

Molecular Simulation Analysis of Polyurethane Molecular Structure under External Electric Field




Abstract
1. Introduction
2. Method
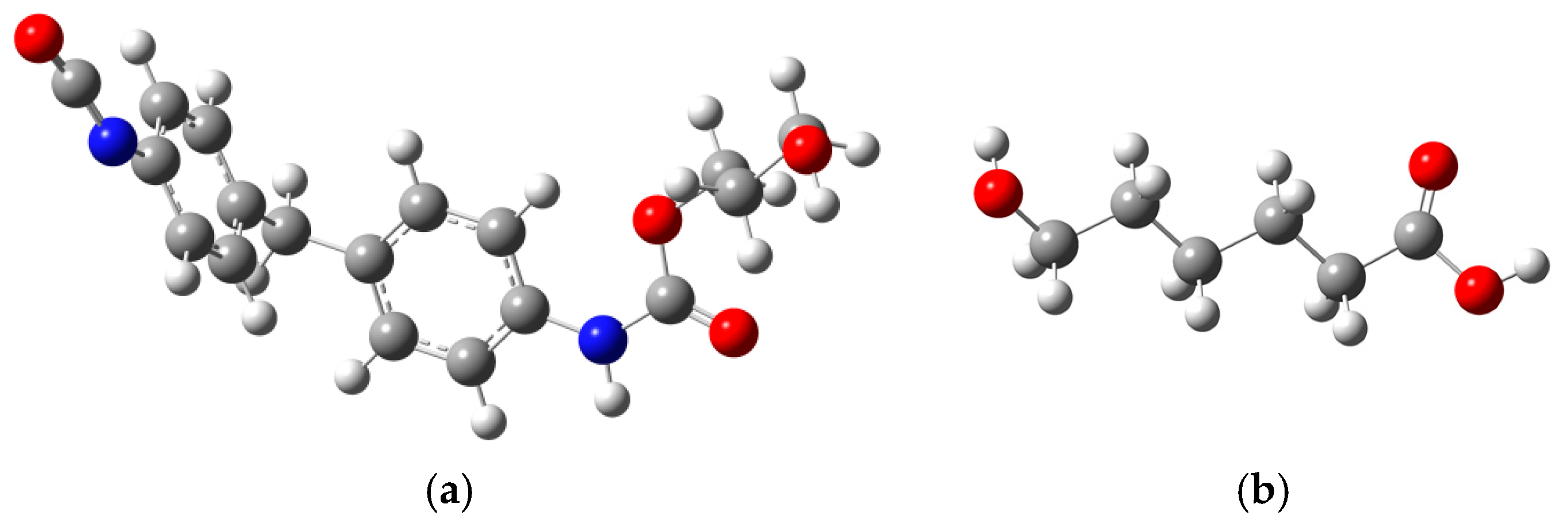
2.1. Model Construction
2.2. Theoretical Calculations and Methods
3. Simulation Results and Discussion
3.1. Effect of External Electric Field on Molecular Dipole Moment, Energy, and Polarizability
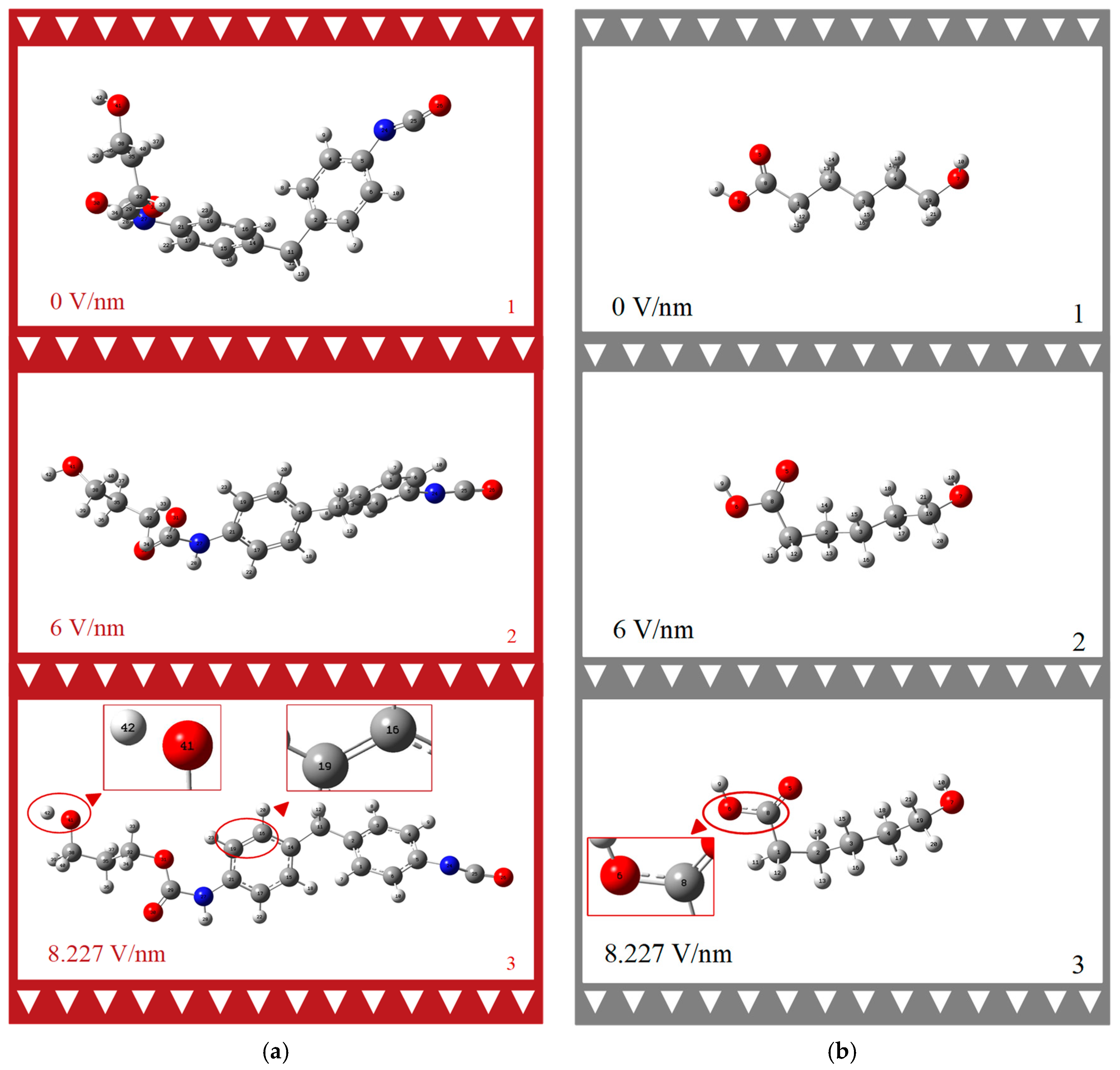
3.2. Effect of External Electric Field on the Geometry of the Molecule
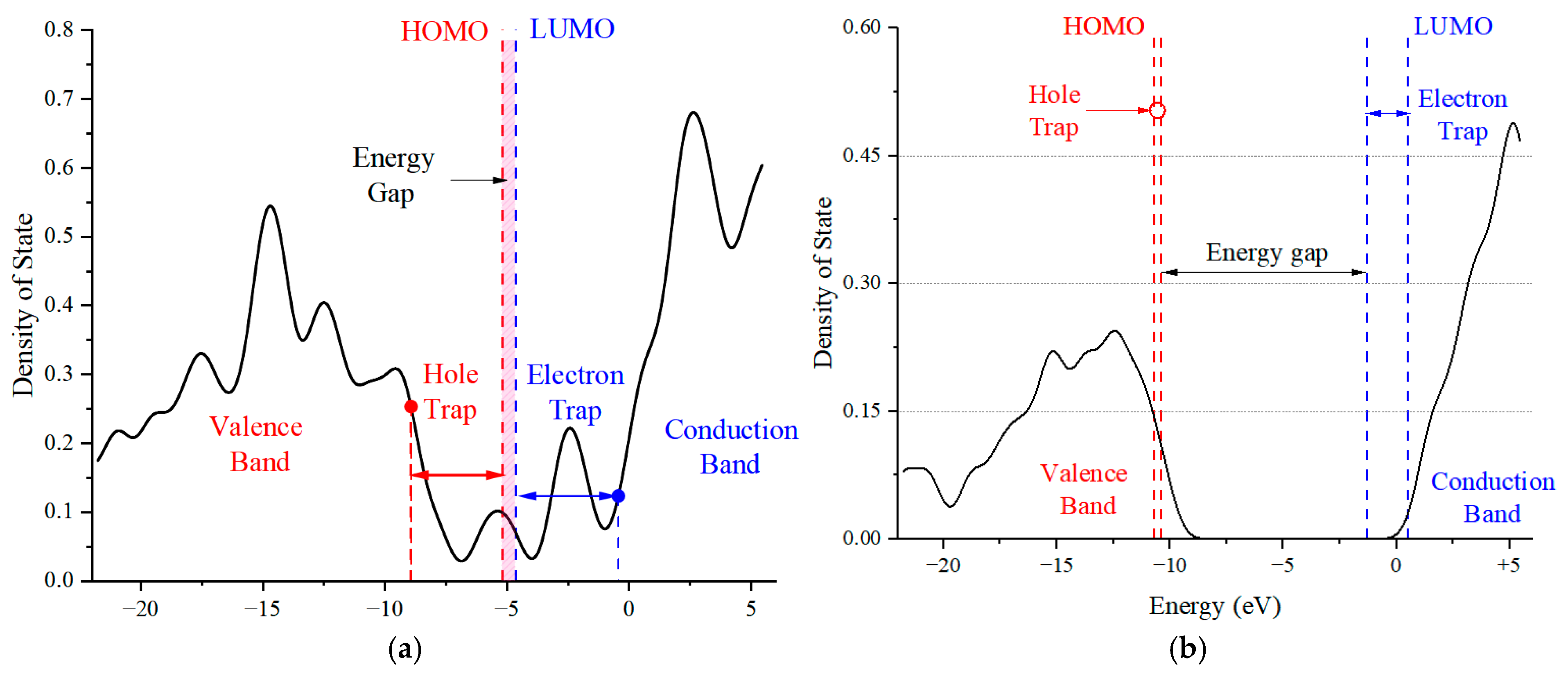
3.3. Effect of External Electric Field on Molecular Front Orbitals Space Charge Properties of HS and SS of PU
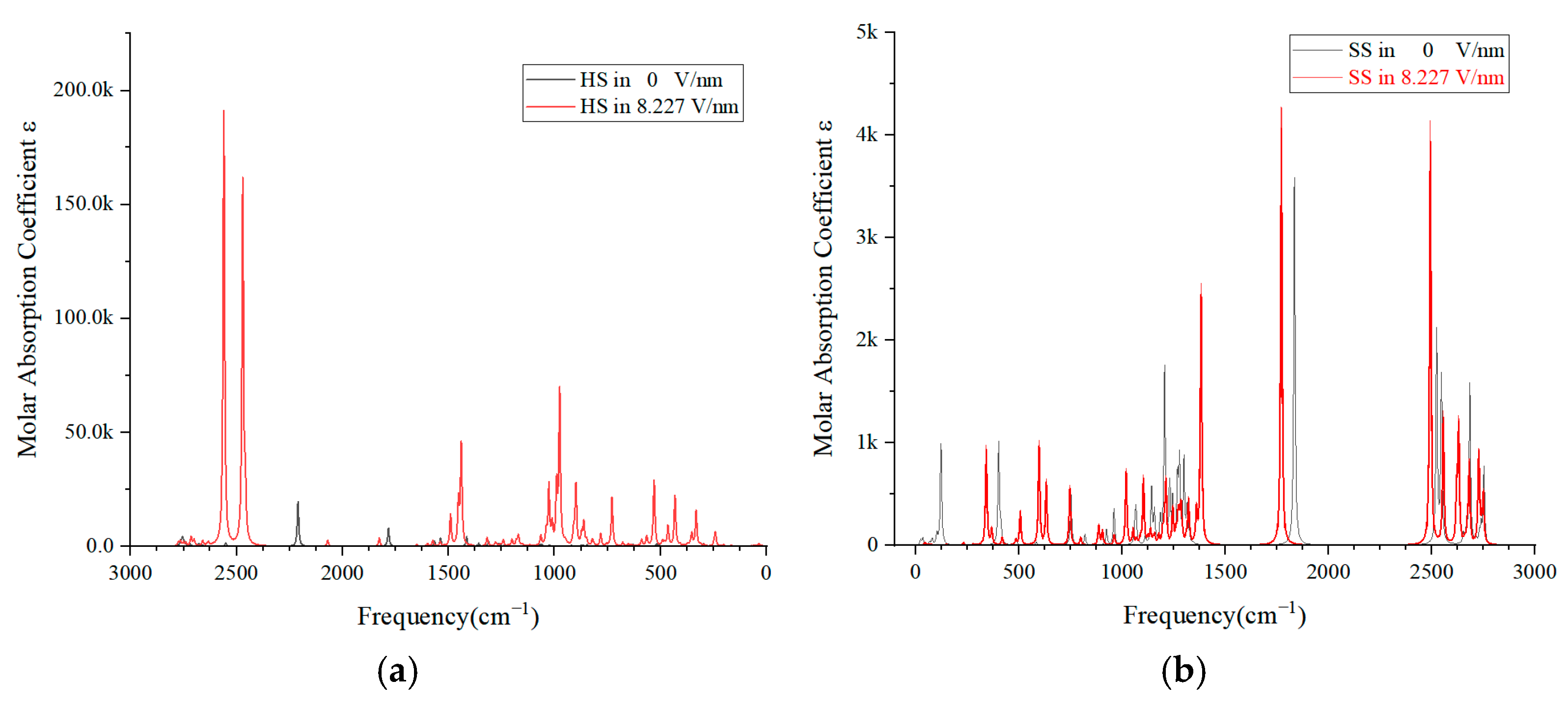
3.4. Molecular IR Spectra under Different Electric Field Intensities
4. Conclusions
- (1)
- The pronounced rise in polarizability and dipole moment of the HS molecules at specific electric field intensities indicates their intrinsic instability, which in turn affects the overall stability of PU materials. This insight is fundamental to grasping the structural dynamics within PU chains and underscores the importance of material optimization for improved stability.
- (2)
- Our examination of molecular bonds and dihedral angles has revealed the distinct mechanical and electrical stabilities between SSs and HSs. The greater electrical stability of SS molecules, even amidst their mechanical vulnerability, suggests opportunities for material modification. This could particularly enhance PU’s electrical properties through targeted alterations to terminal hydroxyl or hydrogen atoms.
- (3)
- The notable reduction in the energy gap of the HS molecules with increasing electric field intensity signals their increased risk of dielectric breakdown. The finding that a higher proportion of SS molecules can mitigate this effect offers a strategic pathway for developing PU materials with superior insulation properties, which could enhance the reliability of power equipment.
- (4)
- The IR spectral analysis confirms the pivotal role of HS molecules in the degradation of PU materials under electric fields. The spectral variations provide a predictive tool for non-destructive testing, which could significantly decrease labor costs and increase the efficiency of engineering practices.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Honarkar, H. Waterborne polyurethanes: A review. J. Disper. Sci. Technol. 2017, 39, 507–516. [Google Scholar] [CrossRef]
- Wei, H.; Wang, X.; Liu, W.; Dong, S.; Dai, T.; Luo, F.; Li, Z.; Tan, H.; Li, J. Investigating the Structure and Properties of Polyurethane Hydrogels with Varying Soft and Hard Segments. J. Polym. Sci. 2024, 62, 3979–3991. [Google Scholar] [CrossRef]
- Jing, X.; Li, X.; Di, Y.; Zhao, Y.; Wang, J.; Kang, M.; Li, Q. Effect of the Amide Units in Soft Segment and Urea Units in Hard Segment on Microstructures and Physical Properties of Polyurethane Elastomer. Polymer 2021, 233, 124205. [Google Scholar] [CrossRef]
- Lulu, Y.; Tao, X.; Peng, C. Preparation of Waterborne Polyurethane Membranes with Different Soft Segments. J. Polym. Mater. 2018, 34, 143–148. [Google Scholar]
- Kwiatkowski, K.; Nachman, M. The Abrasive Wear Resistance of the Segmented Linear Polyurethane Elastomers Based on a Variety of Polyols as Soft Segments. Polymers 2017, 9, 705. [Google Scholar] [CrossRef] [PubMed]
- Kojio, K.; Nozaki, S.; Takahara, A.; Yamasaki, S. Influence of chemical structure of hard segments on physical properties of polyurethane elastomers: A review. J. Polym. Res. 2020, 27, 140. [Google Scholar] [CrossRef]
- Lu, P.; Huang, S.; Shen, Y.; Zhou, C.; Shao, L. Mechanical Performance Analysis of Polyurethane-Modified Asphalt Using Molecular Dynamics Method. Polym. Eng. Sci. 2021, 61, 2323–2338. [Google Scholar] [CrossRef]
- Da Silva, F.I.; Sa, E.; de Matos, J.M.E. Polyurethanes Obtained by Reacting 1,6-HDI with Monoglycerides from Babassu Oil: A DFT Study. J. Braz. Chem. Soc. 2020, 31, 1949–1954. [Google Scholar] [CrossRef]
- Subramani, S.; Lee, J.; Kim, J.; Cheong, I. One-Pack Cross-Linkable Waterborne Methyl Ethyl Ketoxime-Blocked Polyurethane/Clay Nanocomposite Dispersions. Macromol. Res. 2005, 13, 418–426. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Z.; Fang, T.; Zhang, M.; Xu, Y.; Liu, J.; Tan, X. Preparation and Corrosion Resistance of Polyaniline and Waterborne Polyurethane Composite Coating Film. Polym. Advan Technol. 2024, 35, e6307. [Google Scholar] [CrossRef]
- Tang, M.; Huang, L.; Wang, J.; Guan, D. Research Progress of Blade Coatings in Wind Turbines. IOP Conf. Ser. Mat. Sci. Eng. 2019, 542, 012061. [Google Scholar] [CrossRef]
- Vryonis, O.; Laudani, A.A.; Andritsch, T.; Golosnoy, I.O.; Vaughan, A.S. Lightning Protection of Wind Turbine Blades—How Supersizing Has Created New Challenges for Nanodielectrics Research. IEEE Electr. Insul. Mag. 2021, 37, 6–20. [Google Scholar] [CrossRef]
- Wen, T.; Luo, S.; Yang, C. Ionic Conductivity of Polymer Electrolytes Derived from Various Diisocyanate-Based Waterborne Polyurethanes. Polymer 2000, 41, 6755–6764. [Google Scholar] [CrossRef]
- Willocq, B.; Khelifa, F.; Odent, J.; Lemaur, V.; Yang, Y.; Leclere, P.; Cornil, J.; Dubois, P.; Urban, M.W.; Raquez, J.-M. Mechanistic Insights on Spontaneous Moisture-Driven Healing of Urea-Based Polyurethanes. ACS Appl. Mater. Interfaces 2019, 11, 46176–46182. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gao, Y.; Chen, B.; Crespi, V.H.; van Duin, A.C.T. Prediction of a Novel Electromechanical Response in Polar Polymers with Rigid Backbones: Contrasting Furan-Derived Nanothreads to Poly(Vinylidene Fluoride). Nano Lett. 2024, 24, 9195–9201. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Su, K.-H.; Fan, H.-Q.; Wen, Z.-Y. Structure and Electric Properties of Poly(Vinylidene Fluoride–Tetrafluoroethylene) Copolymer Studied with Density Functional Theory. Polymer 2007, 48, 7145–7155. [Google Scholar] [CrossRef]
- Irigoyen, M.; Matxain, J.M.; Ruiperez, F. Combined DFT and MD Simulation Protocol to Characterize Self-Healing Properties in Disulfide-Containing Materials: Polyurethanes and Polymethacrylates as Case Studies. Front. Mater. 2022, 9, 859482. [Google Scholar] [CrossRef]
- Hanna, J.N.; Nziko, V.d.P.N.; Ntie-Kang, F.; Mbah, J.A.; Toze, F.A.A. The Use of Minimal Topological Differences to Inspire the Design of Novel Tetrahydroisoquinoline Analogues with Antimalarial Activity. HELIYON 2021, 7. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis ofmolecular surface based onimproved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Yildirim, E.; Yurtsever, M.; Wilkes, G.L.; Yilgor, I. Effect of Intersegmental Interactions on the Morphology of Segmented Polyurethanes with Mixed Soft Segments: A Coarse-Grained Simulation Study. Polymer 2016, 90, 204–214. [Google Scholar] [CrossRef]
- Lorenzini, R.G.; Kline, W.M.; Wang, C.C.; Ramprasad, R.; Sotzing, G.A. The Rational Design of Polyurea & Polyurethane Dielectric Materials. Polymer 2013, 54, 3529–3533. [Google Scholar]
- Anashkin, I.P.; Klinov, A.V.; Davletbaeva, I.M. Molecular Simulation of Pervaporation on Polyurethane Membranes. Membranes 2023, 13, 128. [Google Scholar] [CrossRef]
- Park, S.; Moon, J.; Kim, B.; Cho, M. Multi-Scale Coarse-Grained Molecular Dynamics Simulation to Investigate the Thermo-Mechanical Behavior of Shape-Memory Polyurethane Copolymers. Polymer 2021, 213, 123228. [Google Scholar] [CrossRef]
- Wang, L.-F. Studies on Fluorinated Polyurethanes by X-ray Diffraction and Density Functional Theory Calculations with Periodic Boundary Conditions. Polymer 2007, 48, 7414–7418. [Google Scholar] [CrossRef]
- Saha, J.K.; Rahman, M.M.; Haq, M.B.; Al Shehri, D.A.; Jang, J. Theoretical and Experimental Studies of Hydrogen Bonded Dihydroxybenzene Isomers Polyurethane Adhesive Material. Polymers 2022, 14, 1701. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hasneen, A.; Jo, N.J.; Kim, H.I.; Lee, W.-K. Properties of Waterborne Polyurethane Adhesives with Aliphatic and Aromatic Diisocyanates. J. Adhes. Sci. Technol. 2011, 25, 2051–2062. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.; Chen, S.; Ji, F. Theoretical Study of Hydrogen Bonding Interactions on MDI-Based Polyurethane. J. Mol. Model. 2010, 16, 1391–1399. [Google Scholar] [CrossRef]
- Karunarathna, B.; Jayakody, R.S.; Karunanayake, L.; Govender, K.K. Computational development and validation of a representative MDI-BDO–based polyurethane hard segment model. J. Mol. Model. 2021, 27, 37. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, R.-Y.; Ying, W.-B.; Hu, H.; Wang, Y.-B.; Guo, Y.-Q.; Wang, W.-Q.; Tang, Z.-B.; Zhu, J. Polyether-Polyester and HMDI Based Polyurethanes: Effect of PLLA Content on Structure and Property. Chin. J. Polym. Sci. 2019, 37, 1152–1161. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; Li, Q.; Wang, J.; Kang, M.; Wang, X.; Xiang, H. Synthesis and Characterization of Waterborne Polyurethane Adhesive from MDI and HDI. J. Appl. Polym. Sci. 2008, 110, 1396–1402. [Google Scholar] [CrossRef]
- Tan, T.; Siew, W.H.; Han, L.; Given, M.; McKendry, C.; Liggat, J.; Li, Q.; He, J. Self-Healing of Electrical Damage in Microphase-Separated Polyurethane Elastomers with Robust Dielectric Strength Utilizing Dynamic Hydrogen Bonding Networks. ACS Appl. Polym. Mater. 2023, 5, 7132–7143. [Google Scholar] [CrossRef]
- Risti, I.; Cakic, S.; Vuki, N.; Teofilovi, V.; Tanasi, J.; Pili, B. The Influence of Soft Segment Structure on the Properties of Polyurethanes. Polymers 2023, 15, 3755. [Google Scholar] [CrossRef] [PubMed]
- Mirhosseini, M.M.; Haddadi-Asl, V.; Khordad, R. Molecular Dynamics Simulation, Synthesis and Characterization of Polyurethane Block Polymers Containing PTHF/PCL Mixture as a Soft Segment. Polym. Bull. 2022, 79, 643–661. [Google Scholar] [CrossRef]
- Li, Y.; Pang, Z.; Zheng, H.; Huang, J.; Huang, B.; Shi, M. Electrical Properties of CF3SO2F Insulating Gas Based on Density Functional Theory. IEEE Trans. Dielectr. Electr. Insul. 2024, 31, 297–303. [Google Scholar] [CrossRef]
- Barman, S.K.; Yang, M.; Parsell, T.H.; Green, M.T.; Borovik, A.S. Semiempirical Method for Examining Asynchronicity in Metal–Oxido-Mediated C–H Bond Activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2108648118. [Google Scholar] [CrossRef]
- Zhu, S.; Lempesis, N.; In’t Veld, P.J.; Rutledge, G.C. Molecular Simulation of Thermoplastic Polyurethanes under Large Compressive Deformation. Macromolecules 2018, 51, 9306–9316. [Google Scholar] [CrossRef]
- Demir, P.; Akman, F. Molecular Structure, Spectroscopic Characterization, HOMO and LUMO Analysis of PU and PCL Grafted onto PEMA-Co-PHEMA with DFT Quantum Chemical Calculations. J. Mol. Struct. 2017, 1134, 404–415. [Google Scholar] [CrossRef]
- Zhuo, L.-G.; Liao, W.; Yu, Z.-X. A Frontier Molecular Orbital Theory Approach to Understanding the Mayr Equation and to Quantifying Nucleophilicity and Electrophilicity by Using HOMO and LUMO Energies. Asian J. Org. Chem. 2012, 1, 336–345. [Google Scholar] [CrossRef]
- Xiang, D.; Liu, L.; Liang, Y. Effect of Hard Segment Content on Structure, Dielectric and Mechanical Properties of Hydroxyl-Terminated Butadiene-Acrylonitrile Copolymer-Based Polyurethane Elastomers. Polymer 2017, 132, 180–187. [Google Scholar] [CrossRef]
- Souza, J.D.C.; de Azevedo Junior, R.V.; Vinhas, G.M.; Lima, N.B.D.; Brito, A.M.S.S. Evaluation of performance of a polyurethane sensing phase by gibbs energy of solvation and infrared spectroscopy for determination of Toluene IN water. Quim. Nova 2018, 41, 394–399. [Google Scholar] [CrossRef]
- Pang, Z.; Li, Y.; Zheng, H.; Qin, R. Microscopic Mechanism of Electrical Aging of PVDF Cable Insulation Material. Polymers 2023, 15, 1286. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electric Field Intensity [V/nm] | HS | SS | |
|---|---|---|---|
| D1 (C21, C14, C2, C5) [°] | D2 (O31, C32, C35, C38) [°] | D3 (C8, C1, C2, C3) [°] | |
| 0 | −2.43 | −172.83 | 175.03 |
| 1.2855 | −3.42 | −164.25 | 172.38 |
| 2.571 | 3.31 | −58.68 | 168.46 |
| 3.8565 | 4.18 | −58.55 | 163.34 |
| 5.142 | 7.00 | −61.31 | 154.79 |
| 6.4275 | 8.19 | −59.53 | 140.53 |
| 7.713 | −7.93 | −42.24 | 114.99 |
| 8.2272 (0.016 a.u.) | −22.03 | −177.97 | 64.63 |
| HH Molecule | SS Molecule | ||||||
|---|---|---|---|---|---|---|---|
| HOMO | LUMO | HOMO | LUMO | ||||
| O41 | 54.52% | C25 | 29.31% | O5 | 63.64% | O7 | 42.23% |
| C38 | 13.19% | O41 | 15.67% | O6 | 13.73% | C19 | 39.04% |
| C25 | 6.84% | O26 | 14.75% | C1 | 8.50% | H10 | 6.27% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, Z.; Huang, S.; Li, Y.; Zhang, Y.; Qin, R. Molecular Simulation Analysis of Polyurethane Molecular Structure under External Electric Field. Molecules 2024, 29, 4329. https://doi.org/10.3390/molecules29184329
Pang Z, Huang S, Li Y, Zhang Y, Qin R. Molecular Simulation Analysis of Polyurethane Molecular Structure under External Electric Field. Molecules. 2024; 29(18):4329. https://doi.org/10.3390/molecules29184329
Chicago/Turabian StylePang, Zhiyi, Shangshi Huang, Yi Li, Yiyi Zhang, and Rui Qin. 2024. "Molecular Simulation Analysis of Polyurethane Molecular Structure under External Electric Field" Molecules 29, no. 18: 4329. https://doi.org/10.3390/molecules29184329
APA StylePang, Z., Huang, S., Li, Y., Zhang, Y., & Qin, R. (2024). Molecular Simulation Analysis of Polyurethane Molecular Structure under External Electric Field. Molecules, 29(18), 4329. https://doi.org/10.3390/molecules29184329