

On the Simulation of Photoreactions Using Restricted Open-Shell Kohn–Sham Theory


Abstract
1. Introduction
2. Results and Discussions
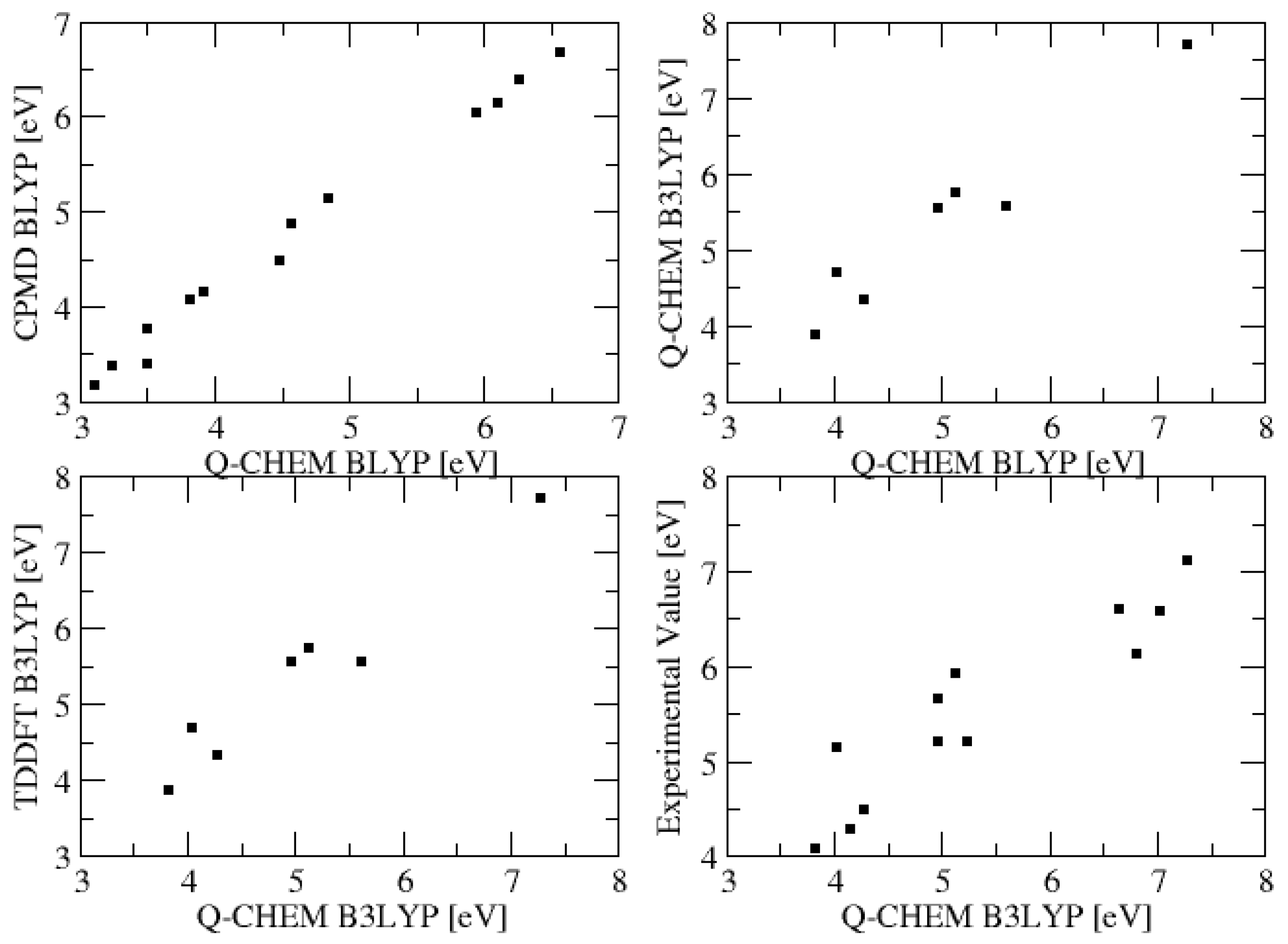
2.1. Excitation Energies and Orbitals
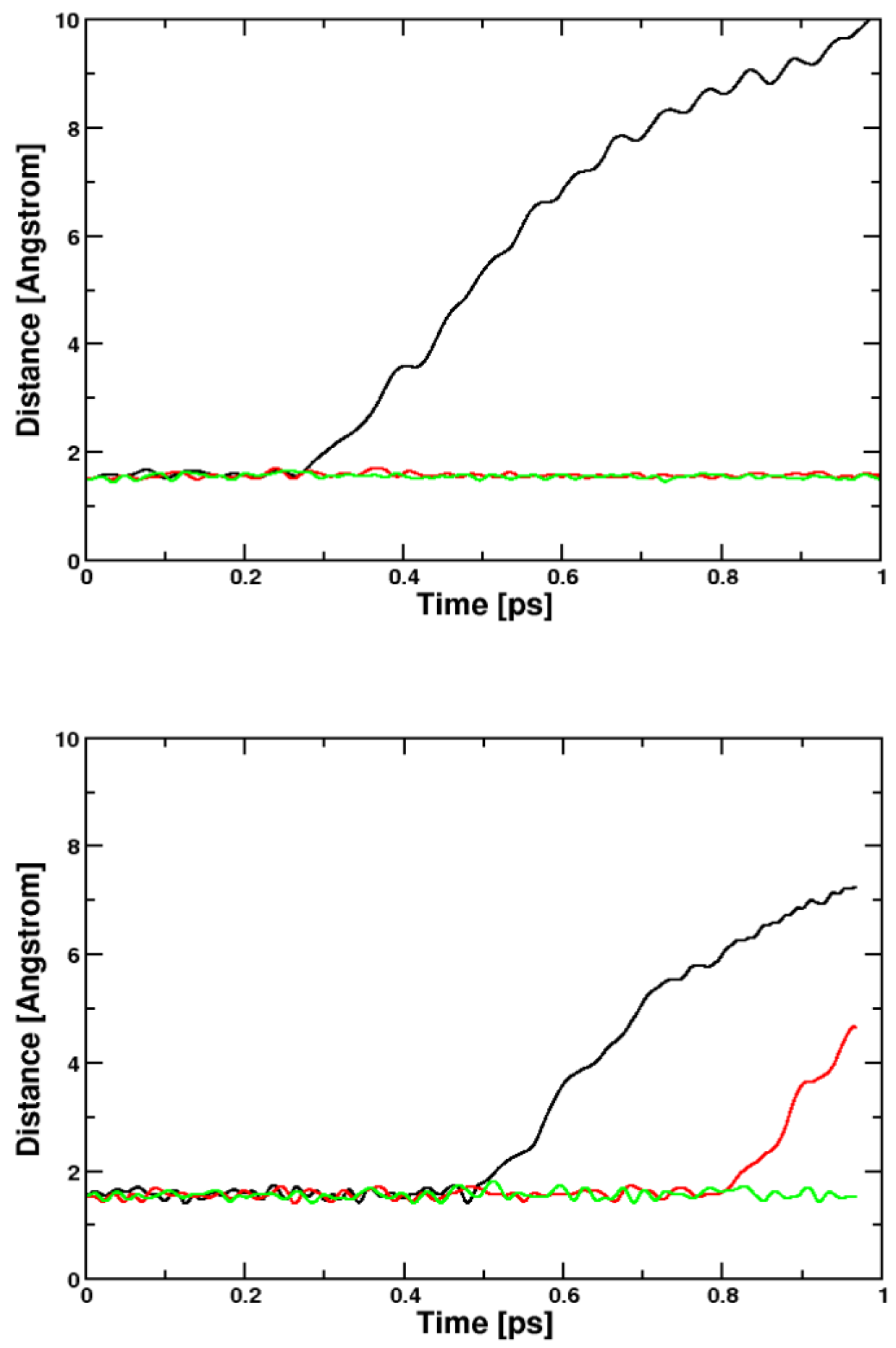
2.2. Molecular Dynamics
2.2.1. Ethene
2.2.2. Toluol and Chlorine
2.2.3. Silver Citrate
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIMD | Ab-Initio Molecular Dynamics |
| BLYP | Becke-Lee-Yang-Parr density functional |
| BOMD | Born-Oppenheimer Molecular Dynamics |
| CASSCF | Complete Active Space Self-Consistent Field |
| CPMD | Car-Parrinello Molecular Dynamics |
| GGA | Generalized Gradient Approximation |
| SCF | Self-Consistent Field |
References
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Farad. Soc. 1950, 9, 14. [Google Scholar] [CrossRef]
- Bernardi, F.; De, S.; Olivucci, M.; Robb, M.A. The Mechanism of Ground-State-Forbidden Photochemical Pericyclic Reactions: Evidence for Real Conical Intersections. J. Am. Chem. Soc. 1990, 112, 1737. [Google Scholar] [CrossRef]
- Bernardi, F.; Olivucci, M.; Robb, M.A. Potential energy surface crossings in organic photochemistry. Chem. Soc. Rev. 1996, 25, 321. [Google Scholar] [CrossRef]
- Frank, I. Classical motion of the nuclei in a molecule: A concept without alternatives. Chem. Sel. 2020, 5, 1872. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Stuchebrukhov, A.A. Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev. 2010, 110, 6939. [Google Scholar] [CrossRef] [PubMed]
- Tully, J.C. Molecular dynamics with electronic transitions. J. Chem. Phys. 1990, 93, 1061. [Google Scholar] [CrossRef]
- Domcke, W.; Yarkony, D.R. Role of Conical Intersections in Molecular Spectroscopy and Photoinduced Chemical Dynamics. Annu. Rev. Phys. Chem. 2012, 63, 325. [Google Scholar] [CrossRef]
- Curchod, B.F.E.; Martínez, T.J. Ab Initio Nonadiabatic Quantum Molecular Dynamics. Chem. Rev. 2018, 118, 3305. [Google Scholar] [CrossRef]
- Nelson, T.R.; White, A.J.; Bjorgaard, J.A.; Sifain, A.E.; Zhang, Y.; Nebgen, B.; Fernandez-Alberti, S.; Mozyrsky, D.; Roitberg, A.E.; Tretiak, S. Non-adiabatic Excited-State Molecular Dynamics: Theory and Applications for Modeling Photophysics in Extended Molecular Materials. Chem. Rev. 2020, 120, 2215. [Google Scholar] [CrossRef]
- Westermayr, J.; Gastegger, M.; Vörös, D.; Panzenboeck, L.; Joerg, F.; González, L.; Marquetand, P. Deep learning study of tyrosine reveals that roaming can lead to photodamage. Nat. Chem. 2022, 14, 914. [Google Scholar] [CrossRef]
- Schulte, M.; Frank, I. Restricted open-shell Kohn-Sham theory: N unpaired electrons. Chem. Phys. 2010, 373, 283. [Google Scholar] [CrossRef]
- Pople, J.A.; Gill, P.M.W.; Handy, N.C. Spin-Unrestricted Character of Kohn-Sham Orbitals for Open-Shell Systems. Int. J. Quantum Chem. 1995, 56, 303. [Google Scholar] [CrossRef]
- Frank, I.; Hutter, J.; Marx, D.; Parrinello, M. Molecular dynamics in low-spin excited states. J. Chem. Phys. 1998, 108, 4060. [Google Scholar] [CrossRef]
- Filatov, M.; Shaik, S. Spin-restricted density functional approach to the open-shell problem. Chem. Phys. Lett. 1998, 288, 689. [Google Scholar] [CrossRef]
- Gräfenstein, J.; Cremer, D.; Kraka, E. Density functional theory for open-shell singlet biradicals. Chem. Phys. Lett. 1998, 288, 593. [Google Scholar] [CrossRef]
- Grimm, S.; Nonnenberg, C.; Frank, I. Restricted open-shell Kohn-Sham theory for π-π* transitions. I. Polyenes, cyanines, and protonated imines. J. Chem. Phys. 2003, 119, 11574. [Google Scholar] [CrossRef]
- Hirao, K.; Nakatsuji, H. General SCF operator satisfying correct variational condition. J. Chem. Phys. 1973, 59, 1457. [Google Scholar] [CrossRef]
- Goedecker, S.; Umrigar, C.J. Critical assessment of the self-interaction-corrected-local-density-functional method and its algorithmic implementation. Phys. Rev. A 1997, 55, 1765. [Google Scholar] [CrossRef]
- Filatov, M. Spin-restricted ensemble-referenced Kohn–Sham method: Basic principles and application to strongly correlated ground and excited states of molecules. WIREs Comput. Mol. Sci. 2014, 5, 146. [Google Scholar] [CrossRef]
- Hait, D.; Head-Gordon, M. Orbital Optimized Density Functional Theory for Electronic Excited States. J. Phys. Chem. Lett. 2021, 12, 4517. [Google Scholar] [CrossRef]
- Röhrig, U.; Guidoni, L.; Laio, A.; Frank, I.; Röthlisberger, U. A molecular spring for vision. J. Am. Chem. Soc. 2004, 126, 15328. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Bräuchle, C.; Frank, I. Light-driven unidirectional rotation in a molecule: ROKS simulation. Chem. Phys. Chem. 2005, 6, 1943. [Google Scholar] [CrossRef] [PubMed]
- Kunze, L.; Hansen, A.; Grimme, S.; Mewes, J.M. PCM-ROKS for the description of charge-transfer states in solution: Singlet-triplet gaps with chemical accuracy from open-shell Kohn-Sham reaction-field calculations. J. Phys. Chem. Lett. 2021, 12, 8470. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. 1984, 52, 997. [Google Scholar] [CrossRef]
- Casida, M.E.; Salahub, D.R. Asymptotic correction approach to improving approximate exchange-correlation potentials: Time-dependent density-functional theory calculations of molecular excitation spectra. J. Chem. Phys. 2000, 113, 8918. [Google Scholar] [CrossRef]
- Shao, Y.; Head-Gordon, M.; Krylov, A.I. The spin–flip approach within time-dependent density functional theory: Theory and applications to diradicals. J. Chem. Phys. 2003, 118, 4807. [Google Scholar] [CrossRef]
- Wang, F.; Ziegler, T. Time-dependent density functional theory based on a noncollinear formulation of the exchange-correlation potential. J. Chem. Phys. 2004, 121, 12191. [Google Scholar] [CrossRef]
- Lee, S.; Filatov, M.; Lee, S.; Choi, C.H. Eliminating spin-contamination of spin-flip time dependent density functional theory within linear response formalism by the use of zeroth-order mixed-reference (MR) reduced density matrix. J. Chem. Phys. 2018, 149, 104101. [Google Scholar] [CrossRef]
- Jacquemin, D.; Wathelet, V.; Perpete, E.A.; Adamo, C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420. [Google Scholar] [CrossRef]
- Marian, C. Spin–orbit coupling and intersystem crossing in molecules. WIREs Comput. Mol. Sci. 2012, 2, 187. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, N.; Goebl, J.; Lu, Z.; Yin, Y. A Systematic Study of the Synthesis of Silver Nanoplates: Is Citrate a ”Magic” Reagent? J. Am. Chem. Soc. 2011, 133, 18931. [Google Scholar] [CrossRef] [PubMed]
- Bastus, N.G.; Merkoci, F.; Piella, J.; Puntes, V. Synthesis of Highly Monodisperse Citrate-Stabilized Silver Nanoparticles of up to 200 nm: Kinetic Control and Catalytic Properties. Chem. Mater. 2014, 26, 2836. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Hutter, J.; Alavi, A.; Deutsch, T.; Bernasconi, M.; Goedecker, S.; Marx, D.; Tuckerman, M.; Parrinello, M. CPMD Version 4.3. Copyright IBM Corp 1990–2015, Copyright MPI für Festkörperforschung Stuttgart 1997–2001. Available online: https://github.com/CPMD-code/CPMD/releases/tag/4.3 (accessed on 18 September 2024).
- Becke, A. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle–Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S. Accurate Description of van der Waals Complexes by Density Functional Theory Including Empirical Corrections. J. Comput. Chem. 2004, 25, 1463. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Boero, M.; Parrinello, M.; Terakura, K.; Weiss, H. Car-Parrinello study of Ziegler-Natta heterogeneous catalysis: Stability and destabilization problems of the active site models. Mol. Phys. 2002, 100, 2935–2940. [Google Scholar] [CrossRef]
- Becke, A. A New Mixing of Hartree-Fock and Local-Density Functional Theories. J. Chem. Phys. 1993, 98, 1372. [Google Scholar] [CrossRef]
- Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package. J. Chem. Phys. 2021, 155, 084801. [CrossRef]
- Wang, L.P.; Titov, A.; McGibbon, R.; Liu, F.; Pande, V.S.; Martínez, T.J. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 2014, 6, 1044. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CPMD BLYP | Q-Chem BLYP | Q-Chem B3LYP | TDDFT/B3LYP [29] | Exp. [13] |
|---|---|---|---|---|---|
| vertical excitation energies | |||||
| Formaldehyde | 3.501 | 3.761 | 3.828 | 3.87 | 4.07 |
| Acetaldehyde | 3.826 | 4.069 | 4.155 | - | 4.28 |
| Acetone | 3.916 | 4.156 | 4.274 | 4.34 | 4.48 |
| Formamide | 5.186 | 5.587 | 5.622 | 5.55 | 5.65 |
| Acetamide | 5.106 | 5.516 | 5.608 | 5.57 | - |
| Formaldimine | 4.576 | 4.873 | 4.970 | - | 5.0–5.4 |
| Acetaldimine | 4.846 | 5.127 | 5.240 | - | 5.0–5.4 |
| Ethene | 6.569 | 6.668 | 7.274 | 7.71 | 7.11 |
| Propene | 6.270 | 6.383 | 7.021 | - | 6.58 |
| 1-Butene | 5.941 | 6.050 | 6.646 | - | 6.61 |
| 2-Butene | 6.103 | 6.145 | 6.804 | - | 6.13 |
| Butadiene | 4.488 | 4.477 | 5.126 | 5.75 | 5.93 |
| Hexatriene | 3.502 | 3.387 | 4.036 | 4.69 | 5.15 |
| Propenal | 3.244 | 3.372 | 3.535 | - | - |
| Pentadienal | 3.106 | 3.161 | 3.355 | - | - |
| adiabatic excitation energies | |||||
| Formaldehyde | 3.151 | 3.362 | 3.352 | - | 3.50 |
| Acetaldehyde | 3.474 | 3.554 | 3.552 | - | 3.69 |
| Acetone | 3.567 | 3.613 | 3.627 | - | 4.73 |
| Formamide | 4.601 | 4.053 | 4.220 | - | - |
| Acetamide | 4.582 | 3.951 | 4.143 | - | - |
| Formaldimine | 3.791 | 2.010 | 3.003 | - | - |
| Acetaldimine | 4.034 | 3.160 | 3.127 | - | - |
| Ethene | 4.542 | 2.942 | 2.887 | - | - |
| Propene | 2.923 | 2.941 | 2.901 | - | - |
| 1-Butene | 4.368 | 2.831 | 2.788 | - | - |
| 2-Butene | 4.392 | 2.898 | 2.871 | - | - |
| Butadiene | 3.914 | 4.004 | 2.508 | - | - |
| Hexatriene | 3.148 | 2.308 | 2.306 | - | - |
| Propenal | 2.923 | 2.659 | 3.088 | - | - |
| Pentadienal | 2.768 | 2.324 | 2.369 | - | - |
| Sim. Number | AIMD Method | Equil. Temp. [K] | CPU Time Exc. State [CPU min per ps] | Cell Size [Å3] | Reaction |
|---|---|---|---|---|---|
| CPMD code | |||||
| Chlorine + toluene | |||||
| 1 | BOMD/BLYP | 300 | 193,667 | 1000 | dissociation |
| 2 | CPMD/BLYP | 300 | 3267 | 1000 | dissociation |
| 3 | CPMD/BLYP | 600 | 3583 | 1000 | dissociation |
| 4 | CPMD/BLYP | 900 | 3483 | 1000 | dissociation |
| Trisilver citrate | |||||
| 5 | BOMD/BLYP | 600 | 350,000 | 1728 | dissociation + proton transfer |
| 6 | BOMD/BLYP | 600 | 342,737 | 2744 | dissociation (2 times) |
| 7 | BOMD/BLYP | 600 | 450,788 | 2197 | dissociation + proton transfer |
| 8 | CPMD/BLYP | 600 | 11,925 | 1728 | - |
| 9 | CPMD/BLYP | 600 | 15,569 | 2744 | dissociation + proton transfer |
| 10 | CPMD/BLYP | 600 | 13,250 | 3375 | - |
| Trisilver citrate + 8 water molecules | |||||
| 11 | BOMD/BLYP | 600 | 424,016 | 2744 | - |
| Q-Chem code | |||||
| Chlorine + toluene | |||||
| 12 | BOMD/B3LYP | 300 | 10,131 | - | dissociation |
| Trisilver citrate | |||||
| 13 | BOMD/BLYP | 300 | 38,637 | - | dissociation + proton transfer |
| 14 | BOMD/B3LYP | 300 | 52,482 | - | dissociaton (2 times) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Büchel, R.; Álvarez, L.; Grage, J.; Maniscalco, D.; Frank, I. On the Simulation of Photoreactions Using Restricted Open-Shell Kohn–Sham Theory. Molecules 2024, 29, 4509. https://doi.org/10.3390/molecules29184509
Büchel R, Álvarez L, Grage J, Maniscalco D, Frank I. On the Simulation of Photoreactions Using Restricted Open-Shell Kohn–Sham Theory. Molecules. 2024; 29(18):4509. https://doi.org/10.3390/molecules29184509
Chicago/Turabian StyleBüchel, Ralf, Luis Álvarez, Jan Grage, Dominykas Maniscalco, and Irmgard Frank. 2024. "On the Simulation of Photoreactions Using Restricted Open-Shell Kohn–Sham Theory" Molecules 29, no. 18: 4509. https://doi.org/10.3390/molecules29184509
APA StyleBüchel, R., Álvarez, L., Grage, J., Maniscalco, D., & Frank, I. (2024). On the Simulation of Photoreactions Using Restricted Open-Shell Kohn–Sham Theory. Molecules, 29(18), 4509. https://doi.org/10.3390/molecules29184509