
The Chemistry of Selenosilanes: A Topic Overview



Department of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3-13, 50019 Sesto Fiorentino, FI, Italy
*
Author to whom correspondence should be addressed.
Molecules 2024, 29(19), 4595; https://doi.org/10.3390/molecules29194595
Submission received: 6 September 2024
/
Revised: 23 September 2024
/
Accepted: 24 September 2024
/
Published: 27 September 2024
(This article belongs to the Special Issue Organoselenium Compounds: Synthesis, Catalysis and Biological Activities)
Abstract
:Selenium-containing molecules represent a valuable class of compounds with a variety of applications in chemical and biological fields. Selenated reagents are used as intermediates to introduce functional groups (e.g., double bonds) onto different substrates or in the synthesis of various selenated derivatives. Among the variety of selenium-containing reagents, silyl selenides are frequently used to transfer a selenated moiety due to the smooth functionalization of the Se-Si bond, which allows for the generation of selenium nucleophilic species under mild conditions. While the use of the analogous sulfur nucleophiles, namely silyl sulfides, has been widely explored, a relatively limited number of reports on selenosilanes have been provided. This contribution will focus on the application of selenosilanes as nucleophiles in a variety of organic transformations, as well as under radical and redox conditions. The use of silyl selenides to prepare metal complexes and as selenium precursors of materials for atomic layer deposition will also be discussed.

1. Introduction
Selenated compounds represent an interesting class of molecules, which are attracting increasing interest for their application in different fields of chemistry [1,2]. They are employed in organic synthesis, in biochemistry (for example, as antioxidants, anticancers and antimicrobials), inorganic chemistry and materials science. Different methods of introducing a selenated moiety into different substrates have been described, and among them, methodologies based on the reactivity of silyl derivatives represent an efficient alternative approach [3,4]. Silyl selenides, in fact, can be regarded as the synthetic equivalents of the corresponding hydrogenated compounds (i.e., RSeSiMe3 = RSeH; (Me3Si)2Se = H2Se), but they are more stable and safe and, hence, are easier to prepare, store, handle and measure. In addition, trimethylsilyl selenides, together with trimethylsilyl sulfides, can also be used as soft silylating agents, as well as in the protection of carbonyl groups.
2. Synthesis of Selenosilanes
Some silyl selenides are commercially available—for example, PhSeSiMe3—or can be prepared by different methods [5], mainly depending on the group on the selenium atom. A typical approach for the synthesis of (phenylseleno)trimethylsilane, PhSeSiMe3 2a or (methylseleno)trimethylsilane, MeSeSiMe3 2b is the reduction of the parent diselenides 1a,b under suitable reducing conditions (Scheme 1) [6,7,8,9,10,11,12,13,14].
The preparation of butylseleno silanes and oligosilanes was reported by Herzog through the reaction of BuSeLi (obtained from BuLi and Se0) with differently substituted chlorosilanes MexPhySiCl(4-x-y) [10].
Bis(trimethylsilyl)selenide (Me3Si)2Se (hexamethyldisilaselenane, HMDSS) 3a is synthesized by treating Se(0) with Li(0) [15,16] or with lithium triethylborohydride [17] followed by the addition of the chlorosilane. Silyl selenides with larger groups (Et3Si, tBuMe2Si) than Me3Si were prepared from Na/Se(0) and the suitable chloroalkylsilane (Scheme 2) [18].
3. Selenosilanes in Chemical Synthesis
3.1. Silyl Selenides in the Nucleophilic Substitutions on Organic Substrates
3.1.1. Reaction with Halogens or Halogenated Compounds
The functionalization of the Si-Se bond under mild conditions enabled the nucleophilic transfer of the seleno moiety onto a variety of organic substrates. In this context, (phenylseleno)trimethylsilane 2a has been widely used in different organic reactions. Detty and Seidler [19] reported the reaction with halogens to afford silyl halides. The reaction with Cl2 was performed in a solvent (CCl4 or 1,2-dichlorobenzene), while Br2 and I2 were used under neat conditions (Scheme 3).
Diphenyldiselenide 1a was formed as a by-product which can be reused to prepare the (phenylseleno)silane. Trialkylsilyl halides were also obtained by the reaction of xenon difluoride with selenosilanes, providing RSe-F intermediates, which were then treated with acetylenes to give selenated fluoro-olefines (Scheme 4) [11].
The treatment of bis(trimethylsilyl)selenide 3a with n-BuLi and alkylation with alkyl halides to provide silyl selenides 2 were reported by Segi and co-workers (Scheme 5, equation a) [20]. Further reaction of silyl selenides 2 with n-BuLi and alkyl or acyl halides gave unsymmetrical selenides 5 (Scheme 5, equation a).
Based on a slightly modified procedure, Corrigan described the reaction of [LiSeSiMe3] 4 with propargyl bromides to obtain propargyl selenoethers 6 by nucleophilic substitution (Scheme 5, equation b) [21]. Characterization by NMR spectroscopy and mass spectrometry were also provided. The reaction of different selenosilanes 2,3a with tropylium bromide 7 led to 1-cyclohepta-2,4,6-trienyl-selanes 8a,b, which were characterized by 1H, 13C and 77Se NMR data, including 1J(77Se-13C) measurement. The formation of 8b can be explained through the generation of the tropylium-selenosilane intermediate (C7H7SeSiMe3), which then reacts with 7 to provide selenide 8b. DFT calculations were also reported (Scheme 6) [22].
3.1.2. Reaction with Benzyl and Allylic Alcohols
As reported by Abe, Harayama and co-workers, (phenylseleno)trimethylsilane 2a in combination with a Lewis acid was used as an efficient nucleophile in the direct conversion of benzyl alcohols into benzyl selenides 9 (Scheme 7) [8]. Compared to other Lewis acids (e.g., ZnI2, TiCl4, AlCl3, BF3.Et2O), better results were obtained with AlBr3, and higher yields were observed when the PhSeSiMe3:AlBr3 system was used at a 1:1 ratio.
When a non-benzylic hydroxy group, such as 2-phenyl ethanol, was reacted, no formation of the expected phenyl(2-phenyl)ethyl selenide was observed, while cinnamyl alcohol afforded the benzoselenane derivative 10a, formed through a [3,3]-sigma tropic rearrangement of the initially formed selenide PhSeCH2CH=CHPh. (Methylseleno)trimethylsilane 2b behaved as a less efficient nucleophile under the same conditions, leading to the corresponding methyl selenides in rather low yields. The behavior of allylic alcohols was general, as reported by Abe et al. upon reacting differently substituted allylic alcohols with PhSeSiMe3/AlBr3 to give selenochroman derivatives 10 through a one-pot reaction (Scheme 8) [23].
3.1.3. Reaction with Acetates and Ethers
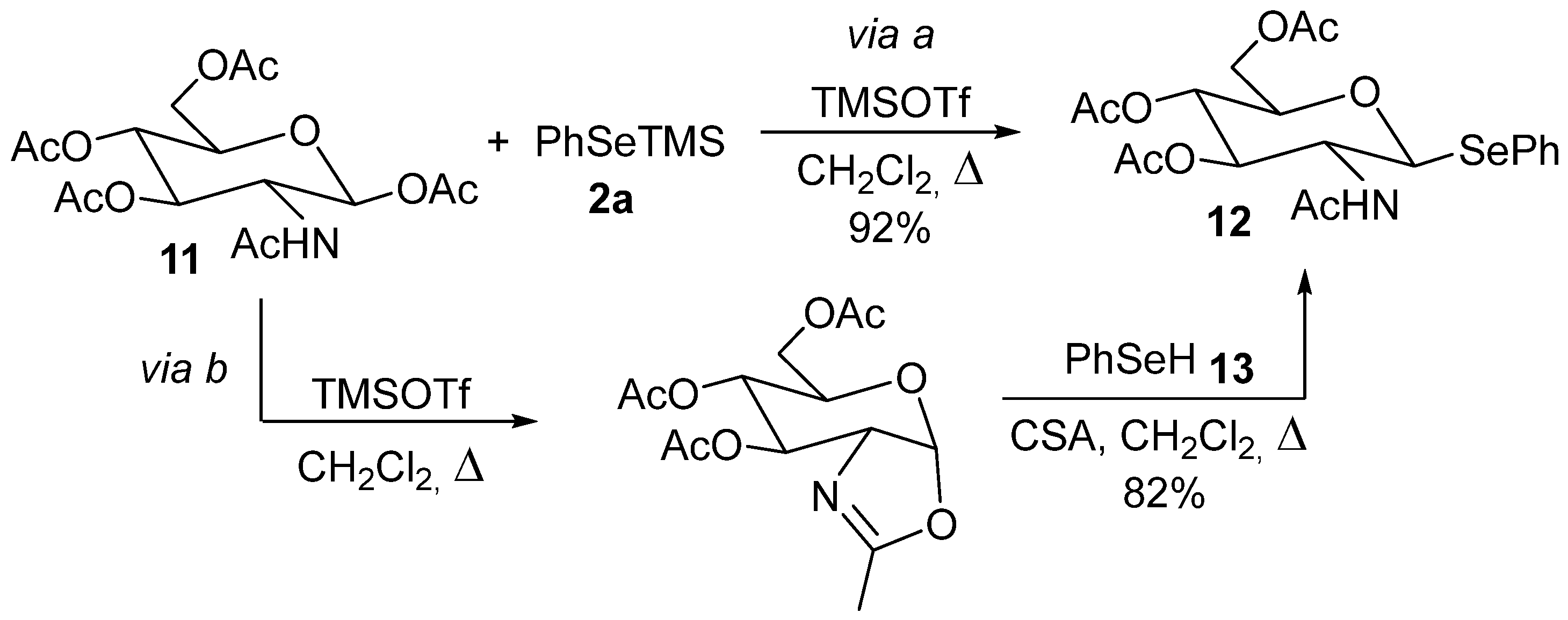
Selenoglycosides represent important intermediates for the synthesis of carbohydrate derivatives. Gallagher and co-workers reported the direct selenoglycosidation of peracetylated amino sugars 11 (galactosamine, mannosamine and glucosamine derivatives) by treatment with (phenylseleno)trimethylsilane 2a and silyl triflate, providing the corresponding anomeric selenides 12, as precursors of α-C-glycosides, after the substitution of an acetoxy group (Scheme 9, via a) [24]. Interestingly, when benzeneselenol 13 was used instead of the selenosilane, in the presence of camphorsulfonic acid (CSA), the formation of the selenoglycosides 12 required a two-step procedure (via oxazoline) (Scheme 9, via b).
In their study on the preparation of amino sugars from 1,2-oxazines, Pfrengle and Reissig reported the reaction of PhSeTMS 2a (as well as of PhSTMS) to synthesize syn- and anti-isomers of a 2-phenylseleno (or 2-phenylthio) substituted 1,3-dioxolanes 14 (Scheme 10) [25]. The phenylthio derivative was demonstrated as a precursor of amino sugar derivatives, obtained by a stereodivergent synthesis.
3.1.4. Reaction of Selenosilanes with Sulfurated Organic Substrates
β-Heterosubstituted nitroalkenes represent useful synthons in organic chemistry [27]. Taking into account that the sulfinyl group is a good living group, Abe, Harayama and co-workers reported the reaction of β-sulfinyl nitroalkenes 18 with Se-nucleophiles to prepare β-seleno-α,β-unsaturated nitroalkenes 19 [27]. It was found that phenyl selenolate PhSeNa was not able to provide the expected vinyl selenides, while benzeneselenol PhSeH, generated in situ from the corresponding (phenylseleno)trimethylsilane 2a in methanol, was efficient as a nucleophile. Therefore, the reaction of unsaturated sulfoxides with PhSeSiMe3 2a and MeOH led to the desired phenyl selenides 19 through a clean addition-elimination reaction (Scheme 12). (Methylseleno)trimethylsilane was also efficient in preparing related methyl selenides in good yields.
N-Thiophthalimides 20 were used as electrophilic sulfur-transfer reagents with bis(trimethylsilyl)selenide 3a under TBAF catalysis to give variously functionalized disulfides 21 (Scheme 13) [28]. The formation of a selenotrisulfide 22 could be proposed as a possible intermediate, which then provides the substituted disulfides after the elimination of elemental selenium.
3.1.5. Ring Opening of Heterocyclic Rings (O, S, N) by Selenosilanes
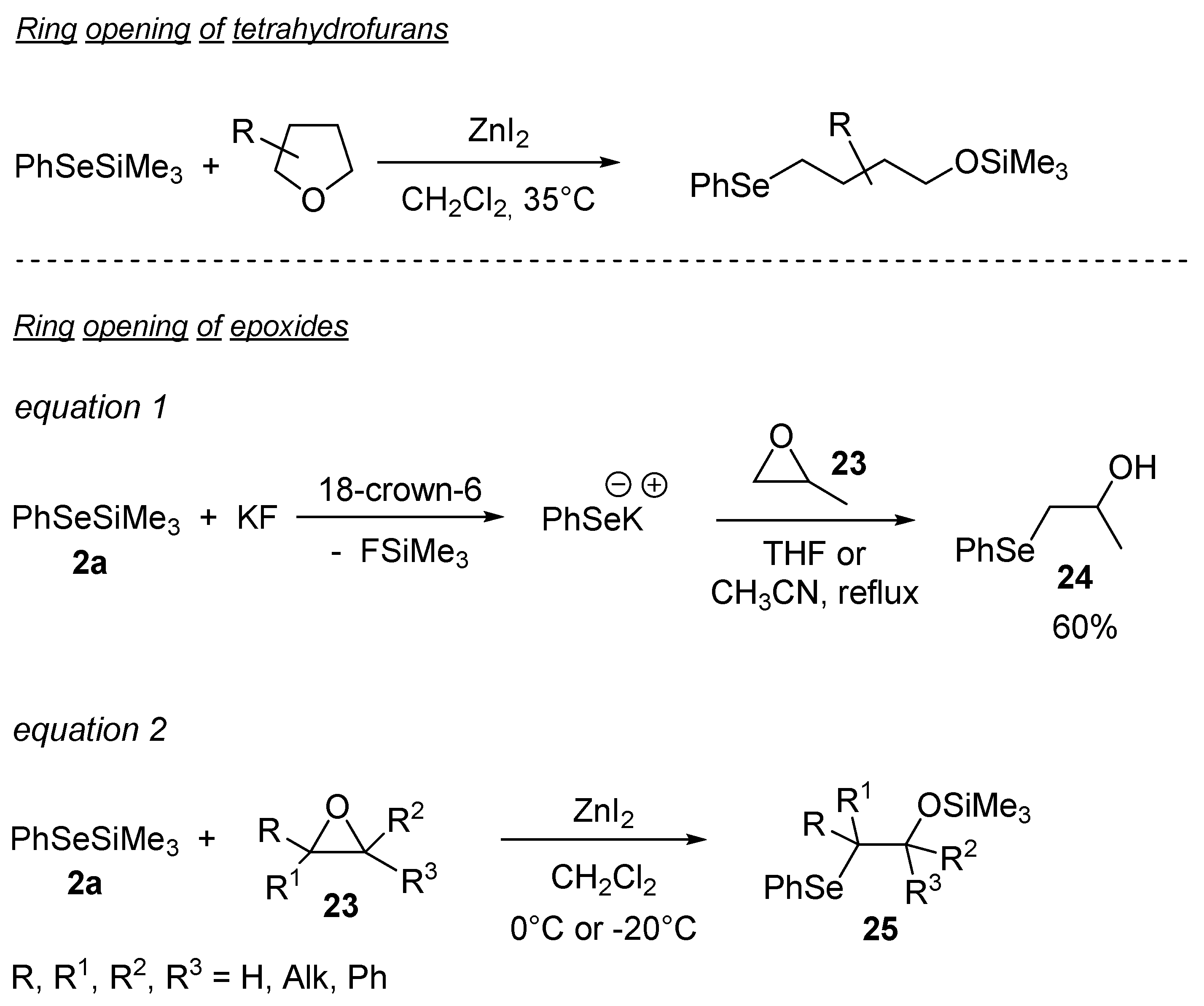
Besides the reaction reported by Crimmins (Scheme 11) [26] and by Murai, Sonoda et al. [29] on the ring opening of tetrahydrofurans by PhSeTMS (Scheme 14 up), most of the examples with oxygenated heterocycles concern the opening of epoxides by selenosilanes. Epoxides 23, as well as thiiranes and aziridines, are rather reactive species towards nucleophiles for the high strain of the three-membered ring. In this context, β-hydroxy selenides 24 are interesting compounds which find application as versatile intermediates for a variety of synthetic transformations and for their different chemical and biological properties [30]. The nucleophilic substitution of silyl selenides onto epoxides, therefore, represents a convenient method to access this class of bifunctionalized molecules. The ring opening of epoxides 23 by (phenylseleno)trimethylsilane 2a, as a potassium phenylselenide source, in the presence of KF/18-crown-6, was described by Detty to obtain β-hydroxy selenides 24 (Scheme 14, equation 1) [31]. The reaction was also efficient with other organic substrates, such as α,β-unsaturated carbonyls, lactones, esters and halides. Sonoda and Murai reported the reaction under ZnI2 catalysis to provide β-siloxyalkyl phenyl selenides 25 (Scheme 14, equation 2) [32].
Enantioselective desymmetrization represents a powerful method to achieve chiral compounds. Organocatalyzed reactions for desymmetrization of epoxides and aziridines with a variety of heteronucleophiles have been quite recently reviewed by Wang [33]. Tiecco and co-workers reported the asymmetric ring opening of meso-epoxides (meso-23) by (phenylseleno)silanes, under salen(Cr)complexes catalysis, to afford various optically active acyclic and cyclic β-hydroxy selenides (S,S)-24 (Scheme 15) [34]. The enantioselectivity of the process depends on the structure of the starting oxirane.
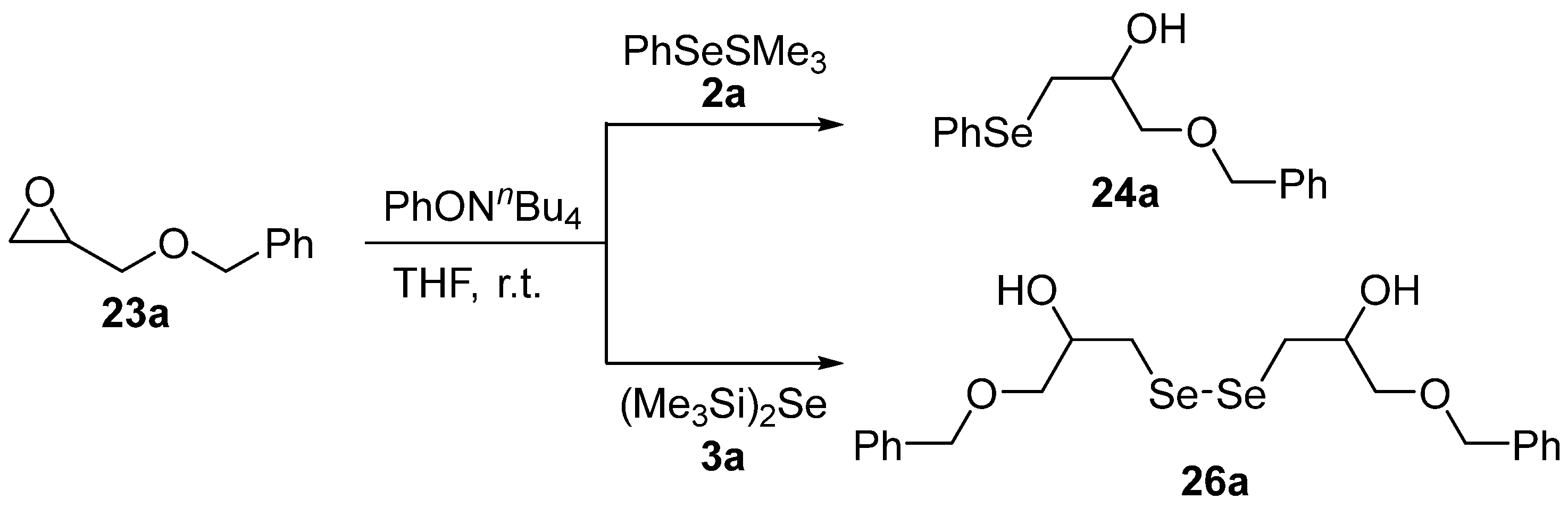
We reported that (phenylseleno)trimethylsilane 2a efficiently reacted with benzylglycidol 23a under PhONnBu4 catalysis, leading to β-phenylselenoalcohol 24a (Scheme 16), while when bis(trimethylsilyl)selenide 3a was used, the formation of the corresponding β-hydroxydiselenide 26a was observed (Scheme 16) [35].
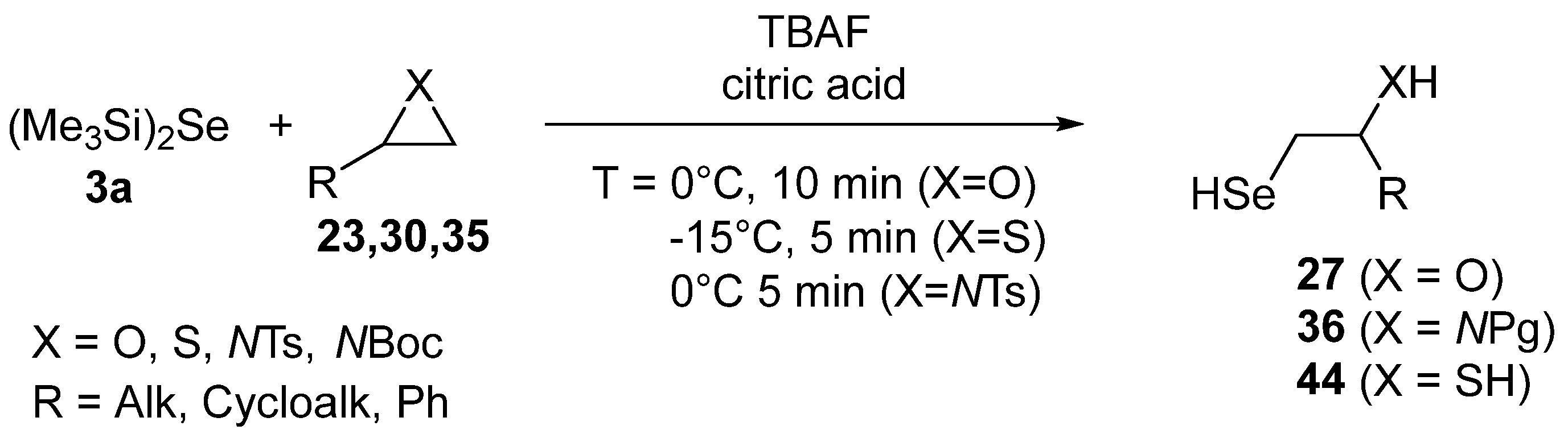
Furthermore, the β-hydroxyselenol 27, prepared by the reaction of epoxides with HMDSS [36] and treated with a suitable bromo ester, behaved as the precursor of the six-membered chalcogen-containing heterocycles, such as 6-substituted 2-hydroxy 1,4-oxaselenolanes 28, obtained as a mixture of stereoisomers (Scheme 17) [37].
The reaction of bis(trimethylsilyl)selenide 3a (HMDSS) was also performed with thiiranes under TBAF catalysis. Depending on the reaction conditions, from thiiranes 30, 3,7-disubstituted-1,2,5-dithiaselenepanes 31 were regioselectively obtained (Scheme 18, equation 1) [38], reasonably formed by the intramolecular oxidative ring closure of the β-mercapto selenide intermediate 29. When suitable fatty acid ester-substituted thiiranes 33 of glycidol were reacted with HMDSS/TBAF to afford the bis-silyl intermediate 32, which was treated with fatty acid acyl chlorides in situ, a regioselective one-pot synthesis of mixed sulfur and selenium isosters of triacyl glycerols 34 was achieved (Scheme 18, equation 2) [39]. The physico-chemical properties of these novel fatty acid chalcogeno esters were determined and compared to those of the fully oxygenated triglycerides.
Our group also found that a N-protected aziridine 35a reacted with HMDSS 3a to provide a regio- and enantioselective synthesis of the 1,2-amino selenol 36a (together with the corresponding diselenide) as a precursor of the 2,4-disubstituted 1,3-selenazolidine 37 upon treatment with aldehydes (Scheme 19) [40].
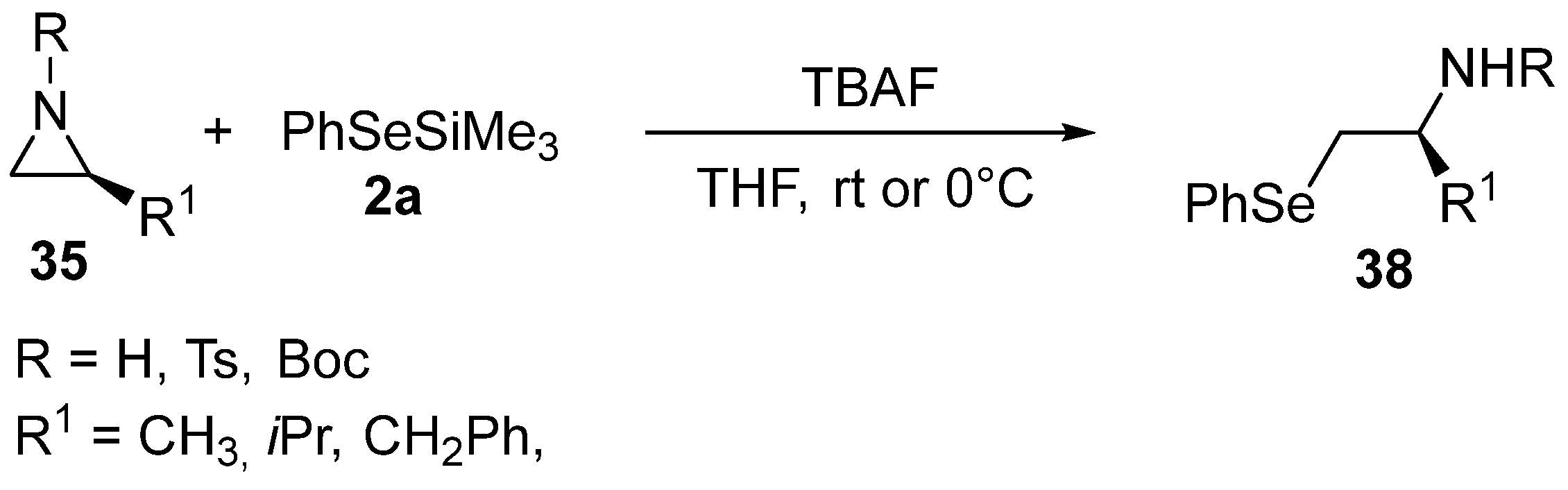
The regio- and enatioselective ring opening of activated (R = Ts, Boc) and unactivated (R = H) aziridines 35 was also described with (phenylseleno)trimethylsilane 2a under metal-free conditions, enabling the synthesis of chiral enantioenriched N-protected and unprotected β-arylseleno amines 38 (Scheme 20) [41].
In 2011, Della Sala and co-workers reported the first example of the organocatalyzed desymmetrization of meso-N-acylaziridines (meso-35) with selenosilanes, promoted by the chiral phosphoric acid (R)-VAPOL, leading to β-N-acyl-substituted phenyl selenides 39 (Scheme 21) [42].
The silyl selenide with the more sterically hindered silyl group (SiMe2t-Bu) showed poor reactivity and required a longer reaction time (4–12 days) with respect to the (phenylseleno)trimethylsilane. Better results were obtained when a mixture of PhSeSiMe3/PhSeH was used, leading to enantioenriched amine derivatives in a shorter time, high yields and high enantioselectivity. However, it was demonstrated that the silyl-nucleophile was necessary, as the reaction of the same aziridine with (R)-VAPOL and the selenol alone gave the product with 45% ee (97% ee with PhSeSiMe3/PhSeH). In 2013, the desymmetrization of meso-aziridines was re-investigated by Della Sala [43]. It was found that when VAPOL was treated with HCl to have the metal-free chiral phosphoric acid as an organocatalyst, the ring-opening products were formed as racemates in low yields. On the other hand, using a mixture 1:1 of calcium and magnesium phosphate salts of VAPOL, the amine derivatives were obtained in high yields and high ee. Therefore, the metal-free phosphoric acid is not an effective catalyst to promote the desymmetrization of meso-aziridines with silyl nucleophiles. The earlier reported results could then be attributed to the action of Ca and Mg phosphate salts, present as unexpected impurities in VAPOL, which could act through a dual Lewis acid–base activation.
As a further step in this study of the behavior of silyl selenides, our group reported the regio- and enantioselective ring opening of epoxides 23, thiiranes 30 and aziridines 35 by bis(trimethylselenide) 3a, with TBAF as a catalyst, providing convenient and general access to a variety of β-substituted diselenides 26, 40–41 (Scheme 22, equation 1) and selenides 42, 32, 43 (Scheme 22, equation 2) through a fine-tuning of the reaction conditions (ratio of reagents, temperature) [44]. The antioxidant catalytic activity of these compounds was also evaluated, with some of them showing a significant glutathione peroxidase (GPx)-like activity. Furthermore, some derivatives proved to be non-toxic, showing no effect on cell viability. The cytotoxicity of selected β-hydroxy selenides was likewise investigated on normal human dermal fibroblasts [45].
In addition, the ring opening of the three-membered heterocycles by (phenylselenotrimethyl)silane was efficiently performed in a variety of ionic liquids, able to act as reaction media and, in some cases, also as catalysts, leading to β-functionalized selenides [46].
Extending the scope of this methodology, when strained heterocycles were reacted with bis(trimethylsilyl)sulfide/TBAF under strictly controlled conditions (equivalents of TBAF, time, T), direct access to β-hydroxy, β-mercapto and β-amino alkyl selenols 27, 36, 44 was obtained, arising from a regioselective nucleophilic attack on the less hindered side of the heterocycle (Scheme 23) [36]. Interestingly, the ring-opening reaction of enantioenriched substrates provided the synthesis of chiral non-racemic selenols. Taking into account their propensity to be oxidized to diselenides, the β-substituted selenols displayed an unexpected stability, which can be attributed to the hydrogen bond interaction between the selenol and the hydroxy moieties, as indicated by the ab initio DF calculations on selected model systems.
3.2. Reaction with C=O Containing Compounds
3.2.1. Reaction with Aldehydes and Ketones
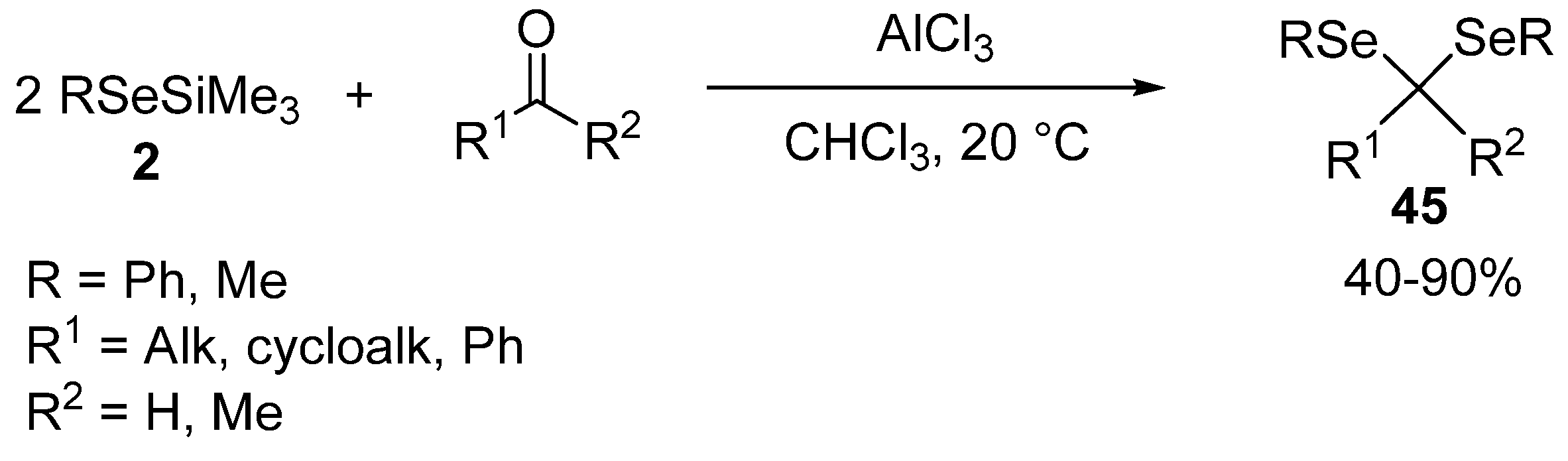
Selenosilanes were efficiently reacted with carbonyl compounds to provide selenoacetals 45, which are used as valuable reagents in organic reactions. Krief et al. reported the selenoacetalization of aldehydes and ketones with selenosilanes under acidic conditions (Scheme 24) [12,47]. Based on the oxygenophilic character of silicon, silylselenides were expected to react without any catalysts. Differently from what was observed with the sulfurated analogs of RSSiMe3 (and also with selenoboranes), it was found that the cleavage of the Se-Si bond required an acid catalyst to give the selenoacetalization. Better results were obtained by the in situ formation of RSeSiMe3 (prepared by diselenide/LiAlH4/ClSiMe3) followed by the addition of the carbonyl compound under Lewis acid catalysis.
The use of silylselenides avoids employing selenols, which are known to be rather unstable. This is important in particular for methylselenol, also because of its high volatility and bad smell. Our group reported the reaction of bis(trimethylsilyl)selenide 3a with aldehydes and acylsilanes, which in the presence of CoCl2.6H2O afforded selenoaldehydes 46 and selenoacylsilanes 47, isolated as Diels–Alder cycloadducts 48, 49 (Scheme 25) [48].
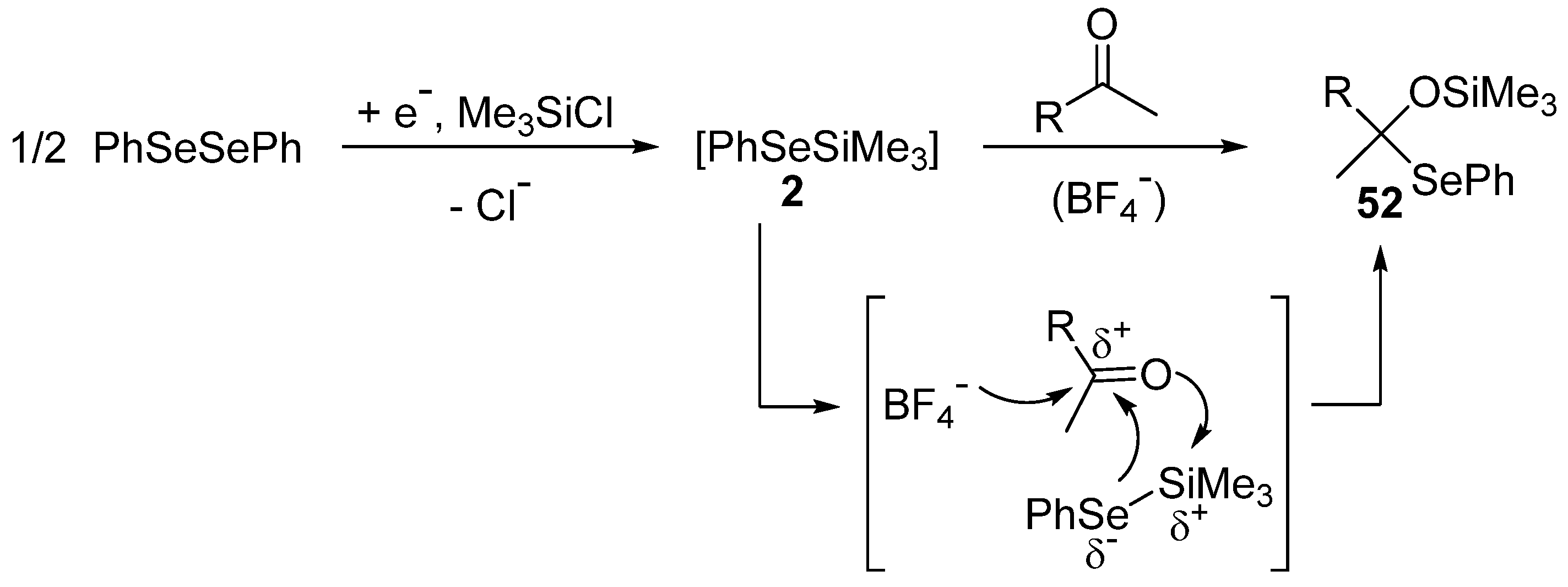
Selenenylation of α,β-unsaturated carbonyls by electrolysis with diaryl diselenides and chlorotrimethylsilane was reported by Torii and co-workers [49]. Aryl selenide anions 50 are electrochemically generated using a Pt electrode in a methanolic solution and treated with the enone, the diselenide and Me3SiCl (Scheme 26). The reaction affords the β-seleno substituted carbonyl compounds 51 through the mechanism proposed in Scheme 26. Aryl selenols 13 are formed in situ as precursors of the selenated adducts 51. Furthermore, the addition of the chlorosilane was crucial since without this reagent or by using less than one equivalent, only the starting material was recovered.
A similar reaction was reported by Jouikov et al. when dealing with the cathodic reduction of diselenides (and disulfides) to form PhSe− (or PhS−) anions, which in the presence of trimethylchlorosilane gave the corresponding (trimethylsilyl)selenides 2 (and sulfides) in good yields (Scheme 27) [14]. Treatment with carbonyl compounds resulted in the formation of silyl ethers 52 of hemiseleno- (or hemithio-) ketals and acetals. It was also found that the functionalization of carbonyls was preferably performed using silylselenides in a one-pot procedure without their isolation.
When a mixed dichalcogenide PhSSePh was reduced under electrolytic conditions, the rate of the SN2 reaction of the PhSe- anion on the Si-Cl bond was faster than the attack of the PhS− anion. This is in agreement with the stronger nucleophilic character of the selenolate anion compared to the thiolate due to the larger size of Se (ionic radius = 198 pm vs. 184 pm for sulfur) and, therefore, the greater localization of the negative charge on Se in the PhSe− species.
Sonoda and co-workers reported the hydrosilylation of carbonyl compounds under radical conditions to obtain silyl ethers 53, formed through the treatment of carbonyls with (phenylseleno)trimethylsilane 2a, tributylstannyl hydride and AIBN (Scheme 28) [50]. On the basis of the proposed mechanism, a silyl radical 54a is formed in situ by the activation of Se-Si bonds with the stannyl radical 55.
3.2.2. Reaction with Acyl Chlorides
Silyl selenides were also reacted with acyl chlorides, leading to a selective synthesis of selenolesters, selenoanhydrides and diacylselenides depending on the type of the selenosilane used and on the stoichiometric ratio of the reagents. Treatment of acyl chlorides, under TBAF catalysis, with (phenylseleno)trimethylsilane 2a, led to selenolesters 56 (Scheme 29, equation 1), while when bis(trimethylsilyl)selenide 3a was reacted in a 2:1 or 1:1 ratio, selective access to selenoanhydrides 57 or diacyl diselenides 58, respectively, was achieved (Scheme 29, equation 2) [51]. 77Se NMR chemical shifts were also reported, showing typical values for these classes of selenated compounds.
Besides (phenylseleno)trimethylsilane, Corrigan and Taher reported the reaction of a variety of acyl chlorides with organoselenosilanes containing two TMSSe- groups, such as 1,1’-Fe(η5-C5H4SeTMS)2, 1,4-TMSSe-C6H4-SeTMS and 4,4’-TMSSe-(C6H4)2-SeTMS, to afford ferrocenyl- and alkyl/aryl-diselenoesters 59–61 (Scheme 30) [52].
3.3. Reduction of Oxides of the Group 16 Elements (S, Se, Te)
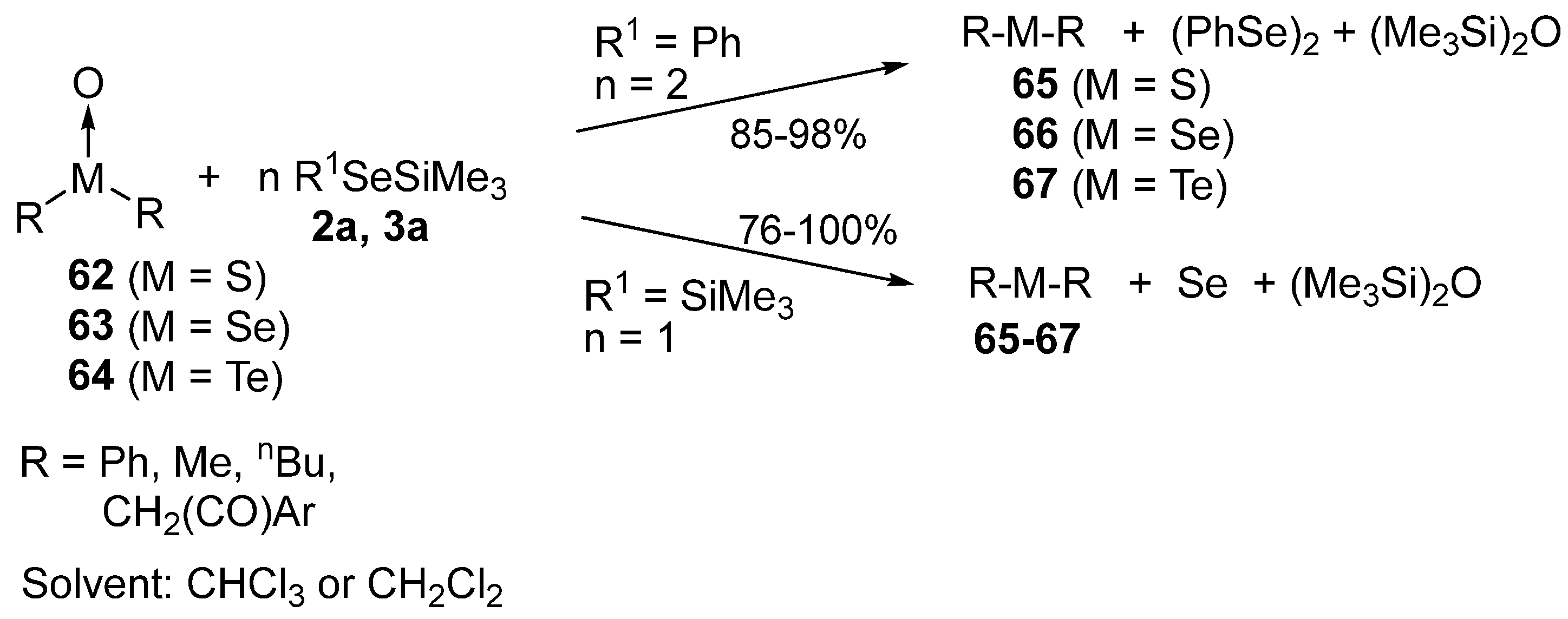
Detty reported the use of (phenylseleno)trimethylsilane 2a [53] and bis(trimethylsilyl)selenide 3a [16] to reduce, under mild conditions, sulfoxides 62, selenoxides 63 and telluroxides 64 to the corresponding sulfides, selenides and tellurides 65–67 in high yields (Scheme 31).
This method is compatible with different functional groups, such as ketones, phenols, alcohols, olefins, sulfones and nitro derivatives. Based on the proposed mechanism, onium species R2M+(OSiMe3) 68 and R2M+(SePh) 69 should be involved in this transformation, as depicted in Scheme 32.
3.4. Reactivity of Selenosilanes under Radical Conditions
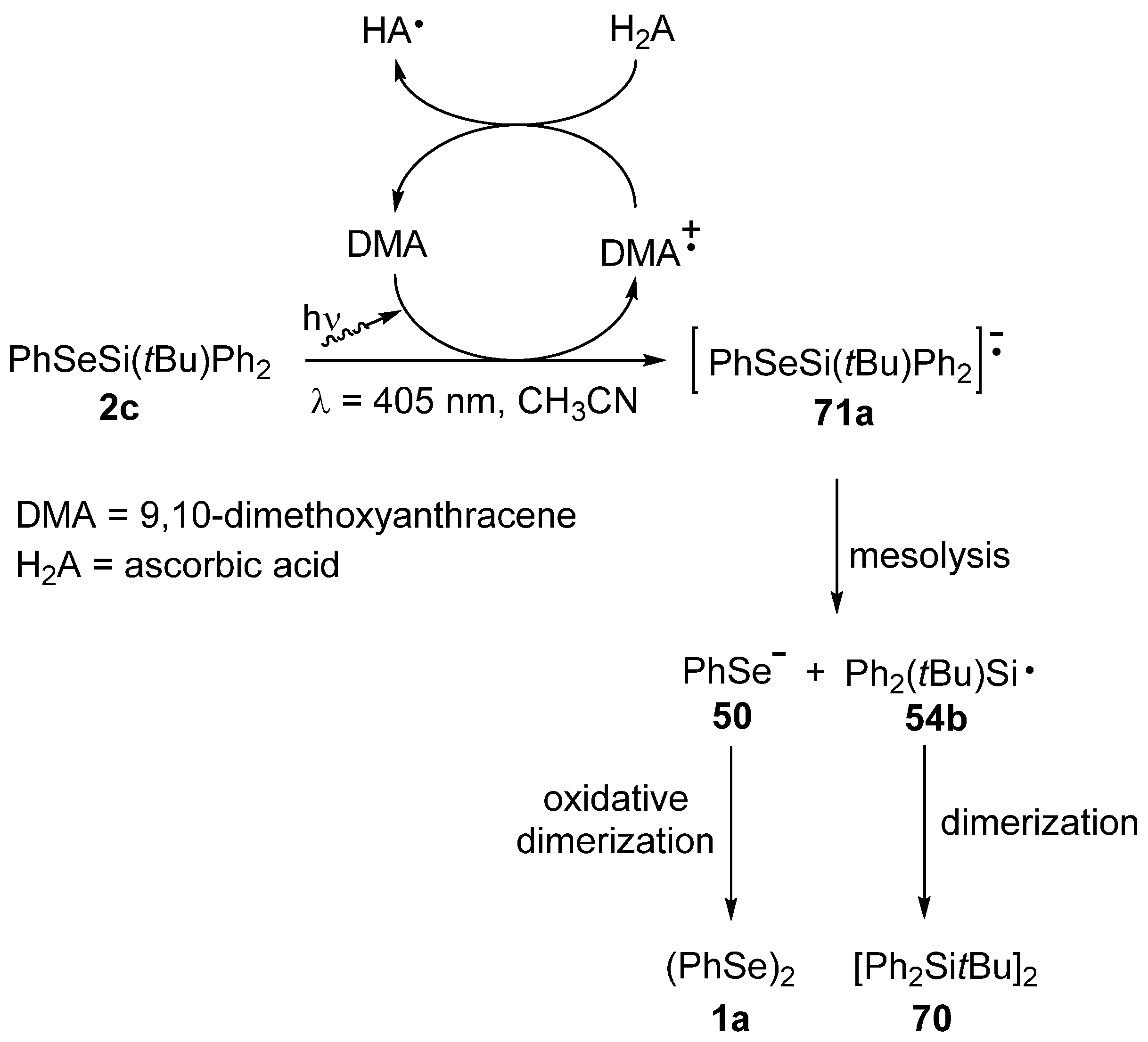
Pandey, Mittal and co-workers investigated the PET (photosensitized electron transfer) promoted activation of selenosilanes to afford radical ions, as well as their fragmentation (mesolysis). t-Butyldiphenyl(phenylseleno)silane 2c was selected for its appreciable stability to study the PET reductive activation of Se-Si bonds using a suitable photosystem to generate the radical anion 71a (Scheme 33) [54,55].
The formation of the dimers 1a,70 could be rationalized through the mesolysis of the primary radical ion 71a to form the phenylselenide anion 50 (transient adsorption at λmax = 490 nm) and the silyl radical 54b (transient adsorption at λmax = 440 nm), which undergo dimerization. It can be assumed that the fragmentation of 71a is driven by the electronegativity difference between silicon and selenium. This efficient dissociation allowed us to consider selenosilanes as silyl radical equivalents, whose chemical behavior in bimolecular group transfer (BMGT) radical reactions was studied, as well as in intermolecular radical chain transfer addition reactions [54]. For example, for evaluating the use of selenosilanes for BMGT radical reactions, a mixture of compounds 2 and 72 was irradiated together with DMN - 1,5-dimethoxynaphthalene (as an electron donor) and ascorbic acid (as a co-oxidant), which provided the cyclization products 73 (major) and 74 (minor) (Scheme 34).
The reaction was extended to the cyclization of bromoallyl ethers and bromopropargyl ethers and also provided substituted tetrahydrofuran derivatives.
3.5. Metal-Selenium Cluster Compounds
Fenske and co-workers reported the reaction of bis(trimethylsilyl)selenide 3a with a phosphane ligand (e.g., dpph = Ph2P(CH2)6PPh2 or dppe = Ph2P(CH2)5PPh2) and [Me2SAuCl] to prepare different gold complexes with chalcogenide bridges, as for example in complexes 75 and 76 in Scheme 35 [56]. The structure of some gold–selenium compounds was determined by X-ray diffraction. For instance, complexes 75 and 76 crystallize in the monoclinic space group P21/c and C2/c, respectively, with four molecules per unit cell.
Fenske also investigated the thermal properties (TGA, DSC) of two series of copper selenide clusters, including structures 77 and 78 depicted in Scheme 36, obtained by the reaction of (Me3Si)2Se with CuX and PEt2Ph or PEt3 [57].
Within a study of the C–F activation in Ni-complexes, Radius et al. investigated the selective replacement of the fluoride ligand by a variety of nucleophiles in the trans-[Ni(iPr2Im)2(F)(C6F5)] complex 79, which was selected as a model compound. The reaction with silyl chalcogenides (RESiMe3, E = S, Se; R = Ph, nPr, iPr) provided the corresponding sulfurated and selenated complexes 80 with the elimination of fluorotrimethylsilane, favored by the formation of the strong Si-F bond (Scheme 37) [58].
The complexes with nPr or iPr groups adopt a square planar geometry, as evidenced by single-crystal X-ray analysis. It was found that the Ni-S bond length is slightly shorter than the distance in the related complex [Ni(PnBu3)2(SC6F5)(C6F5)] and that the Ni-Se distance was rather unusual.
3.6. Silyl Selenides as Se-Precursors for Atomic Layer Deposition (ALD)
In 2009, Pore and co-workers reported the use of bis(trialkylsilyl)selenides (as well as of silyl tellurides) as precursors to obtain metal selenides 81 for atomic layer deposition (ALD) [59], a useful technique to deposit ultra-thin films of a few nanometres in a precise and controlled way for various applications, such as in semiconductors and other nanoscale devices [60]. Silyl selenides are volatile, thermally stable and very reactive towards metal compounds and are thus suitable to produce selenated materials. Compared to alkyl selenides, selenosilanes react more efficiently for the elimination of ligands of the metal precursors. This behavior can be ascribed to the formation of a bond between silicon (hard Lewis acid) and the harder base upon an exchange reaction with metal chlorides. High temperatures are necessary to cause sufficient evaporation of the metal precursors (Scheme 38).
Different combinations of metal precursors and silyl compounds can be used. Besides metal chlorides, growth experiments using Cu(II) pivalate and (Et3Si)2Se to obtain copper selenides showed that the stoichiometry between CuSe and Cu2Se could be controlled, depending on the deposition temperature.
Bureš and co-workers investigated the behavior of silyl selenides (R3Si)2Se bearing different alkyl groups on the silicon (R = Me, Et, iPr, tBuMe2), evidencing good volatility and stability. The trimethylsilyl substituted silylselenide, in combination with MoCl5 as a Mo precursor, was efficiently used for the deposition of MoSe2, while the tert-butyldimethylsilyl derivative evidenced no atomic layer deposition because of its significant stability [61]. The synthesis of various cyclic silylselenides 82−86, obtained by in situ treatment of M2Se (M = Li, Na) with suitable chlorosilanes, was also reported by Bureš et al. (Scheme 39) [62].
Their thermal behavior was studied by TGA and DSC. TGA showed that they have very good volatility with complete evaporation, while DSC measurements evidenced evaporation without decomposition. The thermal properties mainly depend on the ring size and the number of Si/Se atoms in the ring. The cyclic selenosilanes were evaluated as Se precursors for atomic layer deposition. Combined with MoCl5, some of them evidenced a sufficiently fast reaction with metal precursors to permit their application in ALD.
4. Conclusions
Selenated compounds represent an interesting class of molecules for their different applications in many fields, such as organic chemistry, inorganic chemistry, materials science, medicinal chemistry and biology. Therefore, methods which allow a mild and general preparation of selenium-containing compounds have received increasing interest. In this regard, selenosilanes have been demonstrated to be efficient reagents that introduce selenated groups on a variety of substrates. The mild functionalization of the Si-Se bond allows silyl selenides to behave as synthetic equivalents of the analogous hydrogenated compounds, but are more stable, less toxic and easier to handle. Thus, differently substituted selenosilanes found a growing number of applications in organic synthesis as versatile nucleophiles towards a variety of organic substrates, being able to undergo chemo-, regio- and stereoselective transformations. Furthermore, selenosilanes are also used in reducing processes and radical reactions, as well as in the preparation of metal clusters and as Se-precursors of metal selenides for atomic layer deposition.
Author Contributions
Conceptualization, A.C. and D.T.; curation of the literature, A.C. and D.T.; writing—original draft preparation, A.C. and D.T.; revision of the manuscript, A.C. and D.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
We thank MUR-Italy, “Dipartimenti di Eccellenza 2023–2027” allocated to the Department of Chemistry “Ugo Schiff” (DICUS 2.0) of the University of Florence.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lenardão, E.J.; Santi, C.; Sancineto, L. New Frontiers in Organoselenium Compounds; Springer: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Wirth, T. (Ed.) Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2012; ISBN 978-3-527-32944-1. [Google Scholar]
- Armitage, D.A. The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; John Wiley & Sons: Chichester, UK, 1998; Volume 2, Chapter 31; pp. 1869–1894. ISBN 978-0-471-96757-6. [Google Scholar]
- Tanini, D.; Capperucci, A.; Menichetti, S. Nucleophilic Chalcogen-containing Reagents. In Chalcogen Chemistry; Lippolis, V., Santi, C., Lenardão, E.J., Braga, A.L., Eds.; Royal Society of Chemistry: London, UK, 2023; Chapter 12; pp. 307–321. [Google Scholar]
- Baker, A.; Wirth, T. Silyl Sulfides and Selenides. Sci. Synth. Updates 2017, 1, 189–202. [Google Scholar] [CrossRef]
- Miyoshi, N.; Ishii, H.; Kondo, K.; Murai, S.; Sonoda, N. Convenient Syntheses of Phenyl Trimethylsilyl Selenide and Benzeneselenol. Synthesis 1979, 1979, 300–301. [Google Scholar] [CrossRef]
- Detty, M.R. Trimethylsilyl Iodide. Preparation from and Catalytic Behavior with Phenylselenotrimethylsilane. Tetrahedron Lett. 1978, 43, 4189–4192. [Google Scholar] [CrossRef]
- Abe, H.; Yamasaki, A.; Harayama, T. Direct Conversion of a Benzylic Hydroxy Group into a Selenenyl Group Using the Phenyl Trimethylsilyl Selenide-Aluminum Bromide Combination. Chem. Pharm. Bull. 1998, 46, 1311–1313. [Google Scholar] [CrossRef]
- Pandey, G.; Poleshwar Rao, K.S.S. A New Dimension in Radical Chain Group Transfer Reaction by Photosensitized Electron Transfer (PET) Reductive Activation of PhSeSiR3. Angew. Chem. Int. Ed. Engl. 1996, 34, 2669–2671. [Google Scholar] [CrossRef]
- Herzog, U. Synthesis and NMR Investigation of Selenobutyl Substituted Silanes and Oligosilanes. J. Prakt. Chem. 2000, 342, 379–388. [Google Scholar] [CrossRef]
- Poleschner, H.; Heydenreich, M.; Schilde, U. Reactions of RSe-EMe3 (E = Si, Ge, Sn, Pb) with XeF2 -RSe–F Equivalents in the Fluoroselenenylation of Acetylenes. Eur. J. Inorg. Chem. 2000, 2000, 1307–1313. [Google Scholar] [CrossRef]
- Clarembeau, M.; Cravador, A.; Dumont, W.; Hevesi, L.; Krief, A.; Lucchetti, J.; Van Ende, D. Synthesis of selenoacetals. Tetrahedron 1985, 41, 4793–4812. [Google Scholar] [CrossRef]
- Kuciński, K.; Gruszczyński, M.; Hreczycho, G. Ru-catalyzed Formation of Thiosilanes and Selenosilanes using Dichalcogenides as a User-Friendly Alternative to Thiols and Selenols. ChemCatChem 2022, 14, e202200961. [Google Scholar] [CrossRef]
- Jouikov, V.; Grigorieva, L. Competitive electrochemical thio- and selenenylation of chlorosilanes. Electrochim. Acta 1996, 41, 2489–2491. [Google Scholar] [CrossRef]
- Syper, L.; Mlochowski, J. Lithium diselenide in aprotic medium—A convenient reagent for synthesis of organic diselenides. Tetrahedron 1988, 44, 6119–6130. [Google Scholar] [CrossRef]
- Drake, J.E.; Glavineevski, B.M.; Hemmings, R.T.; Henderson, H.E. Silyl and Germyl Selenides and Tellurides. In Inorganic Syntheses; Bush, D.H., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1980; Volume XX, Chapter 6; pp. 171–176. ISBN 0-471-07715-1. [Google Scholar]
- Detty, M.R.; Seidler, M.D. Bis(trialkylsilyl) Chalcogenides. 1. Preparation and Reduction of Group 6A Oxides. J. Org. Chem. 1982, 47, 1354–1356. [Google Scholar] [CrossRef]
- Hatanpää, T.; Pore, V.; Ritala, M.; Leskelä, M. Alkylsilyl Compounds of Selenium and Tellurium: New Precursors for ALD. ECS Trans. 2009, 25, 609–616. [Google Scholar] [CrossRef]
- Detty, M.R.; Seidler, M.D. Silyl Halides from (Phenylseleno)silanes. Reaction with Oxiranes and Alcohols to Give Hydrolytically Stable Silyl Ethers. J. Org. Chem. 1981, 46, 1283–1292. [Google Scholar] [CrossRef]
- Segi, M.; Kato, M.; Nakajima, T.; Suga, S.; Sonoda, N. A Convenient One-pot Synthesis of Unsymmetrical Selenium Compounds Using Bis(trimethylsilyl) Selenide. Chem. Lett. 1989, 1989, 1009–1012. [Google Scholar] [CrossRef]
- Taher, D.; Wallbank, A.I.; Turner, E.A.; Cuthbert, H.L.; Corrigan, J.F. Alk-2-ynyl Trimethylsilyl Chalcogenoethers by Nucleophilic Substitution of Propargyl Bromides. Eur. J. Inorg. Chem. 2006, 2006, 4616–4620. [Google Scholar] [CrossRef]
- Wrackmeyer, B.; García Hernández, Z.; Herberhold, M. 1-Cyclohepta-2,4,6-trienyl-selanes—a 77Se NMR study: Indirect nuclear 77Se-13C spin-spin coupling constants and application of density functional theory (DFT) calculations. Magn. Reson. Chem. 2007, 45, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Yamasaki, A.; Koshiba, N.; Takeuchi, Y.; Harayama, T. One-Pot Conversion of Allyl Alcohols into Selenochroman Derivatives. Chem. Pharm. Bull. 2001, 49, 1223–1225. [Google Scholar] [CrossRef]
- Grant, L.; Liu, Y.; Walsh, K.E.; Walter, D.S.; Gallagher, T. Galacto, Gluco, Manno, and Disaccharide-Based C-Glycosides of 2-Amino-2-deoxy Sugars. Org. Lett. 2002, 4, 4623–4625. [Google Scholar] [CrossRef]
- Pfrengle, F.; Reissig, H.-U. Internally Protected Amino Sugar Equivalents from Enantiopure 1,2-Oxazines: Synthesis of Variably Configured Carbohydrates with C-Branched Amino Sugar Units. Chem. Eur. J. 2010, 16, 11915–11925. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Hauser, E.B. Synthesis of Crossed [2 + 2] Photocycloadducts: A Novel Approach to the Synthesis of Bridged Bicyclic Alkenes. Org. Lett. 2000, 2, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Fujii, H.; Yamasaki, A.; Kinome, Y.; Takeuchi, Y.; Harayama, T. Preparation of β-Seleno-α,β-Unsaturated Nitroalkenes Via A Sulfur-Selenium Exchange Reaction. Synth. Commun. 2000, 30, 543–549. [Google Scholar] [CrossRef]
- Viglianisi, C.; Bonardi, C.; Ermini, E.; Capperucci, A.; Menichetti, S.; Tanini, D. Selenosilane-Promoted Selective Mild Transformation of N-Thiophthalimides into Symmetric Disulfides. Synthesis 2019, 51, 1819–1824. [Google Scholar] [CrossRef]
- Miyoshi, N.; Hatayama, Y.; Ryu, I.; Kambe, N.; Murai, T.; Murai, S.; Sonoda, N. Ring Opening of Tetrahydrofurans with Phenyl Trimethylsilyl Selenide Catalyzed by Moist Zinc Iodide. Synthesis 1988, 1988, 175–177. [Google Scholar] [CrossRef]
- Dapkekar, A.B.; Satyanarayana, G. Electrochemical selenofunctionalization of unactivated alkenes: Access to β-hydroxyselenides. Org. Biomol. Chem. 2024, 22, 1775–1781. [Google Scholar] [CrossRef]
- Detty, M.R. Phenylselenotrimethylsilane. A novel source of phenylselenide anion. Tetrahedron Lett. 1978, 51, 5087–5090. [Google Scholar] [CrossRef]
- Miyoshi, N.; Kondo, K.; Murai, S.; Sonoda, N. Synthesis of β-siloxyalkyl phenyl selenides by the reaction of phenyl trimethylsilyl selenide with epoxides. Chem. Lett. 1979, 8, 909–912. [Google Scholar] [CrossRef]
- Wang, P.-A. Organocatalyzed enantioselective desymmetrization of aziridines and epoxides. Beilstein J. Org. Chem. 2013, 9, 1677–1695. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Marini, F.; Sternativo, S.; Del Verme, F.; Santi, C.; Bagnoli, L.; Temperini, A. Synthesis of enantiomerically enriched β-hydroxy selenides by catalytic asymmetric ring opening of meso-epoxides with (phenylseleno)silanes. Tetrahedron 2008, 64, 3337–3342. [Google Scholar] [CrossRef]
- Capperucci, A.; Tiberi, C.; Pollicino, S.; Degl’Innocenti, A. Tetrabutylammonium phenoxide induced reaction of silyl nucleophiles. Tetrahedron Lett. 2009, 50, 2808–2810. [Google Scholar] [CrossRef]
- Tanini, D.; Tiberi, C.; Gellini, C.; Salvi, P.R.; Capperucci, A. A Straightforward Access to Stable β-Functionalized Alkyl Selenols. Adv. Synth. Catal. 2018, 360, 3367–3375. [Google Scholar] [CrossRef]
- Capperucci, A.; Salles, C.; Scarpelli, S.; Tanini, D. Selective access to sulfurated and selenated heterocycles by intramolecular cyclization of β-substituted sulfides and selenides. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 172–174. [Google Scholar] [CrossRef]
- Capperucci, A.; Tanini, D.; Borgogni, C.; DegľInnocenti, A. Thiosilane- and Organoselenosilane-Mediated Novel Access to 3,7-Disubstituted-1,2,5-trithiepanes and -1,2,5-dithiaselenepanes. Heteroat. Chem. 2014, 25, 678–683. [Google Scholar] [CrossRef]
- Tanini, D.; D’Esopo, V.; Tatini, D.; Ambrosi, M.; Lo Nostro, P.; Capperucci, A. Selenated and Sulfurated Analogues of Triacyl Glycerols: Selective Synthesis and Structural Characterization. Chem. Eur. J. 2020, 26, 2719–2725. [Google Scholar] [CrossRef]
- Tanini, D.; Barchielli, G.; Benelli, F.; Degl’Innocenti, A.; Capperucci, A. Aziridines Ring Opening by Silyl Chalcogenides: A Stereoselective Access to Polyfunctionalized Molecules as Precursor of Sulfurated and Selenated Heterocycles. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1265–1270. [Google Scholar] [CrossRef]
- Tanini, D.; Borgogni, C.; Capperucci, A. Mild and selective silicon-mediated access to enantioenriched 1,2-mercaptoamines and β-amino arylchalcogenides. New J. Chem. 2019, 43, 6388–6393. [Google Scholar] [CrossRef]
- Senatore, M.; Lattanzi, A.; Santoro, S.; Santi, C.; Della Sala, G. A general phosphoric acid-catalyzed desymmetrization of meso-aziridines with silylated selenium nucleophiles. Org. Biomol. Chem. 2011, 9, 6205–6207. [Google Scholar] [CrossRef]
- Della Sala, G. Studies on the true catalyst in the phosphate-promoted desymmetrization of meso-aziridines with silylated nucleophiles. Tetrahedron 2013, 69, 50–56. [Google Scholar] [CrossRef]
- Tanini, D.; Degl’Innocenti, A.; Capperucci, A. Bis(trimethylsilyl)selenide in the Selective Synthesis of β-Hydroxy, β-Mercapto, and β-Amino Diorganyl Diselenides and Selenides Through Ring Opening of Strained Heterocycles. Eur. J. Org. Chem. 2015, 2015, 357–369. [Google Scholar] [CrossRef]
- Capperucci, A.; Coronnello, M.; Salvini, F.; Tanini, D.; Dei, S.; Teodori, E.; Giovannelli, L. Synthesis of functionalised organochalcogenides and in vitro evaluation of their antioxidant activity. Bioorg. Chem. 2021, 110, 104812. [Google Scholar] [CrossRef] [PubMed]
- Tanini, D.; Pecchi, T.; Ignat’ev, N.V.; Capperucci, A. Ionic Liquids-Assisted Ring Opening of Three-Membered Heterocycles with Thio- and Seleno-Silanes. Catalysts 2022, 12, 1259. [Google Scholar] [CrossRef]
- Dumont, W.; Krief, A. Synthesis of Diselenoacetals and O-(Trimethylsilyl)monoselenoacetals. Angew. Chem. Int. Ed. Engl. 1977, 16, 540–541. [Google Scholar] [CrossRef]
- Degl’Innocenti, A.; Capperucci, A.; Acciai, M.; Tiberi, C. Silicon-Mediated Synthesis of Selenoaldehydes and Selenoacylsilanes and Their Hetero Diels-Alder Reactions. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 1621–1626. [Google Scholar] [CrossRef]
- Torii, S.; Inokuchi, T.; Hasegawa, N. A direct arylselenenylation of enones with diaryl diselenides by a electroreductive procedure. Chem. Lett. 1980, 1980, 639–640. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Kajimoto, H.; Kotani, K.; Nishida, T.; Sonoda, N. Reaction of Carbonyl Compounds with Trialkylsilyl Phenylselenide and Tributylstannyl Hydride under Radical Conditions. J. Org. Chem. 2002, 67, 5696–5700. [Google Scholar] [CrossRef] [PubMed]
- Capperucci, A.; Degl’Innocenti, A.; Tiberi, C. Organoselenosilane-Mediated Selective Mild Access to Selenolesters, Selenoanhydrides and Diacyl Diselenides. Synlett 2011, 2011, 2248–2252. [Google Scholar] [CrossRef]
- Taher, D.; Corrigan, J.F. Aryl(trimethylsilyl)selenides as Reagents for the Synthesisof Mono- and Diselenoesters. Organometallics 2011, 30, 5943–5952. [Google Scholar] [CrossRef]
- Detty, M.R. Mild Red uctions of Oxides of the Group 6a Elements Sulfur, Selenium, and Tellurium with (Phenylseleno)trimethylsilane. J. Org. Chem. 1979, 44, 4528–4531. [Google Scholar] [CrossRef]
- Pandey, G.; Poleswara Rao, K.S.S.; Palit, D.K.; Mittal, J.P. Generation and Mesolysis of PhSeSiR3]•-: Mechanistic Studies by Laser Flash Photolysis and Application for Bimolecular Group Transfer Radical Reactions. J. Org. Chem. 1998, 63, 3968–3978. [Google Scholar] [CrossRef]
- Pandey, G.; Gadre, S.R. Generation and Mesolytic Dynamics of Organoselenane and Selenosilane Radical Ions: Development of Mechanistically Interesting and Synthetically Useful Chemistry. Acc. Chem. Res. 2004, 37, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Olkowska-Oetzel, J.; Sevillano, P.; Eichhöfer, A.; Fenske, D. Binary and Ternary Cluster Complexes Containing Gold-Selenium, Gold-Indium-Selenium and Gold-Gallium-Tellurium—Synthesis and Structures of [Au5Se2(PPh3)4]Cl, [(Au3Se)2{Ph2P(CH2)6PPh2}3]Cl2, [Au10Se4{Ph2P(CH2)5PPh2}4]InCl5, [Au4(SeInCl3)2{Ph2P(CH2)5PPh2}2], [Au2(TeGaCl3){Ph2P(CH2)6PPh2}]2 and [Au8Se4In{Ph2P(CH2)2PPh2}4](InCl4)3. Eur. J. Inorg. Chem. 2004, 2004, 1100–1106. [Google Scholar] [CrossRef]
- Cave, D.; Corrigan, J.F.; Eichhöfer, A.; Fenske, D.; Kowalchuk, C.M.; Rösner, H.; Scheer, P. Investigation of the Thermal Properties of a Series of Copper Selenide Cluster Molecules. J. Clust. Sci. 2007, 18, 157–172. [Google Scholar] [CrossRef]
- Schaub, T.; Backes, M.; Radius, U. Square-Planar (Pentafluorophenyl)nickel(II) Complexes by Derivatization of a C–F Activation Product. Eur. J. Inorg. Chem. 2008, 2008, 2680–2690. [Google Scholar] [CrossRef]
- Pore, V.; Hatanpää, T.; Ritala, M.; Leskelä, M. Atomic Layer Deposition of Metal Tellurides and Selenides Using Alkylsilyl Compounds of Tellurium and Selenium. J. Am. Chem. Soc. 2009, 131, 3478–3480. [Google Scholar] [CrossRef] [PubMed]
- George, S.M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef]
- Charvot, J.; Zazpe, R.; Macak, J.M.; Bureš, F. Organoselenium Precursors for Atomic Layer Deposition. ACS Omega 2021, 6, 6554–6558. [Google Scholar] [CrossRef]
- Charvot, J.; Pokorný, D.; Zazpe, R.; Krumpolec, R.; Pavliňák, D.; Hromádko, L.; Přikryl, J.; Rodriguez-Pereira, J.; Klikar, M.; Jelínková, V.; et al. Cyclic Silylselenides: Convenient Selenium Precursors for Atomic Layer Deposition. ChemPlusChem 2020, 85, 576–579. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 2.
Preparation of bis(trialkylsilyl)selenides 3.

Scheme 3.
Reaction of 2a with halogens.
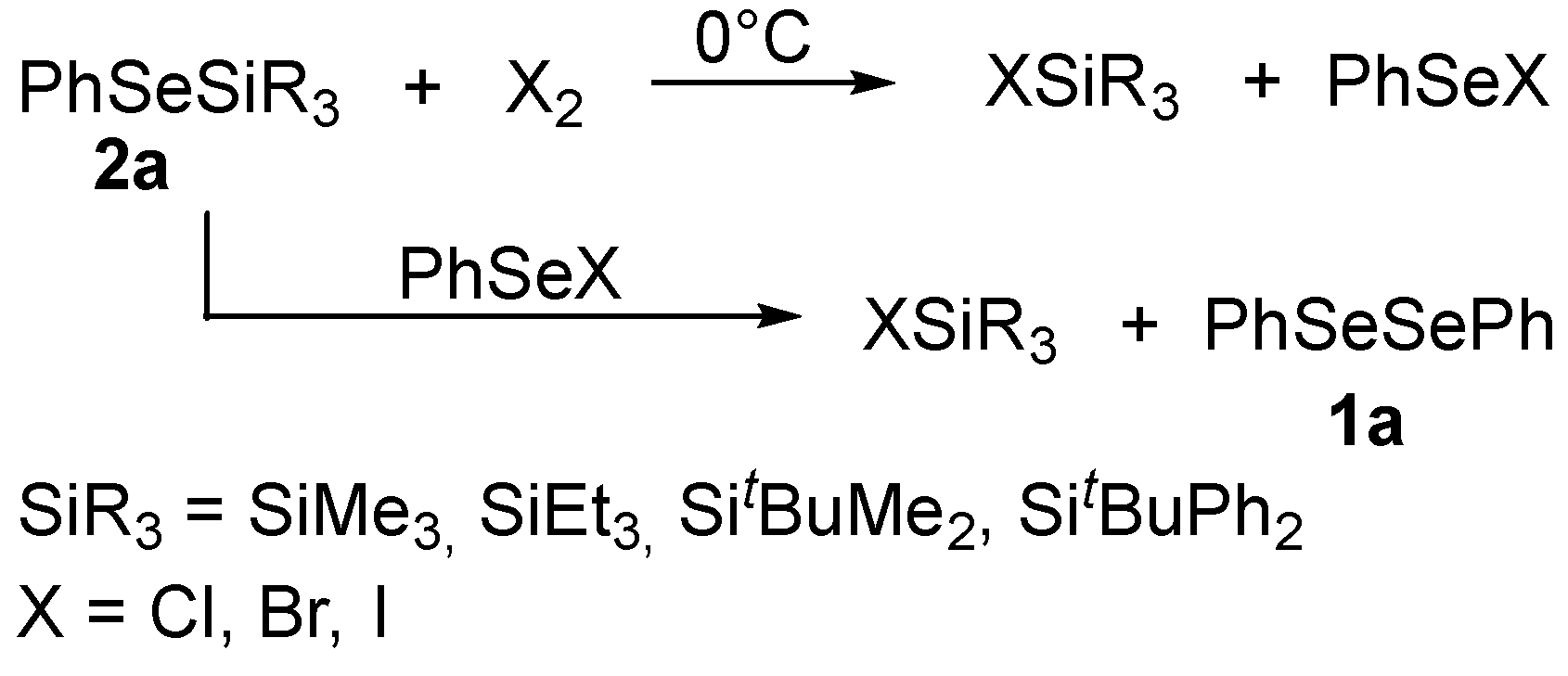
Scheme 4.
Reaction of xenon difluoride with silylselenides.

Scheme 5.
Synthesis of alkyl silyl selenides 5 (eq. a) and propargyl selenoethers 6 (eq. b).

Scheme 6.
Synthesis of 1-cyclohepta-2,4,6-trienyl-selanes 8.

Scheme 7.
Synthesis of benzyl selenides 9.

Scheme 8.
Synthesis of selenochroman derivatives 10.

Scheme 9.
Direct selenoglycosidation of 2-N-acetamido-sugars 11 with PhSeSiMe3 2a (via a) or PhSeH 13 (via b).
Scheme 9.
Direct selenoglycosidation of 2-N-acetamido-sugars 11 with PhSeSiMe3 2a (via a) or PhSeH 13 (via b).
Scheme 10.
Synthesis of phenylseleno- and phenylthio-substituted 1,2-oxazine derivatives 14.

Scheme 11.
Phenylseleno derivatives as precursor of exocyclic alkenes.

Scheme 12.
Synthesis of β-selenated nitroalkenes 19.

Scheme 13.
Selenosilane-promoted synthesis of disulfides from N-thiophthalimides.

Scheme 14.
Reaction of PhSeSiMe3 with oxygenated heterocycles.
Scheme 15.
Enantioselective ring opening of meso-epoxides by (phenylseleno)silanes.

Scheme 16.
Ring opening of epoxide 23a by silyl selenides under tetrabutylammonium phenoxide catalysis.
Scheme 16.
Ring opening of epoxide 23a by silyl selenides under tetrabutylammonium phenoxide catalysis.
Scheme 17.
Synthesis of six-membered seleno-heterocycles 28.

Scheme 18.
Synthesis of mixed chalcogeno compounds by ring opening of thiiranes 30 and 33 with HMDSS.
Scheme 18.
Synthesis of mixed chalcogeno compounds by ring opening of thiiranes 30 and 33 with HMDSS.

Scheme 19.
Reaction of bis(trimethylsilyl)selenide with aziridine 35a.

Scheme 20.
Ring opening of protected and unprotected aziridines by PhSeSiMe3.
Scheme 21.
Organocatalyzed desymmetrization of meso-aziridines.

Scheme 22.
Selenosilane-induced selective synthesis of diselenides and selenides.

Scheme 23.
Synthesis of β-substituted selenols.
Scheme 24.
Synthesis of selenoacetals and selenoketals.
Scheme 25.
Synthesis of selenoaldehydes and selenoacylsilanes.

Scheme 26.
Aryl selenenylation of enones with diselenides/Me3SiCl by electroreductive procedure.
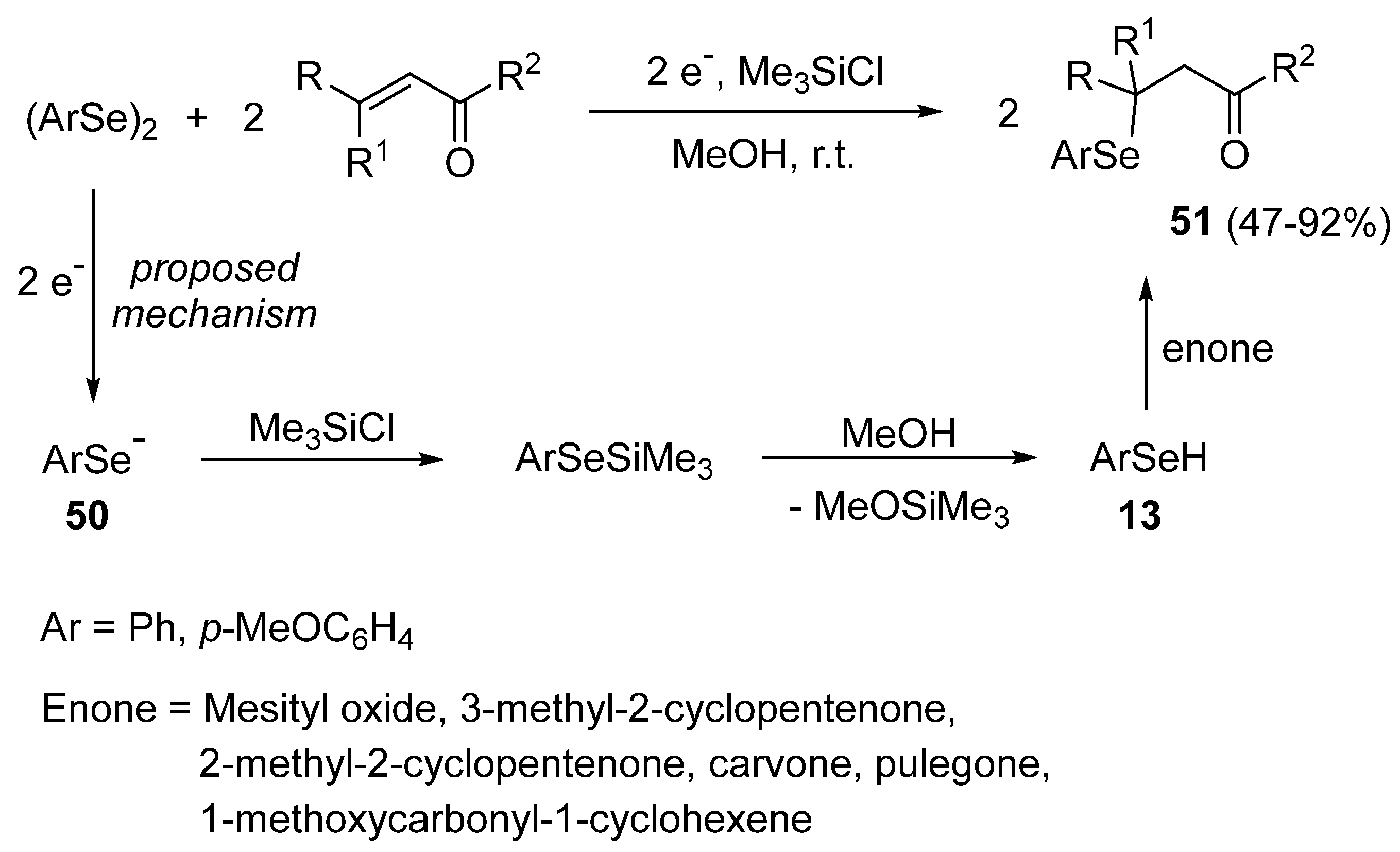
Scheme 27.
Electrochemical reduction of diphenyldiselenide in the presence of Me3SiCl and carbonyls.
Scheme 27.
Electrochemical reduction of diphenyldiselenide in the presence of Me3SiCl and carbonyls.
Scheme 28.
Hydrosilylation of carbonyls with the PhSeSiMe3/Bu3SnH/AIBN system.

Scheme 29.
Selective access to selenolesters 56, selenoanhydrides 57 and diacyl diselenides 58.

Scheme 30.
Synthesis of ferrocenyl- and alkyl/arylselenoesters 59−61.

Scheme 31.
Reduction of S-, Se- and Te-oxides by selenosilanes.
Scheme 32.
Plausible mechanism for the reduction of chalcogen-oxides.

Scheme 33.
PET activation of 2c to radical anion 71a and formation of dimers by mesolysis.
Scheme 34.
Selenosilanes as BMGT reagents in cyclization reactions.

Scheme 35.
Examples of preparation of some gold–selenium complexes with selenosilanes.

Scheme 36.
Examples of synthesis of copper selenide cluster molecules.
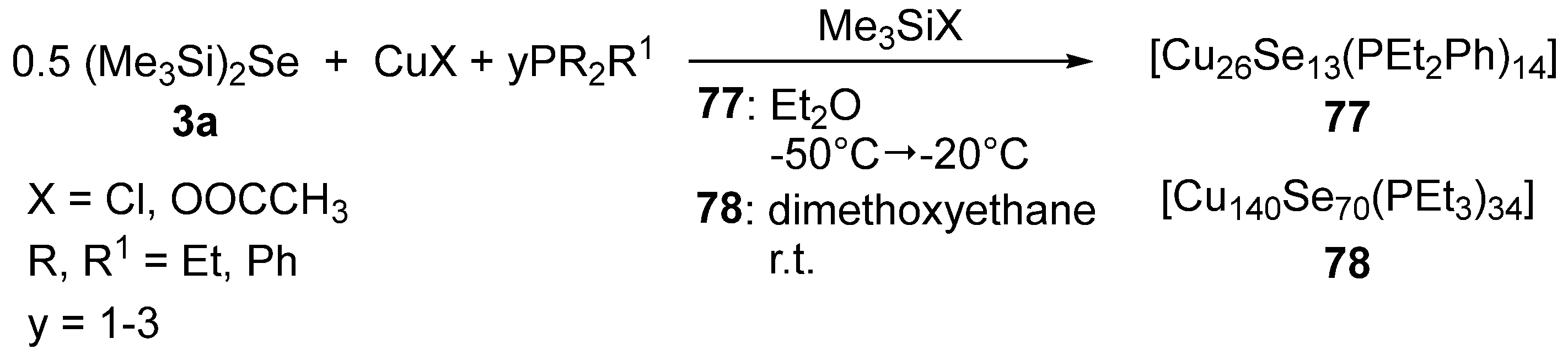
Scheme 37.
Reaction of complex trans-79 with silyl chalcogenides.

Scheme 38.
Reaction of silyl selenides with metal chlorides.

Scheme 39.
Preparation of cyclic silylselenides.
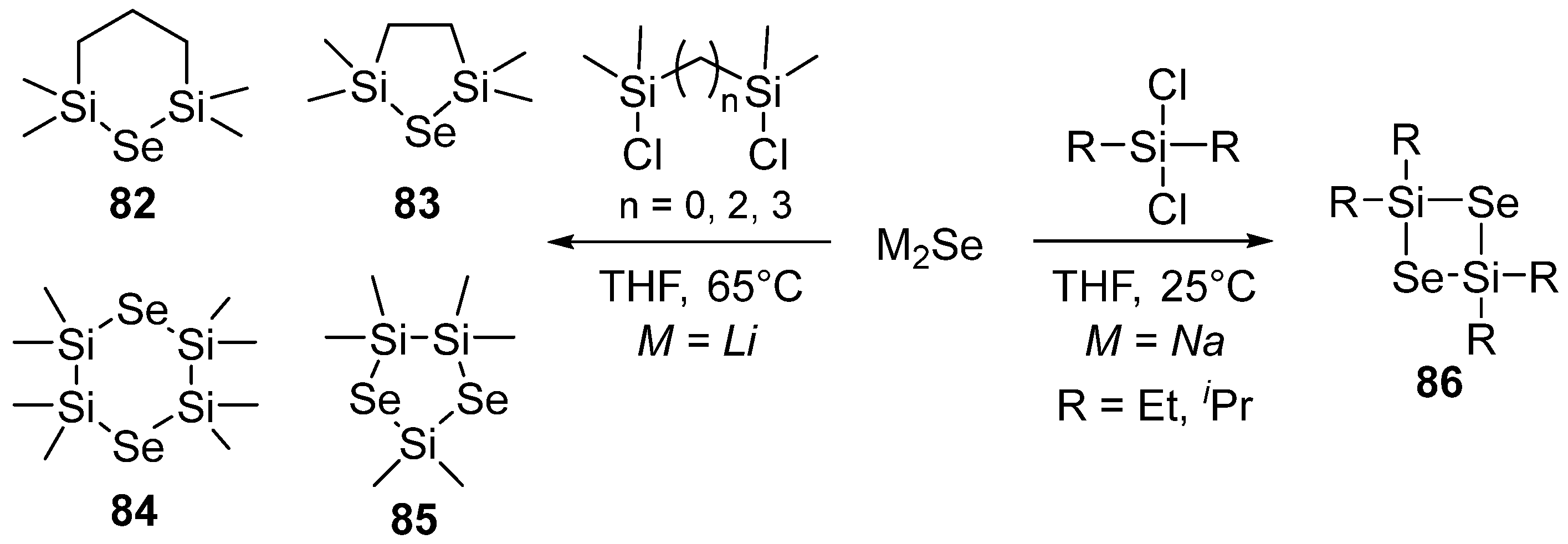
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tanini, D.; Capperucci, A. The Chemistry of Selenosilanes: A Topic Overview. Molecules 2024, 29, 4595. https://doi.org/10.3390/molecules29194595
AMA Style
Tanini D, Capperucci A. The Chemistry of Selenosilanes: A Topic Overview. Molecules. 2024; 29(19):4595. https://doi.org/10.3390/molecules29194595
Chicago/Turabian StyleTanini, Damiano, and Antonella Capperucci. 2024. "The Chemistry of Selenosilanes: A Topic Overview" Molecules 29, no. 19: 4595. https://doi.org/10.3390/molecules29194595