
Computational Exploration of the Mechanism of Action of a Sorafenib-Containing Ruthenium Complex as an Anticancer Agent for Photoactivated Chemotherapy

Abstract
:1. Introduction
2. Results and Discussion
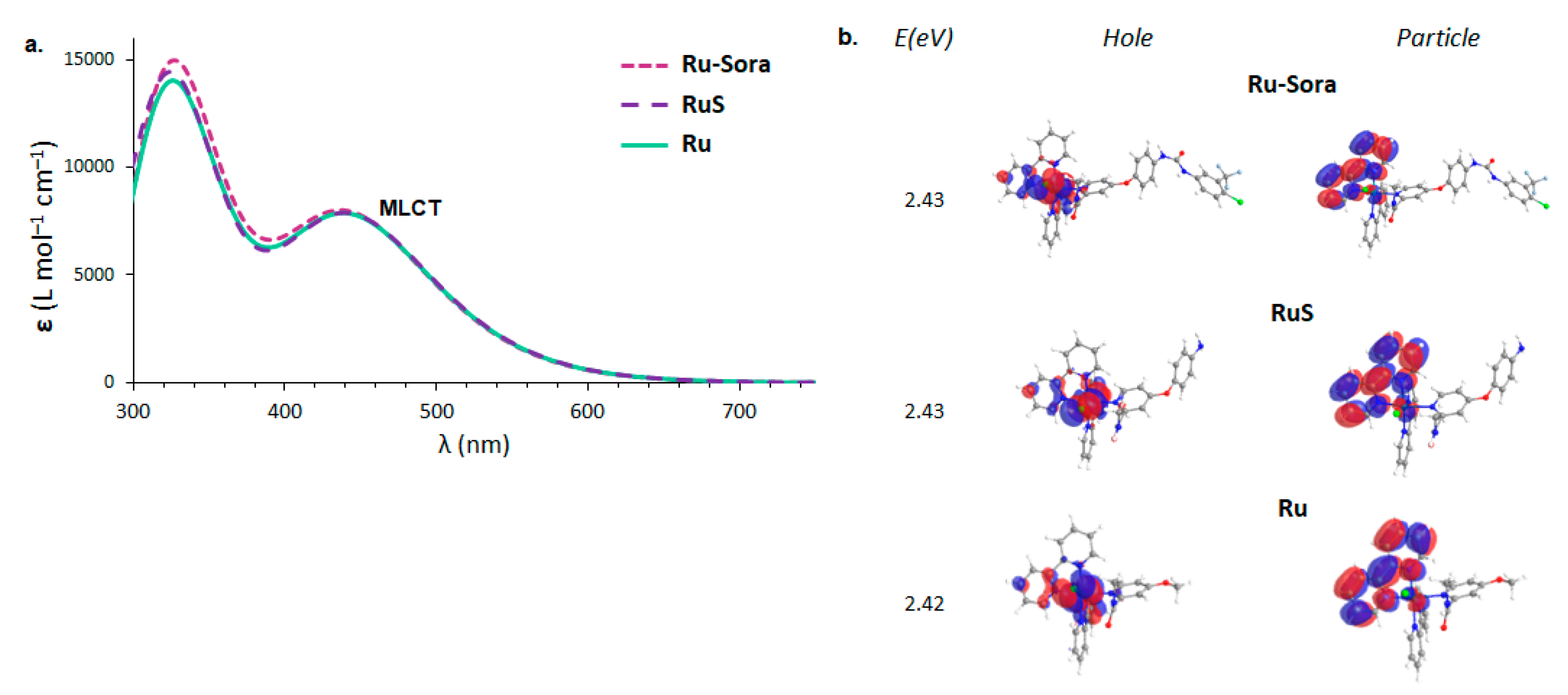
2.1. Structural and Electronic Properties
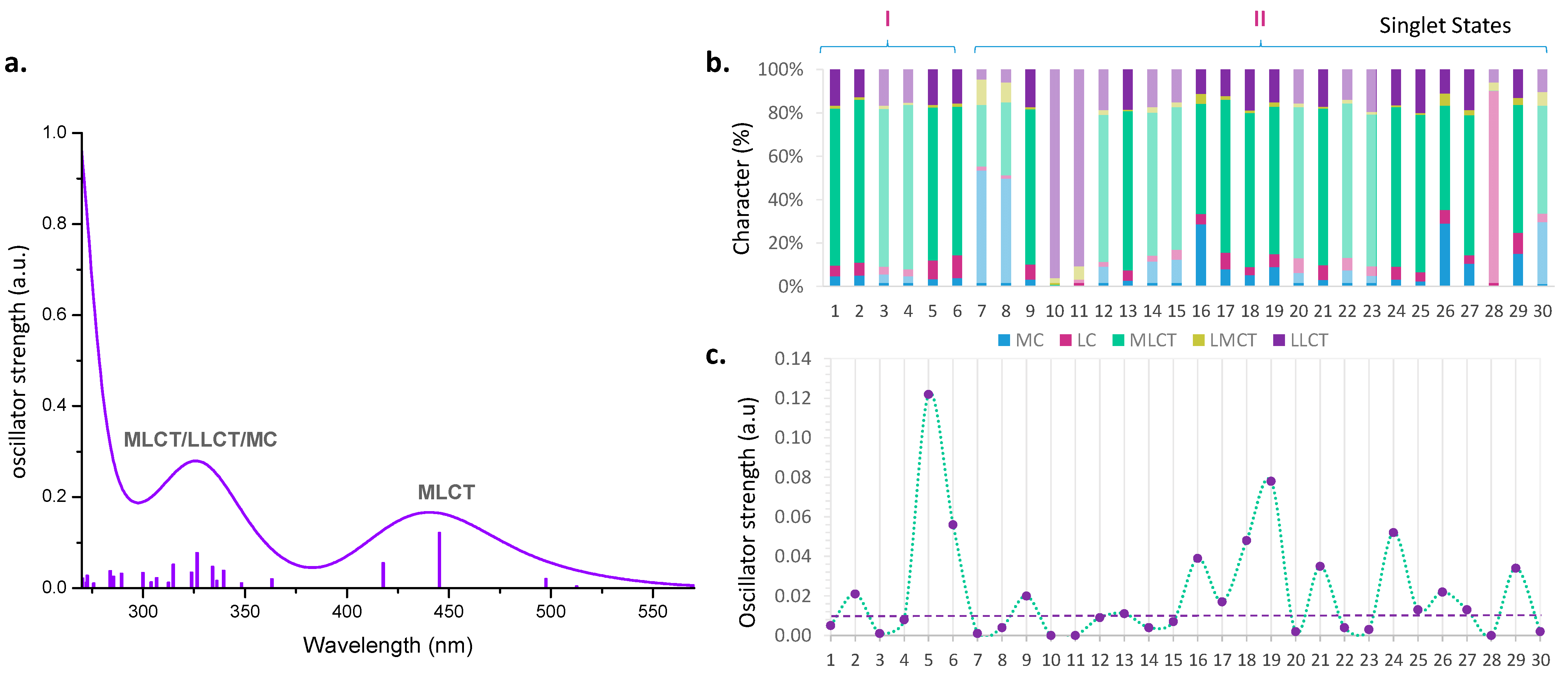
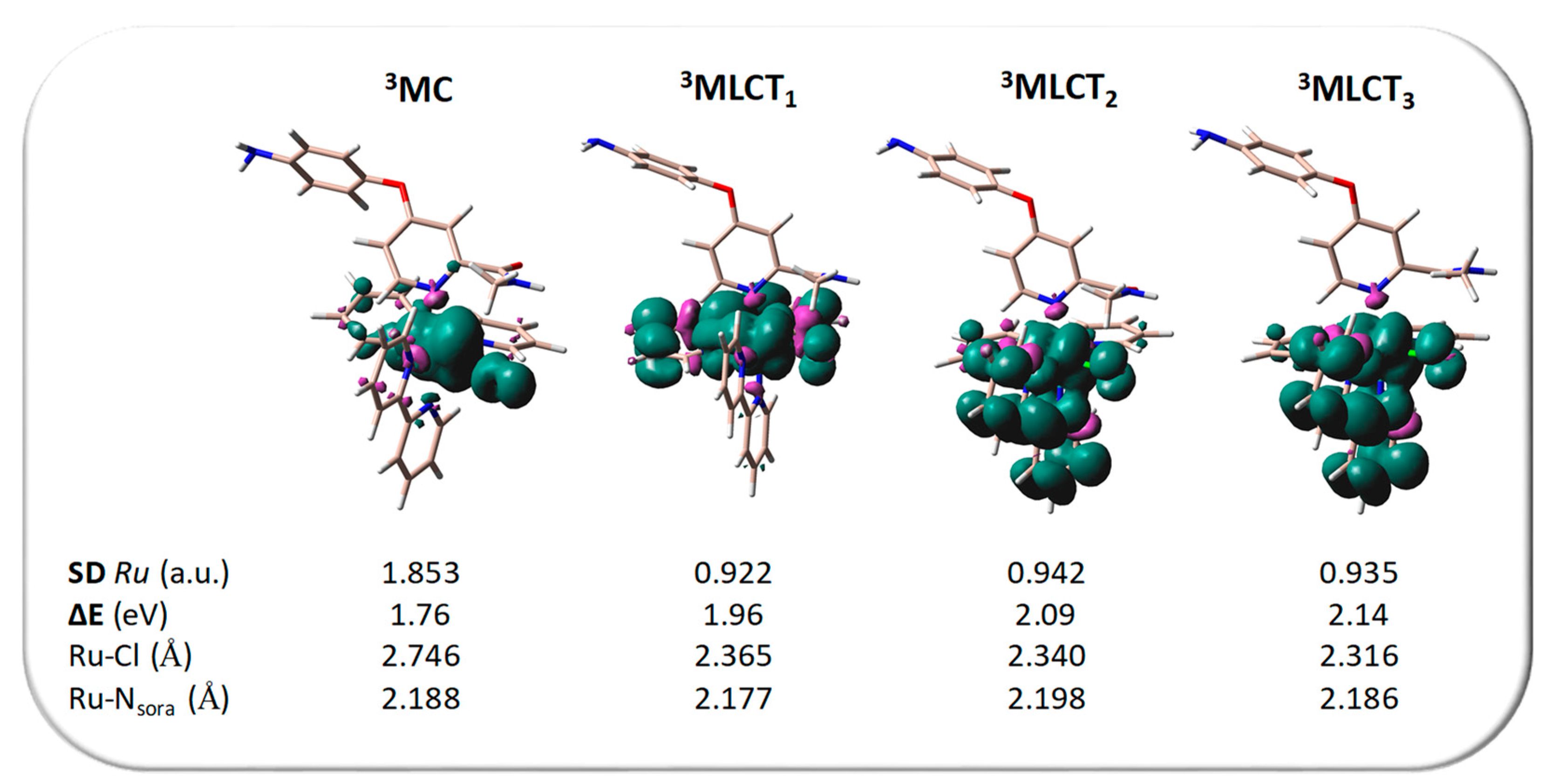
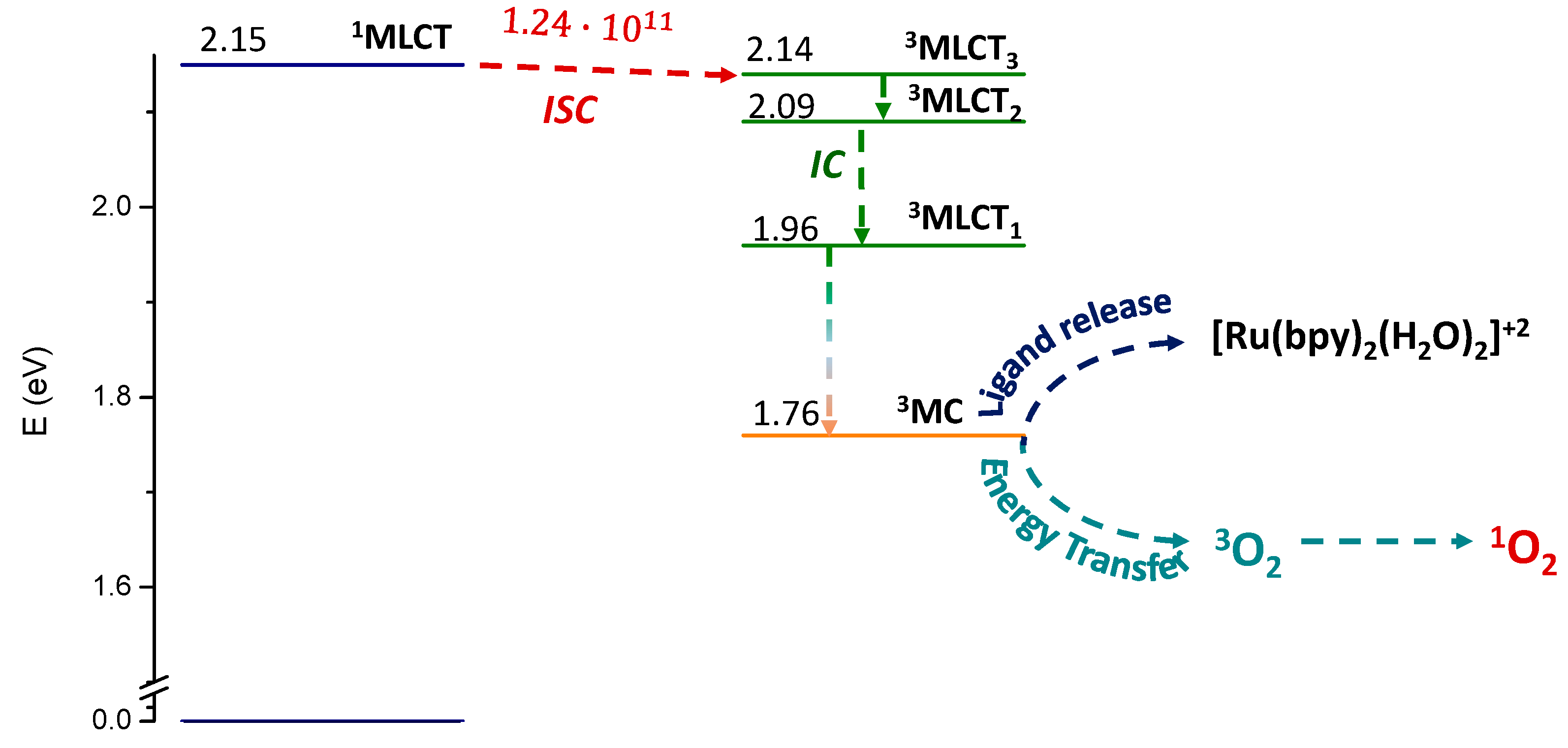
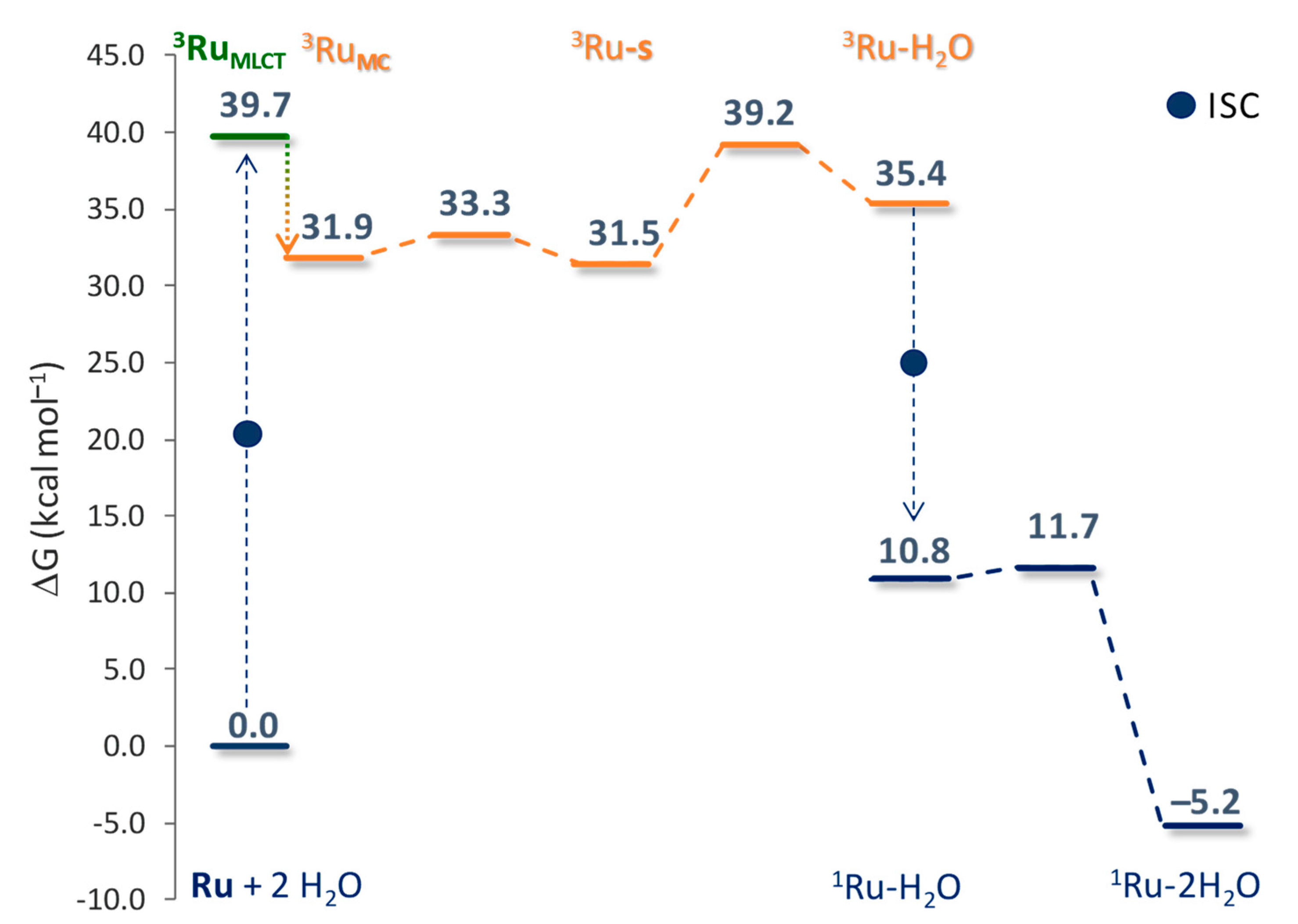
2.2. Photodynamic Processes
2.3. Sorafenib Release Mechanism
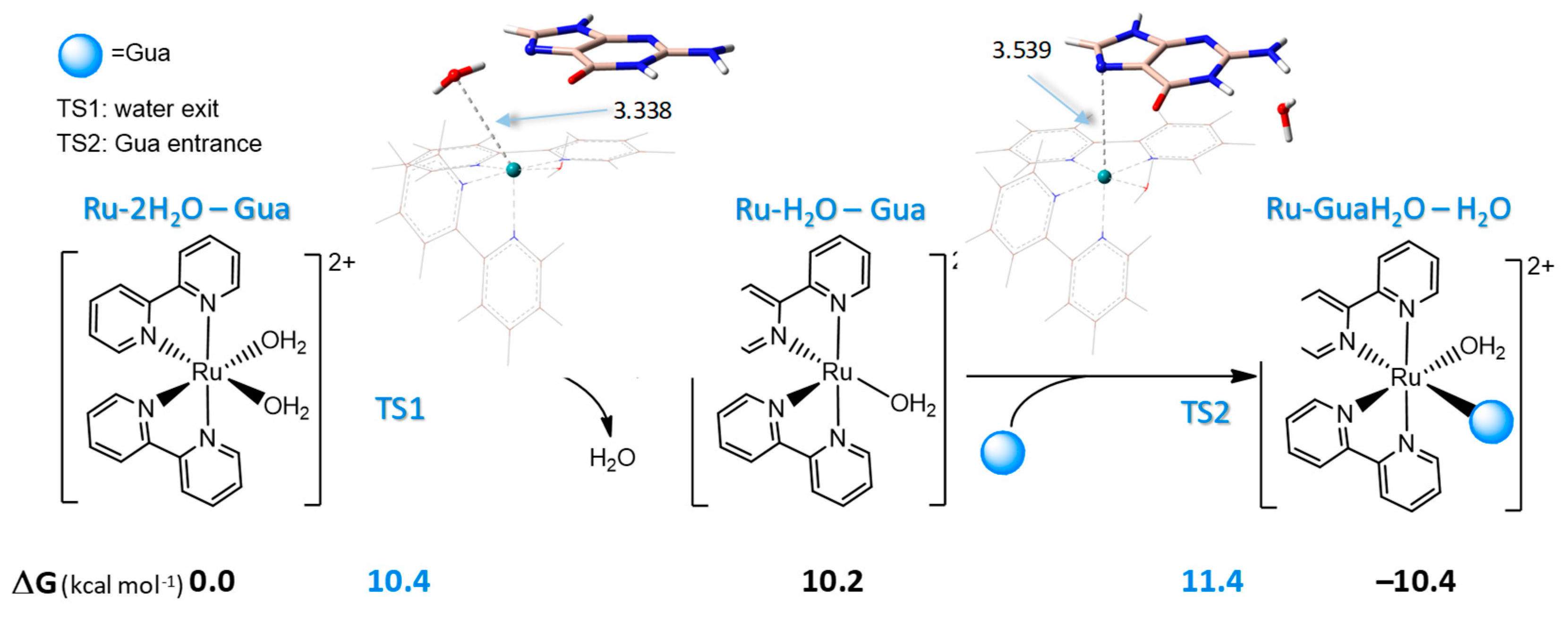
2.4. Interaction of Ru-2H2O with DNA
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and Photodynamic Therapy: Mechanisms, Monitoring and Optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA A Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New Photosensitizers for Photodynamic Therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Barretta, P.; Scoditti, S.; Belletto, D.; Ponte, F.; Vigna, V.; Mazzone, G.; Sicilia, E. Ruthenium Complexes Bearing Nile Red Chromophore and One of Its Derivative: Theoretical Evaluation of PDT-Related Properties. J. Comput. Chem. 2024, 45, 2034–2041. [Google Scholar] [CrossRef]
- Ponte, F.; Scopelliti, D.M.; Sanna, N.; Sicilia, E.; Mazzone, G. How Computations Can Assist the Rational Design of Drugs for Photodynamic Therapy: Photosensitizing Activity Assessment of a Ru(II)-BODIPY Assembly. Molecules 2022, 27, 5635. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Cao, B.; Zhong, M.; Liu, M.; Xiong, X.; Zou, T. Organogold(III) Complexes Display Conditional Photoactivities: Evolving From Photodynamic into Photoactivated Chemotherapy in Response to O2 Consumption for Robust Cancer Therapy. Angew. Chem. Int. Ed. 2022, 61, e202212689. [Google Scholar] [CrossRef] [PubMed]
- Hakkennes, M.L.A.; Meijer, M.S.; Menzel, J.P.; Goetz, A.-C.; Van Duijn, R.; Siegler, M.A.; Buda, F.; Bonnet, S. Ligand Rigidity Steers the Selectivity and Efficiency of the Photosubstitution Reaction of Strained Ruthenium Polypyridyl Complexes. J. Am. Chem. Soc. 2023, 145, 13420–13434. [Google Scholar] [CrossRef]
- Toupin, N.; Steinke, S.J.; Nadella, S.; Li, A.; Rohrabaugh, T.N., Jr.; Samuels, E.R.; Turro, C.; Sevrioukova, I.F.; Kodanko, J.J. Photosensitive Ru(II) Complexes as Inhibitors of the Major Human Drug Metabolizing Enzyme CYP3A4. J. Am. Chem. Soc. 2021, 143, 9191–9205. [Google Scholar] [CrossRef]
- Huang, C.; Liang, C.; Sadhukhan, T.; Banerjee, S.; Fan, Z.; Li, T.; Zhu, Z.; Zhang, P.; Raghavachari, K.; Huang, H. In-Vitro and In-Vivo Photocatalytic Cancer Therapy with Biocompatible Iridium(III) Photocatalysts. Angew. Chem. Int. Ed. 2021, 60, 9474–9479. [Google Scholar] [CrossRef]
- Farrer, N.J.; Salassa, L.; Sadler, P.J. Photoactivated Chemotherapy (PACT): The Potential of Excited-State d-Block Metals in Medicine. Dalton Trans. 2009, 10690–10701. [Google Scholar] [CrossRef]
- Schatzschneider, U. Photoactivated Biological Activity of Transition-Metal Complexes. Eur. J. Inorg. Chem. 2010, 2010, 1451–1467. [Google Scholar] [CrossRef]
- Farrer, N.J.; Woods, J.A.; Salassa, L.; Zhao, Y.; Robinson, K.S.; Clarkson, G.; Mackay, F.S.; Sadler, P.J. A Potent Trans-Diimine Platinum Anticancer Complex Photoactivated by Visible Light. Angew. Chem. Int. Ed. 2010, 49, 8905–8908. [Google Scholar] [CrossRef] [PubMed]
- Imberti, C.; Zhang, P.; Huang, H.; Sadler, P.J. New Designs for Phototherapeutic Transition Metal Complexes. Angew. Chem. Int. Ed. 2020, 59, 61–73. [Google Scholar] [CrossRef]
- Shi, H.; Carter, O.W.L.; Ponte, F.; Imberti, C.; Gomez-Gonzalez, M.A.; Cacho-Nerin, F.; Quinn, P.D.; Parker, J.E.; Sicilia, E.; Huang, H.; et al. A Photodynamic and Photochemotherapeutic Platinum-Iridium Charge-Transfer Conjugate for Anticancer Therapy. Angew. Chem. Int. Ed. 2024, 63, e202400476. [Google Scholar] [CrossRef]
- Steinke, S.J.; Gupta, S.; Piechota, E.J.; Moore, C.E.; Kodanko, J.J.; Turro, C. Photocytotoxicity and Photoinduced Phosphine Ligand Exchange in a Ru(II) Polypyridyl Complex. Chem. Sci. 2022, 13, 1933–1945. [Google Scholar] [CrossRef]
- Denison, M.; Garcia, S.P.; Ullrich, A.; Podgorski, I.; Gibson, H.; Turro, C.; Kodanko, J.J. Ruthenium-Cathepsin Inhibitor Conjugates for Green Light-Activated Photodynamic Therapy and Photochemotherapy. Inorg. Chem. 2024, 63, 7973–7983. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Xiao, C.; Li, Z.; Yang, X. Engineering Nanomedicine for Glutathione Depletion-Augmented Cancer Therapy. Chem. Soc. Rev. 2021, 50, 6013–6041. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, 1904197. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Lai, Y.; Lu, N.; Luo, S.; Wang, H.; Zhang, P. A Photoactivated Sorafenib-Ruthenium(II) Prodrug for Resistant Hepatocellular Carcinoma Therapy through Ferroptosis and Purine Metabolism Disruption. J. Med. Chem. 2022, 65, 13041–13051. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, P.S.; Bhattarai, H.K. Singlet Oxygen, Photodynamic Therapy, and Mechanisms of Cancer Cell Death. J. Oncol. 2022, 2022, 7211485. [Google Scholar] [CrossRef]
- Österman, T.; Persson, P. Excited State Potential Energy Surfaces of Bistridentate RuII Complexes—A TD-DFT Study. Chem. Phys. 2012, 407, 76–82. [Google Scholar] [CrossRef]
- Alcover-Fortuny, G.; Wu, J.; Caballol, R.; de Graaf, C. Quantum Chemical Study of the Interligand Electron Transfer in Ru Polypyridyl Complexes. J. Phys. Chem. A 2018, 122, 1114–1123. [Google Scholar] [CrossRef]
- Li, Y.; Fan, X.-W.; Wang, J.; Kong, C.-P.; Chen, J.; Wang, S.-P.; Li, H.-C.; Bai, F.-Q.; Zhang, H.-X. Comparative Study on the Photophysical Properties between Carbene-Based Fe (II) and Ru (II) Complexes. Appl. Organomet. Chem. 2020, 34, e5821. [Google Scholar] [CrossRef]
- Butera, V.; Mazzone, G.; Detz, H. Dinuclear Ruthenium(II)-Pyrrolide Complexes Linked by Different Organic Units as PDT Photosensitizers: Computational Study of the Linker Influence on the Photophysical Properties. ChemPhotoChem 2022, 6, e202200094. [Google Scholar] [CrossRef]
- Escudero, D.; González, L. RASPT2/RASSCF vs. Range-Separated/Hybrid DFT Methods: Assessing the Excited States of a Ru(II)Bipyridyl Complex. J. Chem. Theory Comput. 2012, 8, 203–213. [Google Scholar] [CrossRef]
- Elias, M.G.; Mehanna, S.; Elias, E.; Khnayzer, R.S.; Daher, C.F. A Photoactivatable Chemotherapeutic Ru(II) Complex Bearing Bathocuproine Ligand Efficiently Induces Cell Death in Human Malignant Melanoma Cells through a Multi-Mechanistic Pathway. Chem.-Biol. Interact. 2021, 348, 109644. [Google Scholar] [CrossRef]
- Spiegel, M.; Adamo, C. Tuning the Photophysical Properties of Ru(II) Photosensitizers for PDT by Protonation and Metallation: A DFT Study. J. Phys. Chem. A 2023, 127, 3625–3635. [Google Scholar] [CrossRef]
- Plasser, F. TheoDORE: A Toolbox for a Detailed and Automated Analysis of Electronic Excited State Computations. J. Chem. Phys. 2020, 152, 084108. [Google Scholar] [CrossRef]
- Zeng, C.; Li, Y.; Zheng, H.; Ren, M.; Wu, W.; Chen, Z. Nature of Ultrafast Dynamics in the Lowest-Lying Singlet Excited State of [Ru(Bpy)3]2+. Phys. Chem. Chem. Phys. 2024, 26, 6524–6531. [Google Scholar] [CrossRef] [PubMed]
- Dabbish, E.; Mazzone, G.; Russo, N.; Sicilia, E. Mechanism of Action of the Curcumin Cis-Diammineplatinum(II) Complex as a Photocytotoxic Agent. Inorg. Chem. Front. 2020, 7, 2759–2769. [Google Scholar] [CrossRef]
- Simone, B.C.D.; Mazzone, G.; Russo, N.; Sicilia, E.; Toscano, M. Excitation Energies, Singlet–Triplet Energy Gaps, Spin–Orbit Matrix Elements and Heavy Atom Effects in BOIMPYs as Possible Photosensitizers for Photodynamic Therapy: A Computational Investigation. Phys. Chem. Chem. Phys. 2018, 20, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Hemnani, T.; Parihar, M.S. Reactive oxygen species and oxidative dna damage. Indian J. Physiol. Pharmacol. 1998, 42, 440–452. [Google Scholar] [PubMed]
- Belletto, D.; Ponte, F.; Mazzone, G.; Sicilia, E. A Detailed Density Functional Theory Exploration of the Photodissociation Mechanism of Ruthenium Complexes for Photoactivated Chemotherapy. Dalton Trans. 2024, 53, 8243–8253. [Google Scholar] [CrossRef] [PubMed]
- Kayanuma, M. Photosubstitution Reaction of a Bidentate Ligand in a Ru(II) Complex in Aqueous Solution. Comput. Theor. Chem. 2022, 1213, 113745. [Google Scholar] [CrossRef]
- Soupart, A.; Dixon, I.M.; Alary, F.; Heully, J.-L. DFT Rationalization of the Room-Temperature Luminescence Properties of Ru(Bpy)32+ and Ru(Tpy)22+: 3MLCT–3MC Minimum Energy Path from NEB Calculations and Emission Spectra from VRES Calculations. Theor. Chem. Acc. 2018, 137, 37. [Google Scholar] [CrossRef]
- Yin, C.-W.; Tsai, M.-K.; Chen, Y.J. Low-Temperature Observation of the Excited-State Decay of Ruthenium-(Mono-2,2′:6′,2″-Terpyridine) Ions with Innocent Ligands: DFT Modeling of an 3MLCT–3MC Intersystem Crossing Pathway. ACS Omega 2023, 8, 11623–11633. [Google Scholar] [CrossRef]
- Nisbett, K.; Tu, Y.-J.; Turro, C.; Kodanko, J.J.; Schlegel, H.B. DFT Investigation of Ligand Photodissociation in [RuII(Tpy)(Bpy)(Py)]2+ and [RuII(Tpy)(Me2bpy)(Py)]2+ Complexes. Inorg. Chem. 2018, 57, 231–240. [Google Scholar] [CrossRef]
- Busemann, A.; Flaspohler, I.; Zhou, X.-Q.; Schmidt, C.; Goetzfried, S.K.; van Rixel, V.H.S.; Ott, I.; Siegler, M.A.; Bonnet, S. Ruthenium-Based PACT Agents Based on Bisquinoline Chelates: Synthesis, Photochemistry, and Cytotoxicity. J. Biol. Inorg. Chem. 2021, 26, 667–674. [Google Scholar] [CrossRef]
- Betanzos-Lara, S.; Habtemariam, A.; Clarkson, G.J.; Sadler, P.J. Organometallic Cis-Dichlorido Ruthenium(II) Ammine Complexes. Eur. J. Inorg. Chem. 2011, 2011, 3257–3264. [Google Scholar] [CrossRef]
- Chen, Q.; Cuello-Garibo, J.-A.; Bretin, L.; Zhang, L.; Ramu, V.; Aydar, Y.; Batsiun, Y.; Bronkhorst, S.; Husiev, Y.; Beztsinna, N.; et al. Photosubstitution in a Trisheteroleptic Ruthenium Complex Inhibits Conjunctival Melanoma Growth in a Zebrafish Orthotopic Xenograft Model. Chem. Sci. 2022, 13, 6899–6919. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.; Denning, C.A.; Heidary, D.K.; Wachter, E.; Nease, L.A.; Ruiz, J.; Glazer, E.C. Ruthenium-Containing P450 Inhibitors for Dual Enzyme Inhibition and DNA Damage. Dalton Trans. 2017, 46, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Bešker, N.; Coletti, C.; Marrone, A.; Re, N. Binding of Antitumor Ruthenium Complexes to DNA and Proteins: A Theoretical Approach. J. Phys. Chem. B 2007, 111, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Ponte, F.; Belletto, D.; Leonetti, R.; Sanna, N.; Scoditti, S.; Mazzone, G.; Sicilia, E. DFT Computational Analysis of the Mechanism of Action of Ru(II) Polypyridyl Complexes as Photoactivated Chemotherapy Agents: From Photoinduced Ligand Solvolysis to DNA Binding. Inorg. Chem. 2024. submitted. [Google Scholar]
- Barretta, P.; Ponte, F.; Scoditti, S.; Vigna, V.; Mazzone, G.; Sicilia, E. Computational Analysis of the Behavior of BODIPY Decorated Monofunctional Platinum(II) Complexes in the Dark and under Light Irradiation. J. Phys. Chem. A 2022, 126, 7159–7167. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, G.; Scoditti, S.; Caligiuri, R.; Ricciardi, L.; Sicilia, E.; Lupo, M.G.; Rimoldi, I.; Godbert, N.; La Deda, M.; Ionescu, A.; et al. Cytotoxicity of Alizarine versus Tetrabromocathecol Cyclometalated Pt(II) Theranostic Agents: A Combined Experimental and Computational Investigation. Inorg. Chem. 2022, 61, 7188–7200. [Google Scholar] [CrossRef] [PubMed]
- Belletto, D.; Ponte, F.; Sanna, N.; Scoditti, S.; Sicilia, E. G-Quadruplex DNA Selective Targeting for Anticancer Therapy: A Computational Study of a Novel PtII Monofunctional Complex Activated by Adaptive Binding. Dalton Trans. 2023, 52, 13517–13527. [Google Scholar] [CrossRef] [PubMed]
- Šebesta, F.; Burda, J.V. Study on Electronic Properties, Thermodynamic and Kinetic Parameters of the Selected Platinum(II) Derivatives Interacting with Guanine. J. Inorg. Biochem. 2017, 172, 100–109. [Google Scholar] [CrossRef]
- Alberto, M.E.; Butera, V.; Russo, N. Which One among the Pt-Containing Anticancer Drugs More Easily Forms Monoadducts with G and A DNA Bases? A Comparative Study among Oxaliplatin, Nedaplatin, and Carboplatin. Inorg. Chem. 2011, 50, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; GaussView 5.0.; Wallingford, E.U.A., Ed.; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted Ab Initio Pseudopotentials for the Rare Earth Elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Butera, V.; Detz, H. Hydrogenation of CO2 to Methanol by the Diphosphine–Ruthenium(II) Cationic Complex: A DFT Investigation to Shed Light on the Decisive Role of Carboxylic Acids as Promoters. Catal. Sci. Technol. 2021, 11, 3556–3567. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-Dependent Density Functional Theory within the Tamm–Dancoff Approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Account. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof Exchange-Correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Barretta, P.; Ponte, F.; Scoditti, S.; Mazzone, G. Computational Assessment of Novel Ruthenium Phenoxazine-Based Complexes as Photosensitizers in Photodynamic Therapy. Eur. J. Inorg. Chem. 2024, e202400309. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Fortino, M.; Collini, E.; Bloino, J.; Pedone, A. Unraveling the Internal Conversion Process within the Q-Bands of a Chlorophyll-like-System through Surface-Hopping Molecular Dynamics Simulations. J. Chem. Phys. 2021, 154, 094110. [Google Scholar] [CrossRef]
- Fortino, M.; Cozza, C.; Bonomi, M.; Pietropaolo, A. Multi-Replica Biased Sampling for Photoswitchable π-Conjugated Polymers. J. Chem. Phys. 2021, 154, 174108. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Ru]+ | 3[Ru]+ | O2 | |
|---|---|---|---|
| VEA a | −2.49 | −4.52 | −2.09 |
| VIP a | 5.17 | 3.13 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barretta, P.; Ponte, F.; Escudero, D.; Mazzone, G. Computational Exploration of the Mechanism of Action of a Sorafenib-Containing Ruthenium Complex as an Anticancer Agent for Photoactivated Chemotherapy. Molecules 2024, 29, 4298. https://doi.org/10.3390/molecules29184298
Barretta P, Ponte F, Escudero D, Mazzone G. Computational Exploration of the Mechanism of Action of a Sorafenib-Containing Ruthenium Complex as an Anticancer Agent for Photoactivated Chemotherapy. Molecules. 2024; 29(18):4298. https://doi.org/10.3390/molecules29184298
Chicago/Turabian StyleBarretta, Pierraffaele, Fortuna Ponte, Daniel Escudero, and Gloria Mazzone. 2024. "Computational Exploration of the Mechanism of Action of a Sorafenib-Containing Ruthenium Complex as an Anticancer Agent for Photoactivated Chemotherapy" Molecules 29, no. 18: 4298. https://doi.org/10.3390/molecules29184298
APA StyleBarretta, P., Ponte, F., Escudero, D., & Mazzone, G. (2024). Computational Exploration of the Mechanism of Action of a Sorafenib-Containing Ruthenium Complex as an Anticancer Agent for Photoactivated Chemotherapy. Molecules, 29(18), 4298. https://doi.org/10.3390/molecules29184298