
In Silico Molecular Modeling of Four New Afatinib Derived Molecules Targeting the Inhibition of the Mutated Form of BCR-ABL T315I

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
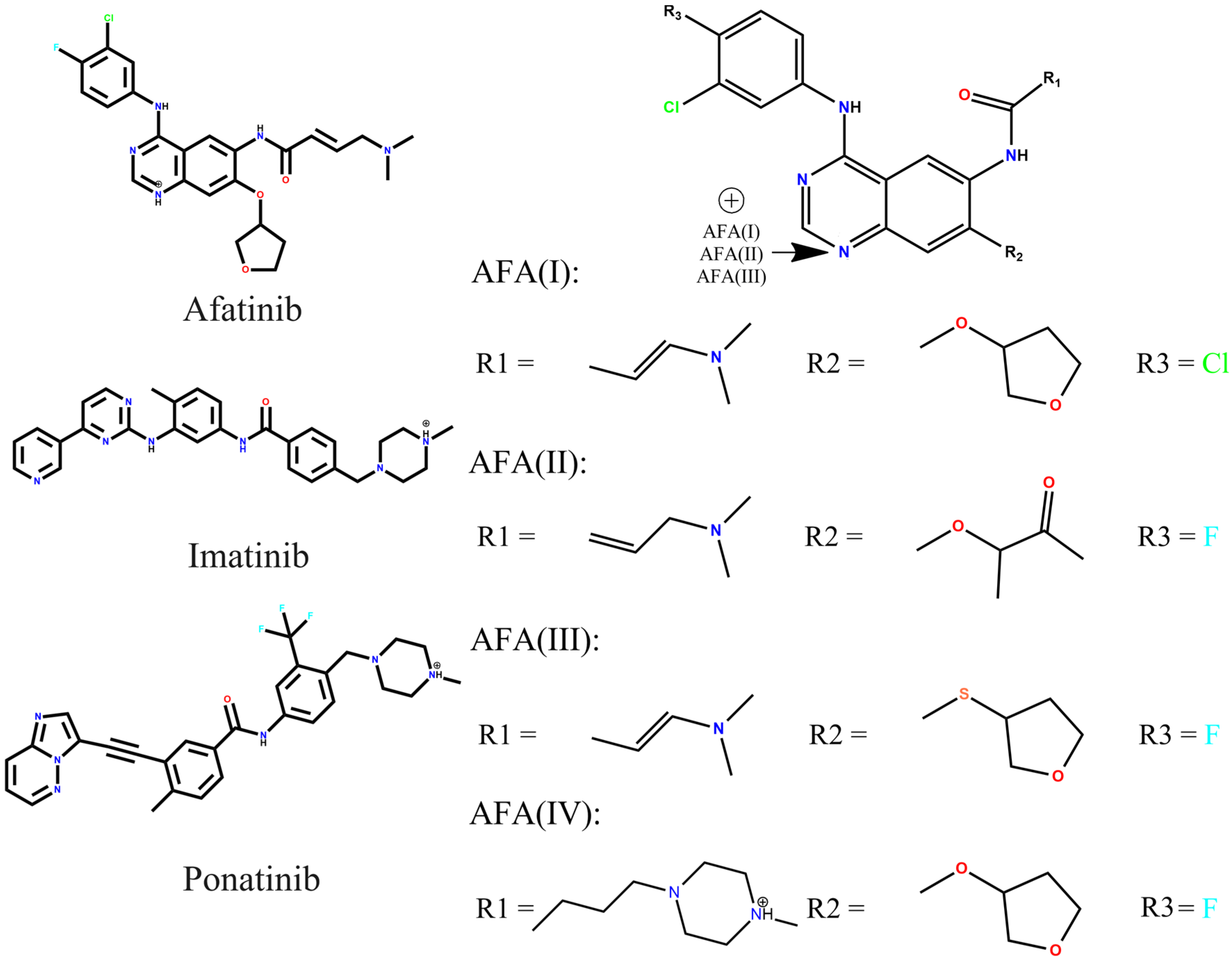
2.1. Electronic Structure, Frontier Molecular Orbitals
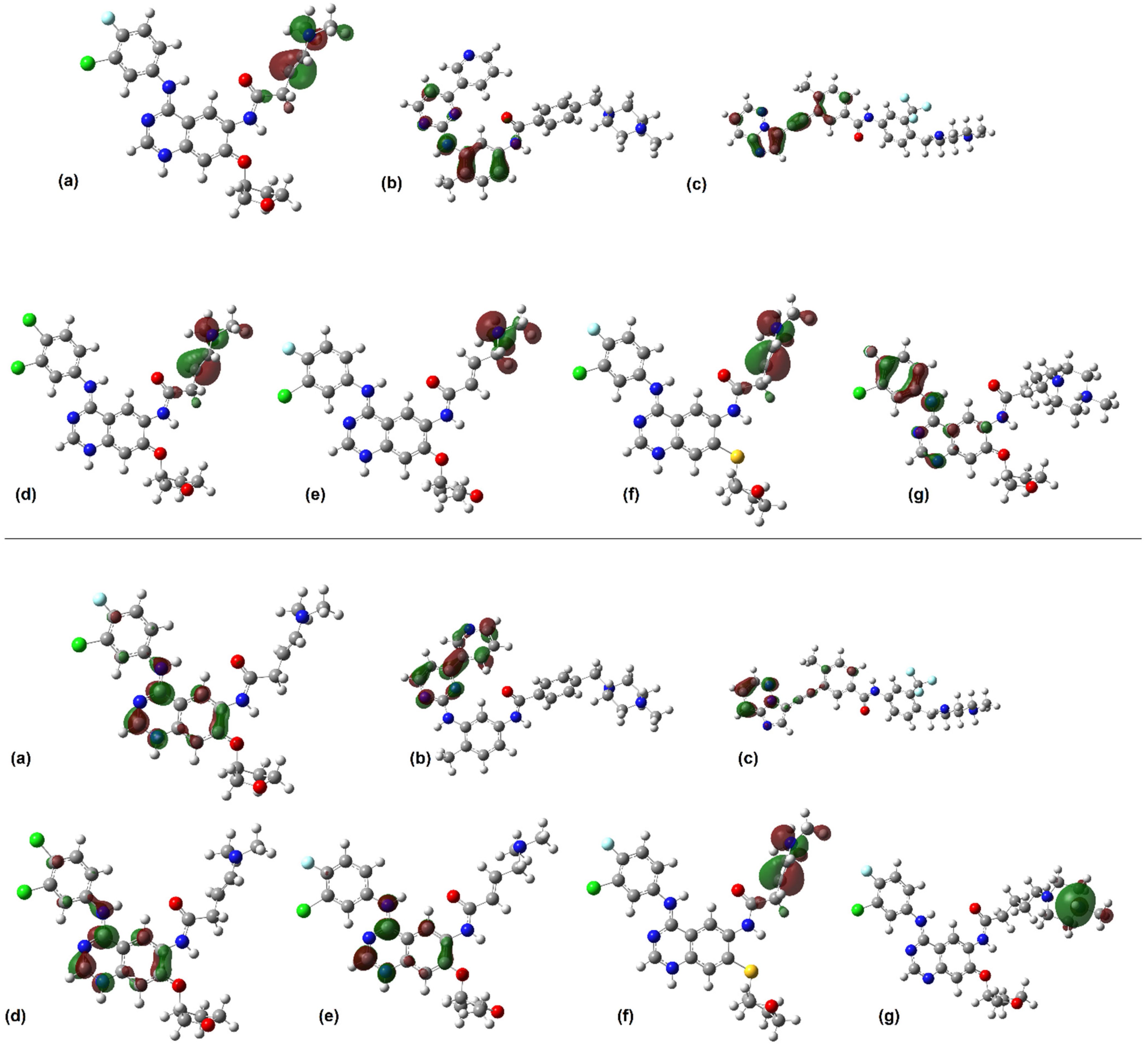
2.2. Fukui Functions
- f0: This function describes the tendency of electrons to flow in and out of a specific region in the molecular structure. This function can be used to indicate regions associated with radical attacks in molecular entities.
- f+: This function represents nucleophilic attack potential. Regions within a molecule that concentrate this function are where electrons are most likely to be donated, indicating high reactivity towards nucleophiles.
- f−: This function represents the electrophilic attack susceptibility, identifying the moieties where electrons are most likely to be accepted.
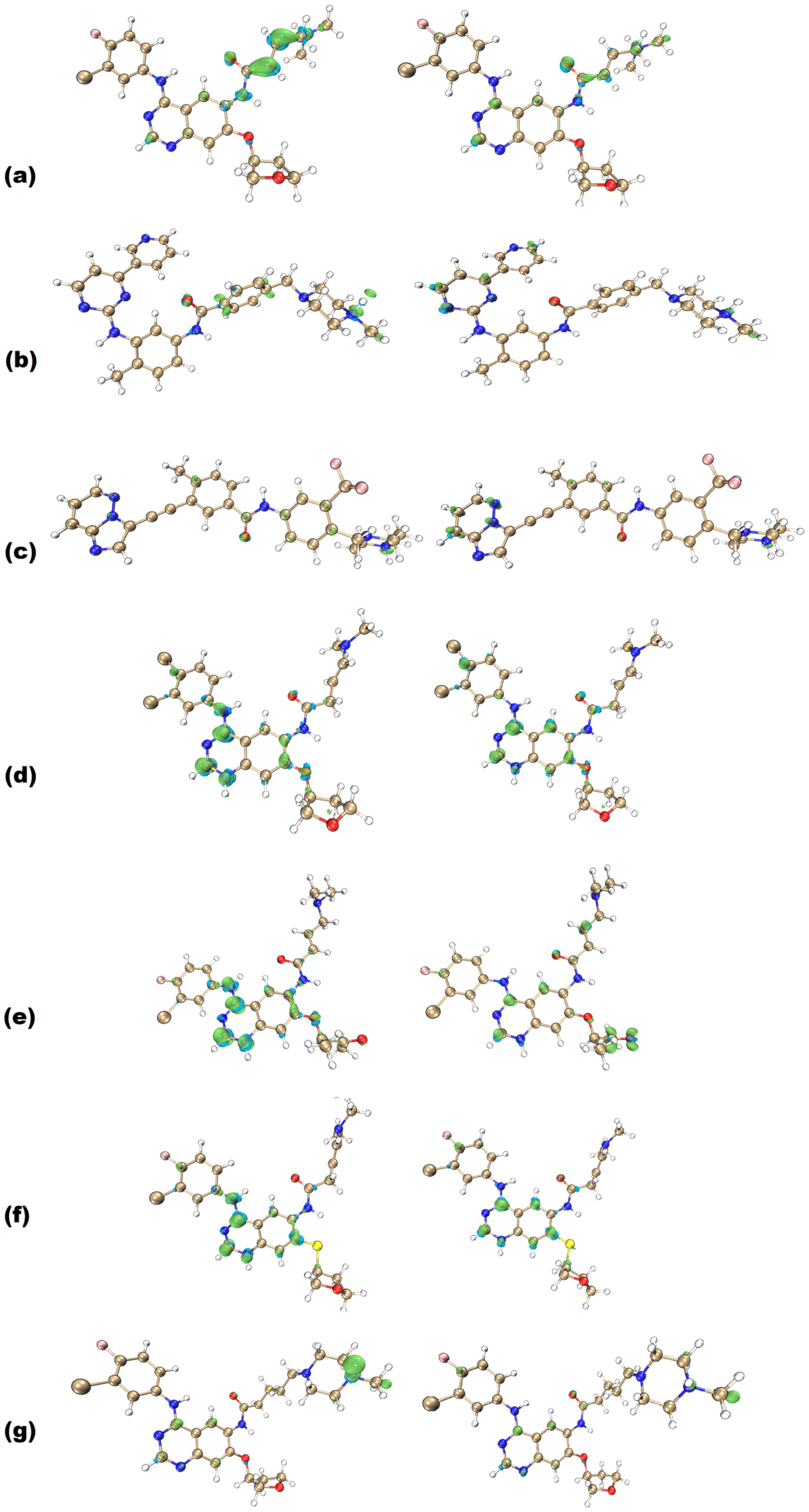
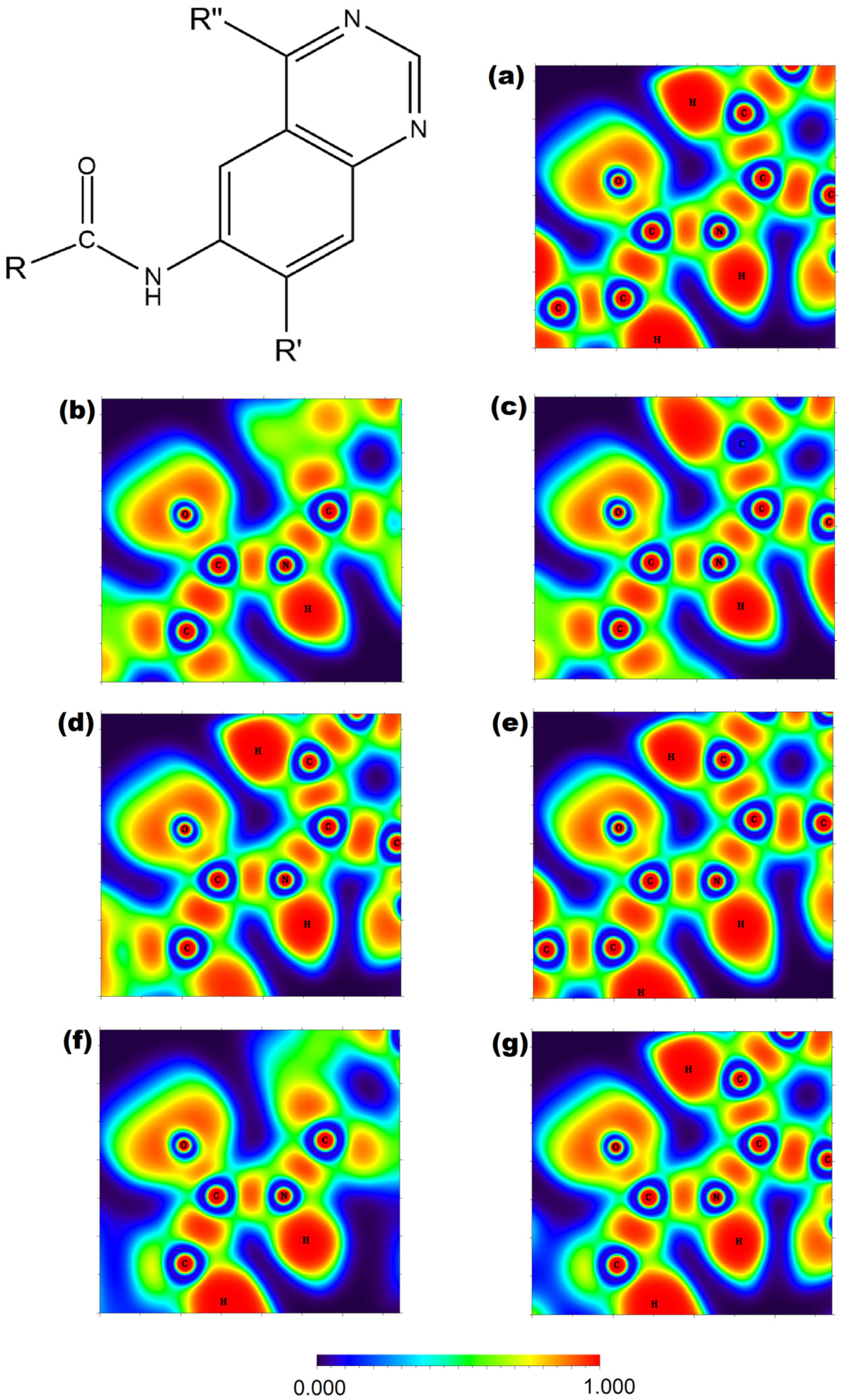
2.3. Electron Localization Function Analysis
2.4. ADMET Evaluation
2.5. Molecular Docking Studies
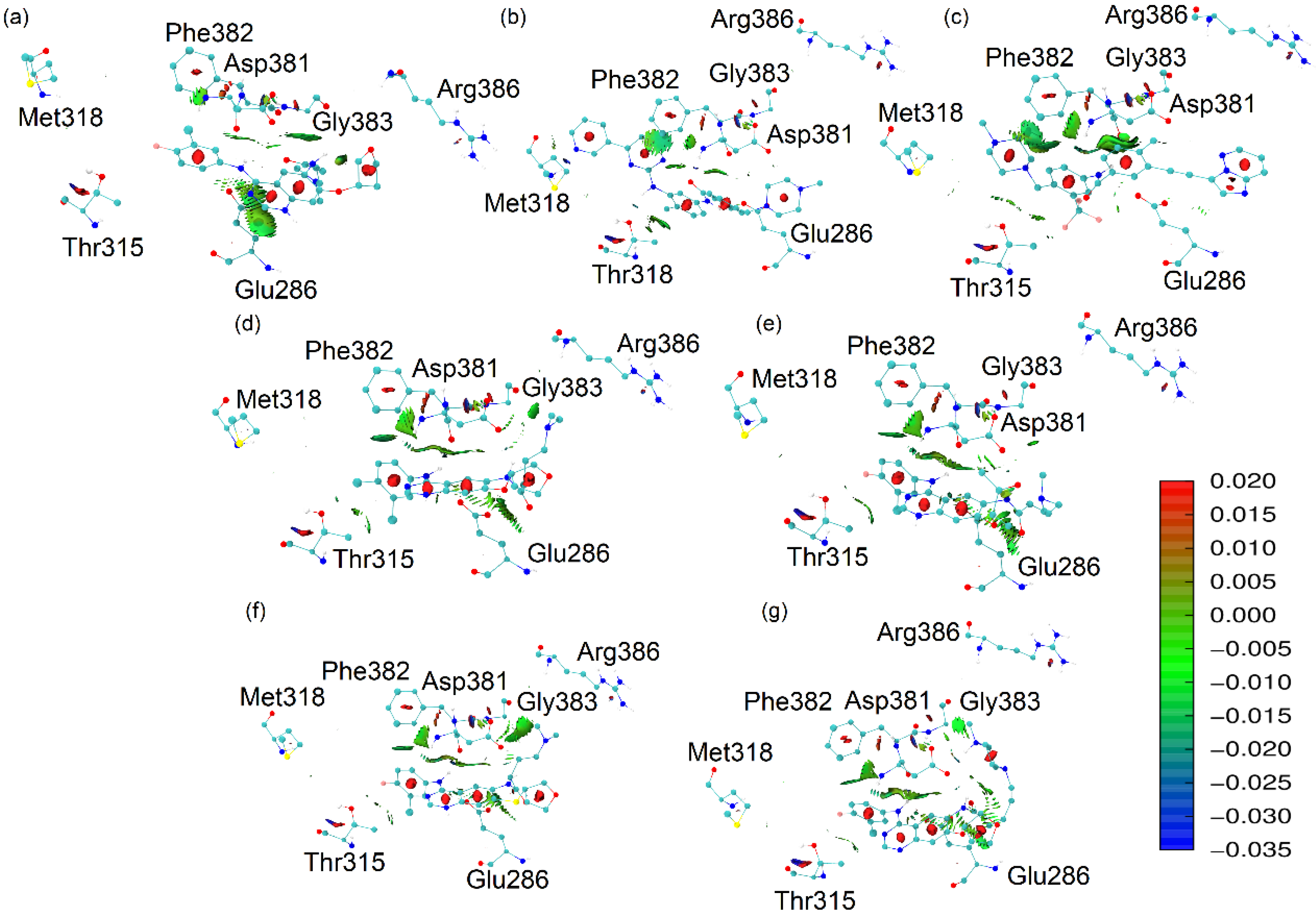
2.6. Non-Covalent Interactions Index
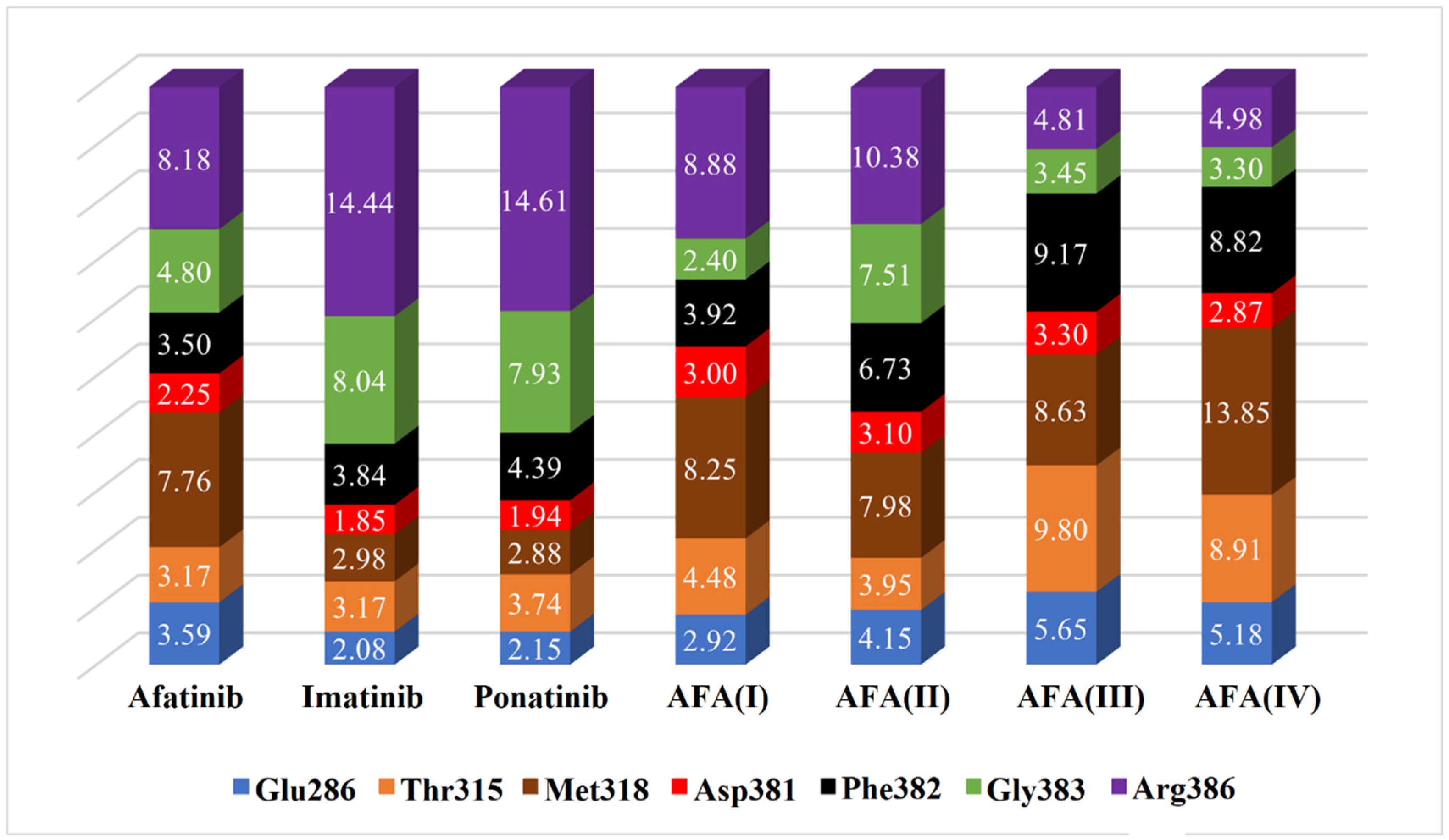
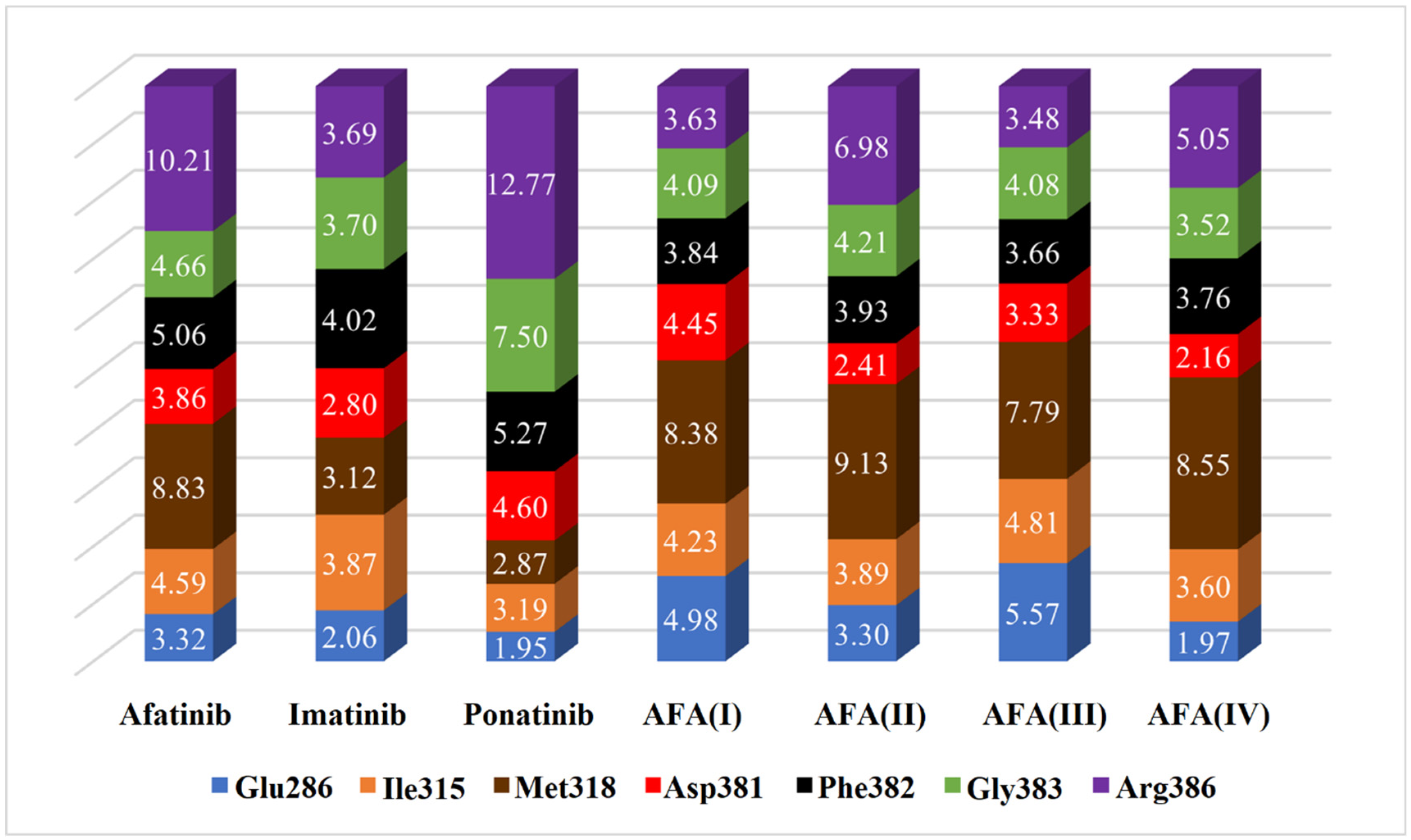
2.7. Heat Map
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C.L. Chronic Myeloid Leukemia. N. Engl. J. Med. 1999, 340, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg, B.P.; Menias, C.O.; Lubner, M.G.; Mellnick, V.M.; Pickhardt, P.J. Splenomegaly: A Combined Clinical and Radiologic Approach to the Differential Diagnosis. Gastroenterol. Clin. N. Am. 2018, 47, 643–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Elghawy, O.; Kurpiel, B.R.; Douvas, M.G. Diagnosis and Management of Atypical Chronic Myeloid Leukemia with a t(2;13)(Q33;Q12) Translocation. Case Rep. Hematol. 2022, 2022, 4628183. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kalaycio, M. Accelerated Phase CML: Outcomes in Newly Diagnosed vs. Progression From Chronic Phase. Curr. Hematol. Malig. Rep. 2016, 11, 86–93. [Google Scholar] [CrossRef]
- Dutcher, J.P.; Wiernik, P.H. Accelerated and Blastic Phase of Chronic Myeloid Leukemia. Curr. Treat. Options Oncol. 2000, 1, 51–62. [Google Scholar] [CrossRef]
- How, J.; Venkataraman, V.; Hobbs, G.S. Blast and Accelerated Phase CML: Room for Improvement. Hematol. Am. Soc. Hematol. Educ. Progr. 2021, 2021, 122. [Google Scholar] [CrossRef] [PubMed]
- Chopra, R.; Pu, Q.Q.; Elefanty, A.G. Biology of BCR-ABL. Blood Rev. 1999, 211–229. [Google Scholar] [CrossRef]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Proetel, U.; Müller, M.C.; Hanfstein, B.; Schreiber, A.; Fabarius, A.; Pfirrmann, M.; Schnittger, S.; et al. Younger Patients with Chronic Myeloid Leukemia Do Well in Spite of Poor Prognostic Indicators: Results from the Randomized CML Study IV. Ann. Hematol. 2014, 93, 71. [Google Scholar] [CrossRef]
- Osman, A.E.G.; Deininger, M.W. Chronic Myeloid Leukemia: Modern Therapies, Current Challenges and Future Directions. Blood Rev. 2021, 49, 100825. [Google Scholar] [CrossRef]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia Chromosome in Leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic Myeloid Leukemia: 2018 Update on Diagnosis, Therapy and Monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic Myeloid Leukemia: The Paradigm of Targeting Oncogenic Tyrosine Kinase Signaling and Counteracting Resistance for Successful Cancer Therapy. Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pendergast, A.M.; Muller, A.J.; Havlik, M.H.; Maru, Y.; Witte, O.N. BCR Sequences Essential for Transformation by the BCR-ABL Oncogene Bind to the ABL SH2 Regulatory Domain in a Non-Phosphotyrosine-Dependent Manner. Cell 1991, 66, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Bentz, M.; Cabot, G.; Moos, M.; Speicher, M.; Ganser, A.; Lichter, P.; Dohner, H. Detection of Chimeric BCR-ABL Genes on Bone Marrow Samples and Blood Smears in Chronic Myeloid and Acute Lymphoblastic Leukemia by in Situ Hybridization. Blood 1994, 83, 1922–1928. [Google Scholar] [CrossRef]
- Elrayess, R.; Abdel Aziz, Y.M.; Elgawish, M.S.; Elewa, M.; Yassen, A.S.A.A.; Elhady, S.S.; Elshihawy, H.A.; Said, M.M. Discovery of Potent Dual Egfr/Her2 Inhibitors Based on Thiophene Scaffold Targeting H1299 Lung Cancer Cell Line. Pharmaceuticals 2021, 14, 9. [Google Scholar] [CrossRef]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The Biology of Chronic Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL Suppresses Autophagy through ATF5-Mediated Regulation of MTOR Transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef]
- Stein, S.J.; Baldwin, A.S. NF-B Suppresses ROS Levels in BCR-ABL+ Cells to Prevent Activation of JNK and Cell Death. Oncogene 2011, 30, 4557–4566. [Google Scholar] [CrossRef]
- Reddy, E.P.; Aggarwal, A.K. The Ins and Outs of Bcr-Abl Inhibition. Genes Cancer 2012, 3, 447–454. [Google Scholar] [CrossRef]
- Pricl, S.; Fermeglia, M.; Ferrone, M.; Tamborini, E. T315I-Mutated Bcr-Abl in Chronic Myeloid Leukemia and Imatinib: Insights from a Computational Study. Mol. Cancer Ther. 2005, 4, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Pandrala, M.; Bruyneel, A.A.N.; Hnatiuk, A.P.; Mercola, M.; Malhotra, S.V. Designing Novel BCR-ABL Inhibitors for Chronic Myeloid Leukemia with Improved Cardiac Safety. J. Med. Chem. 2022, 65, 10898–10919. [Google Scholar] [CrossRef]
- Pereira, W.A.; Nascimento, É.C.M.; Martins, J.B.L. Electronic and Structural Study of T315I Mutated Form in DFG-out Conformation of BCR-ABL Inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 9774–9788. [Google Scholar] [CrossRef]
- Nascimento, É.C.M.; de A. Nascimento, L.; Benicio, L.F.M.A.; Alcântara, J.L.L.; de Pereira, W.A.; Martins, J.B.L. Electronic and Structural Insights of BCR-ABL Inhibitors Under LMC Treatment Perspective. In Research Topics in Bioactivity, Environment and Energy: Experimental and Theoretical Tools; Taft, C.A., de Lazaro, S.R., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 389–404. ISBN 978-3-031-07622-0. [Google Scholar]
- Dobrovic, A.; Peters, G.B.; Ford, J.H. Review: Molecular Analysis of the Philadelphia Chromosome. Chromosoma 1991, 100, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Henkes, M.; van der Kuip, H.; Aulitzky, W.E. Therapeutic Options for Chronic Myeloid Leukemia: Focus on Imatinib (Glivec®, GleevecTM). Ther. Clin. Risk Manag. 2008, 4, 163–187. [Google Scholar] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Muresan, B.; Mamolo, C.; Cappelleri, J.C.; Leip, E.; Viqueira, A.; Heeg, B. An Indirect Comparison between Bosutinib, Nilotinib and Dasatinib in First-Line Chronic Phase Chronic Myeloid Leukemia. Curr. Med. Res. Opin. 2021, 37, 801–809. [Google Scholar] [CrossRef]
- von Amsberg, G.K. Schafhausen Bosutinib in the Management of Chronic Myelogenous Leukemia. Biol. Targets Ther. 2013, 7, 115. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Huang, H.; Lei, X.; Tang, G.; Cao, X.; Peng, J. Recent Advances in Bcr-Abl Tyrosine Kinase Inhibitors for Overriding T315I Mutation. Chem. Biol. Drug Des. 2021, 97, 649–664. [Google Scholar] [CrossRef]
- Kamasani, S.; Akula, S.; Manga, V.; Duyster, J.; Vudem, D.R.; Kancha, R.K.; Sivan, S.K.; Manga, V.; Duyster, J.; Vudem, D.R.; et al. Computational Analysis of ABL Kinase Mutations Allows Predicting Drug Sensitivity against Selective Kinase Inhibitors. Tumor Biol. 2017, 39, 1010428317701643. [Google Scholar] [CrossRef]
- Zhou, T.; Commodore, L.; Huang, W.S.; Wang, Y.; Thomas, M.; Keats, J.; Xu, Q.; Rivera, V.M.; Shakespeare, W.C.; Clackson, T.; et al. Structural Mechanism of the Pan-BCR-ABL Inhibitor Ponatinib (AP24534): Lessons for Overcoming Kinase Inhibitor Resistance. Chem. Biol. Drug Des. 2011, 77, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q.; et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef]
- Marto, J.P.; Strambo, D.; Livio, F.; Michel, P. Drugs Associated With Ischemic Stroke. Stroke 2021, 52, E646–E659. [Google Scholar] [CrossRef]
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.C.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 Activating Mutations Are Targets for Colorectal Cancer Treatment. Cancer Discov. 2015, 5, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kage, H.; Nagoshi, S.; Toyama, K.; Ohno, Y.; Shinozaki-Ushiku, A.; Nakazaki, K.; Suzuki, H.; Kurokawa, M.; Nagase, T. Dual EGFR and ABL Tyrosine Kinase Inhibitor Treatment in a Patient with Concomitant EGFR-Mutated Lung Adenocarcinoma and BCR-ABL1-Positive CML. Case Rep. Oncol. Med. 2020, 2020, 4201727. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Shatsky, R.; Harbeck, N. Afatinib in the Treatment of Breast Cancer. Expert Opin. Investig. Drugs 2014, 23, 1039–1047. [Google Scholar] [CrossRef]
- Lai, W.V.; Lebas, L.; Barnes, T.A.; Milia, J.; Ni, A.; Gautschi, O.; Peters, S.; Ferrara, R.; Plodkowski, A.J.; Kavanagh, J.; et al. Afatinib in Patients with Metastatic or Recurrent HER2-Mutant Lung Cancers: A Retrospective International Multicentre Study. Eur. J. Cancer 2019, 109, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Dungo, R.T.; Keating, G.M. Afatinib: First Global Approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef]
- Rocha, K.M.L.; Nascimento, É.C.M.; Martins, J.B.L. Investigation on the Interaction Behavior of Afatinib, Dasatinib, and Imatinib Docked to the BCR-ABL Protein. J. Mol. Model. 2021, 27, 309. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Chang, C.-F.; Coumar, M.S.; Chen, P.-Y.; Kuo, F.-M.; Chen, C.-H.; Li, M.-C.; Lin, W.-H.; Kuo, P.-C.; Wang, S.-Y.; et al. Drug-like Property Optimization: Discovery of Orally Bioavailable Quinazoline-Based Multi-Targeted Kinase Inhibitors. Bioorg. Chem. 2020, 98, 103689. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Alkhoja, O.A.; Mohamed, W.R. Design, Synthesis and Antitumor Activity of Novel Pyrazolo[3,4-d]Pyrimidine Derivatives as EGFR-TK Inhibitors. Bioorg. Chem. 2016, 66, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Khodair, A.I.; Alsafi, M.A.; Nafie, M.S. Synthesis, Molecular Modeling and Anti-Cancer Evaluation of a Series of Quinazoline Derivatives. Carbohydr. Res. 2019, 486, 107832. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Xie, L.; Wang, J.; Xu, X.; Zhang, Z.; Shi, J.; Le, X.; Hong, J. Discovery of New Quinazoline Derivatives as Irreversible Dual EGFR/HER2 Inhibitors and Their Anticancer Activities—Part 1. Bioorg. Med. Chem. Lett. 2019, 29, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Xie, L.; Wang, J.; Shi, J.; Hong, J. In Vivo Efficacy Studies of Novel Quinazoline Derivatives as Irreversible Dual EGFR/HER2 Inhibitors, in Lung Cancer Xenografts (NCI-H1975) Mice Models. Bioorg. Chem. 2020, 99, 103790. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauzá, A. Biological Halogen Bonds in Protein–Ligand Complexes: A Combined QTAIM and NCIPlot Study in Four Representative Cases. Org. Biomol. Chem. 2021, 19, 6858–6864. [Google Scholar] [CrossRef]
- İş, Y.S. Elucidation of Ligand/Protein Interactions between BCR-ABL Tyrosine Kinase and Some Commercial Anticancer Drugs Via DFT Methods. J. Comput. Biophys. Chem. 2021, 20, 433–447. [Google Scholar] [CrossRef]
- Banavath, H.N.; Sharma, O.P.; Kumar, M.S.; Baskaran, R. Identification of Novel Tyrosine Kinase Inhibitors for Drug Resistant T315I Mutant BCR-ABL: A Virtual Screening and Molecular Dynamics Simulations Study. Sci. Rep. 2014, 4, 6948. [Google Scholar] [CrossRef]
- Lara-Popoca, J.; Thoke, H.S.; Stock, R.P.; Rudino-Pinera, E.; Bagatolli, L.A. Inductive Effects in Amino Acids and Peptides: Ionization Constants and Tryptophan Fluorescence. Biochem. Biophys. Rep. 2020, 24, 100802. [Google Scholar] [CrossRef]
- Frye, L.; Bhat, S.; Akinsanya, K.; Abel, R. From Computer-Aided Drug Discovery to Computer-Driven Drug Discovery. Drug Discov. Today Technol. 2021, 39, 111–117. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G.; Pucci, R. Electron Density, Kohn-Sham Frontier Orbitals, and Fukui Functions. J. Chem. Phys. 1984, 81, 2862–2863. [Google Scholar] [CrossRef]
- Soverini, S.; Bassan, R.; Lion, T. Treatment and Monitoring of Philadelphia Chromosome-Positive Leukemia Patients: Recent Advances and Remaining Challenges. J. Hematol. Oncol. 2019, 12, 39. [Google Scholar] [CrossRef]
- Mahmoudi Gomari, M.; Rostami, N.; Ghodrati, A.; Hernandez, Y.; Fadaie, M.; Sadegh Eslami, S.; Tarighi, P. Implementation of Docking, Molecular Dynamics and Free Energy to Investigate Drug Potency of Novel BCR-ABLT315I Inhibitors as an Alternative to Ponatinib. Comput. Toxicol. 2021, 20, 100180. [Google Scholar] [CrossRef]
- Tarika, J.D.D.; Dexlin, X.D.D.; Madhanhumar, S.; Jayanthi, D.D.; Beaula, T.J. Tuning the Computational Evaluation of Spectroscopic, ELF, LOL, NCI Analysis and Molecular Docking of Novel Anti COVID-19 Molecule 4-Dimethylamino Pyridinium 3, 5-Dichlorosalicylate. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 259, 119907. [Google Scholar] [CrossRef]
- Tarika, J.D.D.; Dexlin, X.D.D.; Arun, A.; Jayanthi, D.D.; Rathika, A.; Beaula, T.J. Insights into Weak and Covalent Interactions, Reactivity Sites and Pharmacokinetic Studies of 4-Dimethylaminopyridinium Salicylate Monohydrate Using Quantum Chemical Computation Method. Comput. Theor. Chem. 2021, 1206, 113483. [Google Scholar] [CrossRef]
- Mirzaei, M.S.; Taherpour, A.A. Tautomeric Preferences of the Cis and Trans Isomers of Axitinib. Chem. Phys. 2018, 507, 10–18. [Google Scholar] [CrossRef]
- Mendes, J.A.; Merino, P.; Soler, T.; Salustiano, E.J.; Costa, P.R.R.; Yus, M.; Foubelo, F.; Buarque, C.D. Enantioselective Synthesis, DFT Calculations, and Preliminary Antineoplastic Activity of Dibenzo 1-Azaspiro[4.5]Decanes on Drug-Resistant Leukemias. J. Org. Chem. 2019, 84, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.A.; Elkady, H.; Hagras, M.; Husein, D.Z.; Ibrahim, I.M.; Taghour, M.S.; El-Mahdy, H.A.; Ismail, A.; Alsfouk, B.A.; Elkaeed, E.B.; et al. Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies of New Thieno[2,3-d]Pyrimidines Targeting VEGFR-2. RSC Adv. 2023, 13, 23365–23385. [Google Scholar] [CrossRef]
- Sampathkumar, J.; Rajamanickam, R. Synthesis, Crystal Structure, Hirshfeld Surface, QTAIM, NCI-RDG, DFT and Molecular Docking Studies of 4-(Aryl)-1,4-Dihydro-N,1-Dimethyl-6-(Methylthio)-3,5-Dinitropyridin-2-Amines. J. Mol. Struct. 2024, 1299, 137063. [Google Scholar] [CrossRef]
- Almeida, M.O.; Barros, D.A.S.; Araujo, S.C.; Faria, S.H.D.M.; Maltarollo, V.G.; Honorio, K.M. Study on Molecular Structure, Spectroscopic Properties (FTIR and UV–Vis), NBO, QTAIM, HOMO-LUMO Energies and Docking Studies of 5-Fluorouracil, a Substance Used to Treat Cancer. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2017, 184, 169–176. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Arulaabaranam, K.; Mani, G.; Muthu, S. Computational Assessment on Wave Function (ELF, LOL) Analysis, Molecular Confirmation and Molecular Docking Explores on 2-(5-Amino-2- Methylanilino)-4-(3-Pyridyl) Pyrimidine. Chem. Data Collect. 2020, 29, 100525. [Google Scholar] [CrossRef]
- Cortés-Guzmán, F.; Bader, R.F.W. Complementarity of QTAIM and MO Theory in the Study of Bonding in Donor-Acceptor Complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar] [CrossRef]
- Daoui, O.; Elkhattabi, S.; Chtita, S.; Elkhalabi, R.; Zgou, H.; Benjelloun, A.T. QSAR, Molecular Docking and ADMET Properties in Silico Studies of Novel 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl Derivatives Derived from Dimedone as Potent Anti-Tumor Agents through Inhibition of C-Met Receptor Tyrosine Kinase. Heliyon 2021, 7, e07463. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational Pharmacokinetics Report, ADMET Study and Conceptual DFT-Based Estimation of the Chemical Reactivity Properties of Marine Cyclopeptides. ChemistryOpen 2021, 10, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an Entirely in-House Developed Drug Discovery Informatics System. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Joganathan, V.; Norris, J.H. Periocular Manifestations of Afatinib Therapy. Ophthal. Plast. Reconstr. Surg. 2019, 35, E12–E13. [Google Scholar] [CrossRef] [PubMed]
- Ondet, T.; Roux, P.-F.; Monshouwer, M.; Stamatas, G.N. Unlocking the Mechanisms of Cutaneous Adverse Drug Reactions: Activation of the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway by EGFR Inhibitors Triggers Keratinocyte Differentiation and Polarization of Epidermal Immune Responses. JID Innov. Ski. Sci. Mol. Popul. Health 2021, 1, 100009. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Rosti, G.; Iacobucci, I.; Baccarani, M.; Martinelli, G. Choosing the Best Second-Line Tyrosine Kinase Inhibitor in Imatinib-Resistant Chronic Myeloid Leukemia Patients Harboring Bcr-Abl Kinase Domain Mutations: How Reliable Is the IC50? Oncologist 2011, 16, 868–876. [Google Scholar] [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A Novel Multi-Tyrosine Kinase Inhibitor against Human Malignancies. OncoTargets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef]
- Tu, Y.; OuYang, Y.; Xu, S.; Zhu, Y.; Li, G.; Sun, C.; Zheng, P.; Zhu, W. Design, Synthesis, and Docking Studies of Afatinib Analogs Bearing Cinnamamide Moiety as Potent EGFR Inhibitors. Bioorg. Med. Chem. 2016, 24, 1495–1503. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.W.; Wise, S.C.; Kaufman, M.D.; Ahn, Y.M.; Ensinger, C.L.; Haack, T.; Hood, M.M.; Jones, J.; Lord, J.W.; Lu, W.P.; et al. Conformational Control Inhibition of the BCR-ABL1 Tyrosine Kinase, Including the Gatekeeper T315I Mutant, by the Switch-Control Inhibitor DCC-2036. Cancer Cell 2011, 19, 556–568. [Google Scholar] [CrossRef]
- Contreras-García, J.; Boto, R.A.; Izquierdo-Ruiz, F.; Reva, I.; Woller, T.; Alonso, M. A Benchmark for the Non-Covalent Interaction (NCI) Index Or…is It Really All in the Geometry? Theor. Chem. Acc. 2016, 135, 242. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Dennington Keith, T.; Millam, J.R.; Dennington, R.; Keith, T.; Millam, J. GaussView; Version 4.1.2; Gaussian, Inc.: Wallingford, CT, USA, 2007. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, L.A.; Nascimento, É.C.M.; Martins, J.B.L. In Silico Study of Tacrine and Acetylcholine Binding Profile with Human Acetylcholinesterase: Docking and Electronic Structure. J. Mol. Model. 2022, 28, 252. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Nagar, B.; Hantschel, O.; Young, M.A.; Scheffzek, K.; Veach, D.; Bornmann, W.; Clarkson, B.; Superti-Furga, G.; Kuriyan, J. Structural Basis for the Autoinhibition of C-Abl Tyrosine Kinase. Cell 2003, 112, 859–871. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.M.; Nascimento, É.C.M.; Martins, J.B.L.; da Mota, T.H.A.; de Oliveira, D.M.; Gatto, C.C. Crystal Design, Antitumor Activity and Molecular Docking of Novel Palladium(II) and Gold(III) Complexes with a Thiosemicarbazone Ligand. Int. J. Mol. Sci. 2023, 24, 11442. [Google Scholar] [CrossRef]
- Cavalcante, C.D.Q.; da Mota, T.H.A.; de Oliveira, D.M.; Nascimento, É.C.M.; Martins, J.B.L.; Pittella-Silva, F.; Gatto, C.C. Dithiocarbazate Ligands and Their Ni(II) Complexes with Potential Biological Activity: Structural, Antitumor and Molecular Docking Study. Front. Mol. Biosci. 2023, 10, 1146820. [Google Scholar] [CrossRef]
- Gatto, C.C.; Dias, L.M.; Paiva, C.A.; da Silva, I.C.R.; Freire, D.O.; Tormena, R.P.I.; Nascimento, É.C.M.; Martins, J.B.L. Effects of Changing Ions on the Crystal Design, Non-Covalent Interactions, Antimicrobial Activity, and Molecular Docking of Cu(II) Complexes with a Pyridoxal-Hydrazone Ligand. Front. Chem. 2024, 12, 1347370. [Google Scholar] [CrossRef]
- Santiago, P.H.d.O.; Duarte, E.d.A.; Nascimento, É.C.M.; Martins, J.B.L.; Castro, M.S.; Gatto, C.C. A Binuclear Copper(II) Complex Based on Hydrazone Ligand: Characterization, Molecular Docking, and Theoretical and Antimicrobial Investigation. Appl. Organomet. Chem. 2022, 36, e6461. [Google Scholar] [CrossRef]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor–Ligand Molecular Docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y. Comprehensive Assessment of Flexible-Ligand Docking Algorithms: Current Effectiveness and Challenges. Brief. Bioinform. 2018, 19, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Keith, T. AIMAll; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ρ (CO) | (CO) | ρ (NH) | ||
|---|---|---|---|---|
| Afatinib | 0.4017 | −0.2541 | 0.3399 | −1.745 |
| Imatinib | 0.3890 | −0.3640 | 0.3396 | −1.730 |
| Ponatinib | 0.3989 | −0.3147 | 0.3421 | −1.705 |
| AFA(I) | 0.2489 | −0.2940 | 0.3384 | −1.769 |
| (AFA(II)) | 0.3756 | −0.5108 | 0.3485 | −1.806 |
| (AFA(III)) | 0.3940 | −0.3124 | 0.3370 | −1.710 |
| (AFA(IV)) | 0.3991 | −0.3167 | 0.3412 | −1.704 |
| MW (Da) | LogP | # RT | # H-acc | # H-don | TPSA (Å2) | GA | Ro5 | Bioavailability Score | Leadlikeness | Druglikeness | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AFA(I) | 503.40 | 2.92 | 9 | 4 | 3 | 87.22 | High | 1 | 0.56 | 2 | 1.77 |
| AFA(II) | 487.95 | 2.21 | 10 | 6 | 3 | 98.22 | High | 0 | 0.55 | 2 | −0.53 |
| AFA(III) | 503.01 | 2.9 | 9 | 4 | 3 | 103.20 | High | 1 | 0.56 | 2 | 0.35 |
| AFA(IV) | 556.05 | 4.42 | 11 | 7 | 3 | 91.85 | High | 1 | 0.55 | 3 | −0.91 |
| Afatinib | 486.95 | 3.64 | 9 | 5 | 3 | 88.61 | High | 0 | 0.55 | 3 | −3.64 |
| Imatinib | 494.61 | 3.94 | 9 | 5 | 3 | 95.07 | High | 0 | 0.55 | 3 | 4.47 |
| Ponatinib | 535.58 | 3.86 | 8 | 7 | 2 | 65.77 | High | 1 | 0.55 | 3 | 2.10 |
| Molecule | Mutagenicity | Tumorigenicity | Irritability | Reproductive Effects |
|---|---|---|---|---|
| AFA(I) | no effect | no effect | mild | no effect |
| AFA(II) | no effect | no effect | no effect | no effect |
| AFA(III) | no effect | no effect | mild | no effect |
| AFA(IV) | no effect | no effect | no effect | no effect |
| Afatinib | no effect | no effect | mild | no effect |
| Imatinib | no effect | no effect | no effect | no effect |
| Ponatinib | no effect | no effect | no effect | no effect |
| Ligand | 1OPJ | 3QRJ | Experimental IC50 (nM)Wild-Resistant |
|---|---|---|---|
| AFA(I) | −8.7 | −7.3 | N.A. |
| AFA(II) | −8.9 | −8.1 | N.A. |
| AFA(III) | −8.3 | −8.3 | N.A. |
| AFA(IV) | −8.5 | −9.0 | N.A. |
| Afatinib | −8.8 | −7.6 | N.A. |
| Imatinib | −12.8 | −8.9 | 260–6400 [74] |
| Ponatinib | −12.5 | −11.8 | 0.37–2.00 [75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, K.M.L.; Nascimento, É.C.M.; de Jesus, R.C.C.; Martins, J.B.L. In Silico Molecular Modeling of Four New Afatinib Derived Molecules Targeting the Inhibition of the Mutated Form of BCR-ABL T315I. Molecules 2024, 29, 4254. https://doi.org/10.3390/molecules29174254
Rocha KML, Nascimento ÉCM, de Jesus RCC, Martins JBL. In Silico Molecular Modeling of Four New Afatinib Derived Molecules Targeting the Inhibition of the Mutated Form of BCR-ABL T315I. Molecules. 2024; 29(17):4254. https://doi.org/10.3390/molecules29174254
Chicago/Turabian StyleRocha, Kelvyn M. L., Érica C. M. Nascimento, Rafael C. C. de Jesus, and João B. L. Martins. 2024. "In Silico Molecular Modeling of Four New Afatinib Derived Molecules Targeting the Inhibition of the Mutated Form of BCR-ABL T315I" Molecules 29, no. 17: 4254. https://doi.org/10.3390/molecules29174254
APA StyleRocha, K. M. L., Nascimento, É. C. M., de Jesus, R. C. C., & Martins, J. B. L. (2024). In Silico Molecular Modeling of Four New Afatinib Derived Molecules Targeting the Inhibition of the Mutated Form of BCR-ABL T315I. Molecules, 29(17), 4254. https://doi.org/10.3390/molecules29174254