
Orthoflaviviral Inhibitors in Clinical Trials, Preclinical In Vivo Efficacy Targeting NS2B-NS3 and Cellular Antiviral Activity via Competitive Protease Inhibition


Abstract
1. Introduction
2. NS2B-NS3 Functional Analysis
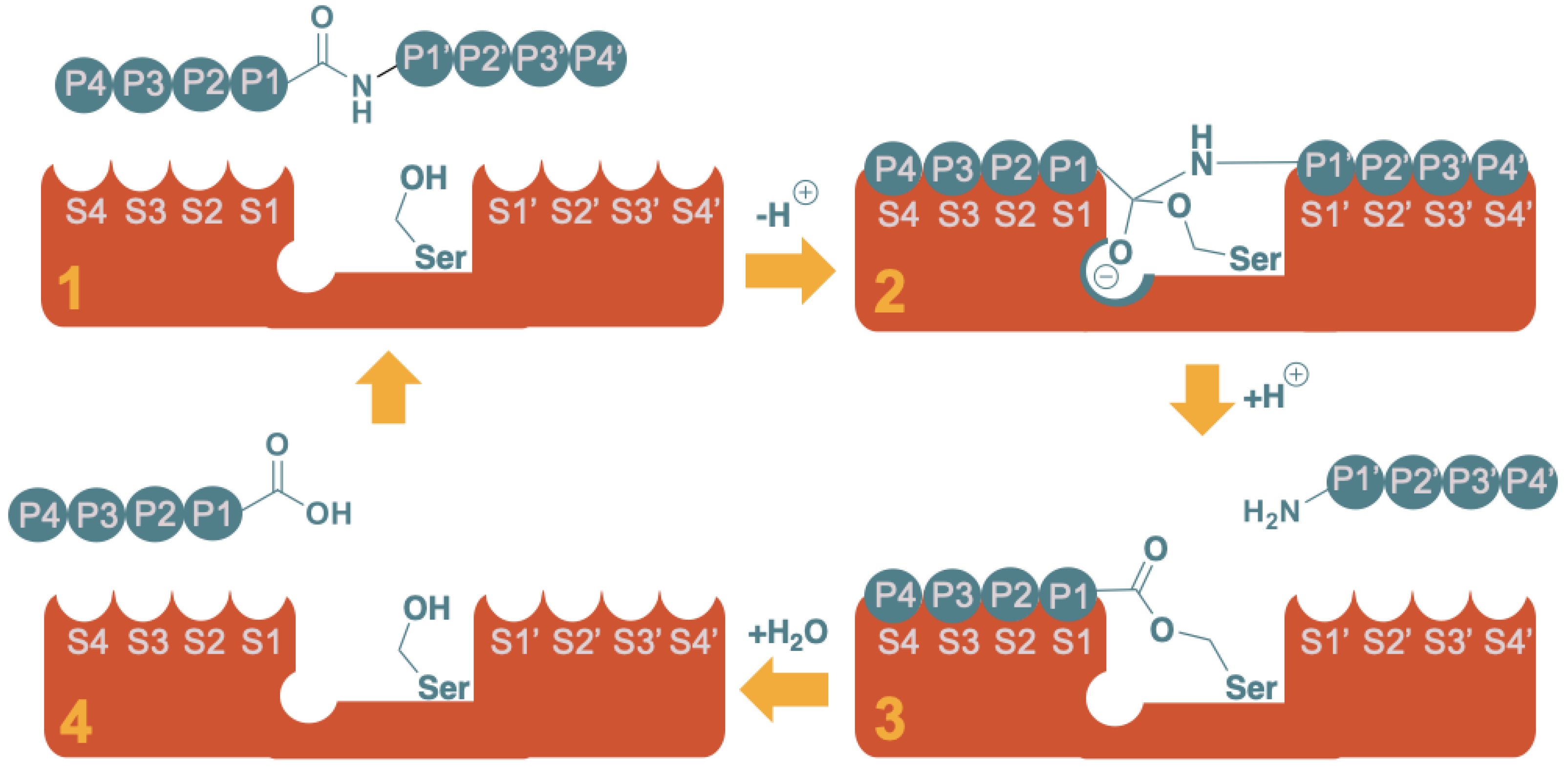
3. Mechanisms of Enzyme Inhibition
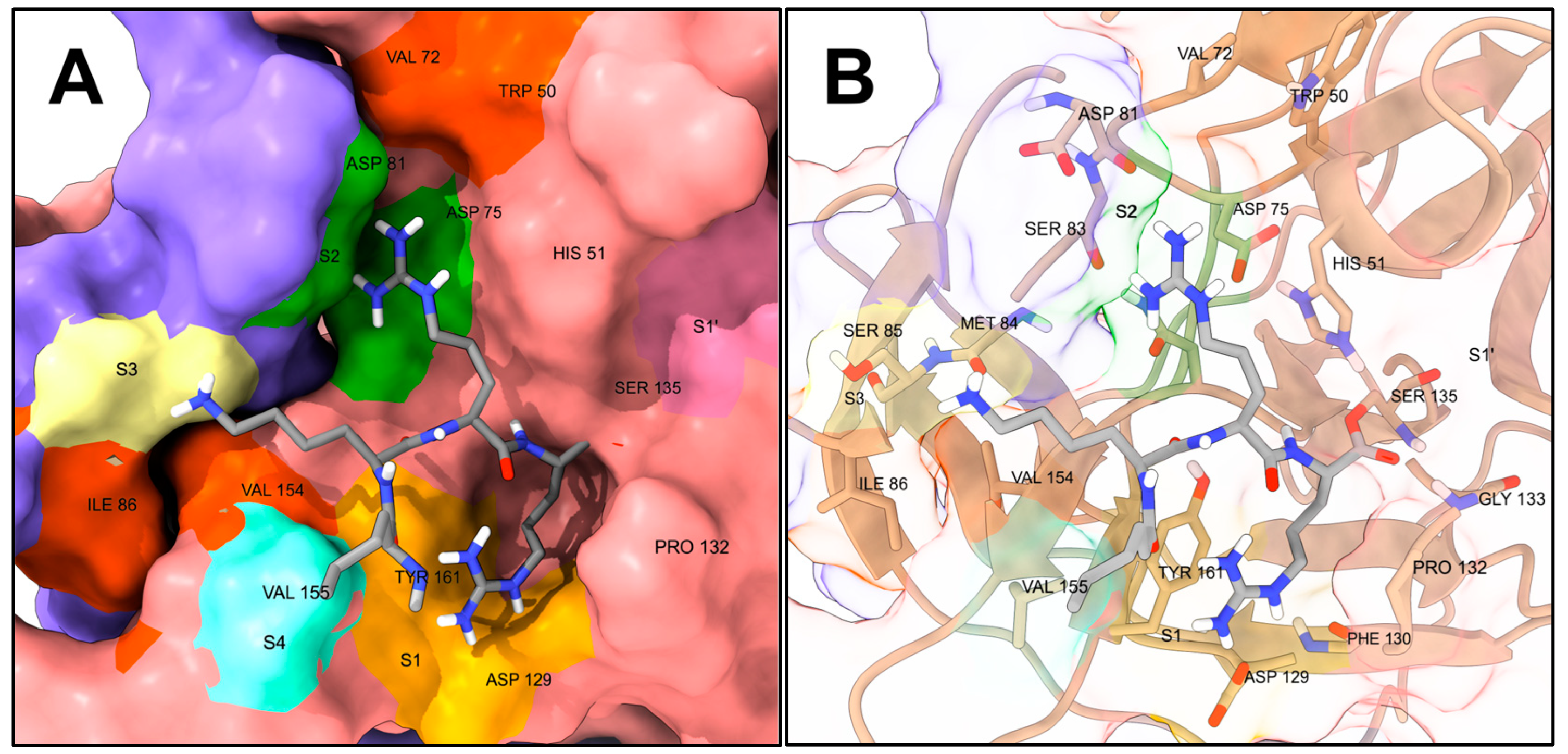
4. NS2B-NS3 Protease Structural Analysis
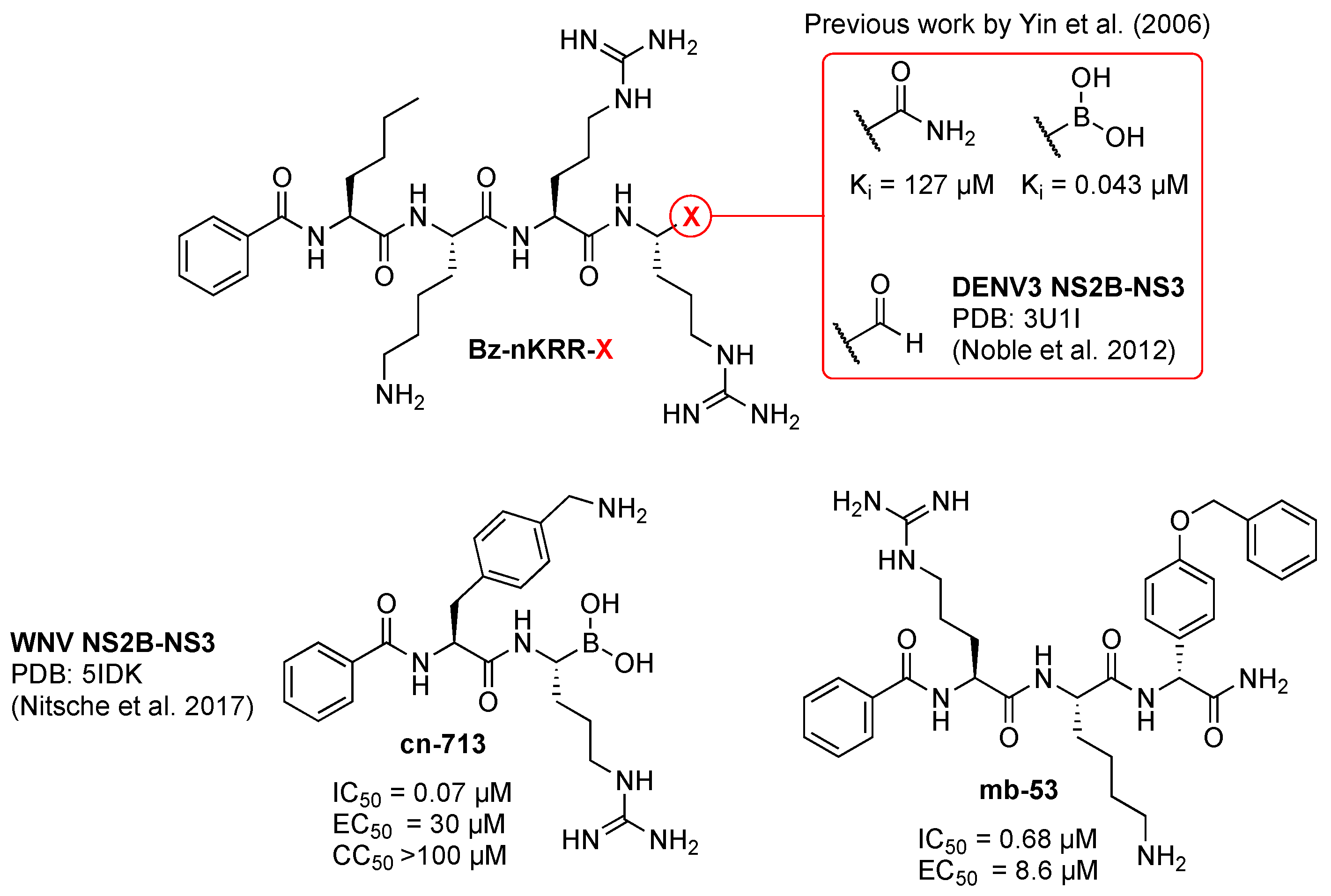
5. NS2B-NS3 Inhibition
6. Small-Molecule Therapeutics in Clinical Trials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preclinical Data | Clinical Data | |||||||
|---|---|---|---|---|---|---|---|---|
| Name | Target | Cellular EC50 Virus (Cell Line) | Peak Viremia Reduction or %SR, Drug Regimen [a], Dosage, Route [b] (Animal Infection model) [c] | Trial Identifier | Phase (Study Type) [d] | Year [e] | Status/Outcome | Ref. |
| JNJ-64281802 | NS3–NS4B | 0.06–1.4 nM DENV1-4 (Multiple) | ≤LLOD, PR, 3 mg/kg, SC (DENV1/2, NHP) ≤LLOD, PR, 2 mg/kg, IP (DENV2 RL, AG129) <1-log, TH (4d p.i.) [f] 60 mg/kg, IP (DENV2 RL, AG129) | NCT05201937 NCT04906980 NCT05048875 NCT05201794 NCT04480736 | I II (wt-TH) II (DHIM-DENV3) II (wt-PR) II (DHIM-DENV1) | 2023 2024 2024 2025 2027 | Safe Terminated [g] Recruiting Recruiting Suspended | [86,87] |
| AT-752 | Viral RNA polymerase | 0.49/0.77 μM [h] DENV2/3 (Huh-7) | ≤1-log, PR, 1000–500 mg/kg, PO (DENV D2Y98P, AG129) | NCT04722627 NCT05366439 NCT05466240 | I I (DHIM) II (wt-TH) | 2021 2023 2023 | Safe Terminated [i] Terminated [i] | [88,89] |
| EYU688 | NS4B | 8–38 nM DENV1-4 (Vero) | 4.3-log, PR, 100 mg/kg, PO (DENV2 RL, AG129) ≤-log, TH (2d p.i.), 30 mg/kg, PO (DENV2 RL, AG129) | NCT06006559 | II (wt-TH) | 2026 | Recruiting | [90] |
| ISLA-101 | Host nuclear import inhibitor, NS5 entry | 1.3–2.4 µM DENV1-4 (Huh-7) | 70% SR, PR, 20 mg/kg, PO (DENV2 S221, AG129) [j] | n.a. | I II (TH and PR) | 2022 2023 [k] | Safe [k] Announced | [91,92] |
| UV-4B | Host ER glucosidases | 2.1–87 µM DENV1-4 (Vero) | 100% SR, PR, 40 mg/kg, PO 90% SR, TH (2d p.i.), 40 mg/kg, PO (DENV2 S221AG129) [l] | NCT02061358 NCT02696291 | I I | 2015 2017 | Safe (1000 mg) Terminated [m] | [93,94] |
| Melatonin | Host anti-inflammatory factors/viral proteins (NS3) | 140–200 µM DENV2 (Huh-7 EA.hy.926, A549, U937) | SR 69%, PR, 500 µg/kg (WNV WN-25, CD1) | NCT05034809 | II (wt-TH) | 2022 | Not yet recruiting | [95,96] |
| Doxycycline | NS2B-NS3 | 40 µM DENV2 (Vero) | n.a. | n.a. n.a. CTRI/2021/09/036661 CTRI/2018/01/011548 | II (wt-TH) II (wt-TH) II (wt-TH) II (wt-TH) | 2015 2022 2023 2019 | Reduction in inflammatory markers n.a. | [97,98,99] |
| Celgosivir | Host ER glucosidases | 0.22–0.65 µM DENV1-4 (BHK-21) | 16.5-fold, PR, 50 mg/kg, PO <1 log, TH (2d p.i.), 50 mg/kg, PO (DENV EDEN2, AG129) | NCT01619969 NCT02569827 | I/II (wt-TH) I/II (wt-TH) | 2013 2019 | No efficacy Withdrawn | [100,101,102] |
| Ivermectin | Host nuclear import inhibitor, NS5 entry | 1.2–1.6 µM DENV1-4 (BHK-21) | n.a. | NCT03432442 NCT02045069 | II (wt-TH) [n] II/III (wt-TH) [n] | 2020 2016 | No efficacy Withdrawn | [103,104] |
| Balapiravir | Viral RNA polymerase | 1.9–11 µM DENV1-4 (Huh-7) | n.a. | NCT01096576 | I (wt-TH) | 2011 | No efficacy | [105] |
| Chloroquine | Virus assembly | 1.7–2.7 µM ZIKV MR766 (Vero) [o] | <LLOD [p], PR, 25 mg/kg, PO 75% [p], TH (1d p.i.), PR, 25 mg/kg, PO 25% [p], TH (2d p.i.), PR, 25 mg/kg, PO (DENV2 NGC, NHP) | NCT00849602 ISRCTN38002730 | I/II (wt-TH) [n] I/II (wt-TH) [n] | 2009 2010 | Unknown No efficacy | [106,107,108,109] |
7. Small-Molecule NS2B-NS3 Protease Inhibitors with In Vivo Efficacy
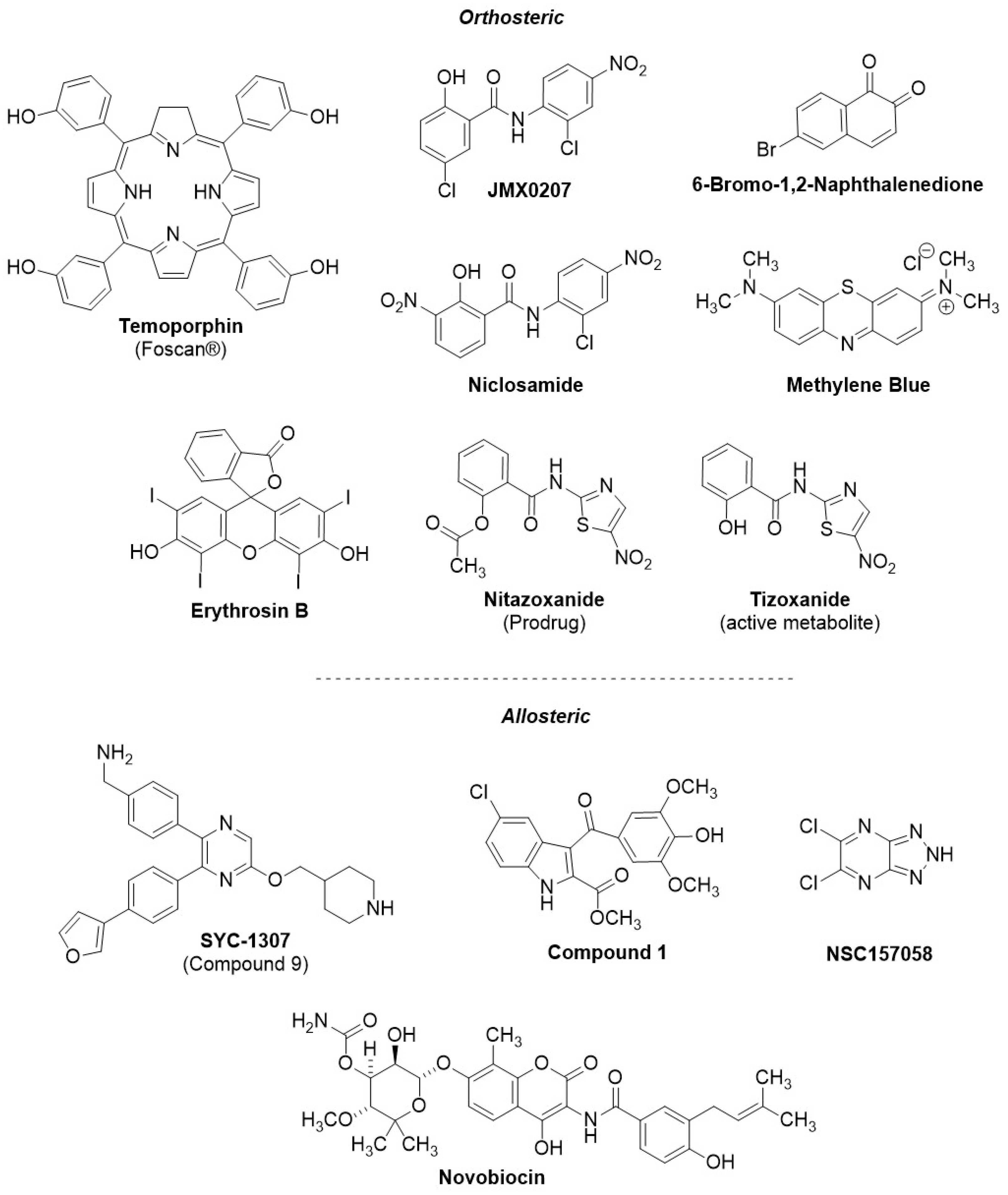
8. Small-Molecule NS2B-NS3 Orthosteric Competitive Inhibitors with Cellular Efficacy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Best, S.M. Flaviviruses. Curr. Biol. 2016, 26, R1258–R1260. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Ronca, S.E.; Ruff, J.C.; Murray, K.O. A 20-year historical review of West Nile virus since its initial emergence in North America: Has West Nile virus become a neglected tropical disease? PLoS Neglected Trop. Dis. 2021, 15, e0009190. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Ko, A.I.; Baud, D. Zika Virus Infection—After the Pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Japanese encephalitis vaccines: WHO position paper-February 2015. Wkly. Epidemiol. Rec. 2015, 90, 69–88. [Google Scholar]
- World Health Organization. Detection and Investigation of Serious Adverse Events Following Yellow Fever Vaccination. Guidance from an Informal Consultation of Experts; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Kubinski, M.; Beicht, J.; Gerlach, T.; Volz, A.; Sutter, G.; Rimmelzwaan, G.F. Tick-borne encephalitis virus: A quest for better vaccines against a virus on the rise. Vaccines 2020, 8, 451. [Google Scholar] [CrossRef]
- Waickman, A.T.; Newell, K.; Endy, T.P.; Thomas, S.J. Biologics for dengue prevention: Up-to-date. Expert Opin. Biol. Ther. 2023, 23, 73–87. [Google Scholar] [CrossRef]
- Dutta, S.K.; Langenburg, T. A Perspective on Current Flavivirus Vaccine Development: A Brief Review. Viruses 2023, 15, 860. [Google Scholar] [CrossRef]
- Malik, S.; Muhammad, K.; Ahsan, O.; Khan, M.; Sah, R.; Waheed, Y. Advances in Zika virus vaccines and therapeutics: A systematic review. Asian Pac. J. Trop. Med. 2024, 17, 97–109. [Google Scholar] [CrossRef]
- Khetarpal, N.; Khanna, I. Dengue Fever: Causes, Complications, and Vaccine Strategies. J. Immunol. Res. 2016, 2016, 6803098. [Google Scholar] [CrossRef]
- Arkin, F. Dengue Vaccine Fiasco Leads to Criminal Charges for Researcher in the Philippines. Available online: https://www.science.org/content/article/dengue-vaccine-fiasco-leads-criminal-charges-researcher-philippines (accessed on 23 August 2024).
- Dengvaxia®. Vaccine Approved for Prevention of Dengue in Europe. Available online: https://www.sanofi.com/en/media-room/press-releases/2018/2018-12-19-12-00-00-1669374 (accessed on 23 August 2024).
- Mallapaty, S. Dengue vaccine poised for roll-out—But concerns linger. Indonesia will be using the jab from next year, although some scientists say the safety data are insufficient. Nature 2022, 611, 434–435. [Google Scholar] [CrossRef]
- Lenharo, M. Dengue is spreading. Can new vaccines and antivirals halt its rise? Nature 2023, 623, 470. [Google Scholar] [CrossRef]
- Takeda Announces Voluntary Withdrawal of, U.S. Biologics License Application (BLA) for Dengue Vaccine Candidate TAK-003. Available online: https://www.takeda.com/newsroom/statements/2023/takeda-announces-voluntary-withdrawal-of-US-biologics-license-application-for-dengue-vaccine-candidate-TAK-003/ (accessed on 23 August 2024).
- Lambach, P.; Orenstein, W.; Wu, J.S. Report from the World Health Organization’s immunization and vaccines related implementation research advisory committee (IVIR-AC) meeting, Geneva, 11–13 September 2023. Vaccine 2023, 42, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Hassan, A.; Farooq, M.; Afzal, S.; Khan, M.A.; Amin, I.; Shahid, M.; Idrees, M.; Shahid, A.A. Dengue Vaccines: Ongoing Challenges and Current Status in the Advancement of Different Candidates. Crit. Rev. Eukaryot. Gene Expr. 2021, 31, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Schroder, K.; White, H.; Fang, N.X.; Stoermer, M.J.; Abbenante, G.; Martin, J.L.; Young, P.R.; Fairlie, D.P. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J. Biol. Chem. 2001, 276, 45762–45771. [Google Scholar] [CrossRef]
- Nitsche, C.; Holloway, S.; Schirmeister, T.; Klein, C.D. Biochemistry and Medicinal Chemistry of the Dengue Virus Protease. Chem. Rev. 2014, 114, 11348–11381. [Google Scholar] [CrossRef]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Yuan, L.; Ross, D.; Johansen, A.; Sands, D.; Stanley, V.; Guemez-Gamboa, A.; Gregor, A.; Evans, T.; et al. Zika virus protease cleavage of host protein septin-2 mediates mitotic defects in neural progenitors. Neuron 2019, 101, 1089–1098. [Google Scholar] [CrossRef]
- Nie, Y.; Deng, D.Q.; Mou, L.; Long, Q.; Chen, J.; Wu, J.H. Dengue Virus 2 NS2B Targets MAVS and IKKε to Evade the Antiviral Innate Immune Response. J. Microbiol. Biotechnol. 2023, 33, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Kakumani, P.K.; Rajgokul, K.S.; Ponia, S.S.; Kaur, I.; Mahanty, S.; Medigeshi, G.R.; Banerjea, A.C.; Chopra, A.P.; Malhotra, P.; Mukherjee, S.K.; et al. Dengue NS3, an RNAi suppressor, modulates the human miRNA pathways through its interacting partner. Biochem. J. 2015, 471, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, C.M.D.; Basavannacharya, C.; Chan, K.W.K.; Chan, S.-A.; Singh, D.; Wei, N.; Phoo, W.W.; Luo, D.; Lescar, J.; Vasudevan, S.G. NS3 helicase from dengue virus specifically recognizes viral RNA sequence to ensure optimal replication. Nucleic Acids Res. 2017, 45, 12904–12920. [Google Scholar] [CrossRef]
- Serman, T.; Chiang, C.; Liu, G.Q.; Sayyad, Z.; Pandey, S.; Volcic, M.; Lee, H.; Muppala, S.; Acharya, D.; Goins, C.; et al. Acetylation of the NS3 helicase by KAT5γ is essential for flavivirus replication. Cell Host Microbe 2023, 31, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C. Proteases from dengue, West Nile and Zika viruses as drug targets. Biophys. Rev. 2019, 11, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Vasudevan, S.G.; Lescar, J. The flavivirus NS2B–NS3 protease–helicase as a target for antiviral drug development. Antivir. Res. 2015, 118, 148–158. [Google Scholar] [CrossRef]
- van den Elsen, K.; Quek, J.P.; Luo, D. Molecular Insights into the Flavivirus Replication Complex. Viruses 2021, 13, 956. [Google Scholar] [CrossRef]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular Localization and Membrane Topology of the Dengue Virus Type 2 Non-structural Protein 4B*. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef]
- Umareddy, I.; Chao, A.; Sampath, A.; Gu, F.; Vasudevan, S.G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 2006, 87 Pt 9, 2605–2614. [Google Scholar] [CrossRef]
- Yap, T.L.; Xu, T.; Chen, Y.L.; Malet, H.; Egloff, M.P.; Canard, B.; Vasudevan, S.G.; Lescar, J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007, 81, 4753–4765. [Google Scholar] [CrossRef]
- Dong, H.; Chang, D.C.; Xie, X.; Toh, Y.X.; Chung, K.Y.; Zou, G.; Lescar, J.; Lim, S.P.; Shi, P.Y. Biochemical and genetic characterization of dengue virus methyltransferase. Virology 2010, 405, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Pryor, M.J.; Rawlinson, S.M.; Butcher, R.E.; Barton, C.L.; Waterhouse, T.A.; Vasudevan, S.G.; Bardin, P.G.; Wright, P.J.; Jans, D.A.; Davidson, A.D. Nuclear localization of dengue virus nonstructural protein 5 through its importin alpha/beta-recognized nuclear localization sequences is integral to viral infection. Traffic 2007, 8, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Maldonado, T.; Moreno-Herrera, A.; Pujadas, G.; Vázquez-Jiménez, L.K.; González-González, A.; Rivera, G. Recent advances in the development of methyltransferase (MTase) inhibitors against (re)emerging arboviruses diseases dengue and Zika. Eur. J. Med. Chem. 2023, 252, 115290. [Google Scholar] [CrossRef]
- Sinha, S.; Singh, K.; Ravi Kumar, Y.S.; Roy, R.; Phadnis, S.; Meena, V.; Bhattacharyya, S.; Verma, B. Dengue virus pathogenesis and host molecular machineries. J. Biomed. Sci. 2024, 31, 43. [Google Scholar] [CrossRef] [PubMed]
- van den Elsen, K.; Chew, B.L.A.; Ho, J.S.; Luo, D. Flavivirus nonstructural proteins and replication complexes as antiviral drug targets. Curr. Opin. Virol. 2023, 59, 101305. [Google Scholar] [CrossRef]
- Rodenhuis-Zybert, I.A.; Wilschut, J.; Smit, J.M. Dengue virus life cycle: Viral and host factors modulating infectivity. Cell. Mol. Life Sci. 2010, 67, 2773–2786. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G. Serine Protease. In Biochemistry, 4th ed.; Wiley: Hoboken, NJ, USA, 2011; p. 525. [Google Scholar]
- Radisky, E.S.; Lee, J.M.; Lu, C.-J.K.; Koshland, D.E. Insights into the serine protease mechanism from atomic resolution structures of trypsin reaction intermediates. Proc. Natl. Acad. Sci. USA 2006, 103, 6835–6840. [Google Scholar]
- Nussinov, R.; Tsai, C.-J. The different ways through which specificity works in orthosteric and allosteric drugs. Curr. Pharm. Des. 2012, 18, 1311. [Google Scholar] [CrossRef]
- Hauske, P.; Ottmann, C.; Meltzer, M.; Ehrmann, M.; Kaiser, M. Allosteric Regulation of Proteases. ChemBioChem 2008, 9, 2920–2928. [Google Scholar] [CrossRef]
- Pesaresi, A. Mixed and non-competitive enzyme inhibition: Underlying mechanisms and mechanistic irrelevance of the formal two-site model. J. Enzym. Inhib. Med. Chem. 2023, 38, 2245168. [Google Scholar] [CrossRef]
- Pedersen, C.N.; Yang, F.Y.; Ita, S.; Xu, Y.B.; Akunuri, R.; Trampari, S.; Neumann, C.M.T.; Desdorf, L.M.; Schiott, B.; Salvino, J.M.; et al. Cryo-EM structure of the dopamine transporter with a novel atypical non-competitive inhibitor bound to the orthosteric site. J. Neurochem. 2024. early view. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.-R.; Kalodimos, C.G. Allosteric inhibition through suppression of transient conformational states. Nat. Chem. Biol. 2013, 9, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Q.; Yin, M.-M.; Song, P.-J.; He, X.-H.; Liu, Y.; Jiang, F.-L. Thermodynamics, Kinetics and Mechanisms of Noncompetitive Allosteric Inhibition of Chymotrypsin by Dihydrolipoic Acid-Coated Gold Nanoclusters. Langmuir 2020, 36, 6447–6457. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Ekka, M.K.; Kaushik, A.; Pandya, V.; Singh, R.P.; Banerjee, S.; Mittal, M.; Singh, V.; Kumaran, S. Substrate-Induced Facilitated Dissociation of the Competitive Inhibitor from the Active Site of O-Acetyl Serine Sulfhydrylase Reveals a Competitive-Allostery Mechanism. Biochemistry 2017, 56, 5011–5025. [Google Scholar] [CrossRef]
- Alphey, M.S.; Pirrie, L.; Torrie, L.S.; Boulkeroua, W.A.; Gardiner, M.; Sarkar, A.; Maringer, M.; Oehlmann, W.; Brenk, R.; Scherman, M.S. Allosteric Competitive Inhibitors of the Glucose-1-phosphate Thymidylyltransferase (RmlA) from Pseudomonas aeruginosa. ACS Chem. Biol. 2013, 8, 387–396. [Google Scholar] [CrossRef]
- Tuley, A.; Fast, W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Noble, C.G.; Seh, C.C.; Chao, A.T.; Shi, P.Y. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 2012, 86, 438–446. [Google Scholar] [CrossRef]
- Agback, T.; Lesovoy, D.; Han, X.; Lomzov, A.; Sun, R.; Sandalova, T.; Orekhov, V.Y.; Achour, A.; Agback, P. Combined NMR and molecular dynamics conformational filter identifies unambiguously dynamic ensembles of Dengue protease NS2B/NS3pro. Commun. Biol. 2023, 6, 1193. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Noble, C.G.; Shi, P.-Y. Structural biology of dengue virus enzymes: Towards rational design of therapeutics. Antivir. Res. 2012, 96, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Samrat, S.K.; Xu, J.; Li, Z.; Zhou, J.; Li, H. Antiviral Agents against Flavivirus Protease: Prospect and Future Direction. Pathogens 2022, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.B.; Behnam, M.A.M.; El Sherif, Y.; Nitsche, C.; Vechi, S.M.; Klein, C.D. Dual inhibitors of the dengue and West Nile virus NS2B–NS3 proteases: Synthesis, biological evaluation and docking studies of novel peptide-hybrids. Bioorg. Med. Chem. 2015, 23, 5748–5755. [Google Scholar] [CrossRef]
- Kühl, N.; Graf, D.; Bock, J.; Behnam, M.A.M.; Leuthold, M.-M.; Klein, C.D. A New Class of Dengue and West Nile Virus Protease Inhibitors with Submicromolar Activity in Reporter Gene DENV-2 Protease and Viral Replication Assays. J. Med. Chem. 2020, 63, 8179–8197. [Google Scholar] [CrossRef]
- Dražić, T.; Kühl, N.; Gottscheber, N.; Hacker, C.N.; Klein, C.D. The spectrum between substrates and inhibitors: Pinpointing the binding mode of dengue protease ligands with modulated basicity and hydrophobicity. Bioorg. Med. Chem. 2021, 48, 116412. [Google Scholar] [CrossRef] [PubMed]
- Kühl, N.; Leuthold, M.M.; Behnam, M.A.M.; Klein, C.D. Beyond Basicity: Discovery of Nonbasic DENV-2 Protease Inhibitors with Potent Activity in Cell Culture. J. Med. Chem. 2021, 64, 4567–4587. [Google Scholar] [CrossRef]
- Nitsche, C.; Schreier, V.N.; Behnam, M.A.M.; Kumar, A.; Bartenschlager, R.; Klein, C.D. Thiazolidinone–Peptide Hybrids as Dengue Virus Protease Inhibitors with Antiviral Activity in Cell Culture. J. Med. Chem. 2013, 56, 8389–8403. [Google Scholar] [CrossRef]
- Nitsche, C.; Zhang, L.; Weigel, L.F.; Schilz, J.; Graf, D.; Bartenschlager, R.; Hilgenfeld, R.; Klein, C.D. Peptide–Boronic Acid Inhibitors of Flaviviral Proteases: Medicinal Chemistry and Structural Biology. J. Med. Chem. 2017, 60, 511–516. [Google Scholar] [CrossRef]
- Nitsche, C.; Klein, C.D. Fluorimetric and HPLC-Based Dengue Virus Protease Assays Using a FRET Substrate. In Antiviral Methods and Protocols; Gong, E.Y., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 221–236. [Google Scholar]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease Inhibitors: Current Status and Future Prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Patel, S.J.; Wang, W.-L.; Wang, G.; Chan, W.-L.; Rao, K.R.R.; Alam, J.; Jeyaraj, D.A.; Ngew, X.; Patel, V.; et al. Peptide inhibitors of dengue virus NS3 protease. Part 1: Warhead. Bioorg. Med. Chem. Lett. 2006, 16, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Patel, S.J.; Wang, W.L.; Chan, W.L.; Ranga Rao, K.R.; Wang, G.; Ngew, X.; Patel, V.; Beer, D.; Knox, J.E.; et al. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 40–43. [Google Scholar] [CrossRef]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Jöst, C.; Nitsche, C.; Scholz, T.; Roux, L.; Klein, C.D. Promiscuity and Selectivity in Covalent Enzyme Inhibition: A Systematic Study of Electrophilic Fragments. J. Med. Chem. 2014, 57, 7590–7599. [Google Scholar] [CrossRef]
- Behnam, M.A.M.; Graf, D.; Bartenschlager, R.; Zlotos, D.P.; Klein, C.D. Discovery of Nanomolar Dengue and West Nile Virus Protease Inhibitors Containing a 4-Benzyloxyphenylglycine Residue. J. Med. Chem. 2015, 58, 9354–9370. [Google Scholar] [CrossRef]
- Kang, C.B.; Keller, T.H.; Luo, D. Zika Virus Protease: An Antiviral Drug Target. Trends Microbiol. 2017, 25, 797–808. [Google Scholar] [CrossRef]
- Behnam, M.A.M.; Klein, C.D. Alternate recognition by dengue protease: Proteolytic and binding assays provide functional evidence beyond an induced-fit. Biochimie 2024, in press. [Google Scholar] [CrossRef]
- Yildiz, M.; Ghosh, S.; Bell, J.A.; Sherman, W.; Hardy, J.A. Allosteric Inhibition of the NS2B-NS3 Protease from Dengue Virus. ACS Chem. Biol. 2013, 8, 2744–2752. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural evidence for regulation and specificity of flaviviral proteases and evolution. Prot. Sci. 2007, 16, 795–806. [Google Scholar] [CrossRef]
- Santos, N.P.; Santos, L.H.; Torquato Quezado de Magalhães, M.; Lei, J.; Hilgenfeld, R.; Salgado Ferreira, R.; Bleicher, L. Characterization of an Allosteric Pocket in Zika Virus NS2B-NS3 Protease. J. Chem. Inf. Model. 2022, 62, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, S.A.; Farhy, C.; Pinto, A.; Huang, C.-T.; Simonetti, N.; Ngono, A.E.; Dewing, A.; Shresta, S.; Pinkerton, A.B.; Cieplak, P.; et al. Characterization of the Zika virus two-component NS2B-NS3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antivir. Res. 2017, 143, 218–229. [Google Scholar] [CrossRef]
- Starvaggi, J.; Previti, S.; Zappalà, M.; Ettari, R. The Inhibition of NS2B/NS3 Protease: A New Therapeutic Opportunity to Treat Dengue and Zika Virus Infection. Int. J. Mol. Sci. 2024, 25, 4376. [Google Scholar] [CrossRef] [PubMed]
- Schul, W.; Liu, W.; Xu, H.Y.; Flamand, M.; Vasudevan, S.G. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J. Infect. Dis. 2007, 195, 665–674. [Google Scholar] [CrossRef]
- Tan, G.K.; Ng, J.K.; Trasti, S.L.; Schul, W.; Yip, G.; Alonso, S. A non mouse-adapted dengue virus strain as a new model of severe dengue infection in AG129 mice. PLoS Neglected Trop. Dis. 2010, 4, e672. [Google Scholar] [CrossRef]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Atkinson, B.; Hall, G.; Watson, R.J.; Bosworth, A.; Bonney, L.C.; Kitchen, S.; Hewson, R. A Susceptible Mouse Model for Zika Virus Infection. PLoS Neglected Trop. Dis. 2016, 10, e0004658. [Google Scholar] [CrossRef]
- Simmons, C.P.; Farrar, J.J.; Nguyen, V.V.C.; Wills, B. Current Concepts: Dengue. N. Engl. J. Med. 2012, 36, 1423–1432. [Google Scholar] [CrossRef]
- Lyke, K.E.; Chua, J.; Koren, M.; Friberg, H.; Gromowski, G.D.; Rapaka, R.R.; Waickman, A.T.; Joshi, S.; Strauss, K.; McCracken, M.K.; et al. Efficacy and immunogenicity following dengue virus-1 human challenge after a tetravalent prime-boost dengue vaccine regimen: An open-label, phase 1 trial. Lancet Infect. Dis. 2024, 24, 896–908. [Google Scholar] [CrossRef]
- Waickman, A.T.; Lu, J.Q.; Fang, H.S.; Waldran, M.J.; Gebo, C.; Currier, J.R.; Ware, L.; Van Wesenbeeck, L.; Verpoorten, N.; Lenz, O.; et al. Evolution of inflammation and immunity in a dengue virus 1 human infection model. Sci. Transl. Med. 2022, 14, abo5019. [Google Scholar] [CrossRef]
- Ackaert, O.; Vanhoutte, F.; Verpoorten, N.; Buelens, A.; Lachau-Durand, S.; Lammens, L.; Hoetelmans, R.; Van Loock, M.; Herrera-Taracena, G. Safety, tolerability, and pharmacokinetics of JNJ-1802, a pan-serotype dengue direct antiviral small molecule, in a phase 1, double-blind, randomized, dose-escalation study in healthy volunteers. Clin. Infect. Dis. 2023, 77, 857–865. [Google Scholar] [CrossRef]
- Goethals, O.; Kaptein, S.J.F.; Kesteleyn, B.; Bonfanti, J.-F.; Van Wesenbeeck, L.; Bardiot, D.; Verschoor, E.J.; Verstrepen, B.E.; Fagrouch, Z.; Putnak, J.R.; et al. Blocking NS3–NS4B interaction inhibits dengue virus in non-human primates. Nature 2023, 615, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Lickliter, J.; Montrond, M.; Ishak, L.; Pietropaolo, K.; James, D.; Belanger, B.; Horga, A.; Hammond, J. First-in-human trial evaluating safety and pharmacokinetics of AT-752, a novel nucleotide prodrug with pan-serotype activity against dengue virus. Antimicrob. Agents Chemother. 2024, 68, e0161523. [Google Scholar] [CrossRef] [PubMed]
- Good, S.S.; Shannon, A.; Lin, K.; Moussa, A.; Julander, J.G.; Colla, P.L.; Collu, G.; Canard, B.; Sommadossi, J.-P. Evaluation of AT-752, a Double Prodrug of a Guanosine Nucleotide Analog with In Vitro and In Vivo Activity against Dengue and Other Flaviviruses. Antimicrob. Agents Chemother. 2021, 65, e00988-21. [Google Scholar] [CrossRef] [PubMed]
- Moquin, S.A.; Simon, O.; Karuna, R.; Lakshminarayana, S.B.; Yokokawa, F.; Wang, F.; Saravanan, C.; Zhang, J.; Day, C.W.; Chan, K.; et al. NITD-688, a pan-serotype inhibitor of the dengue virus NS4B protein, shows favorable pharmacokinetics and efficacy in preclinical animal models. Sci. Transl. Med. 2021, 13, eabb2181. [Google Scholar] [CrossRef]
- Fraser, J.E.; Wang, C.; Chan, K.W.K.; Vasudevan, S.G.; Jans, D.A. Novel dengue virus inhibitor 4-HPR activates ATF4 independent of protein kinase R–like Endoplasmic Reticulum Kinase and elevates levels of eIF2α phosphorylation in virus infected cells. Antivir. Res. 2016, 130, 1–6. [Google Scholar] [CrossRef]
- Fraser, J.E.; Watanabe, S.; Wang, C.; Chan, W.K.K.; Maher, B.; Lopez-Denman, A.; Hick, C.; Wagstaff, K.M.; Mackenzie, J.M.; Sexton, P.M.; et al. A Nuclear Transport Inhibitor That Modulates the Unfolded Protein Response and Provides In Vivo Protection Against Lethal Dengue virus Infection. J. Infect. Dis. 2014, 210, 1780–1791. [Google Scholar] [CrossRef]
- Warfield, K.L.; Plummer, E.M.; Sayce, A.C.; Alonzi, D.S.; Tang, W.; Tyrrell, B.E.; Hill, M.L.; Caputo, A.T.; Killingbeck, S.S.; Beatty, P.R.; et al. Inhibition of endoplasmic reticulum glucosidases is required for in vitro and in vivo dengue antiviral activity by the iminosugar UV-4. Antivir. Res. 2016, 129, 93–98. [Google Scholar] [CrossRef]
- Callahan, M.; Treston, A.M.; Lin, G.; Smith, M.; Kaufman, B.; Khaliq, M.; Evans DeWald, L.; Spurgers, K.; Warfield, K.L.; Lowe, P.; et al. Randomized single oral dose phase 1 study of safety, tolerability, and pharmacokinetics of Iminosugar UV-4 Hydrochloride (UV-4B) in healthy subjects. PLoS Neglected Trop. Dis. 2022, 16, e0010636. [Google Scholar] [CrossRef]
- Morchang, A.; Malakar, S.; Poonudom, K.; Noisakran, S.; Yenchitsomanus, P.-t.; Limjindaporn, T. Melatonin Inhibits Dengue Virus Infection via the Sirtuin 1-Mediated Interferon Pathway. Viruses 2021, 13, 659. [Google Scholar] [CrossRef]
- Bennathan, D.; Maestroni, G.J.M.; Lustig, S.; Conti, A. Protective Effects of Melatonin in Mice Infected with Encephalitis Viruses. Arch. Virol. 1995, 140, 223–230. [Google Scholar] [CrossRef]
- Chong Teoh, T.; Al-Harbi, S.J.; Abdulrahman, A.Y.; Rothan, H.A. Doxycycline Interferes with Zika Virus Serine Protease and Inhibits Virus Replication in Human Skin Fibroblasts. Molecules 2021, 26, 4321. [Google Scholar] [CrossRef]
- Fredeking, T.M.; Zavala-Castro, J.E.; González-Martínez, P.; Moguel-Rodríguez, W.; Sanchez, E.C.; Foster, M.J.; Diaz-Quijano, F.A. Dengue Patients Treated with Doxycycline Showed Lower Mortality Associated to a Reduction in IL-6 and TNF Levels. Recent Pat. Anti-Infect. Drug Discov. 2015, 10, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Kamboj, K.; Pannu, A.K.; Yadav, A.K.; Bhatia, M.; Saroch, A. Role of doxycycline in the treatment of dengue infection: An open-label, randomized, controlled, pilot trial. Asian Pac. J. Trop. Med. 2024, 17, 160–165. [Google Scholar] [CrossRef]
- Rathore, A.P.; Paradkar, P.N.; Watanabe, S.; Tan, K.H.; Sung, C.; Connolly, J.E.; Low, J.; Ooi, E.E.; Vasudevan, S.G. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antivir. Res. 2011, 92, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Chan, K.W.; Dow, G.; Ooi, E.E.; Low, J.G.; Vasudevan, S.G. Optimizing celgosivir therapy in mouse models of dengue virus infection of serotypes 1 and 2: The search for a window for potential therapeutic efficacy. Antivir. Res. 2016, 127, 10–19. [Google Scholar] [CrossRef]
- Low, J.G.; Sung, C.; Wijaya, L.; Wei, Y.; Rathore, A.P.S.; Watanabe, S.; Tan, B.H.; Toh, L.; Chua, L.T.; Hou, Y.; et al. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): A phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect. Dis. 2014, 14, 706–715. [Google Scholar] [CrossRef]
- Tay, M.Y.; Fraser, J.E.; Chan, W.K.; Moreland, N.J.; Rathore, A.P.; Wang, C.; Vasudevan, S.G.; Jans, D.A. Nuclear localization of dengue virus (DENV) 1-4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antivir. Res. 2013, 99, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Suputtamongkol, Y.; Avirutnan, P.; Mairiang, D.; Angkasekwinai, N.; Niwattayakul, K.; Yamasmith, E.; Saleh-Arong, F.A.; Songjaeng, A.; Prommool, T.; Tangthawornchaikul, N.; et al. Ivermectin Accelerates Circulating Nonstructural Protein 1 (NS1) Clearance in Adult Dengue Patients: A Combined Phase 2/3 Randomized Double-blinded Placebo Controlled Trial. Clin. Infect. Dis. 2021, 72, e586–e593. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Tran, C.N.; Phung, L.K.; Duong, K.T.; Huynh Hle, A.; Farrar, J.; Nguyen, Q.T.; Tran, H.T.; Nguyen, C.V.; Merson, L.; et al. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J. Infect. Dis. 2013, 207, 1442–1450. [Google Scholar] [CrossRef]
- Farias, K.J.; Machado, P.R.; de Almeida Junior, R.F.; de Aquino, A.A.; da Fonseca, B.A. Chloroquine interferes with dengue-2 virus replication in U937 cells. Microbiol. Immunol. 2014, 58, 318–326. [Google Scholar] [CrossRef]
- Tricou, V.; Minh, N.N.; Van, T.P.; Lee, S.J.; Farrar, J.; Wills, B.; Tran, H.T.; Simmons, C.P. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Neglected Trop. Dis. 2010, 4, e785. [Google Scholar] [CrossRef]
- Farias, K.J.; Machado, P.R.; Muniz, J.A.; Imbeloni, A.A.; da Fonseca, B.A. Antiviral activity of chloroquine against dengue virus type 2 replication in Aotus monkeys. Viral Immunol. 2015, 28, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.-Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef]
- Pitts, J.D.; Li, P.C.; de Wispelaere, M.; Yang, P.L. Antiviral activity of N-(4-hydroxyphenyl) retinamide (4-HPR) against Zika virus. Antivir. Res. 2017, 147, 124–130. [Google Scholar] [CrossRef]
- Wang, C.; Yang, S.N.Y.; Smith, K.; Forwood, J.K.; Jans, D.A. Nuclear import inhibitor N-(4-hydroxyphenyl) retinamide targets Zika virus (ZIKV) nonstructural protein 5 to inhibit ZIKV infection. Biochem. Biophys. Res. Commun. 2017, 493, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Janssen Announces Promising Antiviral Activity against Dengue in a Phase 2a Human Challenge Model. Available online: https://www.jnj.com/janssen-announces-promising-antiviral-activity-against-dengue-in-a-phase-2a-human-challenge-model (accessed on 28 November 2023).
- Kesteleyn, B.; Bonfanti, J.-F.; Bardiot, D.; De Boeck, B.; Goethals, O.; Kaptein, S.J.F.; Stoops, B.; Coesemans, E.; Fortin, J.; Muller, P.; et al. Discovery of JNJ-1802, a First-in-Class Pan-Serotype Dengue Virus NS4B Inhibitor. J. Med. Chem. 2024, 67, 4063–4082. [Google Scholar] [CrossRef]
- McCormack, C.P.; Goethals, O.; Goeyvaerts, N.; Woot de Trixhe, X.D.; Geluykens, P.; Borrenberghs, D.; Ferguson, N.M.; Ackaert, O.; Dorigatti, I. Modelling the impact of JNJ-1802, a first-in-class dengue inhibitor blocking the NS3-NS4B interaction, on in-vitro DENV-2 dynamics. PLoS Comput. Biol. 2023, 19, e1011662. [Google Scholar] [CrossRef] [PubMed]
- Feracci, M.; Eydoux, C.; Fattorini, V.; Lo Bello, L.; Gauffre, P.; Selisko, B.; Sutto-Ortiz, P.; Shannon, A.; Xia, H.; Shi, P.-Y.; et al. AT-752 targets multiple sites and activities on the Dengue virus replication enzyme NS5. Antivir. Res. 2023, 212, 105574. [Google Scholar] [CrossRef]
- ASX Announcement. Island Provides ISLA-101 Clinical Program Update. Available online: https://www.islandpharmaceuticals.com/site/pdf/67d20e62-2ff1-4788-b2b9-991ca9daed72/ISLA101-Clinical-Program-Update.pdf (accessed on 23 August 2024).
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin--a pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef]
- Paemanee, A.; Hitakarun, A.; Roytrakul, S.; Smith, D.R. Screening of melatonin, α-tocopherol, folic acid, acetyl-L-carnitine and resveratrol for anti-dengue 2 virus activity. BMC Res. Notes 2018, 11, 307. [Google Scholar] [CrossRef]
- Wongchitrat, P.; Yasawong, M.; Jumpathong, W.; Chanmanee, T.; Samutpong, A.; Dangsakul, W.; Govitrapong, P.; Reiter, R.J.; Puthavathana, P. Melatonin inhibits Zika virus replication in Vero epithelial cells and SK-N-SH human neuroblastoma cells. Melatonin Res. 2022, 5, 171–185. [Google Scholar] [CrossRef]
- Moon, J.H.; Hong, J.-M.; Seol, J.W.; Park, B.Y.; Eo, S.K.; Park, S.Y. Melatonin inhibits Japanese encephalitis virus replication and neurotoxicity via calcineurin- autophagy pathways. BMC Neurosci. 2023, 24, 59. [Google Scholar] [CrossRef]
- Rothan, H.A.; Mohamed, Z.; Paydar, M.; Abd Rahman, N.; Yusof, R. Inhibitory effect of doxycycline against dengue virus replication in vitro. Arch. Virol. 2014, 159, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Pambhar, V.; Mathur, N.; Mehta, A.; Mathur, M.; Kumawat, D.C.; Mangalia, R.; Verma, A.; Patyal, A. Effect of doxycycline and doxycycline with carica papaya on thrombocytopenia and leucopenia in acute dengue fever patients. J. Fam. Med. Prim. Care 2022, 11, 3270–3275. [Google Scholar]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef]
- Denolly, S.; Guo, H.B.; Martens, M.; Płaszczyca, A.; Scaturro, P.; Prasad, V.; Kongmanas, K.; Punyadee, N.; Songjaeng, A.; Mairiang, D.; et al. Dengue virus NS1 secretion is regulated via importin-subunit β1 controlling expression of the chaperone GRp78 and targeted by the clinical drug ivermectin. mBio 2023, 14, e01441-23. [Google Scholar] [CrossRef]
- Palacios-Rápalo, S.N.; Farfan-Morales, C.N.; Cordero-Rivera, C.D.; De Jesús-González, L.A.; Reyes-Ruiz, J.M.; Meraz-Ríos, M.A.; Del Ángel, R.M. An ivermectin—Atorvastatin combination impairs nuclear transport inhibiting dengue infection in vitro and in vivo. iScience 2023, 26, 108294. [Google Scholar] [CrossRef]
- Osuna-Ramos, J.; Farfan-Morales, C.; Cordero-Rivera, C.D.; De Jesús-González, L.A.; Reyes-Ruiz, J.M.; Hurtado-Monzón, A.M.; Palacios-Rápalo, S.N.; Ricardo Jiménez-Camacho, R.; Meraz-Ríos, M.A.; Del Ángel, R.M. Cholesterol-Lowering Drugs as Potential Antivirals: A Repurposing Approach against Flavivirus Infections. Viruses 2023, 15, 1465. [Google Scholar] [CrossRef]
- Li, Z.; Brecher, M.; Deng, Y.-Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef]
- Li, Z.; Xu, J.; Lang, Y.; Fan, X.; Kuo, L.; D’Brant, L.; Hu, S.; Samrat, S.K.; Trudeau, N.; Tharappel, A.M.; et al. JMX0207, a Niclosamide Derivative with Improved Pharmacokinetics, Suppresses Zika Virus Infection Both In Vitro and In Vivo. ACS Infect. Dis. 2020, 6, 2616–2628. [Google Scholar] [CrossRef]
- Xu, J.; Shi, P.-Y.; Li, H.; Zhou, J. Broad Spectrum Antiviral Agent Niclosamide and Its Therapeutic Potential. ACS Infect. Dis. 2020, 6, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.M.; Boorgu, D.; Levin, M.; Kaplan, D.L. Niclosamide rescues microcephaly in a humanized in vivo model of Zika infection using human induced neural stem cells. Biol. Open 2018, 7, bio031807. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.C.; HuangFu, W.C.; Tsai, T.T.; Ho, M.R.; Jhan, M.K.; Shen, T.J.; Tseng, P.C.; Wang, Y.T.; Lin, C.F. The antiparasitic drug niclosamide inhibits dengue virus infection by interfering with endosomal acidification independent of mTOR. PLoS Neglected Trop. Dis. 2018, 12, e0006715. [Google Scholar] [CrossRef]
- Li, Z.; Lang, Y.; Sakamuru, S.; Samrat, S.; Trudeau, N.; Kuo, L.; Rugenstein, N.; Tharappel, A.; D’Brant, L.; Koetzner, C.A.; et al. Methylene blue is a potent and broad-spectrum inhibitor against Zika virus in vitro and in vivo. Emerg. Microbes Infect. 2020, 9, 2404–2416. [Google Scholar] [CrossRef]
- Li, Z.; Sakamuru, S.; Huang, R.; Brecher, M.; Koetzner, C.A.; Zhang, J.; Chen, H.; Qin, C.F.; Zhang, Q.Y.; Zhou, J.; et al. Erythrosin B is a potent and broad-spectrum orthosteric inhibitor of the flavivirus NS2B-NS3 protease. Antivir. Res. 2018, 150, 217–225. [Google Scholar] [CrossRef]
- Li, Z.; Xu, J.; Lang, Y.; Wu, X.; Hu, S.; Samrat, S.K.; Tharappel, A.M.; Kuo, L.; Butler, D.; Song, Y.; et al. In vitro and in vivo characterization of erythrosin B and derivatives against Zika virus. Acta Pharm. Sin. B 2022, 12, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Yuan, H.; Rao, J.; Zou, J.; Yang, K.; Peng, G.; Cao, S.; Chen, H.; Song, Y. Identification of a small compound that specifically inhibits Zika virus in vitro and in vivo by targeting the NS2B-NS3 protease. Antivir. Res. 2022, 199, 105255. [Google Scholar] [CrossRef]
- Shi, Z.; Wei, J.; Deng, X.; Li, S.; Qiu, Y.; Shao, D.; Li, B.; Zhang, K.; Xue, F.; Wang, X.; et al. Nitazoxanide inhibits the replication of Japanese encephalitis virus in cultured cells and in a mouse model. Virol. J. 2014, 11, 10. [Google Scholar] [CrossRef]
- Rossignol, J.F. Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014, 110, 94–103. [Google Scholar] [CrossRef]
- Cao, R.Y.; Xu, Y.F.; Zhang, T.H.; Yang, J.J.; Yuan, Y.; Hao, P.; Shi, Y.; Zhong, J.; Zhong, W. Pediatric Drug Nitazoxanide: A Potential Choice for Control of Zika. Open Forum Infect. Dis. 2017, 4, ofx009. [Google Scholar] [CrossRef]
- Yamamoto, K.A.; Blackburn, K.; Goshe, M.B.; Brown, D.T.; Migoswski, E.; Campanhon, I.B.; Moreira, M.F.; Ferreira, D.F.; Soares, M.R. Tizoxanide Antiviral Activity on Dengue Virus Replication. Viruses 2023, 15, 696. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Yao, Y.; Wu, F.; Wu, X.; Zhao, J.; Hua, Y.; Wu, J.; Huo, T.; Lin, Y.-L.; Kneubehl, A.R.; et al. Synthesis, Structure–Activity Relationships, and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B–NS3 Protease. J. Med. Chem. 2021, 64, 2777–2800. [Google Scholar] [CrossRef]
- Yao, Y.; Huo, T.; Lin, Y.-L.; Nie, S.; Wu, F.; Hua, Y.; Wu, J.; Kneubehl, A.R.; Vogt, M.B.; Rico-Hesse, R.; et al. Discovery, X-ray Crystallography and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Am. Chem. Soc. 2019, 141, 6832–6836. [Google Scholar] [CrossRef]
- Coluccia, A.; Puxeddu, M.; Nalli, M.; Wei, C.-K.; Wu, Y.-H.; Mastrangelo, E.; Elamin, T.; Tarantino, D.; Bugert, J.J.; Schreiner, B.; et al. Discovery of Zika Virus NS2B/NS3 Inhibitors That Prevent Mice from Life-Threatening Infection and Brain Damage. ACS Med. Chem. Lett. 2020, 11, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, J.F.; den-Haan, H.; Chik, K.K.; Zhang, A.J.; Chan, C.C.; Poon, V.K.; Yip, C.C.; Mak, W.W.; Zhu, Z.; et al. Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antivir. Res. 2017, 145, 33–43. [Google Scholar] [CrossRef]
- Montes-Grajales, D.; Puerta-Guardo, H.; Espinosa, D.A.; Harris, E.; Caicedo-Torres, W.; Olivero-Verbel, J.; Martínez-Romero, E. In silico drug repurposing for the identification of potential candidate molecules against arboviruses infection. Antivir. Res. 2020, 173, 104668. [Google Scholar] [CrossRef]
- Dražić, T.; Kopf, S.; Corridan, J.; Leuthold, M.M.; Bertoša, B.; Klein, C.D. Peptide-β-lactam Inhibitors of Dengue and West Nile Virus NS2B-NS3 Protease Display Two Distinct Binding Modes. J. Med. Chem. 2020, 63, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, A.; Manzano, M.; Teramoto, T.; Pilankatta, R.; Padmanabhan, R. High-throughput screening for the identification of small-molecule inhibitors of the flaviviral protease. Antivir. Res. 2016, 134, 6–16. [Google Scholar] [CrossRef]
- Shin, H.J.; Kim, M.H.; Lee, J.Y.; Hwang, I.; Yoon, G.Y.; Kim, H.S.; Kwon, Y.C.; Ahn, D.G.; Kim, K.D.; Kim, B.T.; et al. Structure-Based Virtual Screening: Identification of a Novel NS2B-NS3 Protease Inhibitor with Potent Antiviral Activity against Zika and Dengue Viruses. Microorganisms 2021, 9, 545. [Google Scholar] [CrossRef]




| Drug | Virus | NS2B-NS3 IC50 (μM) | Cellular EC50 (μM) | Peak Viremia Reduction or SR % Dosage (Infection Model) | Year [a] | Ref. |
|---|---|---|---|---|---|---|
| Temoporfin | DENV2 | 1.1 ± 0.1 | 0.020 | 2.0-log 1.6 mg/kg, IP (ZIKV GZ01, Balb/c) | 2017 | [127] |
| ZIKV | 0.024 | |||||
| WNV | 0.010 | |||||
| JMX0207 | DENV2 | 8.2 | 0.31 | 0.8-log 20 mg/kg, PO (ZIKV PRVABC59, A129) | 2020 | [128] |
| ZIKV | 0.3 | |||||
| Niclosamide | ZIKV | 12.3 ± 0.6 | 0.48 ± 0.06 | 33% 2–5 mg/kg, PO (DENV PL046, ICR suckling mice) | 2018 | [127,129,130,131] |
| DENV | 21.6 | 0.55 ± 0.05 | ||||
| WNV | 0.54 ± 0.17 | |||||
| Methylene Blue | DENV2 | 8.9 | 0.36 | 1.69-log 100 mg/kg, PO (ZIKV PRVABC59, A129 mice) | 2020 | [132] |
| ZIKV | 0.087–0.2 | |||||
| Erythrosin B | DENV2 | 1.9 | 1.2 | 80% SR 200–400 mg/kg, PO (ZIKV PRVABC59, A129 mice) | 2021 | [133,134] |
| ZIKV | 1.7 | 0.62 | ||||
| WNV | 0.66 | |||||
| 6-Bromo-1,2-Naphthalenedione | ZIKV | 67.47 | 1 | 20–60% SR 100 μL (50 μg), IV (ZIKV MR766, AG129 mice) | 2022 | [135] |
| Nitazoxanide (Prodrug) | ZIKV | 15.9 ± 0.9 | 1.48 ± 0.18 | ~90% SR 100 mg/kg, IG (JEV SH-JEV01, CK) | 2017 | [127,136,137,138,139] |
| Tizoxanide (Active Metabolite) | DENV2 | 0.1 | n.a. | n.a. |
| Drug | Virus | NS2B-NS3 IC50 (μM) | Cellular EC50 (μM) | Peak Viremia Reduction or SR% Dosage (Infection Model) | Year | Ref. |
|---|---|---|---|---|---|---|
| SYC-1307 | ZIKV | 0.20 ± 0.01 | 0.3–0.6 [c] | 96–98% 15 mg/kg, IP (ZIKV FLR, HN16; C57BL/6) | 2019 | [140,141] |
| DENV2 | 0.59 ± 0.02 | |||||
| DENV3 | 0.59 ± 0.06 | |||||
| WNV | 0.78 ± 0.02 | |||||
| Compound 1 | ZIKV | 158 ± 25 | 13.9 ± 0.4 | 100% SR 1 mg/kg, IP (ZIKV Uganda, ICR) | 2022 | [142] |
| Novobiocin | ZIKV | 14.2 ± 1.1 | 42.63 | 100% SR 150 mg/kg, SC [a] (ZIKV PRVABC59, BALB/c [b]) | 2017 | [143,144] |
| NSC157058 | ZIKV | 0.82 | 50 | ~10-fold 30 mg/kg, PO (ZIKV PA259459, SJL mice) | 2017 | [78] |
| WNV | ||||||
| DENV2 |
| Drug | Virus | IC50 (μM) | EC50 (μM) | CC50 (μM) | Cell Line | Year | Ref. |
|---|---|---|---|---|---|---|---|
| Compound 71 | DENV2 | 8.0 | 0.24 | >100 | Huh-7 | 2021 | [61] |
| WNV | 23 | ||||||
| NK-189 | DENV2 | 11 | 0.89 | >50 | Huh-7 | 2020 | [59] |
| WNV | 13 | ||||||
| Compound 11d | DENV2 | 2.5 | 4.1 | >50 | Huh-7 | 2019 | [145] |
| Compound 104 | DENV2 | 0.18 | 3.4 | >100 | Huh-7 | 2015 | [72] |
| WNV | 0.56 | 15.5 | >100 | ||||
| Tolcapone | DENV1 | 1.1 | BHK-21 | 2016 | [146] | ||
| DENV2 | 0.98 | 2.0 | 29 | ||||
| DENV3 | 0.91 | ||||||
| DENV4 | 0.64 | ||||||
| WNV | 0.70 | ||||||
| Compound-C | DENV1 | 4.1 | BHK-21 | 2016 | [146] | ||
| DENV2 | 4.0 | 9.0 | 76 | ||||
| DENV3 | 2.9 | ||||||
| DENV4 | 3.4 | ||||||
| WNV | 2.5 | ||||||
| 6-APNP | DENV4 ZIKV | n.a. 1.5 | 7.1 0.69–5.0 [a] | 35 | Huh-7 | 2021 | [147] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavina, L.; Bouma, M.J.; Gironés, D.; Feiters, M.C. Orthoflaviviral Inhibitors in Clinical Trials, Preclinical In Vivo Efficacy Targeting NS2B-NS3 and Cellular Antiviral Activity via Competitive Protease Inhibition. Molecules 2024, 29, 4047. https://doi.org/10.3390/molecules29174047
Cavina L, Bouma MJ, Gironés D, Feiters MC. Orthoflaviviral Inhibitors in Clinical Trials, Preclinical In Vivo Efficacy Targeting NS2B-NS3 and Cellular Antiviral Activity via Competitive Protease Inhibition. Molecules. 2024; 29(17):4047. https://doi.org/10.3390/molecules29174047
Chicago/Turabian StyleCavina, Lorenzo, Mathijs J. Bouma, Daniel Gironés, and Martin C. Feiters. 2024. "Orthoflaviviral Inhibitors in Clinical Trials, Preclinical In Vivo Efficacy Targeting NS2B-NS3 and Cellular Antiviral Activity via Competitive Protease Inhibition" Molecules 29, no. 17: 4047. https://doi.org/10.3390/molecules29174047
APA StyleCavina, L., Bouma, M. J., Gironés, D., & Feiters, M. C. (2024). Orthoflaviviral Inhibitors in Clinical Trials, Preclinical In Vivo Efficacy Targeting NS2B-NS3 and Cellular Antiviral Activity via Competitive Protease Inhibition. Molecules, 29(17), 4047. https://doi.org/10.3390/molecules29174047