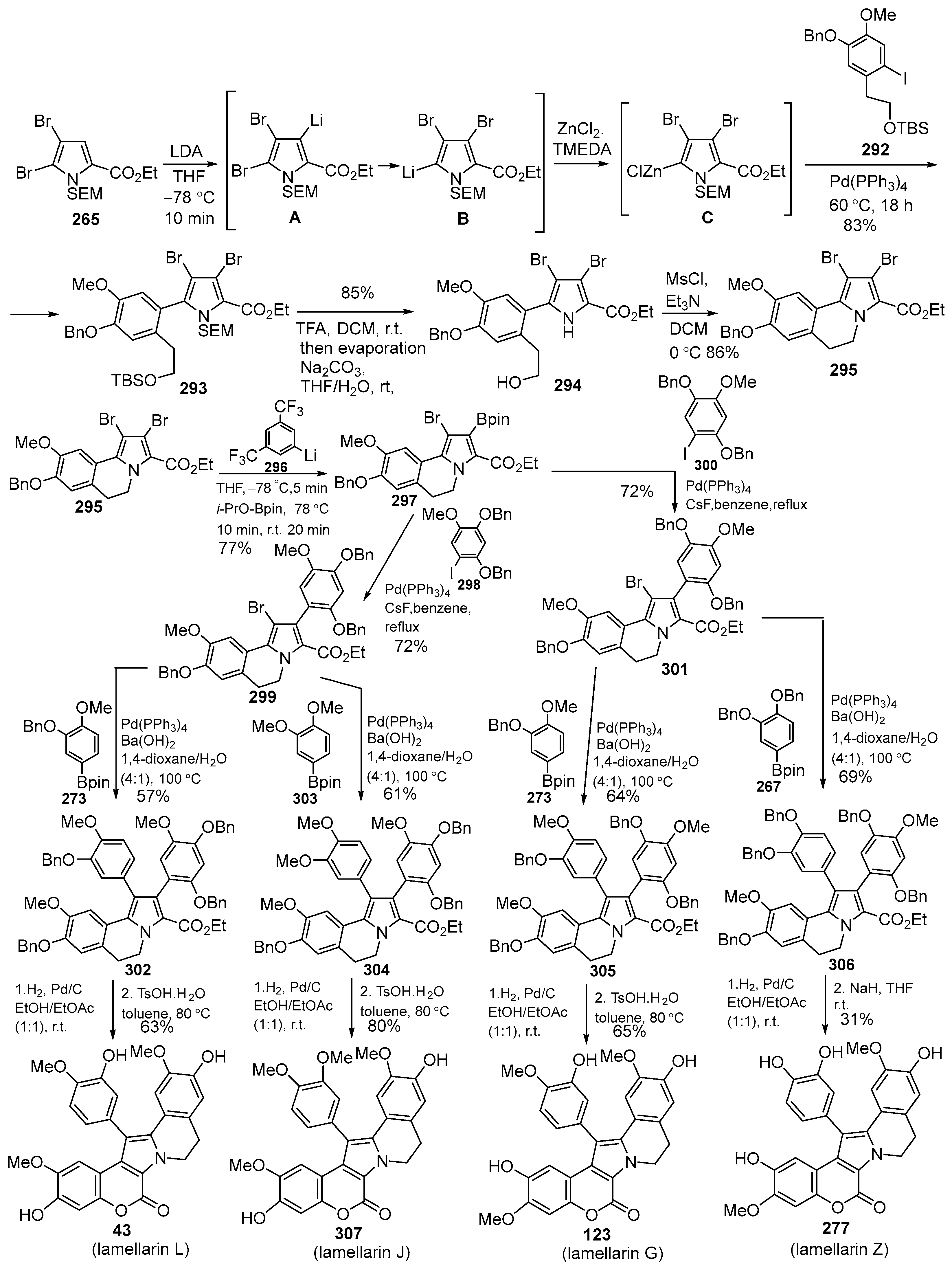


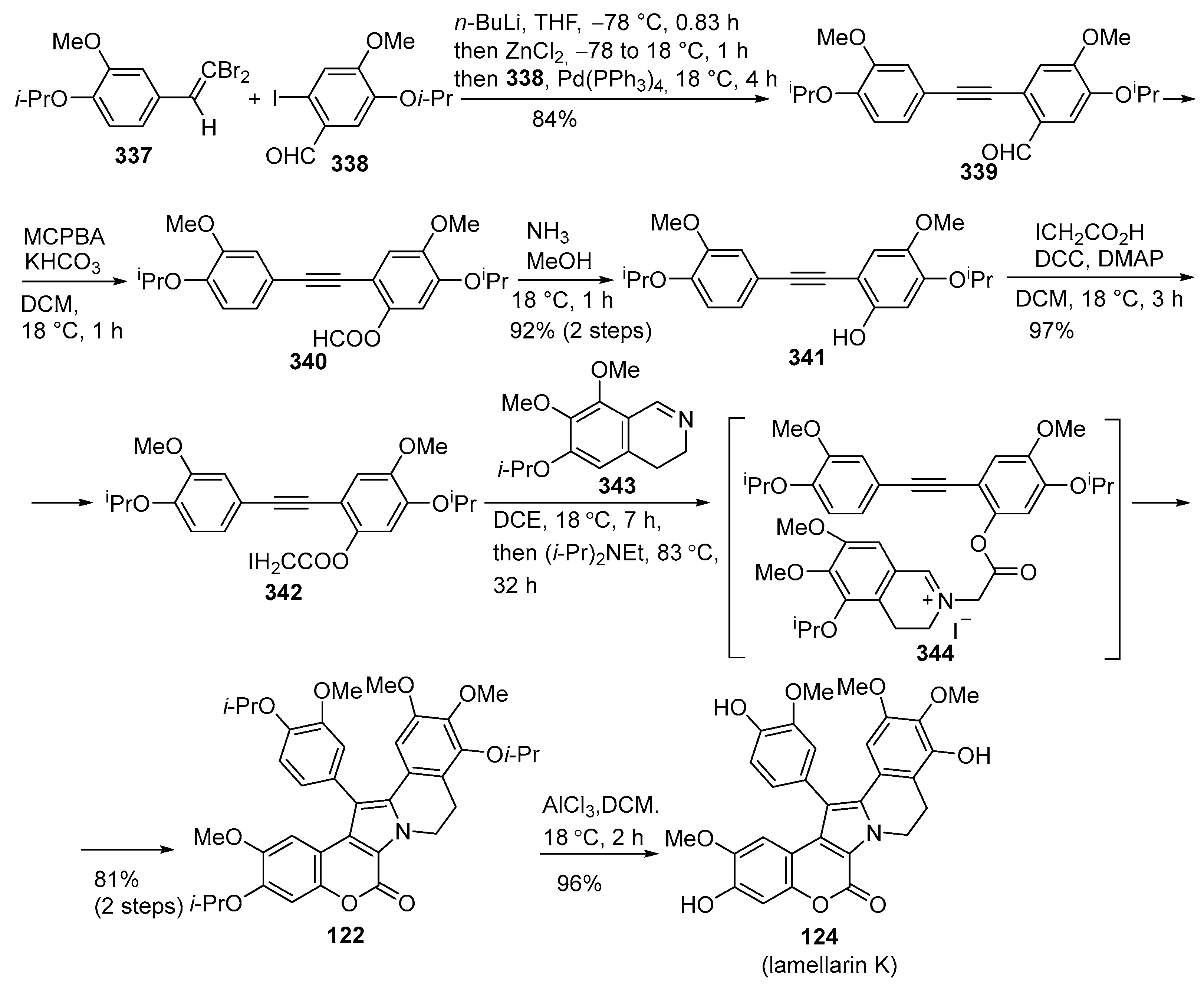
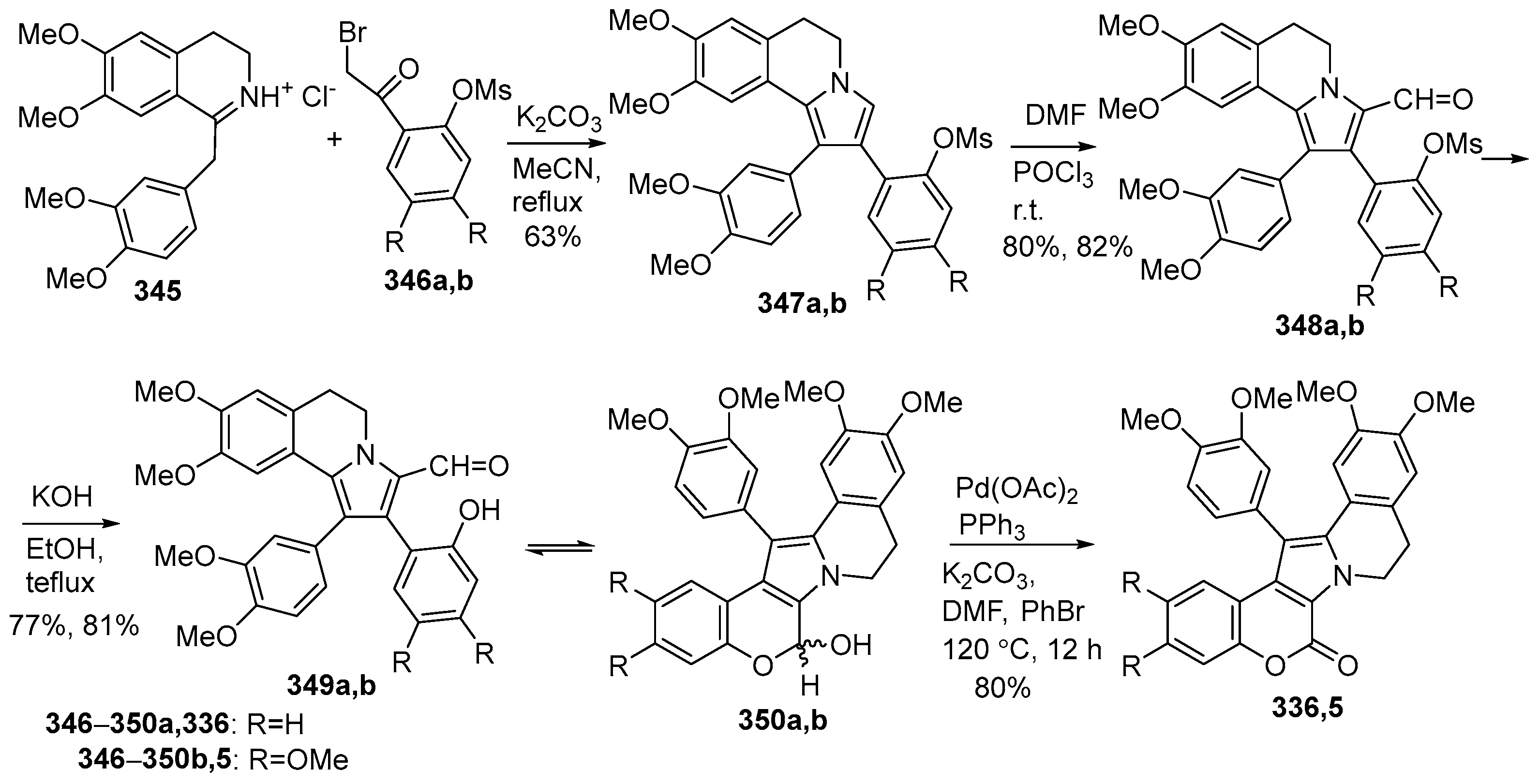





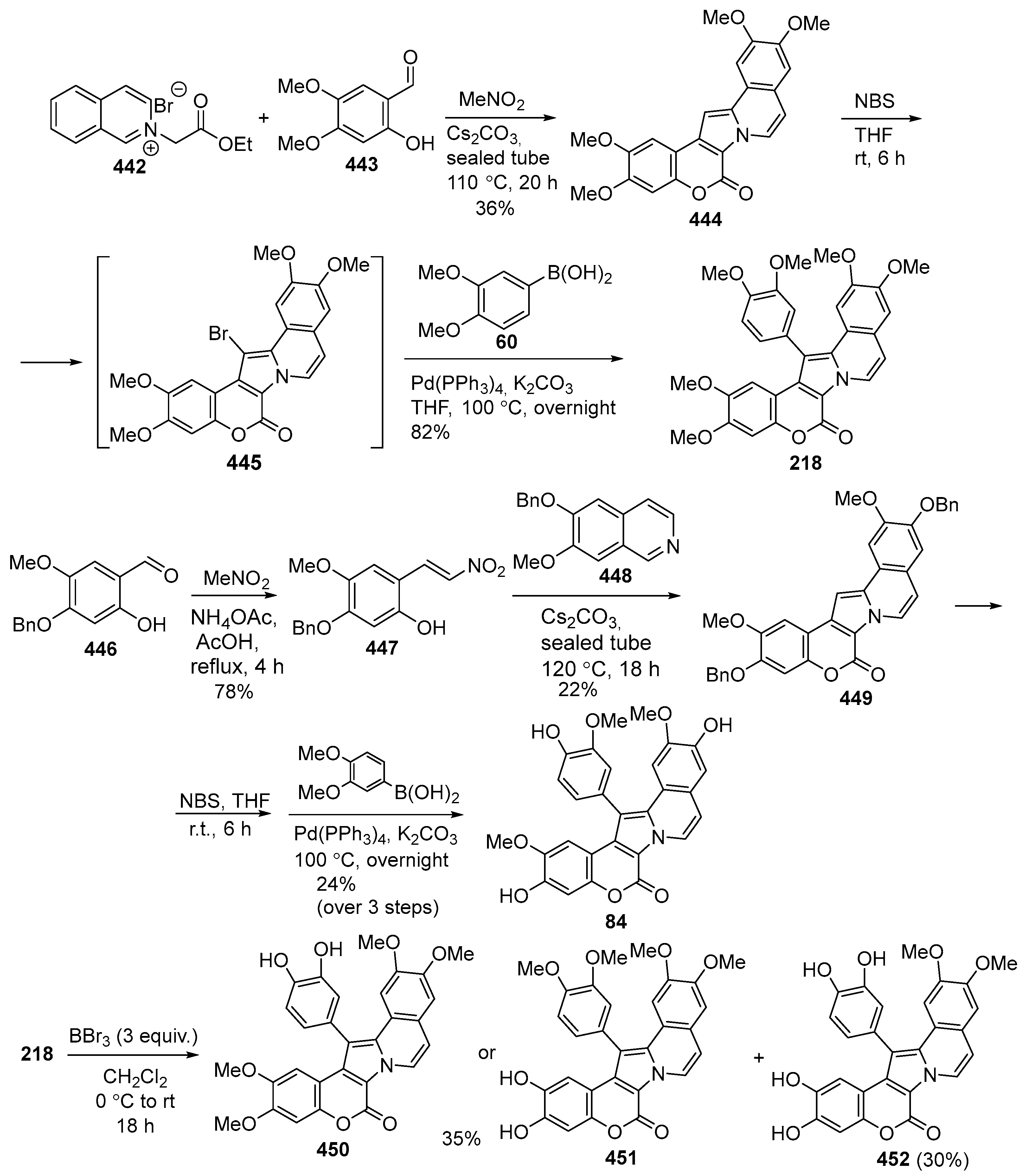
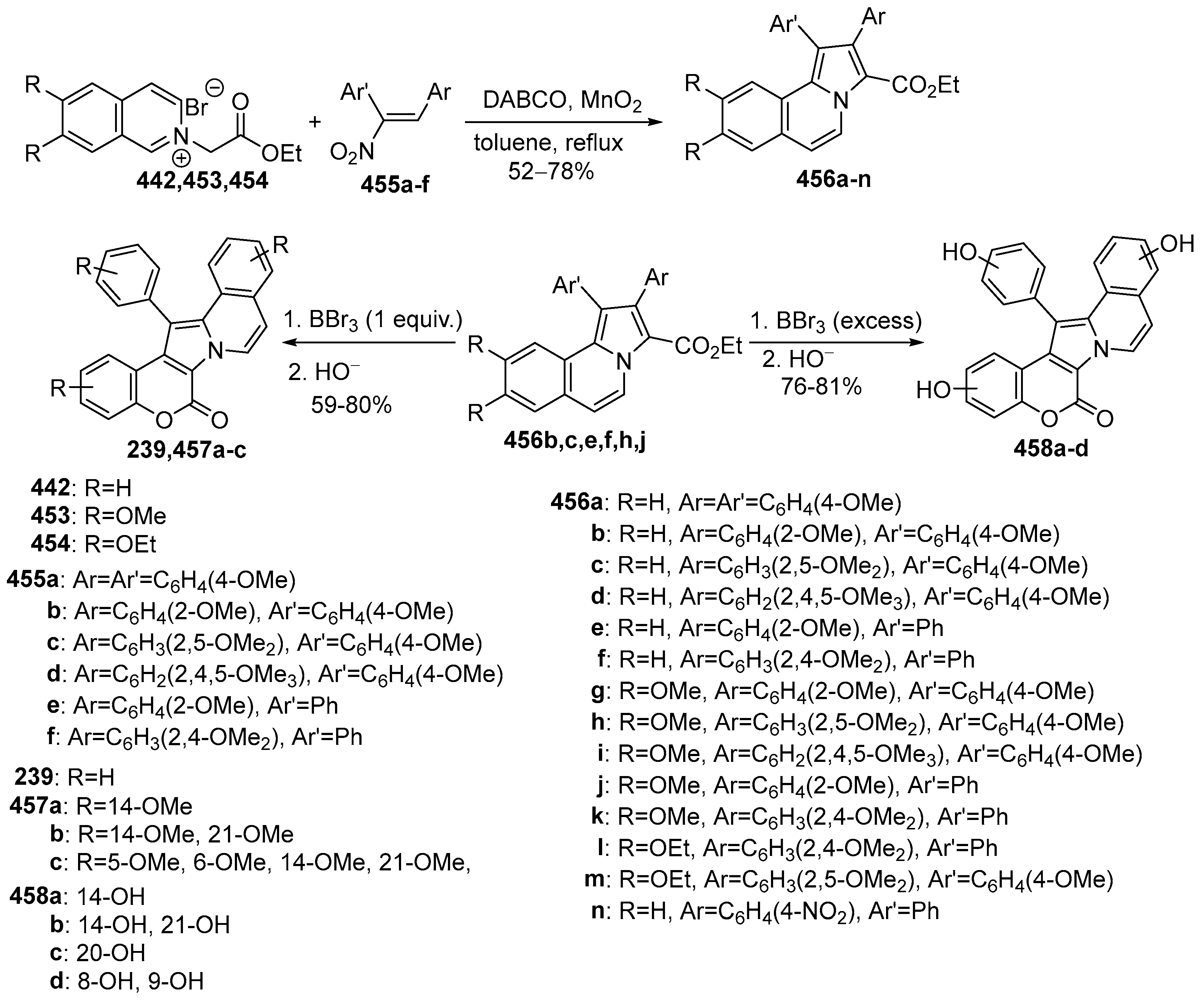


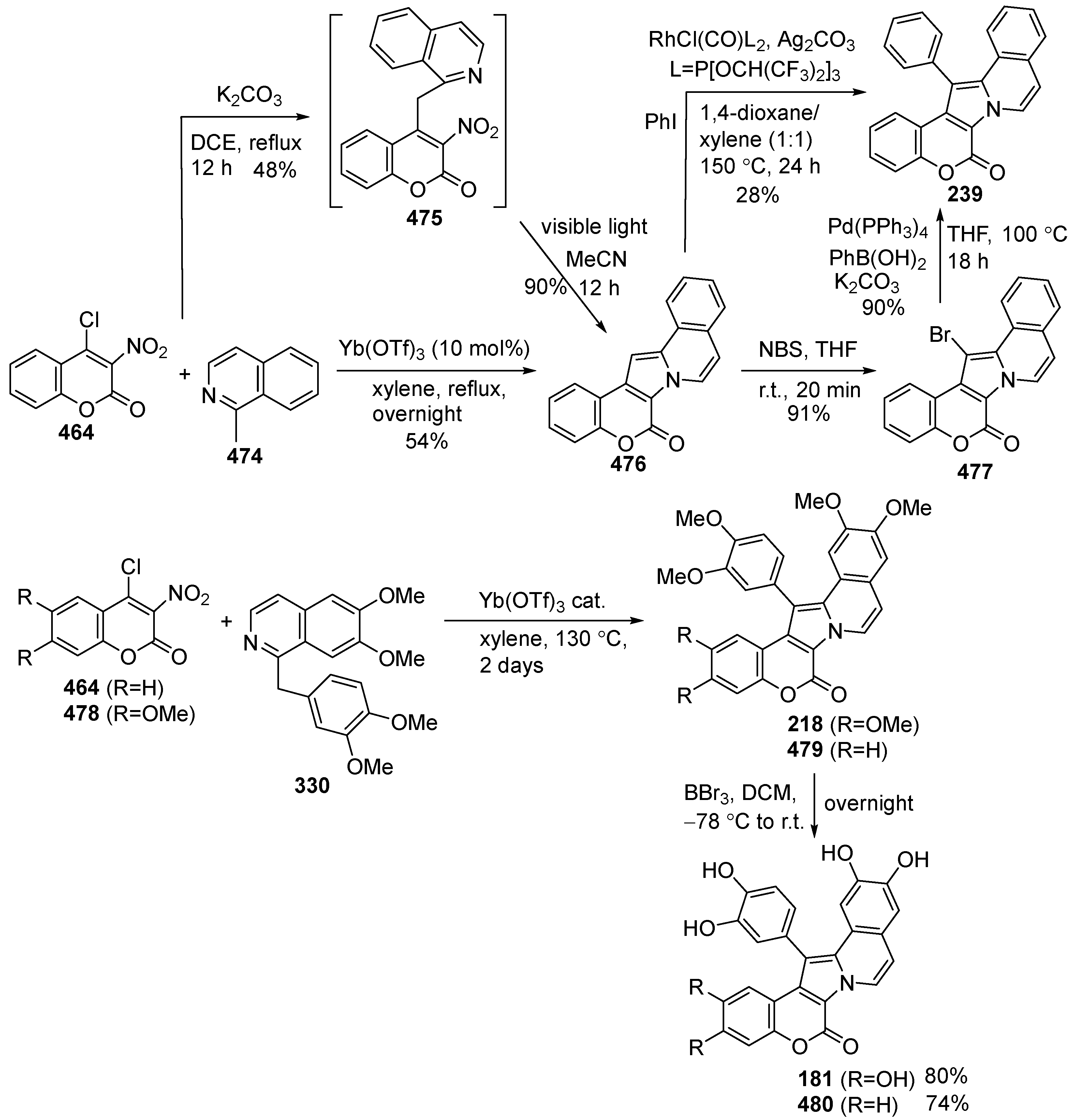
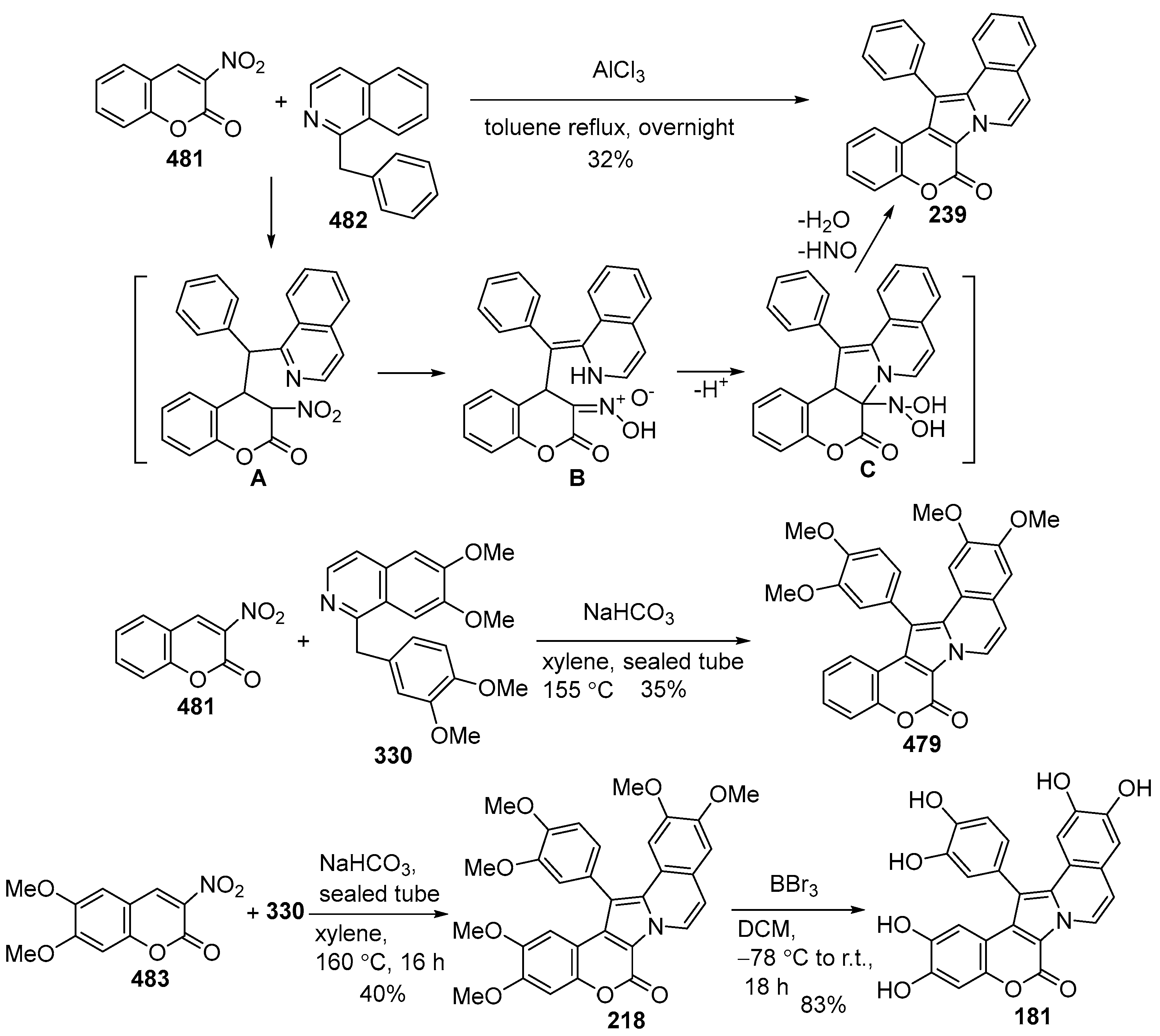















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsiou, V.-P.M.; Antonaki, A.-M.N.; Douka, M.D.; Litinas, K.E. An Overview on the Synthesis of Lamellarins and Related Compounds with Biological Interest. Molecules 2024, 29, 4032. https://doi.org/10.3390/molecules29174032
Mitsiou V-PM, Antonaki A-MN, Douka MD, Litinas KE. An Overview on the Synthesis of Lamellarins and Related Compounds with Biological Interest. Molecules. 2024; 29(17):4032. https://doi.org/10.3390/molecules29174032
Chicago/Turabian StyleMitsiou, Vasiliki-Panagiota M., Anastasia-Maria N. Antonaki, Matina D. Douka, and Konstantinos E. Litinas. 2024. "An Overview on the Synthesis of Lamellarins and Related Compounds with Biological Interest" Molecules 29, no. 17: 4032. https://doi.org/10.3390/molecules29174032