


Synthesis and Properties of α-Phosphate-Modified Nucleoside Triphosphates


Abstract
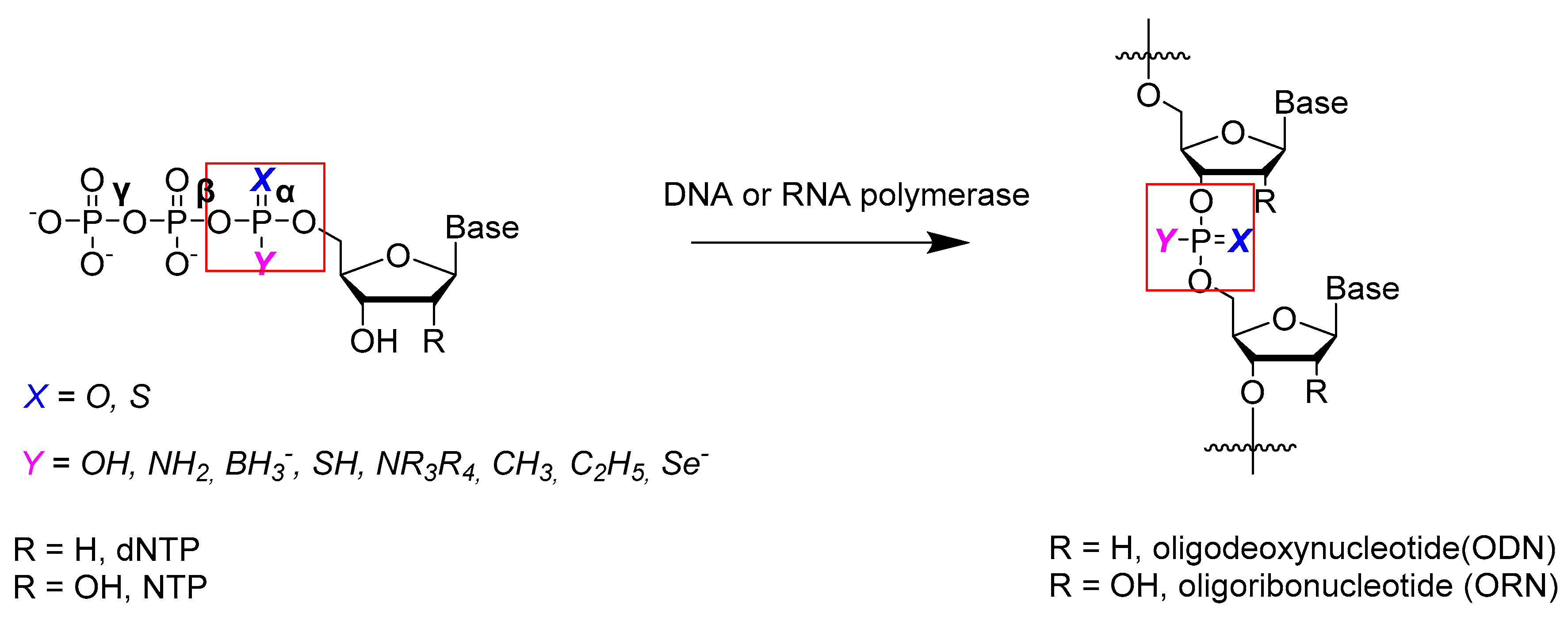
:1. Introduction
2. Chemical Synthesis of Nucleoside 5′-α-P-Modified Triphosphates
2.1. Synthesis of Nucleoside 5′-(α-P-Thio)- and 5′-(α-P-Seleno)triphosphates
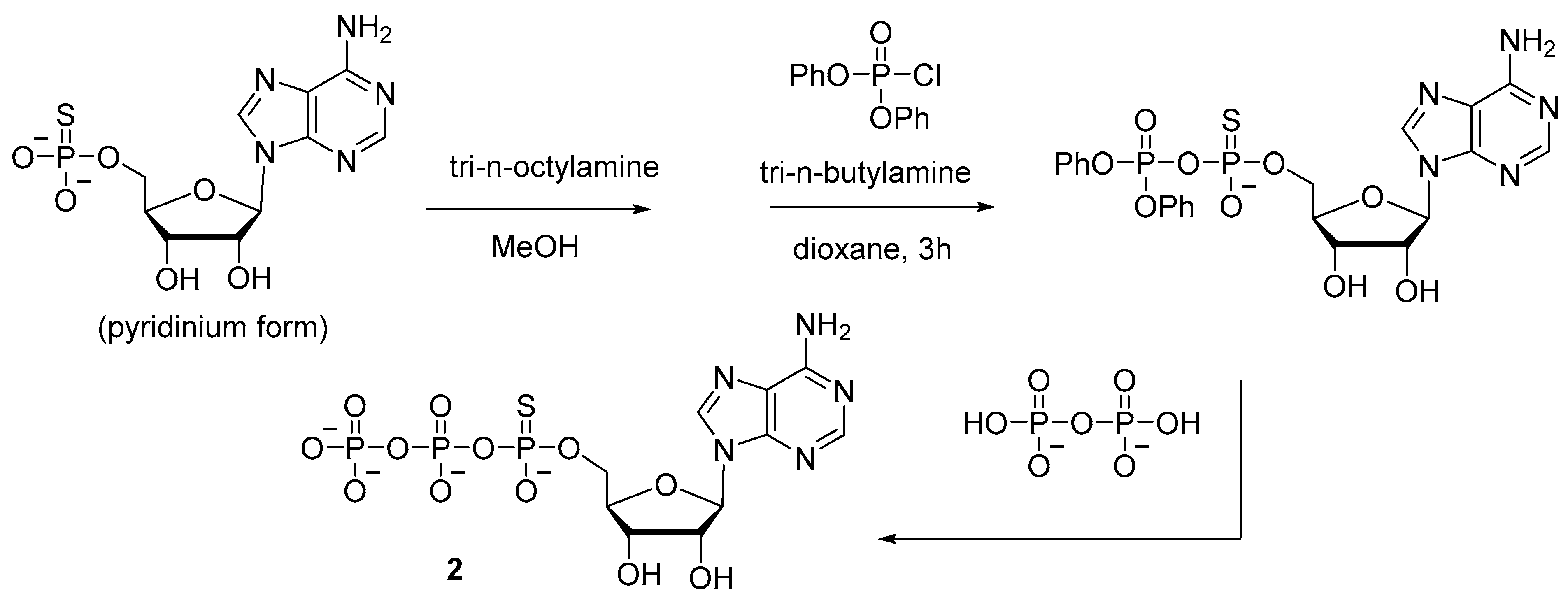
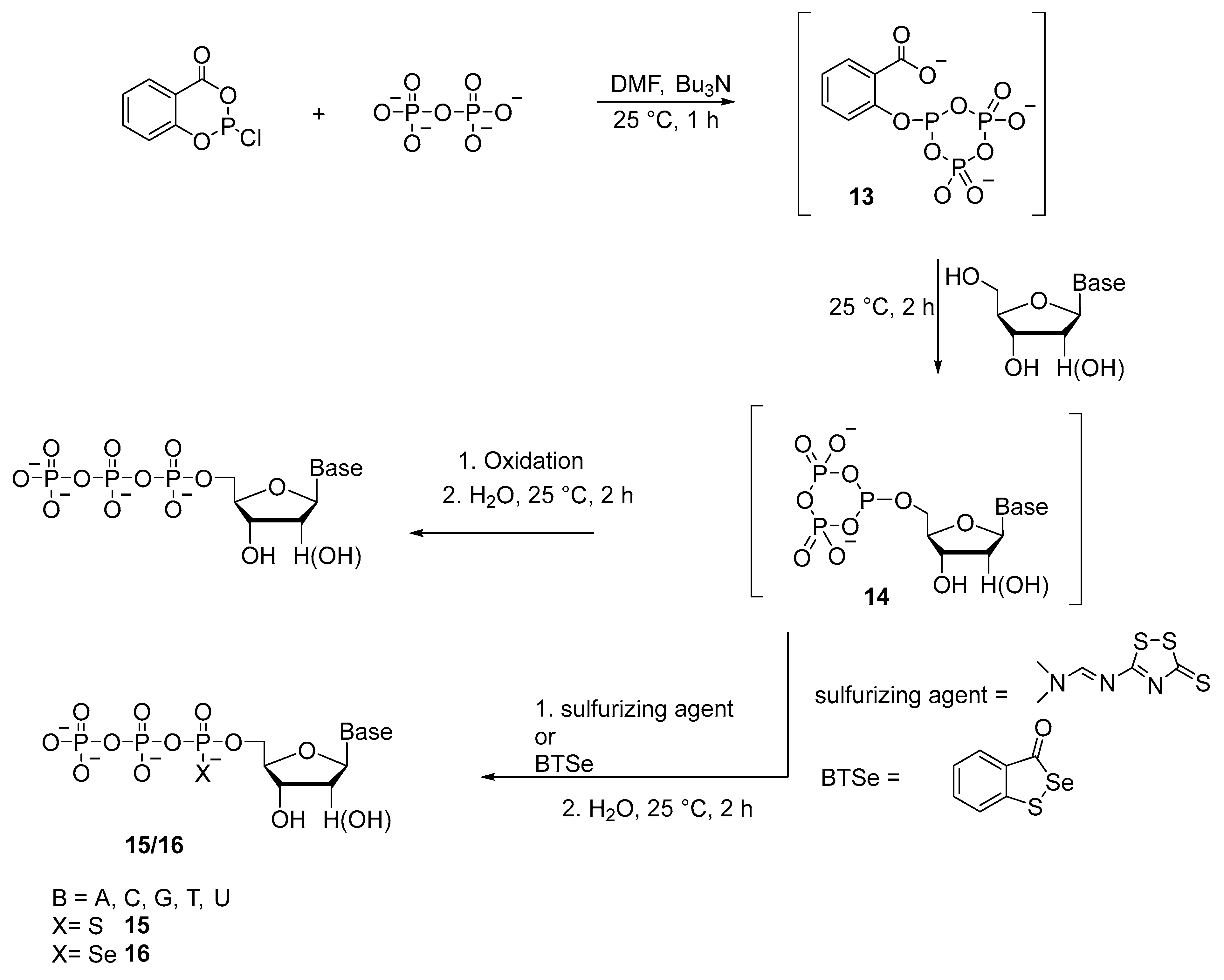
2.1.1. Syntheses via a Nucleophilic Attack of Pyrophosphate on Activated Nucleoside 5′-Phosphorothioates
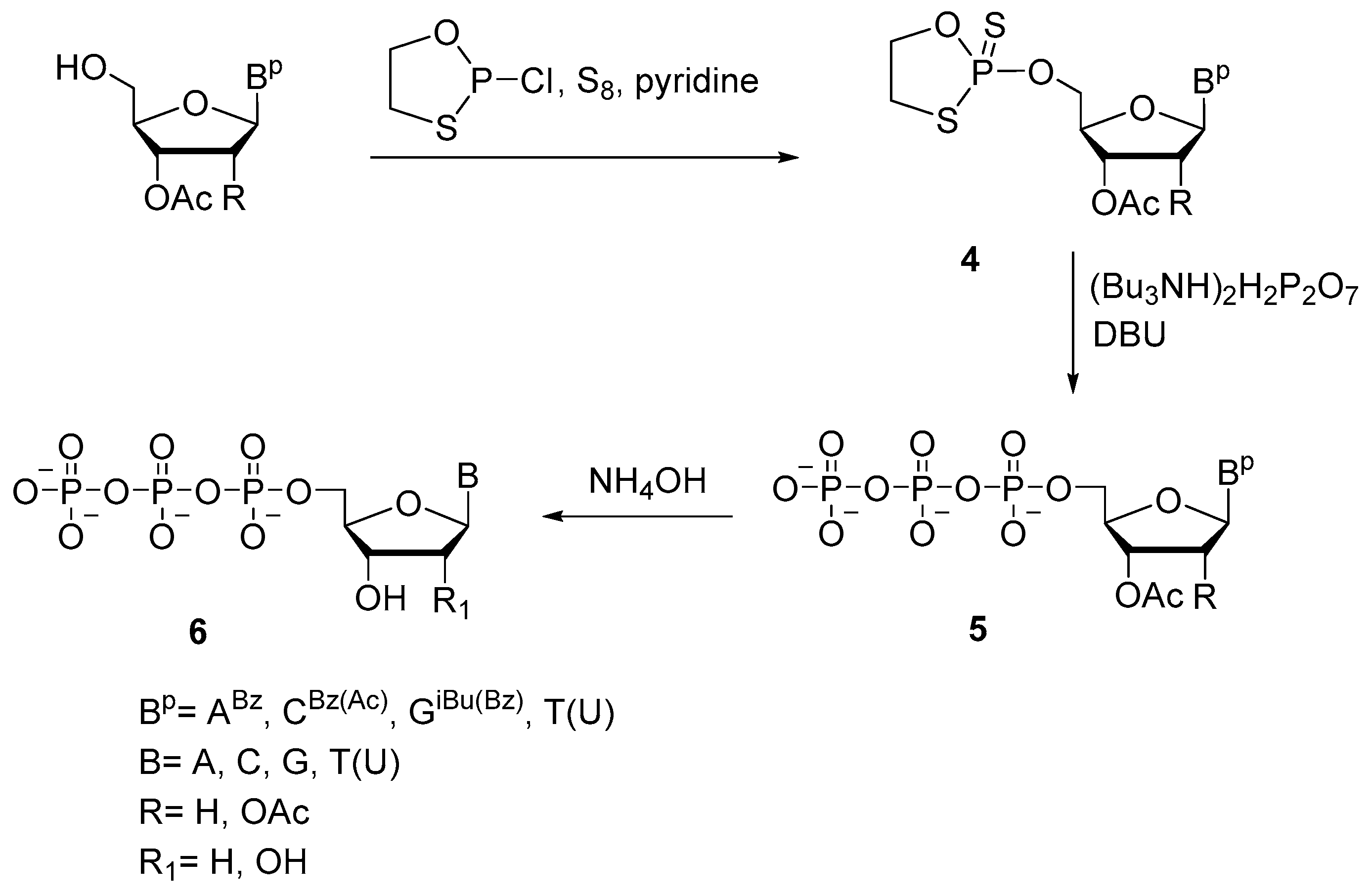
2.1.2. Synthesis of Nucleoside Triphosphates via a Cyclotriphosphite Intermediate (Ludwig–Eckstein Method)
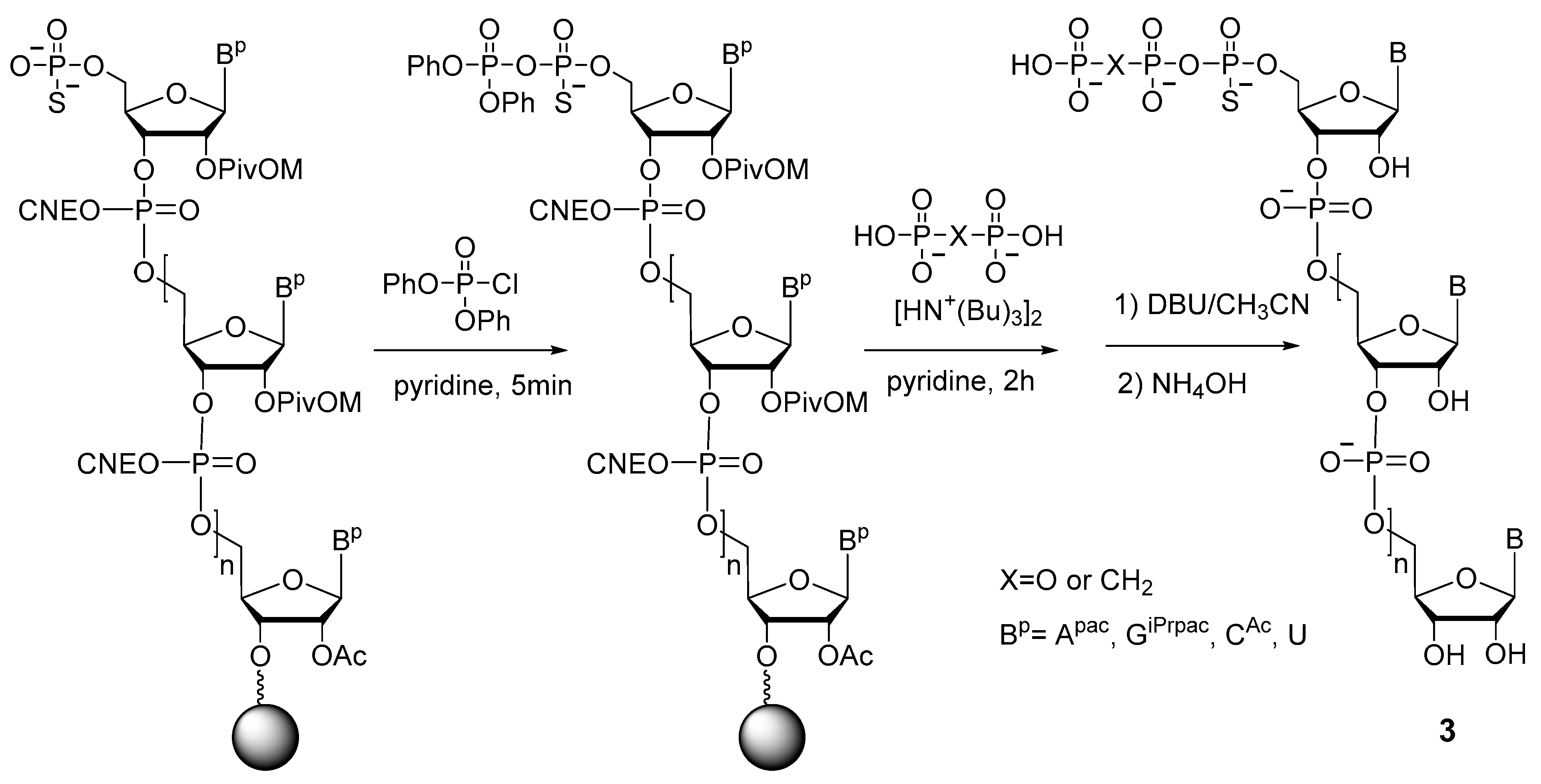
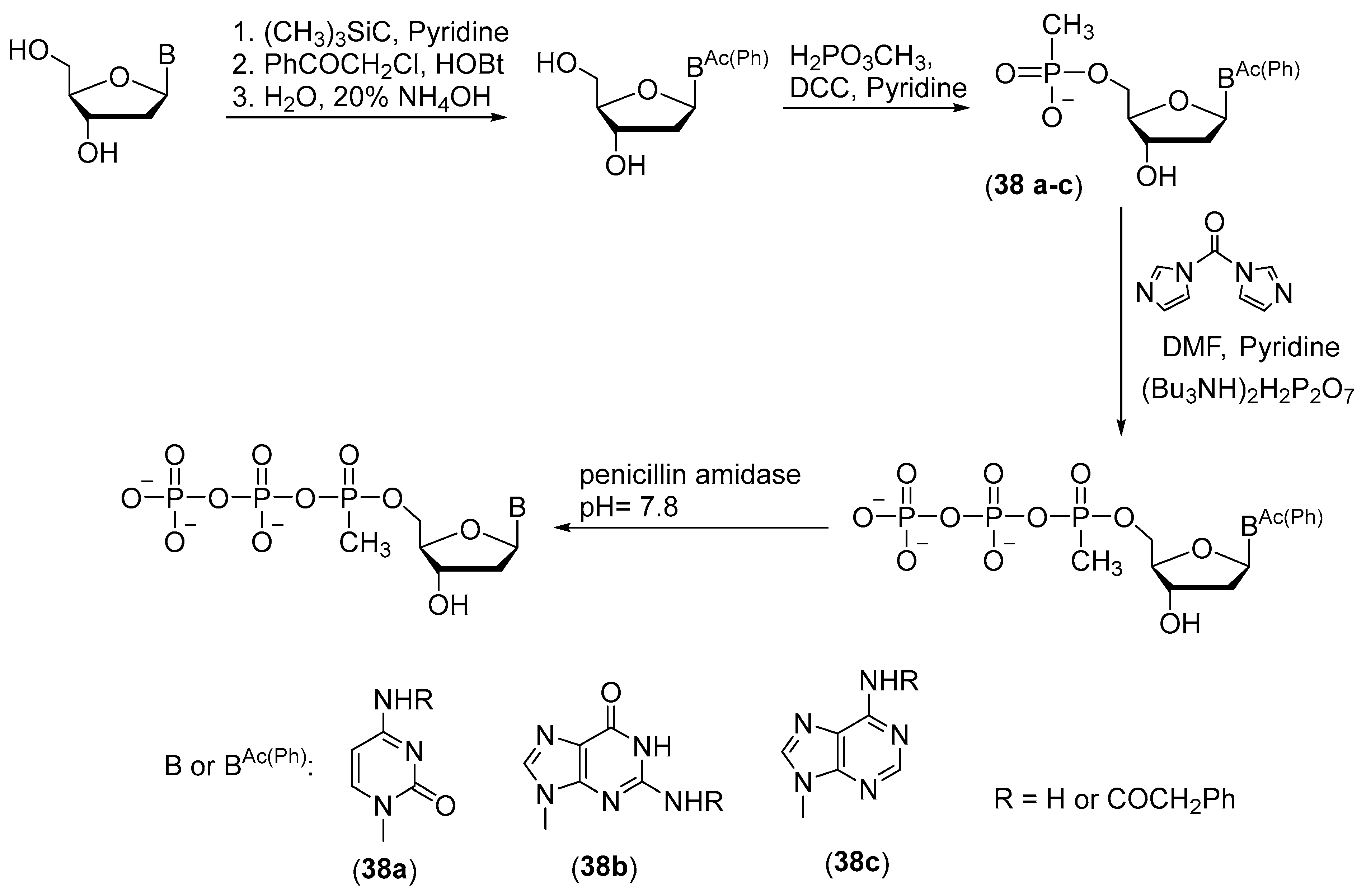
2.1.3. Synthesis by the Amidophosphite Method
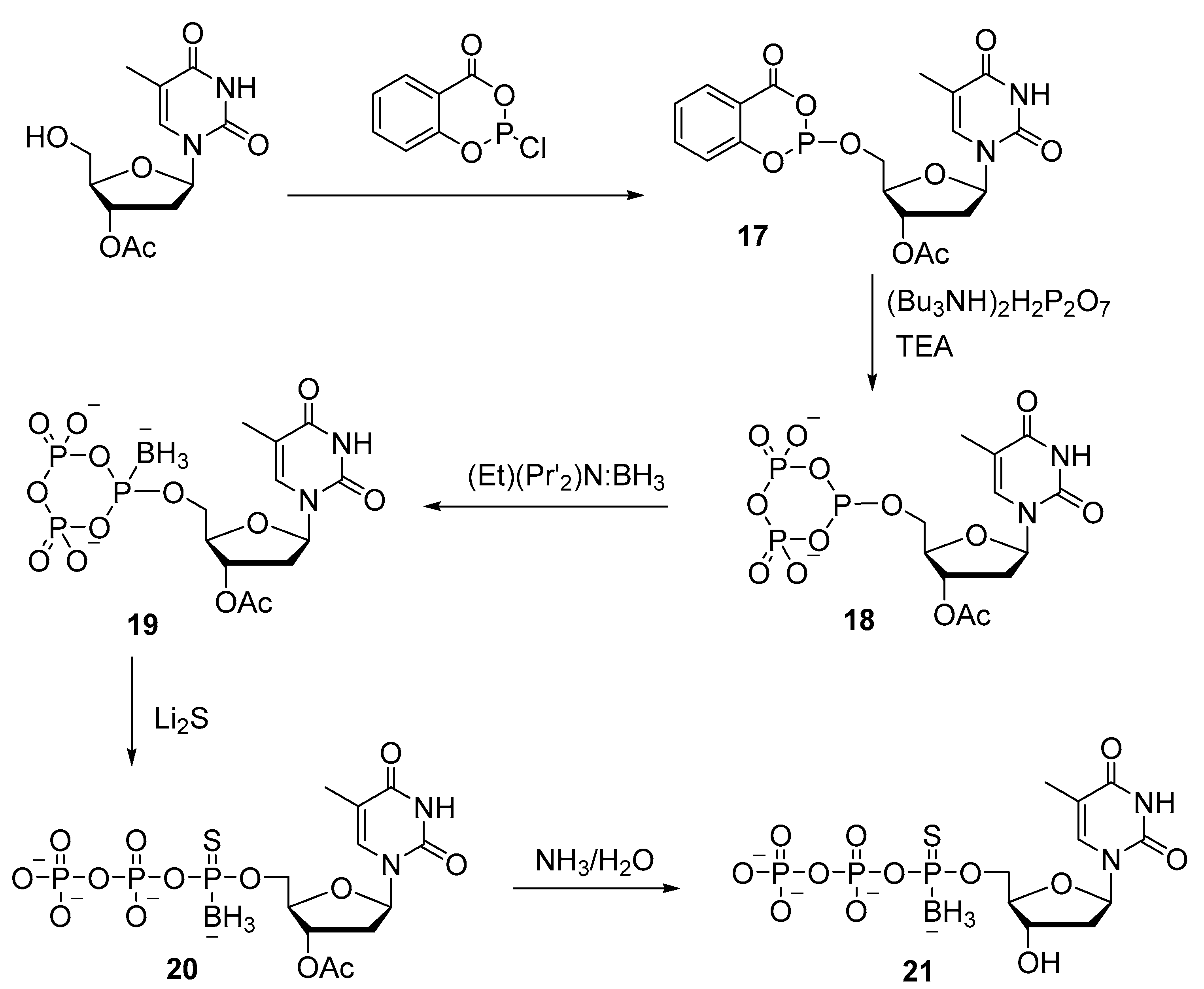
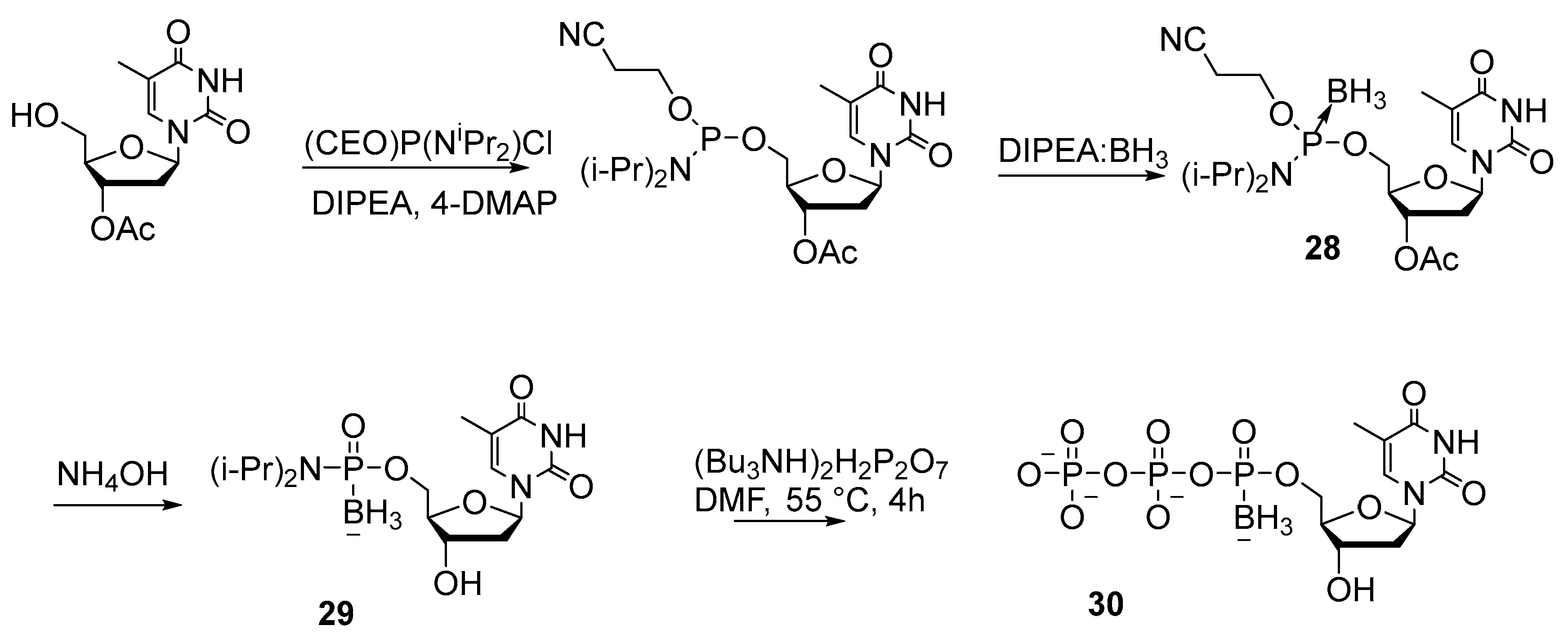
2.2. Synthesis of Nucleoside 5′-(α-P-Borano)triphosphates
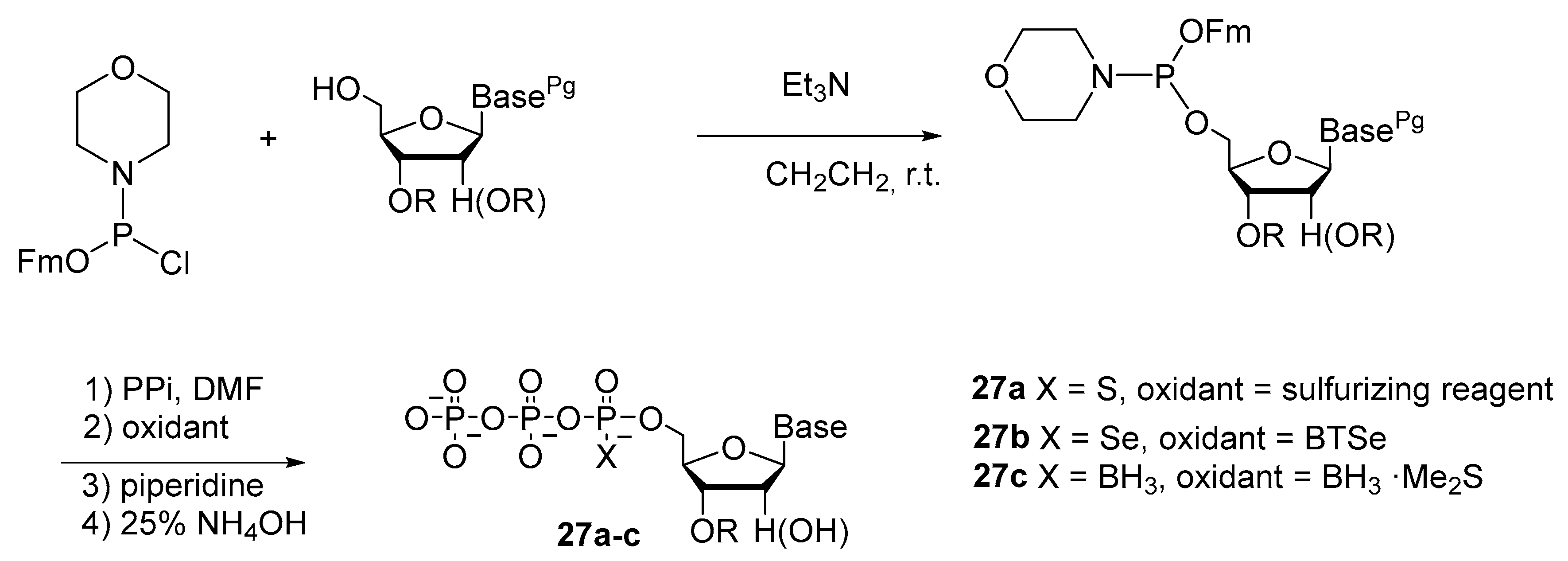
2.2.1. Synthesis by the Amidophosphite Method
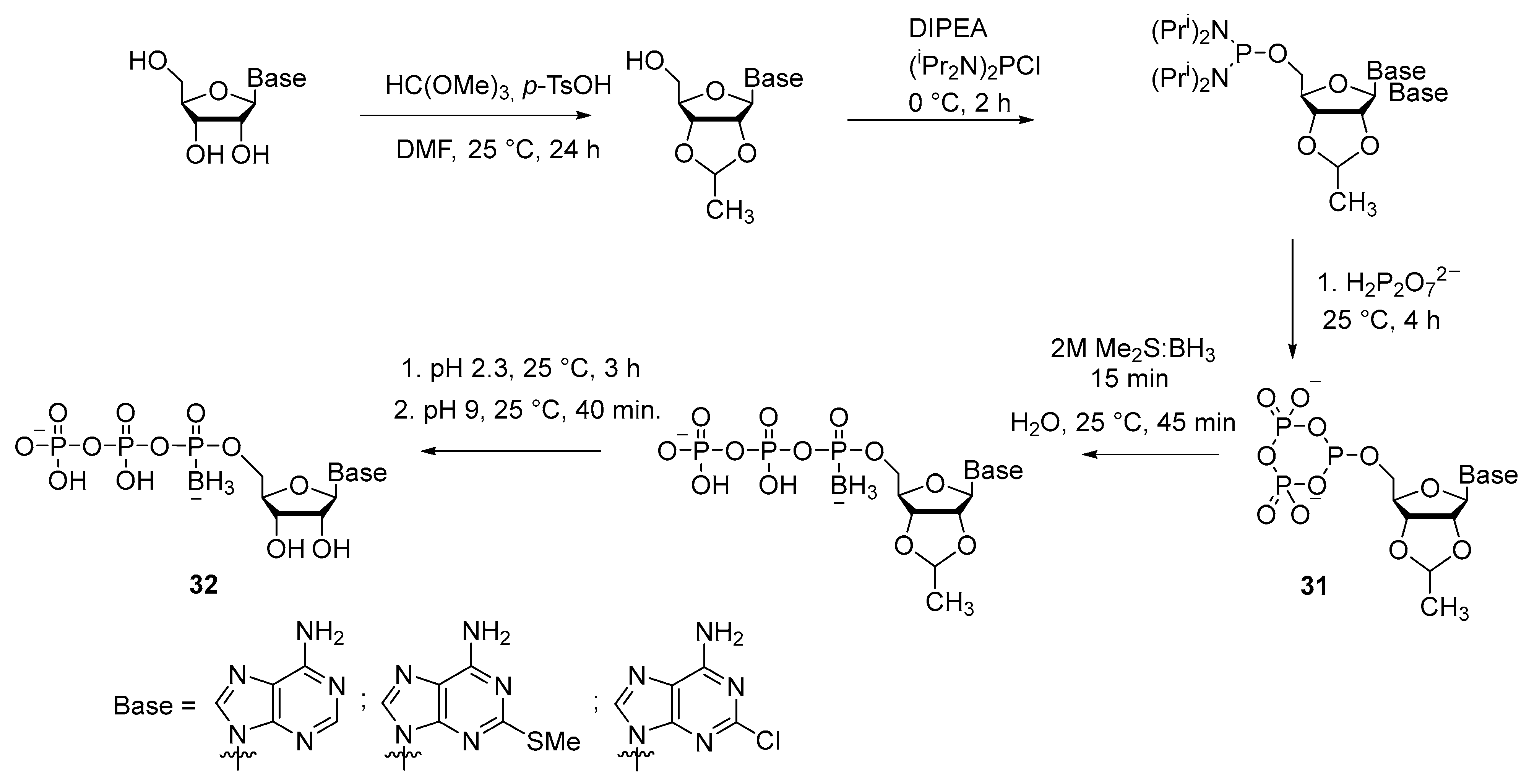
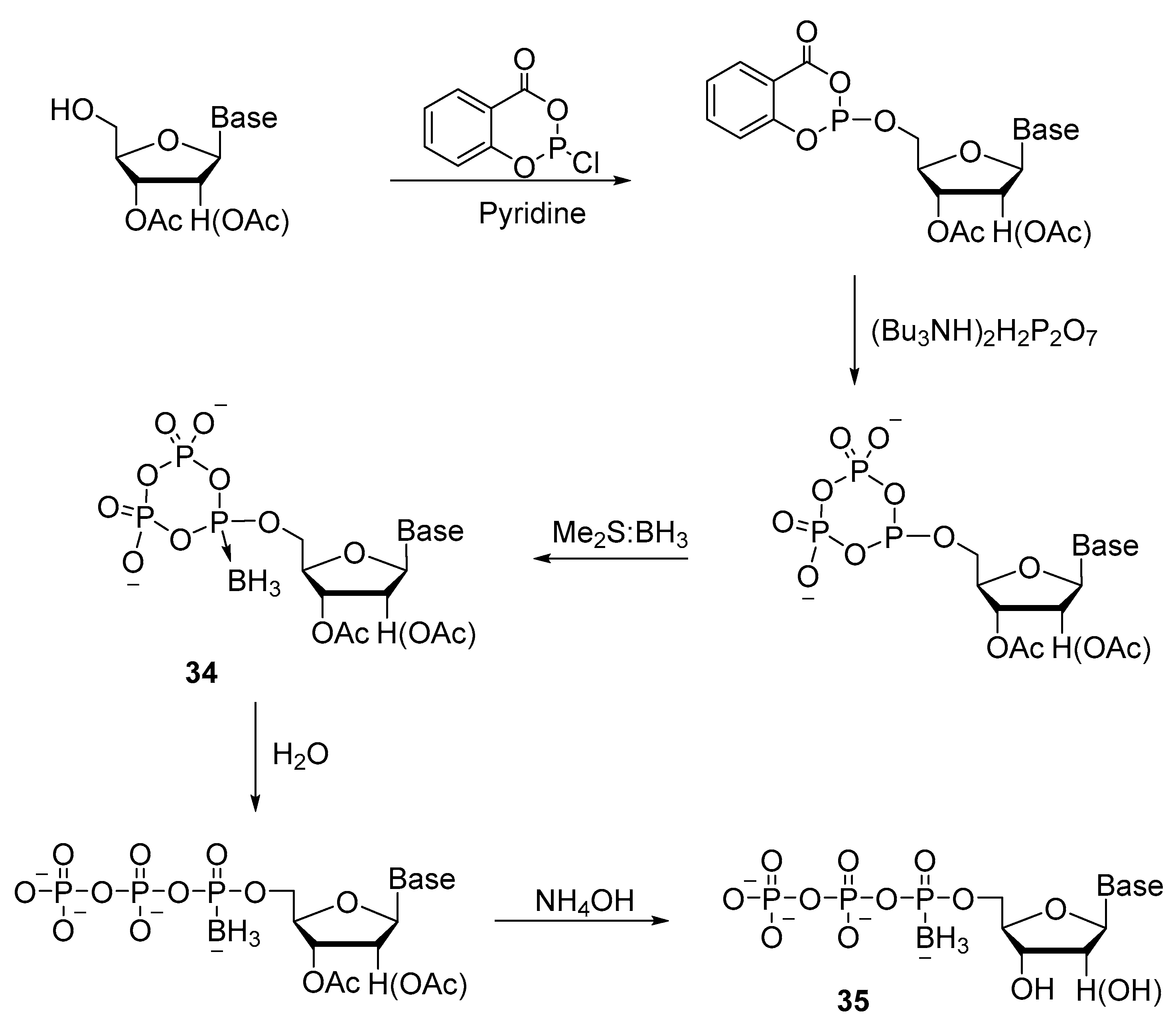
2.2.2. Synthesis through a Cyclotriphosphite Intermediate
2.3. Synthesis of Nucleoside 5′-(α-P-Alkyl)triphosphates
2.4. Synthesis of 5′-(α-P-Amido)triphosphates and 5′-(α-P-Alkylamido)triphosphates of Nucleosides
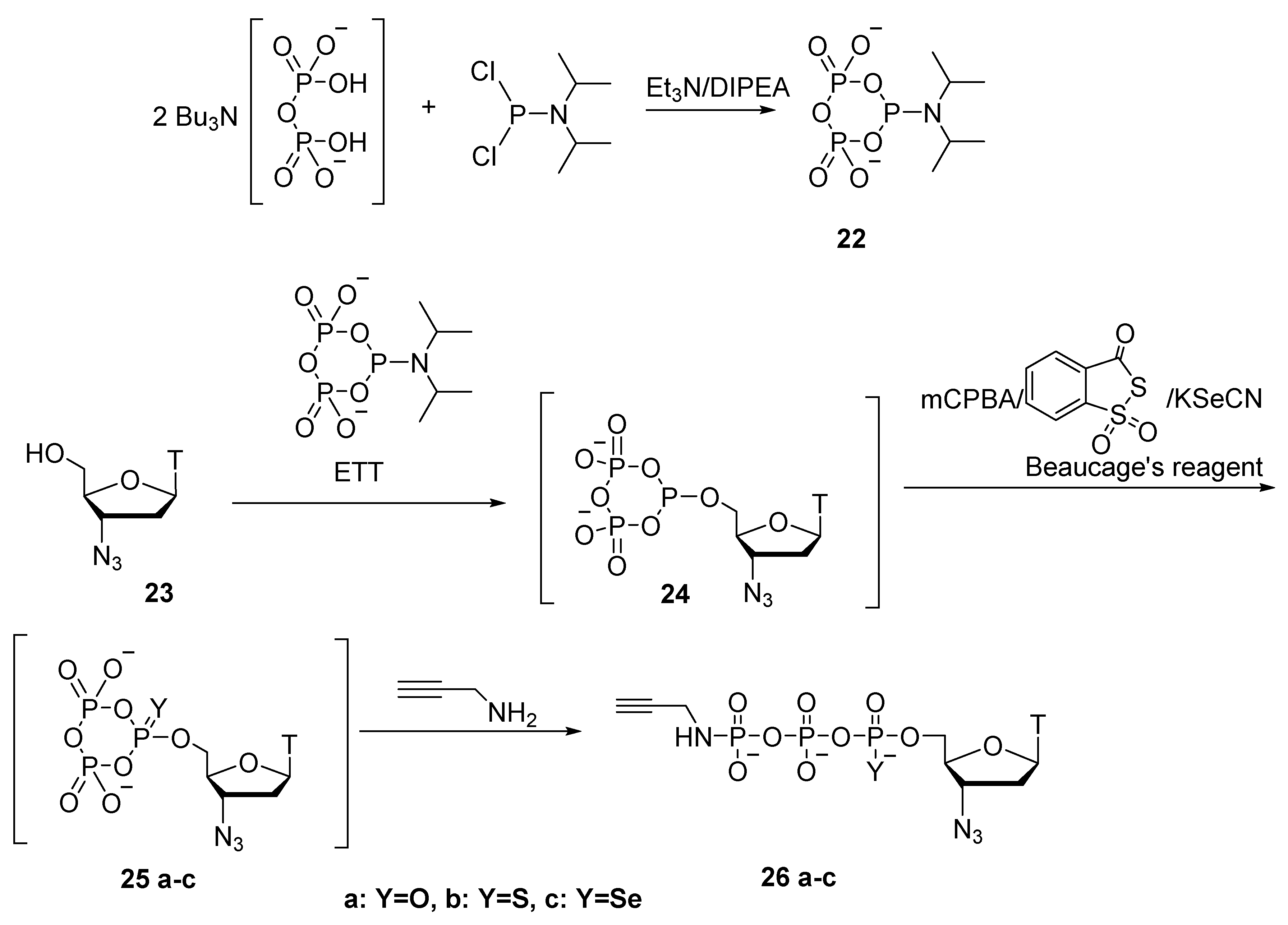
2.5. Synthesis of Nucleoside 5′-(α-P-Imido-R)triphosphates by Means of Organic Azides N3-R
2.6. Summary
3. Substrate Properties of 5′-α-P-Modified Nucleoside Triphosphates and of Oligonucleotides Synthesized from Them in Relation to Nucleic Acid-Processing Enzymes; Applications of α-Phosphate-Modified Nucleoside Triphosphates
3.1. Nucleoside 5′-(α-P-Thio)triphosphates
3.2. Nucleoside 5′-(α-P-Borano)triphosphates
3.3. Nucleoside 5′-(α-P-Alkyl)triphosphates
3.4. Nucleoside 5′-(α-P-Imido)triphosphates
4. Prospects of α-Phosphate-Modified Nucleotides for the Design of Mutant Enzymes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hollenstein, M. Nucleoside Triphosphates-Building Blocks for the Modification of Nucleic Acids. Molecules 2012, 17, 13569–13591. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Sugimoto, N. Molecular Evolution of Functional Nucleic Acids with Chemical Modifications. Molecules 2010, 15, 5423–5444. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Cload, S.T. SELEX with Modified Nucleotides. Curr. Opin. Chem. Biol. 2008, 12, 448–456. [Google Scholar] [CrossRef]
- Martinussen, J.; Willemoës, M.; Kilstrup, M. Nucleotide Metabolism. Compr. Biotechnol. Second. Ed. 2011, 1, 91–107. [Google Scholar] [CrossRef]
- Porter, K.W.; Briley, J.D.; Shaw, B.R. Direct PCR Sequencing with Boronated Nucleotides. Nucleic Acids Res. 1997, 25, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Boyle, N.A.; Fagan, P.; Brooks, J.L.; Prhavc, M.; Lambert, J.; Cook, P.D. 2′,3′-Dideoxynucleoside 5′-β,γ- (Difluoromethylene) Triphosphates with α-P-Thio or α-P-Seleno Modifications: Synthesis and Their Inhibition of HIV-1 Reverse Transcriptase. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1651–1664. [Google Scholar] [CrossRef]
- King, D.J.; Ventura, D.A.; Brasier, A.R.; Gorenstein, D.G. Novel Combinatorial Selection of Phosphorothioate Oligonucleotide Aptamers. Biochemistry 1998, 37, 16489–16493. [Google Scholar] [CrossRef]
- Hu, B.; Wang, Y.; Li, N.; Zhang, S.; Luo, G.; Huang, Z. Highly Convenient and Highly Specific-and-Sensitive PCR Using Se-Atom Modified DNTPs. Chem. Commun. 2021, 57, 57–60. [Google Scholar] [CrossRef]
- Eckstein, F.; Thomson, J.B. Phosphate Analogs for Study of DNA Polymerases. Methods Enzym. 1995, 262, 189–202. [Google Scholar] [CrossRef]
- Dong, Y.Y.; Huang, H.S.; Zhong, R.Y.; Gong, S.S.; Sun, Q. Efficient Synthesis of α-P-Modified Nucleoside Triphosphates and Dinucleoside Tetraphosphates via Linear PVPVPIII-Nucleoside Intermediates. Asian J. Org. Chem. 2023, 12, e202200696. [Google Scholar] [CrossRef]
- Eckstein, F. Nucleoside phosphorothioates. Annu. Rev. Biochem. 1985, 54, 367–402. [Google Scholar] [CrossRef]
- Shaw, B.R.; Dobrikov, M.; Wang, X.; Wan, J.; He, K.; Lin, J.L.; Li, P.; Rait, V.; Sergueeva, Z.A.; Sergueev, D. Reading, Writing, and Modulating Genetic Information with Boranophosphate Mimics of Nucleotides, DNA, and RNA. Ann. N. Y. Acad. Sci. 2003, 1002, 12–29. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Eckstein, F.; Mildvan, A.S.; Koplitz, R.M.; Loeb, L.A. Deoxynucleoside [1-Thio]Triphosphates Prevent Proofreading during in Vitro DNA Synthesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6734–6738. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. Phosphorothioatanaloga von Nucleotiden—Werkzeuge Zur Untersuchung Biochemischer Prozesse. Angew. Chem. 1983, 95, 431–447. [Google Scholar] [CrossRef]
- Espinasse, A.; Lembke, H.K.; Cao, A.A.; Carlson, E.E. Modified Nucleoside Triphosphates in Bacterial Research for in Vitro and Live-Cell Applications. RSC Chem. Biol. 2020, 1, 333–351. [Google Scholar] [CrossRef]
- Caton-Williams, J.; Fiaz, B.; Hoxhaj, R.; Smith, M.; Huang, Z. Convenient Synthesis of Nucleoside 5′-(α-P-Thio)Triphosphates and Phosphorothioate Nucleic Acids (DNA and RNA). Sci. China Chem. 2012, 55, 80–89. [Google Scholar] [CrossRef]
- Burgess, K.; Cook, D. Syntheses of Nucleoside Triphosphates. Chem. Rev. 2000, 100, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Depaix, A.; Périgaud, C.; Peyrottes, S. Recent Trends in Nucleotide Synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef]
- Eckstein, F.; Gindl, H. Synthesis of Nucleoside 5′-Polyphosphorothioates. BBA Sect. Nucleic Acids Protein Synth. 1967, 149, 35–40. [Google Scholar] [CrossRef]
- Eckstein, F.; Goody, R.S. Synthesis and Properties of Diastereoisomers of Adenosine 5′-(O-1-Thiotriphosphate) and Adenosine 5′-(O-2-Thiotriphosphate). Biochemistry 1976, 15, 1685–1691. [Google Scholar] [CrossRef]
- Goody, R.S.; Isakov, M. Simple Synthesis and Separation of the Diastereomers of α-Thio Analogs of Ribo- and Deoxyribo- Di- and Triphosphates. Tetrahedron Lett. 1986, 27, 3599–3602. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Kato, T.; Takenishi, T. Studies of Phosphorylation. III. Selective Phosphorylation of Unprotected Nucleosides. Bull. Chem. Soc. Jpn. 1969, 42, 3505–3508. [Google Scholar] [CrossRef]
- Auer, M.; Kriege, C.; Gautel, M.; Labeit, S.; Sczakiel, G.; Weisz, S.; Goody, R.S. Synthesis and Biological Applications of 2′,3′- Dideoxynucleoside-5′-O-(α-Thio)Triphosphates. Nucleosides Nucleotides 1989, 8, 849–853. [Google Scholar] [CrossRef]
- Thillier, Y.; Sallamand, C.; Baraguey, C.; Vasseur, J.J.; Debart, F. Solid-Phase Synthesis of Oligonucleotide 5′-(α-P-Thio)Triphosphates and 5′-(α-P-Thio)(β,γ-Methylene)Triphosphates. Eur. J. Org. Chem. 2015, 2015, 302–308. [Google Scholar] [CrossRef]
- Misiura, K.; Szymanowicz, D.; Stec, W.J. Synthesis of Nucleoside α-Thiotriphosphates via an Oxathiaphospholane Approach. Org. Lett. 2005, 7, 2217–2220. [Google Scholar] [CrossRef]
- Okruszek, A.; Olesiak, M.; Balzarini, J. The Synthesis of Nucleoside 5′-O-(1,1-Dithiotriphosphates). J. Med. Chem. 1994, 37, 3850–3854. [Google Scholar] [CrossRef]
- Eckstein, F.; Ludwig, J. Rapid and Efficient Synthesis of Nucleoside 5′-O-(1-Thiotriphosphates), 5′-Triphosphates and 2′,3′-Cyclophosphorothioates Using 2-Chloro-4H-1,3,2-Benzodioxaphosphorin-4-One. J. Org. Chem. 1989, 54, 631–635. [Google Scholar] [CrossRef]
- Caton-Williams, J.; Hoxhaj, R.; Fiaz, B.; Huang, Z. Use of a Novel 5′-Regioselective Phosphitylating Reagent for One-Pot Synthesis of Nucleoside 5′-Triphosphates from Unprotected Nucleosides. Curr. Protoc. Nucleic Acid. Chem. 2013, 52, 1.30.1–1.30.21. [Google Scholar] [CrossRef]
- Lin, L.; Caton-Williams, J.; Kaur, M.; Patino, A.M.; Sheng, J.; Punetha, J.; Huang, Z. Facile Synthesis of Nucleoside 5′-(α-P-Seleno)-Triphosphates and Phosphoroselenoate RNA Transcription. RNA 2011, 17, 1932–1938. [Google Scholar] [CrossRef]
- Lin, J.; Shaw, B.R. Synthesis of a Novel Triphosphate Analogue: Nucleoside α-P-Borano, α-P-Thiotriphosphate. Chem. Commun. 2000, 21, 2115–2116. [Google Scholar] [CrossRef]
- Ripp, A.; Singh, J.; Jessen, H.J. Rapid Synthesis of Nucleoside Triphosphates and Analogues. Curr. Protoc. Nucleic Acid. Chem. 2020, 81, e108. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, J.; Shaw, B.R.; Porter, K.; Spielvogel, B.F.; Sood, A. 5′-P-Borane-Substituted Thymidine Monophosphate and Triphosphate. Angew. Chem. Int. Ed. Engl. 1992, 31, 1373–1375. [Google Scholar] [CrossRef]
- Nahum, V.; Zündorf, G.; Lévesque, S.A.; Beaudoin, A.R.; Reiser, G.; Fischer, B. Adenosine 5′-O-(1-Boranotriphosphate) Derivatives as Novel P2Y1 Receptor Agonists. J. Med. Chem. 2002, 45, 5384–5396. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Dobrikov, M.; Liu, H.; Shaw, B.R. Synthesis of Acyclothymidine Triphosphate and α-P-Boranotriphosphate and Their Substrate Properties with Retroviral Reverse Transcriptase. Org. Lett. 2003, 5, 2401–2403. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Hasan, A.; Krzyzanowska, B.; Shaw, B.R. Synthesis and Separation of Diastereomers of Ribonucleoside 5′-(α-P-Borano)Triphosphates. J. Org. Chem. 1998, 63, 5769–5773. [Google Scholar] [CrossRef]
- Krzyzanowska, B.K.; He, K.; Hasan, A.; Shaw, B.R. A Convenient Synthesis of 2’-Deoxyribonucleoside 5’-(α-P- Borano)Triphosphates. Tetrahedron 1998, 54, 5119–5128. [Google Scholar] [CrossRef]
- He, K.; Porter, K.W.; Hasan, A.; Briley, J.D.; Shaw, B.R. Synthesis of 5-Substituted 2’-Deoxycytidine 5’-(α-P-Borano)Triphosphates, Their Incorporation into DNA and Effects on Exonuclease. Nucleic Acids Res. 1999, 27, 1788–1794. [Google Scholar] [CrossRef]
- Wang, G.; Boyle, N.; Chen, F.; Rajappan, V.; Fagan, P.; Brooks, J.L.; Hurd, T.; Leeds, J.M.; Rajwanshi, V.K.; Jin, Y.; et al. Synthesis of AZT 5′-Triphosphate Mimics and Their Inhibitory Effects on HIV-1 Reverse Transcriptase. J. Med. Chem. 2004, 47, 6902–6913. [Google Scholar] [CrossRef]
- Dineva, M.A.; Chakurov, S.; Bratovanova, E.K.; Devedjiev, I.; Petkov, D.D. Complete Template-Directed Enzymatic Synthesis of a Potential Antisense DNA Containing 42 Methylphosphonodiester Bonds. Bioorg. Med. Chem. 1993, 1, 411–414. [Google Scholar] [CrossRef]
- Higuchi, H.; Endo, T.; Kaji, A. Enzymic Synthesis of Oligonucleotides Containing Methylphosphonate Internucleotide Linkages. Biochemistry 1990, 29, 8747–8753. [Google Scholar] [CrossRef]
- Dineva, M.A.; Petkov, D.D. Chemoenzymic Synthesis of P(α)-Methyl Deoxynucleoside Triphosphates. Nucleosides Nucleotides 1996, 15, 1459–1467. [Google Scholar] [CrossRef]
- Arzumanov, A.A.; Dyatkina, N.B. An Alternative Route for Preparation of α-Methylphosphonyl-β,γ-Diphosphates of Thymidine Derivatives. Nucleosides Nucleotides 1994, 13, 1031–1037. [Google Scholar] [CrossRef]
- Arangundy-Franklin, S.; Taylor, A.I.; Porebski, B.T.; Genna, V.; Peak-Chew, S.; Vaisman, A.; Woodgate, R.; Orozco, M.; Holliger, P. A Synthetic Genetic Polymer with an Uncharged Backbone Chemistry Based on Alkyl Phosphonate Nucleic Acids. Nat. Chem. 2019, 11, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Simoncsits, A.; Tomasz, J. A New Type of Nucleoside 5′-Triphosphate Analogue: P1- (Nucleoside 5′-) P1-Amino-Triphosphates. Tetrahedron Lett. 1976, 17, 3995–3998. [Google Scholar] [CrossRef]
- Simoncsits, A.; Tomasz, J. On the Stability of Phosphodiester-Amide Internucleotide Bond. Tetrahedron Lett. 1981, 22, 3905–3908. [Google Scholar] [CrossRef]
- Wyrzykiewicz, K.T.; Dan Cook, P. Process for the Preparation of Alpha Modified Nucleoside Triphosphates and Compounds Therefrom. Patent WO2003008432A1, 16 July 2002. [Google Scholar]
- Staudinger, H.; Meyer, J. Über Neue Organische Phosphorverbindungen III. Phosphinmethylenderivate Und Phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar] [CrossRef]
- Heindl, D. Nucleotide with an Alpha-Phosphate Mimetic. Patent WO2008128686A1, 16 April 2008. [Google Scholar]
- Vasilyeva, S.V.; Kuznetsova, A.A.; Baranovskaya, E.E.; Kuznetsov, N.A.; Lomzov, A.A.; Pyshnyi, D.V. Synthesis of the New Nucleoside 5′-Alpha-Iminophosphates Using Staudinger Reaction. Bioorg. Chem. 2022, 127, 105987. [Google Scholar] [CrossRef]
- Bogachev, V.S. Synthesis of Deoxynucleotide-5’-Triphosphates Using Trifluoroacetic Anhydride as an Activating Reagent. Bioorg. Chem. 1996, 22, 699–705. [Google Scholar]
- Bezold, D.; Dürr, T.; Singh, J.; Jessen, H.J. Cyclotriphosphate: A Brief History, Recent Developments, and Perspectives in Synthesis. Chem.—A Eur. J. 2020, 26, 2298–2308. [Google Scholar] [CrossRef]
- Antonov, K.V.; Esipov, R.S.; Gurevich, A.I.; Chuvikovsky, D.V.; Mikulinskaya, G.V.; Feofanov, S.A.; Miroshnikov, A.I. Chemical and Chemoenzymatic Synthesis of Nucleoside 5′-α- Thiotriphosphates. Russ. J. Bioorg. Chem. 2003, 29, 560–565. [Google Scholar] [CrossRef]
- Burgers, P.M.J.; Eckstein, F. A Study of the Mechanism of DNA Polymerase I from Escherichia Coli with Diastereomeric Phosphorothioate Analogs of Deoxyadenosine Triphosphate. J. Biol. Chem. 1979, 254, 6889–6893. [Google Scholar] [CrossRef] [PubMed]
- Brody, R.S.; Adler, S.; Modrich, P.; Stec, W.J.; Leznikowski, Z.J.; Frey, P.A. Stereochemical Course of Nucleotidyl Transfer Catalyzed by Bacteriophage T7 Induced DNA Polymerase. Biochemistry 1982, 21, 2570–2572. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.D.; Potter, B.V.L.; Eperon, I.C. Stereospecificity of Nucleases towards Phosphorothioate-Substituted RNA: Stereochemistry of Transcription by T7 RNA Polymerase. Nucleic Acids Res. 1987, 15, 4145–4162. [Google Scholar] [CrossRef] [PubMed]
- Hacia, J.G.; Wold, B.J.; Dervan, P.B. Phosphorothioate Oligonucleotide-Directed Triple Helix Formation. Biochemistry 1994, 33, 5367–5369. [Google Scholar] [CrossRef]
- Tang, J.; Roskey, A.; Li, Y.; Agrawal, S. Enzymatic Synthesis of Stereoregular (All Rp) Oligonucleotide Phosphorothioate and Its Properties. Nucleosides Nucleotides 1995, 14, 985–990. [Google Scholar] [CrossRef]
- Matzura, H.; Eckstein, F. A Polyribonucleotide Containing Alternating → P = O and → P = S Linkages. Eur. J. Biochem. 1968, 3, 448–452. [Google Scholar] [CrossRef]
- Eckstein, F.; Schulz, H.H.; Rüterjans, H.; Haar, W.; Maurer, W. Stereochemistry of the Ttransesterification Step of Ribonuclease T1. Biochemistry 1972, 11, 3507–3512. [Google Scholar] [CrossRef]
- Burgers, P.M.J.; Eckstein, F. Absolute Configuration of the Diastereomers of Adenosine 5’-O-(1-Thiotriphosphate): Consequences for the Stereochemistry of Polymerization by DNA-Dependent RNA Polymerase from Escherichia Coli. Proc. Natl. Acad. Sci. USA 1978, 75, 4798–4800. [Google Scholar] [CrossRef]
- Potter, B.V.L.; Connolly, B.A.; Eckstein, F. Synthesis and Configurational Analysis of a Dinucleoside Phosphate Isotopically Chiral at Phosphorus. Stereochemical Course of Penicillium Citrum Nuclease P1 Reaction. Biochemistry 1983, 22, 1369–1377. [Google Scholar] [CrossRef]
- Eckstein, F.; Gish, G. Phosphorothioates in Molecular Biology. Trends Biochem. Sci. 1989, 14, 97–100. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioate Oligodeoxynucleotides: What Is Their Origin and What Is Unique about Them? Antisense Nucleic Acid. Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Hyjek-Składanowska, M.; Vickers, T.A.; Napiórkowska, A.; Anderson, B.A.; Tanowitz, M.; Crooke, S.T.; Liang, X.H.; Seth, P.P.; Nowotny, M. Origins of the Increased Affinity of Phosphorothioate-Modified Therapeutic Nucleic Acids for Proteins. J. Am. Chem. Soc. 2020, 142, 7456–7468. [Google Scholar] [CrossRef]
- Elskens, J.P.; Elskens, J.M.; Madder, A. Chemical Modification of Aptamers for Increased Binding Affinity in Diagnostic Applications: Current Status and Future Prospects. Int. J. Mol. Sci. 2020, 21, 4522. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Akutsu, Y.; Yamasaki, T.; Miyagishi, M.; Kubota, T. Enhanced Affinity of Racemic Phosphorothioate DNA with Transcription Factor SATB1 Arising from Diastereomer-Specific Hydrogen Bonds and Hydrophobic Contacts. Nucleic Acids Res. 2020, 48, 4551–4561. [Google Scholar] [CrossRef]
- Zelikman, V.; Pelletier, J.; Simhaev, L.; Sela, A.; Gendron, F.P.; Arguin, G.; Senderowitz, H.; Sévigny, J.; Fischer, B. Highly Selective and Potent Ectonucleotide Pyrophosphatase-1 (NPP1) Inhibitors Based on Uridine 5′-P α,α -Dithiophosphate Analogues. J. Med. Chem. 2018, 61, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, L.K.; El-Khoury, R.; Thorpe, J.D.; Damha, M.J.; Hollenstein, M. Recent Progress in Non-Native Nucleic Acid Modifications. Chem. Soc. Rev. 2021, 50, 5126–5164. [Google Scholar] [CrossRef]
- Selmi, B.; Boretto, J.; Sarfati, S.R.; Guerreiro, C.; Canard, B. Mechanism-Based Suppression of Dideoxynucleotide Resistance by K65R Human Immunodeficiency Virus Reverse Transcriptase Using an α-Boranophosphate Nucleoside Analogue. J. Biol. Chem. 2001, 276, 48466–48472. [Google Scholar] [CrossRef]
- Cheek, M.A.; Dobrikov, M.I.; Wennefors, C.K.; Xu, Z.; Hashmi, S.N.; Shen, X.; Shaw, B.R. Synthesis and Properties of (Alpha-P-Borano)-Nucleoside 5’-Triphosphate Analogues as Potential Antiviral Agents. Nucleic Acids Symp. Ser. 2008, 52, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Porter, K.; Huang, F.; Shaw, B.R. Boron-Containing Oligodeoxyribonucleotide 14mer Duplexes: Enzymatic Synthesis and Melting Studies. Nucleic Acids Res. 1995, 23, 4495–4501. [Google Scholar] [CrossRef]
- Shaw, B.R.; Sergueev, D.; He, K.; Porter, K.; Summers, J.; Sergueeva, Z.; Rait, V. Boranophosphate Backbone: A Mimic of Phosphodiesters, Phosphorothioates, and Methyl Phosphonates. Methods Enzym. 2000, 313, 226–257. [Google Scholar] [CrossRef]
- Lato, S.M.; Ozerova, N.D.S.; He, K.; Sergueeva, Z.; Shaw, B.R.; Burke, D.H. Boron-Containing Aptamers to ATP. Nucleic Acids Res. 2002, 30, 1401–1407. [Google Scholar] [CrossRef]
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.-G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The Chemistry of Neutron Capture Therapy. Chem. Rev. 1998, 98, 2389–2390. [Google Scholar] [CrossRef]
- Brad Wan, W.; Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667. [Google Scholar] [CrossRef]
- Miller, P.S.; Yano, J.; Yano, E.; Carroll, C.; Jayaraman, K.; Ts’o, P.O.P. Nonionic Nucleic Acid Analogues. Synthesis and Characterization of Dideoxyribonucleoside Methylphosphonates. Biochemistry 1979, 18, 5134–5143. [Google Scholar] [CrossRef] [PubMed]
- Thiviyanathan, V.; Vyazovkina, K.V.; Gozansky, E.K.; Bichenchova, E.; Abramova, T.V.; Luxon, B.A.; Lebedev, A.V.; Gorenstein, D.G. Structure of Hybrid Backbone Methylphosphonate DNA Heteroduplexes: Effect of R and S Stereochemistry. Biochemistry 2002, 41, 827–838. [Google Scholar] [CrossRef]
- Jensen, M.A.; Davis, R.W. Template-Independent Enzymatic Oligonucleotide Synthesis (TiEOS): Its History, Prospects, and Challenges. Biochemistry 2018, 57, 1821–1832. [Google Scholar] [CrossRef]
- Liu, W.; Iwamoto, N.; Marappan, S.; Luu, K.; Tripathi, S.; Purcell-Estabrook, E.; Shelke, J.D.; Shah, H.; Lamattina, A.; Pan, Q.; et al. Impact of Stereopure Chimeric Backbone Chemistries on the Potency and Durability of Gene Silencing by RNA Interference. Nucleic Acids Res. 2023, 51, 4126–4147. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, P.; Liu, Y.; Aduda, V.; Akare, S.; Alam, R.; Andreucci, A.; Boulay, D.; Bowman, K.; Byrne, M.; Cannon, M.; et al. Impact of Guanidine-Containing Backbone Linkages on Stereopure Antisense Oligonucleotides in the CNS. Nucleic Acids Res. 2022, 50, 5401–5423. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.Y.; McLaughlin, S.H.; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science (1979) 2012, 336, 341–344. [Google Scholar] [CrossRef]
- Houlihan, G.; Arangundy-Franklin, S.; Holliger, P. Engineering and Application of Polymerases for Synthetic Genetics. Curr. Opin. Biotechnol. 2017, 48, 168–179. [Google Scholar] [CrossRef]
- Taylor, A.I.; Holliger, P. Selecting Fully-Modified XNA Aptamers Using Synthetic Genetics. Curr. Protoc. Chem. Biol. 2018, 10, e44. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.I.; Pinheiro, V.B.; Smola, M.J.; Morgunov, A.S.; Peak-Chew, S.; Cozens, C.; Weeks, K.M.; Herdewijn, P.; Holliger, P. Catalysts from Synthetic Genetic Polymers. Nature 2015, 518, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.I.; Beuron, F.; Peak-Chew, S.Y.; Morris, E.P.; Herdewijn, P.; Holliger, P. Nanostructures from Synthetic Genetic Polymers. ChemBioChem 2016, 17, 1107–1110. [Google Scholar] [CrossRef] [PubMed]






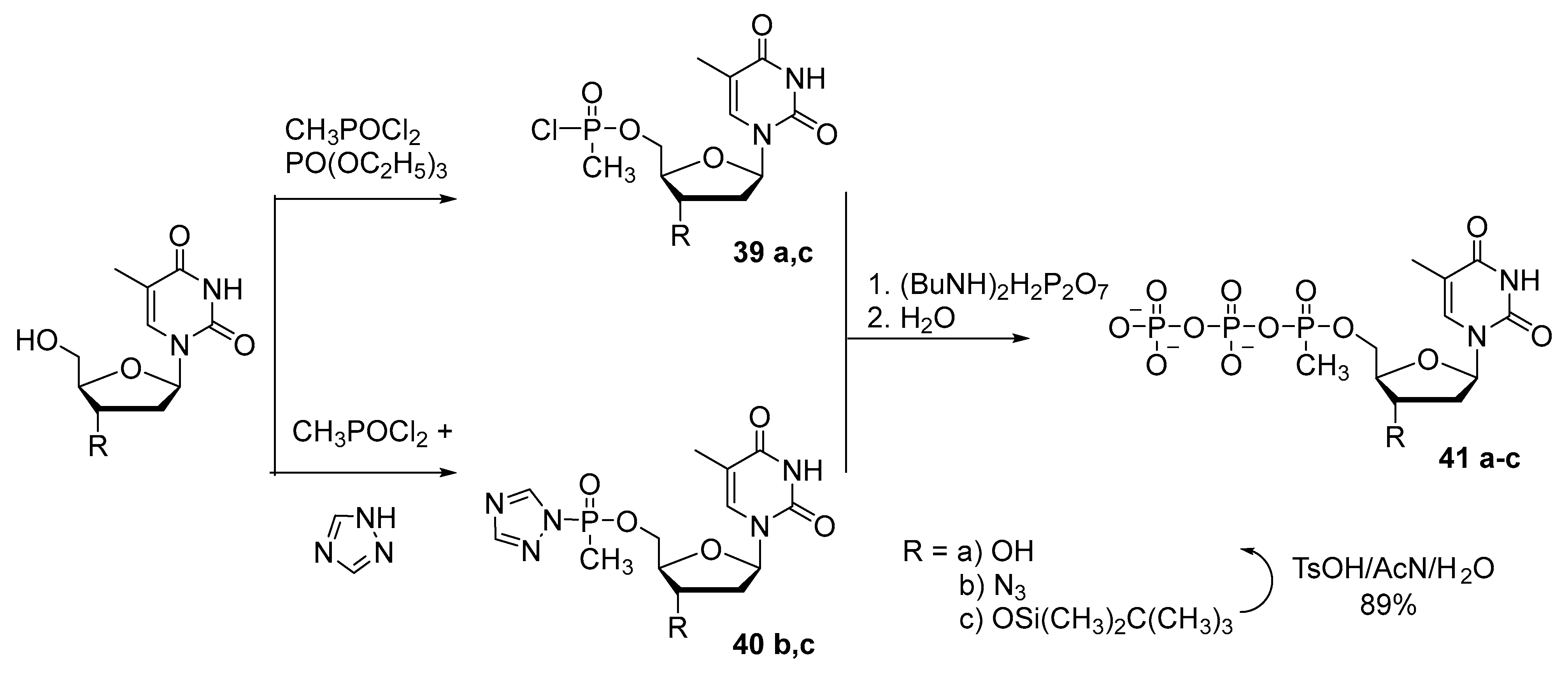
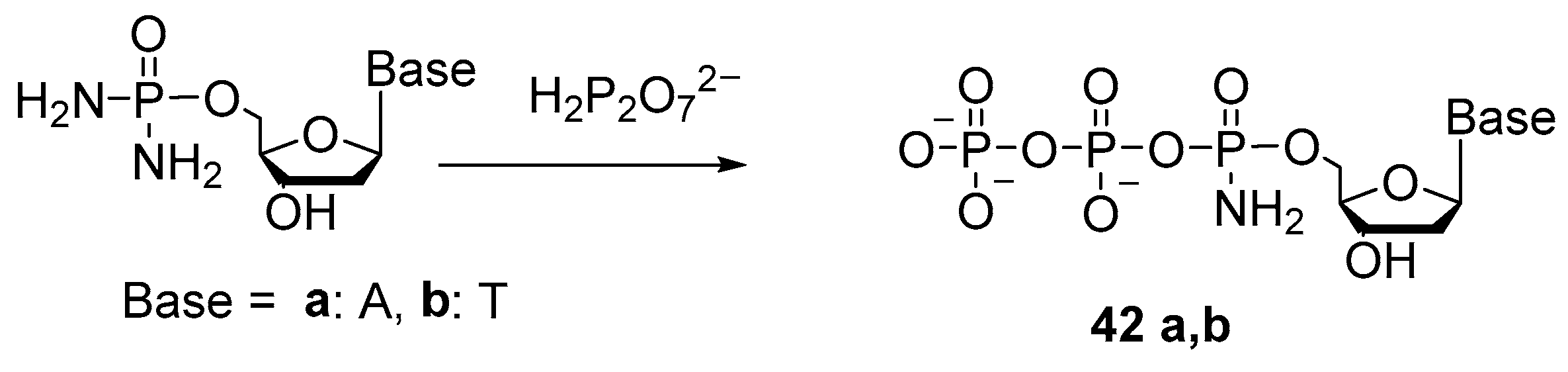


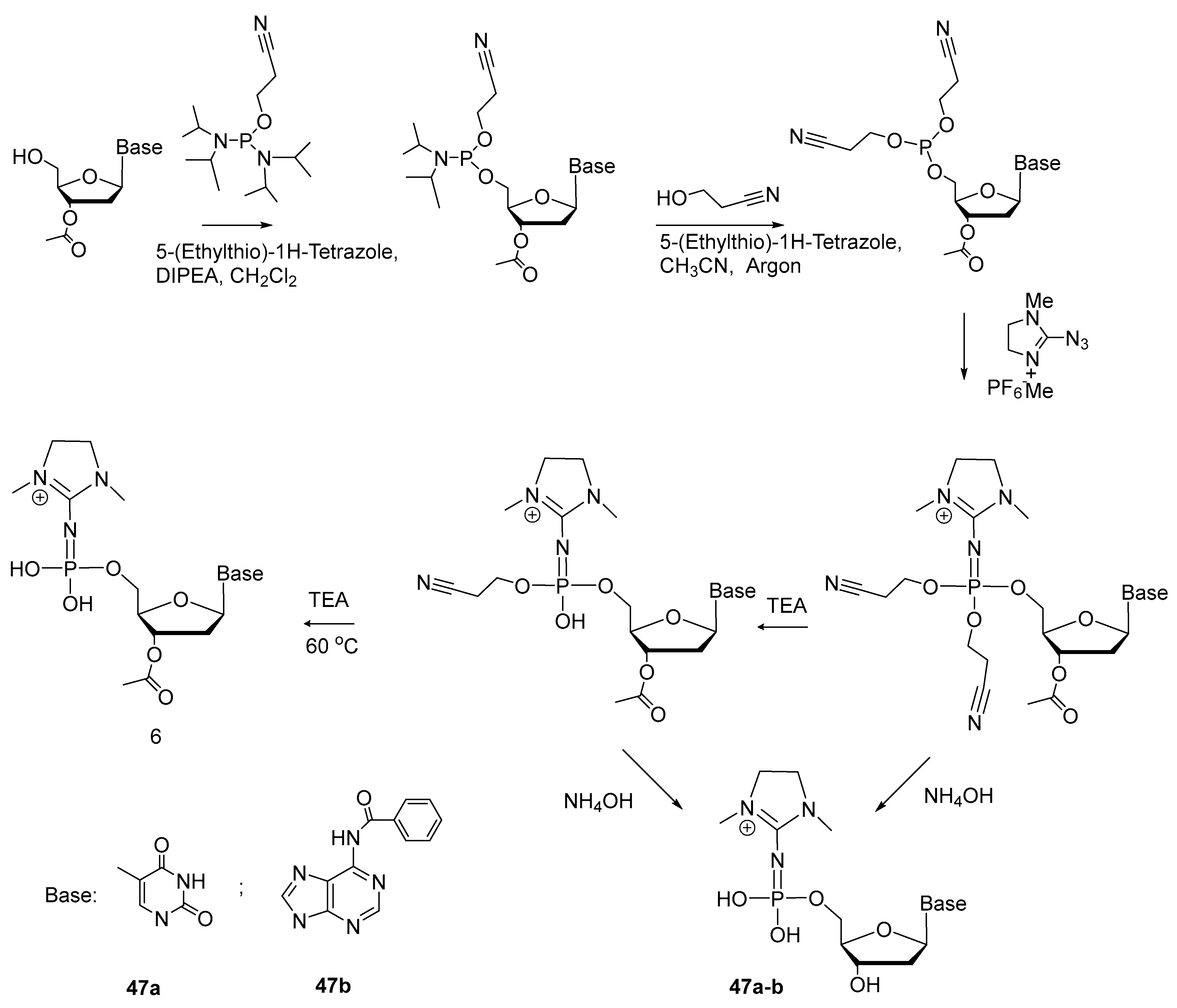
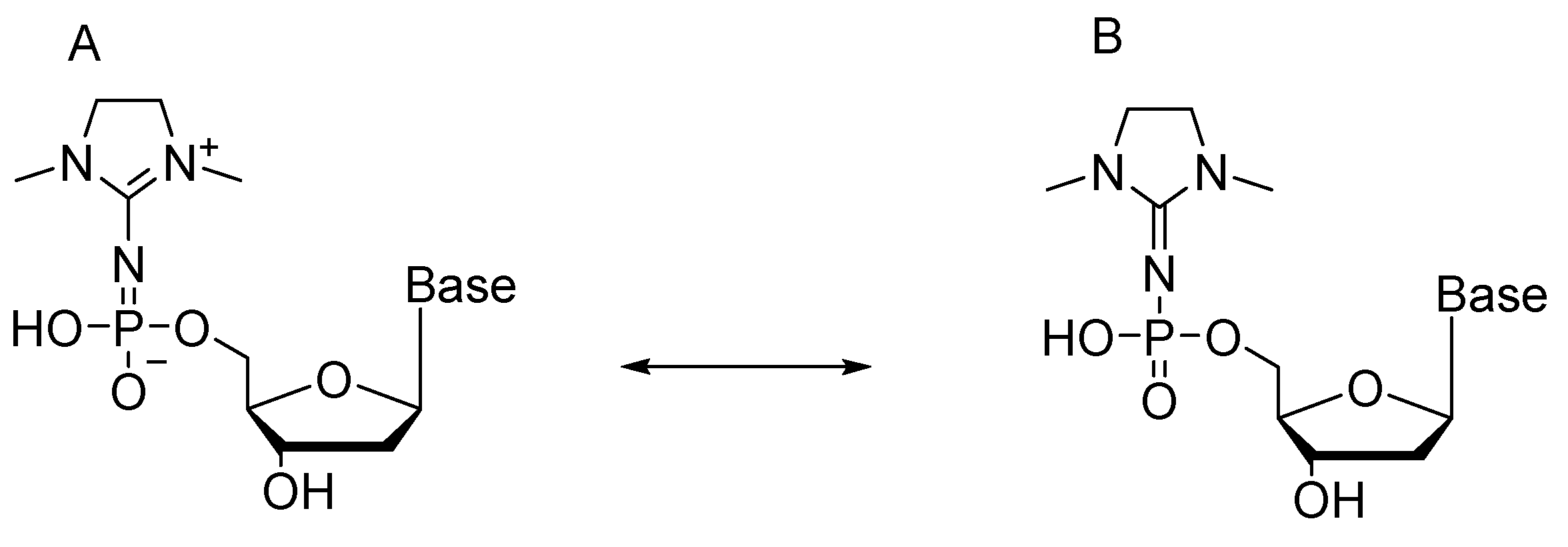



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthetic Approach | 5′-(α-P-Thio)triphosphate (Yield, %) | 5′-(α-P-Seleno)triphosphate (Yield, %) | 5′-(α-P-Borano)triphosphate (Yield, %) | 5′-(α-P-Alkyl)triphosphate (Yield, %) | 5′-(α-P-Amido)triphosphate and 5′-(α-P-Alkylamido)triphosphate (Yield, %) | 5′-(α-P-Imido)triphosphate (Yield, %) |
|---|---|---|---|---|---|---|
| 1. Coupling of pyrophosphate with activated forms of modified NMP | dTTP (5) * [19] ATP (20) [20] ATP (25) [21] ddNTP (15–20) [23] NTP, dNTP (58–78) [25] (α-P-dithio)NTP (12–26) [26] oligonucleotides (30–50) [24] | ATP (31) [25] - | (α-P-methyl)dTTP (20–32) [36,39,40,42] (α-P-methyl)dNTP (80–85) [41] 3′-azido-5′-(α-P-methyl)dTTP (20–30) [36] (α-P-ethyl)dNTP (1–2) [43] | (α-P-amido)dTTP (72) [44] (α-P-amido)dATP (57) [44] | ||
| 2. Cyclic PVPVPIII-nucleoside intermediates | NTP, dNTP (60–75) [24,27] NTP, dNTP (45–60), [16] (α-P-borano, α-P-thio)dTTP (26) [30] 3′-azido-5′-(α-P-thio)dTTP (59) [31] | NTP, dNTP (30–67) [28,29] 3′-azido-5′-[α-P-seleno]dTTP (40) [31] | dNTP (25–43) [36] NTP, dNTP (35–45) [35] Acyclo-TTP (53) [34] [α-P-borano, α-P-thio]dTTP (26) [30] 5-R-dCTP (33–52) [37] ATP (31–43) [33] | (α-P-amido)dTTP [46] (α-P-alkylamido)dGTP [46] | 2′-deoxy, 5′-(α-P-(4-acetamidobenzenesulfonyl)imido)dTTP [48] | |
| 3. Linear PVPVPIII-nucleoside intermediates | NTP, dNTP (66–76) [10] | NTP, dNTP (54–63) [10] | NTP, dNTP (67–70) [10] | |||
| 4. Amidophosphite method | dTTP (30) [32] | |||||
| 5. Staudinger reaction | (α-P-(1,3-dimethylimidazolidin-2-ylidene)dTTP (3) [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novgorodtseva, A.I.; Lomzov, A.A.; Vasilyeva, S.V. Synthesis and Properties of α-Phosphate-Modified Nucleoside Triphosphates. Molecules 2024, 29, 4121. https://doi.org/10.3390/molecules29174121
Novgorodtseva AI, Lomzov AA, Vasilyeva SV. Synthesis and Properties of α-Phosphate-Modified Nucleoside Triphosphates. Molecules. 2024; 29(17):4121. https://doi.org/10.3390/molecules29174121
Chicago/Turabian StyleNovgorodtseva, Alina I., Alexander A. Lomzov, and Svetlana V. Vasilyeva. 2024. "Synthesis and Properties of α-Phosphate-Modified Nucleoside Triphosphates" Molecules 29, no. 17: 4121. https://doi.org/10.3390/molecules29174121
APA StyleNovgorodtseva, A. I., Lomzov, A. A., & Vasilyeva, S. V. (2024). Synthesis and Properties of α-Phosphate-Modified Nucleoside Triphosphates. Molecules, 29(17), 4121. https://doi.org/10.3390/molecules29174121