
Reactions of Cadmium(II) Halides and Di-2-Pyridyl Ketone Oxime: One-Dimensional Coordination Polymers
 , ,
, ,  , , ,
, , ,  , and
, and
Abstract
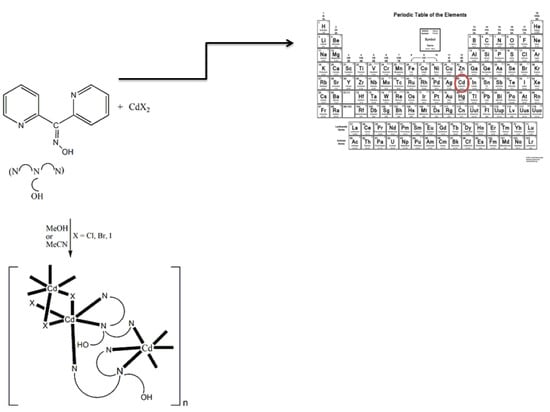
:
1. Introduction
 ), which is commonly produced by the reaction of a carbonyl group (from an aldehyde giving aldoximes and from a ketone resulting in ketoximes) with hydroxylamine, is a classical functional group in organic chemistry. It also plays a significant role in supramolecular chemistry and crystal engineering due to its capability to form several types of H bonds. The landmark of the use of oximes was the gravimetric determination of Ni(II) as the highly insoluble, red bis(dimethylglyoximato)nickel(II) solid [1]. Oxime and oximato metal complexes are central “players” in several aspects of coordination [2,3] and bioinorganic [4,5] chemistry, molecular magnetism [6,7], and homogeneous catalysis [8]. The reactivity of the coordinated oxime group is also of interest; this group has three sites (C, N, and O) for electrophilic or nucleophilic additions, most often promoted/assisted by metal ions [9,10,11]. Such reactions can proceed with rupture or preservation of the {CNO} moiety. In many cases, the oxime group is part of an organic molecule that contains one or more donor sites. Typical examples are the various 2-pyridyl oximes (Figure 1, left; R = a non-donor group). These ligands have been used among other purposes, including the following: (i) the linking (by coordination bonds) of smaller clusters to supramolecular entities [12]; (ii) the synthesis of single-chain magnets (SCMs) [13]; (iii) the isolation of 3d/4f-metal dinuclear and polynuclear complexes [14]; (iv) the “switching on” of single-molecule magnetism (SMM) properties [15]; and (v) the modeling of solvent extraction of Cd(II) from aqueous media using 2-pyridyl ketoxime extractants [16].
), which is commonly produced by the reaction of a carbonyl group (from an aldehyde giving aldoximes and from a ketone resulting in ketoximes) with hydroxylamine, is a classical functional group in organic chemistry. It also plays a significant role in supramolecular chemistry and crystal engineering due to its capability to form several types of H bonds. The landmark of the use of oximes was the gravimetric determination of Ni(II) as the highly insoluble, red bis(dimethylglyoximato)nickel(II) solid [1]. Oxime and oximato metal complexes are central “players” in several aspects of coordination [2,3] and bioinorganic [4,5] chemistry, molecular magnetism [6,7], and homogeneous catalysis [8]. The reactivity of the coordinated oxime group is also of interest; this group has three sites (C, N, and O) for electrophilic or nucleophilic additions, most often promoted/assisted by metal ions [9,10,11]. Such reactions can proceed with rupture or preservation of the {CNO} moiety. In many cases, the oxime group is part of an organic molecule that contains one or more donor sites. Typical examples are the various 2-pyridyl oximes (Figure 1, left; R = a non-donor group). These ligands have been used among other purposes, including the following: (i) the linking (by coordination bonds) of smaller clusters to supramolecular entities [12]; (ii) the synthesis of single-chain magnets (SCMs) [13]; (iii) the isolation of 3d/4f-metal dinuclear and polynuclear complexes [14]; (iv) the “switching on” of single-molecule magnetism (SMM) properties [15]; and (v) the modeling of solvent extraction of Cd(II) from aqueous media using 2-pyridyl ketoxime extractants [16].2. Results and Discussion
2.1. Synthetic Comments
2.2. Description of Structures
2.3. Spectroscopic Characterization in Brief
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Preparation of the Complexes
3.3. Single-Crystal X-ray Crystallography
4. Conclusions in Brief and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tschugaeff, L. Ueber ein neues, empfindliches reagens auf nickel. Ber. Dtsch. Chem. Ges. 1885, 18, S.2728–S.2734. [Google Scholar] [CrossRef]
- Smith, A.G.; Tasker, P.A.; White, D.J. The structures of phenolic oximes and their complexes. Coord. Chem. Rev. 2003, 241, 61–85. [Google Scholar] [CrossRef]
- Tasker, P.A.; Tong, C.C.; Westra, A.N. Co-extraction of cations and anions in base metal recovery. Coord. Chem. Rev. 2007, 251, 1868–1877. [Google Scholar] [CrossRef]
- Gerasimchuk, N.; Maher, T.; Durham, P.; Domasevitch, K.V.; Wilking, J.; Mokhir, A. Tin(IV) cyanoximates: Synthesis, characterization, and cytotoxicity. Inorg. Chem. 2007, 46, 7268–7274. [Google Scholar] [CrossRef] [PubMed]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and oximes. Synthesis, structure, and their key role as NO donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-J.; Zhang, Z.Z.; Lin, S.-B. A review of manganese-based molecular magnets and supramolecular architectures from phenolic oximes. Coord. Chem. Rev. 2015, 289–290, 289–314. [Google Scholar] [CrossRef]
- Chaudhuri, P. Homo- and hetero-polymetallic exchange coupled metal-oximates. Coord. Chem. Rev. 2003, 243, 143–190. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Kukushkin, V.Y.; Haukka, M.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Zinc(II)/ketoxime system as a simple and efficient catalyst for hydrolysis of organonitriles. Inorg. Chem. 2002, 41, 4798–4804. [Google Scholar] [CrossRef]
- Garnovskii, D.A.; Kukushkin, V.Y. Metal-mediated reactions of oximes. Russ. Chem. Rev. 2006, 75, 111–124. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-involving synthesis and reactions of oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef]
- Lada, Z.G.; Soto Beobide, A.; Savvidou, A.; Raptopoulou, C.P.; Psycharis, V.; Voyiatzis, G.A.; Turnbull, M.M.; Perlepes, S.P. A unique copper(II)-assisted transformation of acetylacetone dioxime in acetone that leads to one-dimensional, quinoxaline-bridged coordination polymers. Dalton Trans. 2017, 46, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Mowson, A.M.; Nguyen, T.N.; Abboud, K.A.; Christou, G. Dimeric and tetrameric supramolecular aggregates of single-molecule magnets via carboxylate substitution. Inorg. Chem. 2013, 52, 12320–12322. [Google Scholar] [CrossRef]
- Clérac, R.; Miyasaka, H.; Yamashita, M.; Coulon, C. Evidence for single-chain magnet behavior in a MnIIINiII chain designed with high spin magnetic units: A route to high temperature metastable magnets. J. Am. Chem. Soc. 2002, 124, 12837–12844. [Google Scholar] [CrossRef] [PubMed]
- Lada, Z.G.; Polyzou, C.D.; Nika, V.; Stamatatos, T.C.; Konidaris, K.F.; Perlepes, S.P. Adventures in the coordination chemistry of 2-pyridyl oximes: On the way to 3d/4f-metal coordination clusters. Inorg. Chim. Acta 2022, 539, 120954. [Google Scholar] [CrossRef]
- Stamatatos, T.C.; Foguet-Albiol, D.; Lee, S.C.; Stoumpos, C.C.; Raptopoulou, C.P.; Terzis, A.; Wernsdorfer, W.; Hill, S.O.; Perlepes, S.P.; Christou, G. “Switching on” the properties of single-molecule magnetism in triangular manganese(III) complexes. J. Am. Chem. Soc. 2007, 129, 9484–9499. [Google Scholar] [CrossRef] [PubMed]
- Routzomani, A.; Lada, Z.G.; Angelidou, V.; Raptopoulou, C.P.; Psycharis, V.; Konidaris, K.F.; Chasapis, C.T.; Perlepes, S.P. Confirming the Molecular Basis of the Solvent Extraction of Cadmium(II) Using 2-Pyridyl Oximes through a Synthetic Inorganic Chemistry Approach and a Proposal for More Efficient Extractants. Molecules 2022, 27, 1619. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Ishida, T.; Nogami, T. Structure and magnetic properties of 3d-4f heterometallic complexes containing di-2-pyridyl ketoximate. An approach to single-molecule magnets. Polyhedron 2005, 24, 2588–2592. [Google Scholar] [CrossRef]
- Mori, F.; Ishida, T.; Nogami, T.; Choi, K.Y.; Nojiri, H. Oximate-bridged trinuclear Dy-Cu-Dy complex behaving as a single-molecule magnet and its mechanistic investigation. J. Am. Chem. Soc. 2006, 128, 1440–1441. [Google Scholar] [CrossRef]
- Stemmler, A.J.; Kampf, J.W.; Pecoraro, V.L. Synthesis and Crystal Structure of the First Inverse 12-Metallacrown-4. Inorg. Chem. 1995, 34, 2771–2772. [Google Scholar] [CrossRef]
- Psomas, G.; Stemmler, A.J.; Dendrinou-Samara, C.; Bodwin, J.J.; Schneider, M.; Alexiou, M.; Kampf, J.W.; Kessissoglou, D.P.; Pecoraro, V.L. Preparation of Site-Differentiated Mixed Ligand and Mixed Ligand/Mixed Metal Metallacrowns. Inorg. Chem. 2001, 40, 1562–1570. [Google Scholar] [CrossRef]
- Kumagai, H.; Endo, M.; Kondo, M.; Kawata, S.; Kitagawa, S. Reactions of di-2-pyridyl ketone oxime in the presence of vanadium(III): Crystal structures of the coordination products. Coord. Chem. Rev. 2003, 237, 197–203. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Duca, G.; Siminel, A.V.; Fonari, M.S. Nine Mn(II), Zn(II), and Cd(II) mixed-ligand coordination networks with rigid dicarboxylate and pyridine-n-aldoxime ligands: Impact of the second ligand in the structures’ dimensionality and solvent capacity. Polyhedron 2017, 129, 9–21. [Google Scholar] [CrossRef]
- Coropceanu, E.B.; Croitor, L.; Siminel, A.V.; Fonari, M.S. Preparation, structural characterization and luminescence studies of mono- and binuclear Zn(II) and Cd(II) acetates with pyridine-4-aldoxime and pyridine-4-amidoxime ligands. Polyhedron 2014, 75, 73–80. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Masunov, A.A.; Rivea-Jacquez, H.J.; Siminel, A.V.; Fonari, M.S. Mechanism of Nonlinear Optical Enhancement and Supramolecular Isomerism in 1D Polymeric Zn(II) and Cd(II) Sulfates with Pyridine-4-aldoxime Ligands. J. Phys. Chem. C 2014, 118, 9217–9227. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Masunov, A.E.; Rivera-Jacquez, H.J.; Siminel, A.V.; Zelentsov, V.I.; Datsko, T.Y.; Fonari, M.S. Polymeric Luminescent Zn(II) and Cd(II) Dicarboxylates Decorated by Oxime Ligands: Tuning the Dimensionality and Adsorption Capacity. Cryst. Growth Des. 2014, 14, 3935–3948. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Siminel, A.V.; Masunov, A.E.; Fonari, M.S. From discrete molecules to one-dimensional coordination polymers containing Mn(II), Zn(II) or Cd(II) pyridine-2-aldoxime building unit. Polyhedron 2013, 60, 140–150. [Google Scholar] [CrossRef]
- Shirvan, S.A.; Dezfuli, S.H. Bis(acetato,κO)bis(2-pyridinealdoxime-κ2N, N′)cadmium. Acta Crystallogr. Sect. E 2012, 68, m1080–m1081. [Google Scholar] [CrossRef]
- Papatriantafyllopoulou, C.; Kostakis, G.E.; Raptopoulou, C.P.; Terzis, A.; Perlepes, S.P.; Plakatouras, J.C. Investigation of the MSO4∙XH2O (M = Zn, x = 7; M = Cd, x = 8/3)/methyl 2-pyridyl ketone oxime reaction system: A novel Cd(II) coordination polymer versus mononuclear and dinuclear Zn(II) complexes. Inorg. Chim. Acta 2009, 362, 2361–2370. [Google Scholar] [CrossRef]
- Yan, J.; Liu, G.-X. Tris(phenyl 2-pyridyl ketone oxime- κ2Ν, Ν′)cadmium(II) dinitrate. Acta Crystallogr. Sect. E 2009, 65, m461. [Google Scholar] [CrossRef]
- Mazarakioti, E.C.; Soto Beobide, A.; Angelidou, V.; Efthymiou, C.G.; Terzis, A.; Psycharis, V.; Voyiatzis, G.A.; Perlepes, S.P. Modeling the Solvent Extraction of Cadmium(II) from Aqueous Chloride Solutions by 2-pyridyl Ketoximes: A Coordination Chemistry Approach. Molecules 2019, 24, 2219. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Sommerer, S.O.; Westcott, B.L.; Jircitano, A.J.; Abboud, K.A. The synthesis and structure of two novel metal-di-2-pyridyl ketone oxime dimers. Inorg. Chim. Acta 1995, 238, 149–153. [Google Scholar] [CrossRef]
- Milios, C.J.; Kyritsis, P.; Raptopoulou, C.P.; Terzis, A.; Vicente, R.; Escuer, A.; Perlepes, S.P. Di-2-pyridyl ketone oxime [(py)2CNOH] in manganese carboxylate chemistry: Mononuclear, dinuclear and tetranuclear complexes, and partial transformation of (py)2CNOH to the gem-diolate(2-) derivative of di-2-pyridyl ketone leading to the formation of NO3−. Dalton Trans. 2005, 501–511. [Google Scholar] [CrossRef]
- Goher, M.A.S.; Mautner, F.A. Dimeric and polymeric copper(I) complexes. Synthesis and characterization of copper(I) complexes of di-2-pyridyl ketone oxime (DPKox) and crystal structures of [Cu(DPKox)Cl]2∙2H2O and [Cu(DPKox)(NCS)]n. Polyhedron 1999, 18, 3425–3431. [Google Scholar] [CrossRef]
- Westcott, B.L.; Crundwell, G.; Remesic, M.; Knopf, K.; Chandler, K.; McMaster, J.; Davies, E.S. Crystal structure and magnetic properties of di-copper and di-zinc complexes with di-2-pyridyl ketone oxime. Inorg. Chem. Commun. 2016, 74, 79–81. [Google Scholar] [CrossRef]
- Gökce, H.; Alpaslan, G.; Alasalvar, C. Crystal structure, spectroscopic characterization, DFT computations and molecular docking study of a synthesized Zn(II) complex. J. Coord. Chem. 2019, 72, 1075–1096. [Google Scholar] [CrossRef]
- Holynska, M. Formation of Ni(II) oxime-bridged basket- like complexes and their structural aspects. Curr. Inorg. Chem. 2015, 5, 64–70. [Google Scholar] [CrossRef]
- Dollish, F.R.; Fateley, W.G.; Bentley, F.F. Characteristic Raman Frequencies of Organic Compounds; Wiley: New York, NY, USA, 1974; pp. 134–137. [Google Scholar]
- Adams, D.M. Metal-Ligand and Related Vibrations: A Critical Survey of the Infrared and Raman Spectra of Metallic and Organometallic Compounds; Edward Arnold Publishers: London, UK, 1967; pp. 45, 79. [Google Scholar]
- Jackman, L.M.; Sternhell, S. Applications of Nuclear Magnetic Resonance in Organic Chemistry, 2nd ed.; Pergamon Press: Oxford, UK, 1969; pp. 216, 220. [Google Scholar]
- Thomas, S.; Brühl, I.; Heilmann, D.; Kleinpeter, E. 13C NMR chemical shift calculations for some substituted pyridines: A comparative consideration. J. Chem. Inf. Comput. Sci. 1997, 37, 726–730. [Google Scholar] [CrossRef]
- Oszczapowicz, J. Substituent Effects in the 13C-NMR spectra of six-membered nitrogen heteroatomic compounds. Int. J. Mol. Sci. 2005, 6, 11–17. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C.; Morrill, T.C. Spectrometric Identification of Organic Compounds, 4th ed.; Wiley: New York, NY, USA, 1981; pp. 134, 135, 266, 267. [Google Scholar]
- Batterham, T.J. NMR Spectra of Simple Heterocycles; Wiley: New York, NY, USA, 1973; pp. 8–67. [Google Scholar]
- Kolehmainen, E. NMR Spectroscopy, Heteronuclei, Y-Cd. In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Koppenaal, D.W., Tranter, G.E., Eds.; Elsevier: New York, NY, USA, 2017; pp. 366–374. [Google Scholar]
- Elis, P.D. Cadmium-113 Magnetic Resonance Spectroscopy. Science 1983, 221, 1141–1146. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterization of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Frost, J.M.; Kobera, L.; Pialat, A.; Zhang, Y.; Southern, S.A.; Gabidullin, B.; Bryce, D.L.; Murugesu, M. From discrete molecule, to polymer, to MOF: Mapping the coordination chemistry of CdII using 113Cd solid-state NMR. Chem. Commun. 2016, 52, 10680–10683. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuyzen, M.; Wilkins, C.J. Crystal Structures and Significance of Complexes Formed Between Cadmium Bromide and Dimethyl Sulfoxide. J. Chem. Soc. Dalton Trans. 1993, 2673–2681. [Google Scholar] [CrossRef]
- CrystalClear; Rigaku: The Woodlands, TX, USA; MSC Inc.: The Woodlands, TX, USA, 2005.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Version 2.03; Bruker Analytical X-ray Systems: Madison, WI, USA, 2000. [Google Scholar]
- Sheldrick, G.M. SHELXT-Intergrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Diamond, Crystal and Molecular Structure Visualization, Version 3.1; Crystal Impact: Bonn, Germany, 2018.
- Dey, A.; Jetti, R.K.R.; Boese, R.; Desiraju, G.R. Supramolecular equivalence of halogen, ethynyl and hydroxyl groups. A comparison of the crystal structures of some 4-substituted anilines. Cryst. Eng. Comm. 2003, 5, 248–252. [Google Scholar] [CrossRef]
- Hsu, Y.-F.; Hsu, W.; Wu, C.-J.; Cheng, P.-C.; Yeh, C.-W.; Chang, W.-J.; Chen, J.-D.; Wang, J.-C. Roles of halide anions in the structural diversity of Zn(II) complexes containing the flexible N, N’’-di(4-pyridyl)adipoamide ligand. Cryst. Eng. Comm. 2010, 12, 702–710. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | {[CdCl2(dpkoxH)]∙2H2O}n (1∙2H2O) | {[CdBr2(dpkoxH)]}n (2) | {[CdI2(dpkoxH)]}n (3) |
|---|---|---|---|
| Empirical formula | C11H9CdCl2N3O∙2H2O | C11H9CdBr2N3O | C11H9CdI2N3O |
| Formula weight | 418.54 | 471.43 | 565.41 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group | Pī | C2/c | C2/c |
| Color | Colorless | Colorless | Colorless |
| a, Å | 8.3231(2) | 15.8842(11) | 16.0480(12) |
| b, Å | 9.0416(2) | 9.2829(6) | 9.6812(6) |
| c, Å | 10.4143(2) | 18.0648(12) | 18.7611(13) |
| α, ° | 88.371(1) | 90.00 | 90.00 |
| β, ° | 83.666(1) | 93.572(2) | 93.558(5) |
| γ, ° | 76.119(1) | 90.00 | 90.00 |
| Volume, Å3 | 756.19(3) | 2658.5(3) | 2909.2(3) |
| Z | 2 | 8 | 8 |
| Temperature, °C | −103 | −153 | −93 |
| Radiation, Å | Cu Kα, 1.54178 | Μο Κα, 0.71073 | Cu Κα, 1.54178 |
| Calculated density, g∙cm−3 | 1.838 | 2.356 | 2.582 |
| Absorption coefficient, mm−1 | 14.92 | 7.64 | 45.30 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 17,055, 2607, 2400 | 49,909, 2736, 2684 | 12,775, 2518, 2269 |
| Rint | 0.092 | 0.030 | 0.113 |
| Final R indices [I > 2σ(I)] α | R1 = 0.0641 wR2 = 0.1625 | R1 = 0.0129 wR2 = 0.0325 | R1 = 0.0605 wR2 = 0.1601 |
| Number of parameters | 164 | 164 | 164 |
| Goodness-of-fit on F2 | 1.15 | 1.07 | 1.06 |
| Larger differences in peak and hole (e Å−3) | 1.95/−1.67 | 0.38/−0.46 | 1.30/−1.91 |
| Complex 1∙2H2O a | Complex 2 b | Complex 3 c | |||
|---|---|---|---|---|---|
| Lengths (Å) | |||||
| Cd1-Cl1 | 2.731(2) | Cd1-Br2* | 2.950(1) | Cd1-I2* | 3.290(1) |
| Cd1-Cl2 | 2.520(2) | Cd1-Br2 | 2.643(1) | Cd1-I2 | 2.818(1) |
| Cd1-Cl1′ | 2.539(2) | Cd1-Br1 | 2.607(1) | Cd1-I1 | 2.787(1) |
| Cd1-N1 | 2.314(5) | Cd1-N2 | 2.402(1) | Cd1-N2 | 2.437(7) |
| Cd1-N2 | 2.473(6) | Cd1-N1 | 2.355(1) | Cd-N1 | 2.375(6) |
| Cd1-N3″ | 2.450(5) | Cd1-N3** | 2.486(1) | Cd1-N3** | 2.503(7) |
| Angles (°) | |||||
| Cl1-Cd1-Cl2 | 87.3(1) | Br2*-Cd1-Br2 | 86.7(1) | I2*-Cd1-I2 | 91.3(2) |
| Cl1-Cd1-Cl1′ | 85.9(1) | Br2*-Cd1-Br1 | 95.4(1) | I2*-Cd1-I1 | 93.2(1) |
| Cl1-Cd1-N2 | 76.3(1) | Br2*-Cd1-N2 | 75.8(1) | I2-Cd1-N2 | 71.6(2) |
| Cl1-Cd1-N1 | 101.0(1) | Br2*-Cd1-N1 | 83.1(1) | I2*-Cd1-N1 | 81.6(2) |
| Cl2-Cd1-Cl1′ | 103.2(1) | Br2-Cd1-Br1 | 112.1(1) | I2-Cd1-I1 | 110.0(1) |
| Cl1′-Cd1-N2 | 95.9(2) | Br1-Cd1-N2 | 87.7(1) | I1-Cd1-N2 | 88.8(2) |
| N2-Cd1-N1 | 67.9(2) | N2-Cd1-N1 | 67.9(1) | N2-Cd1-N1 | 67.1(2) |
| N1-Cd1-Cl2 | 96.1(2) | N1-Cd1-Br2 | 92.7(1) | N1-Cd1-I2 | 93.8(1) |
| N3″-Cd1-Cl2 | 95.3(2) | N3**-Cd1-Br2 | 90.7(1) | N3**-Cd1-I2 | 92.4(1) |
| N3″-Cd1-Cl1′ | 85.8(2) | N3**-Cd1-Br1 | 92.8(1) | N3**-Cd1-I1 | 94.8(2) |
| N3″-Cd1-N2 | 103.9(2) | N3**-Cd1-N2 | 100.9(1) | N3**-Cd1-N2 | 101.7(2) |
| N3″-Cd1-N1 | 86.6(2) | N3**-Cd1-N1 | 87.9(1) | N3**-Cd1-N1 | 88.4(2) |
| Cl1-Cd1-N3″ | 171.7(2) | Br2*-Cd1-N3** | 171.0(1) | I2*-Cd1-N3** | 169.5(2) |
| Cl2-Cd1-N2 | 153.9(1) | Br1-Cd1-N1 | 155.2(1) | I1-Cd1-N1 | 155.8(2) |
| Cl1′-Cd1-N1 | 159.9(2) | Br2-Cd1-N2 | 156.7(1) | I2-Cd1-N2 | 155.6(2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stamou, C.; Dechambenoit, P.; Lada, Z.G.; Gkolfi, P.; Riga, V.; Raptopoulou, C.P.; Psycharis, V.; Konidaris, K.F.; Chasapis, C.T.; Perlepes, S.P. Reactions of Cadmium(II) Halides and Di-2-Pyridyl Ketone Oxime: One-Dimensional Coordination Polymers. Molecules 2024, 29, 509. https://doi.org/10.3390/molecules29020509
Stamou C, Dechambenoit P, Lada ZG, Gkolfi P, Riga V, Raptopoulou CP, Psycharis V, Konidaris KF, Chasapis CT, Perlepes SP. Reactions of Cadmium(II) Halides and Di-2-Pyridyl Ketone Oxime: One-Dimensional Coordination Polymers. Molecules. 2024; 29(2):509. https://doi.org/10.3390/molecules29020509
Chicago/Turabian StyleStamou, Christina, Pierre Dechambenoit, Zoi G. Lada, Patroula Gkolfi, Vassiliki Riga, Catherine P. Raptopoulou, Vassilis Psycharis, Konstantis F. Konidaris, Christos T. Chasapis, and Spyros P. Perlepes. 2024. "Reactions of Cadmium(II) Halides and Di-2-Pyridyl Ketone Oxime: One-Dimensional Coordination Polymers" Molecules 29, no. 2: 509. https://doi.org/10.3390/molecules29020509