
An In Vitro RNA Editing-Based Reporter Assay for Transcriptional Activity of Therapeutic Gene in Gene Therapy Products

 ,
,
Abstract
:
1. Introduction
2. Results
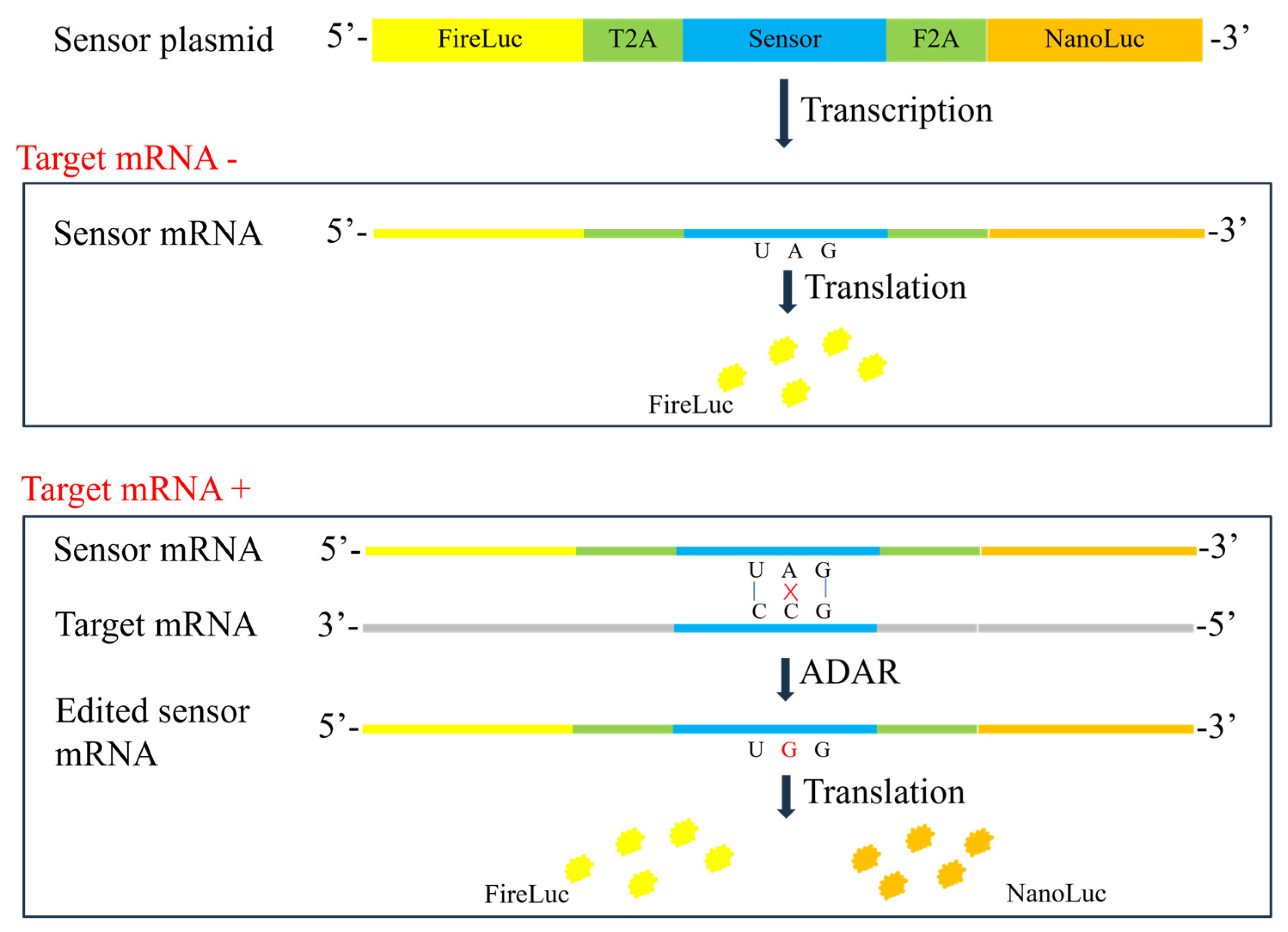
2.1. Construction of Sensors for Specific mRNA
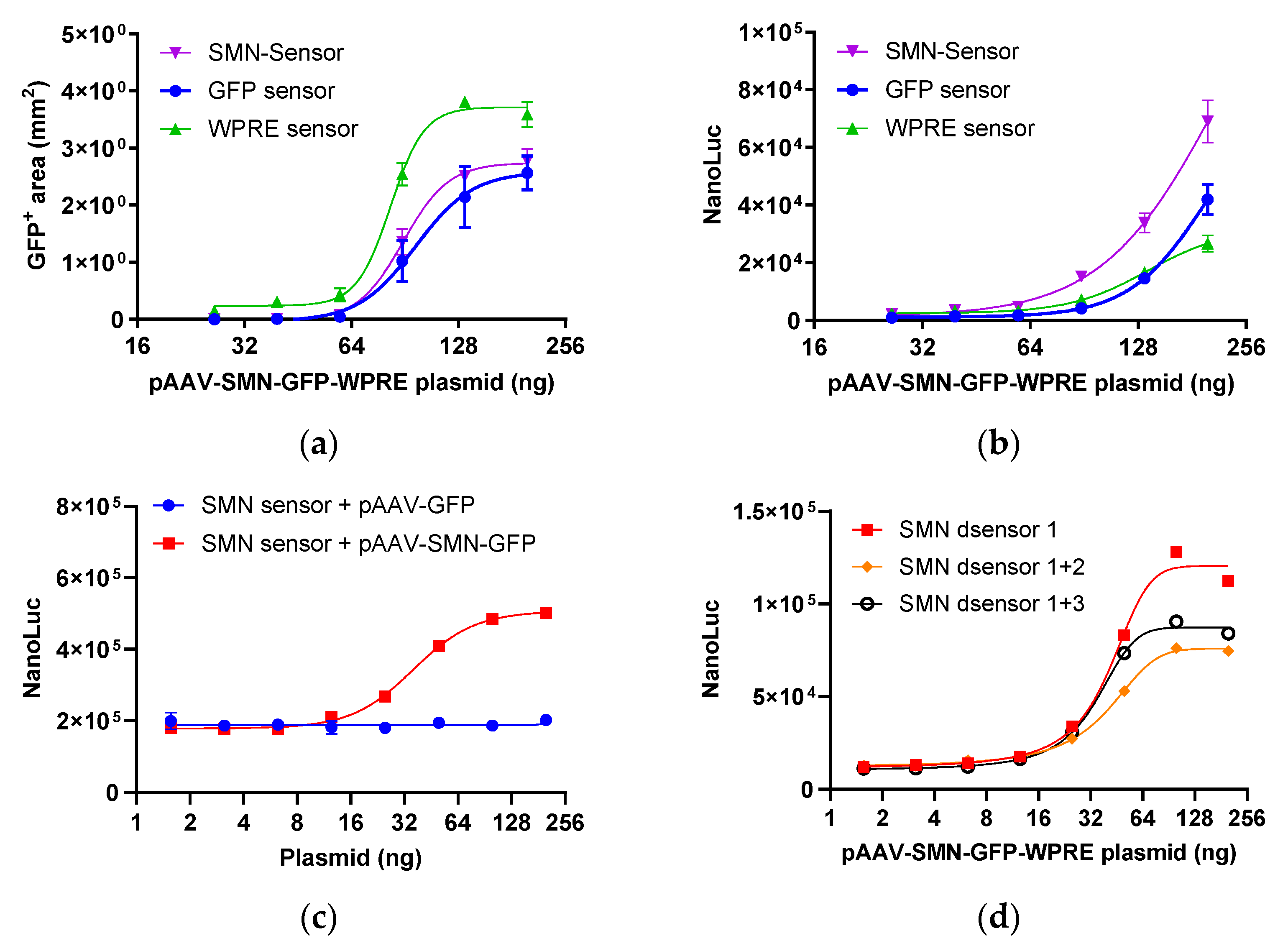
2.1.1. Response of Sensors Targeting Different Regions
2.1.2. SMN Sensors with Double Termination Codons
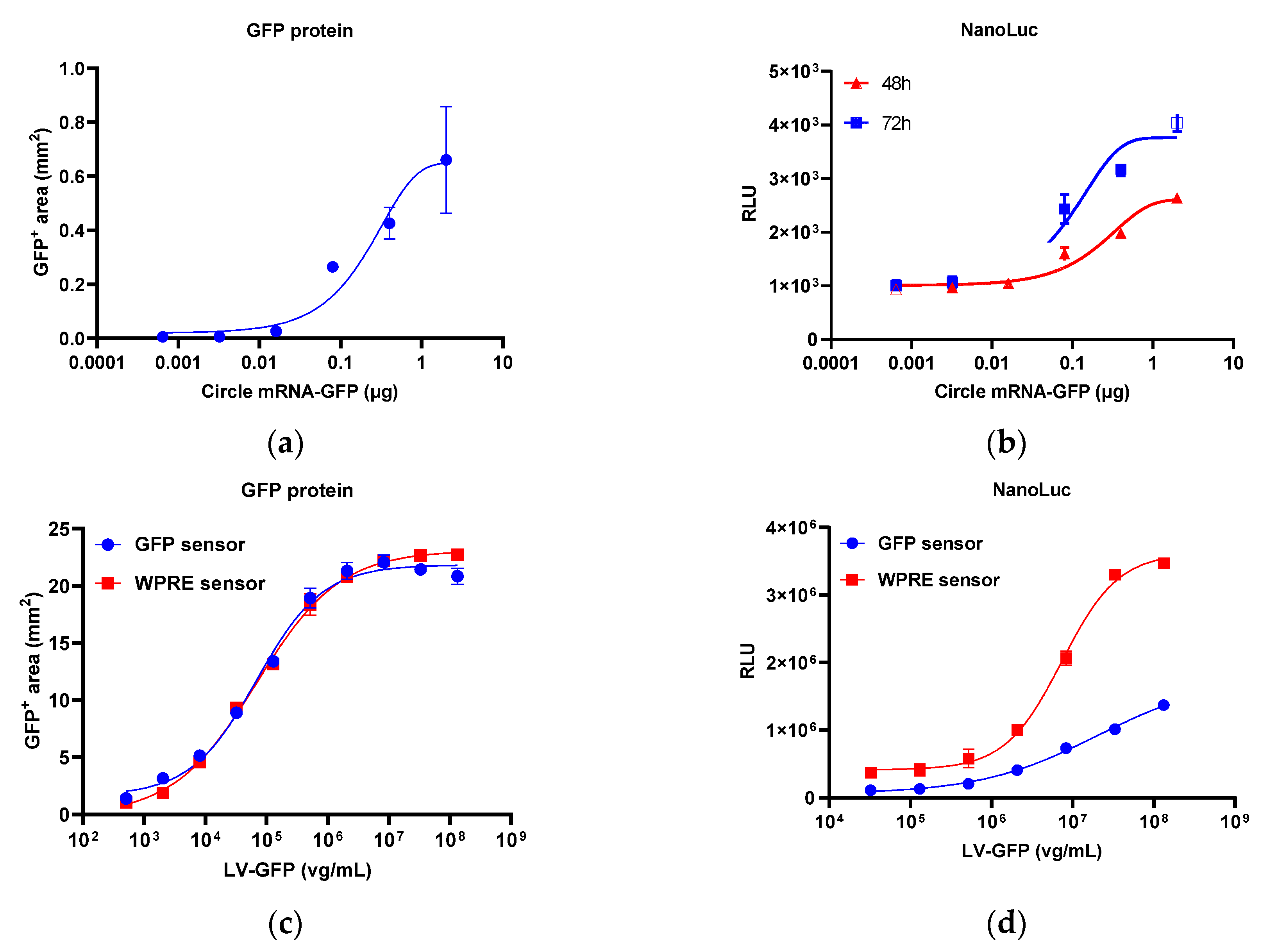
2.2. Effect of Overexpressed ADARs
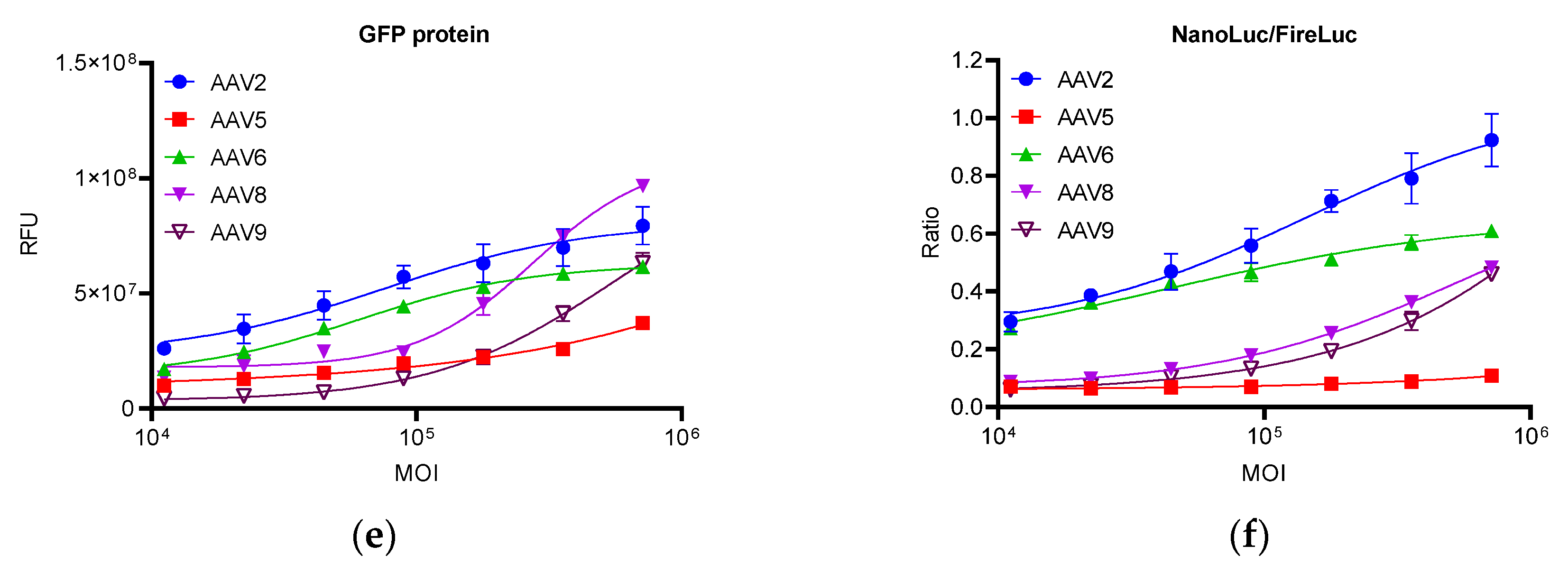
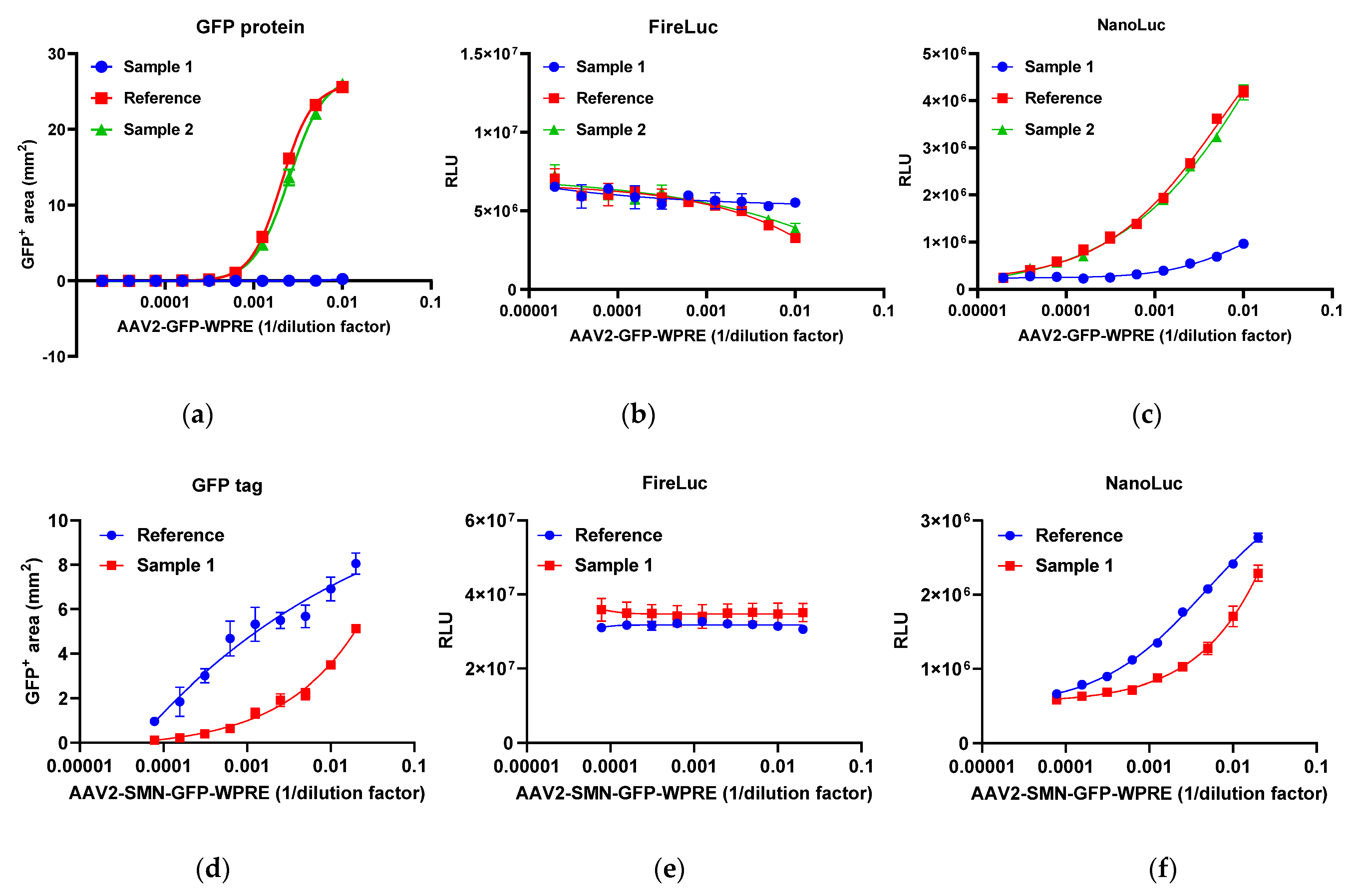
2.3. Response of the Reporter Assay to Different Gene Therapy Vectors
2.4. Transcriptional Activity of rAAV Products Determined Using the Developed Reporter Assay
2.4.1. Specificity of the Reporter Assay
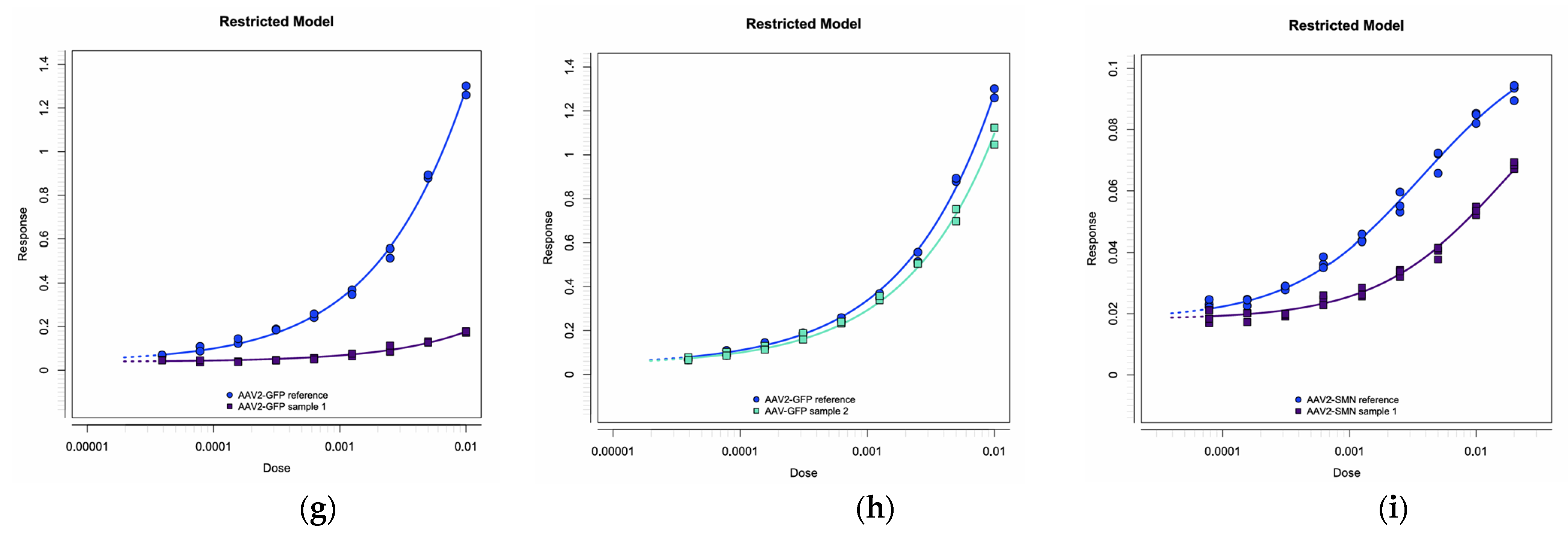
2.4.2. Relative Quantitative Method
2.4.3. Precision, Accuracy, and Linearity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Construction of Sensor Plasmid
4.3. Establishment of Stable-Transfected Sensor Cells
4.4. Transcriptional Activity Assays
4.5. AAV Genomic Titer Determination by dPCR
4.6. Precision, Accuracy, and Linearity Assessment
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, E.; Blomberg, P. Gene therapy—From idea to reality. Lakartidningen 2017, 114, EWYL. [Google Scholar] [PubMed]
- Arjmand, B.; Larijani, B.; Sheikh Hosseini, M.; Payab, M.; Gilany, K.; Goodarzi, P.; Parhizkar Roudsari, P.; Amanollahi Baharvand, M.; Hoseini Mohammadi, N.S. The Horizon of Gene Therapy in Modern Medicine: Advances and Challenges. Adv. Exp. Med. Biol. 2020, 1247, 33–64. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Lee, K. Development of mRNA Vaccines/Therapeutics and Their Delivery System. Mol. Cells. 2023, 46, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zu, H.; Gao, D. Non-Viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef]
- Li, X.; Le, Y.; Zhang, Z.; Nian, X.; Liu, B.; Yang, X. Viral Vector-Based Gene Therapy. Int. J. Mol. Sci. 2023, 24, 7736. [Google Scholar] [CrossRef]
- Wright, J.F. Quality Control Testing, Characterization and Critical Quality Attributes of Adeno-Associated Virus Vectors Used for Human Gene Therapy. Biotechnol. J. 2021, 16, 2000022. [Google Scholar] [CrossRef]
- Aronson, S.J.; Bakker, R.S.; Moenis, S.; van Dijk, R.; Bortolussi, G.; Collaud, F.; Shi, X.; Duijst, S.; Ten Bloemendaal, L.; Ronzitti, G.; et al. A Quantitative In Vitro Potency Assay for Adeno-Associated Virus Vectors Encoding for the UGT1A1 Transgene. Mol. Ther. Methods Clin. Dev. 2020, 18, 250–258. [Google Scholar] [CrossRef]
- Buck, T.M.; Wijnholds, J. Recombinant Adeno-Associated Viral Vectors (rAAV)-Vector Elements in Ocular Gene Therapy Clinical Trials and Transgene Expression and Bioactivity Assays. Int. J. Mol. Sci. 2020, 21, 4197. [Google Scholar] [CrossRef]
- Gao, K.; Wang, J.; Rao, C.; Wu, X. Quality control of recombinant adeno-associated virus type 2/human blood coagulation factor IX. Yao Xue Xue Bao 2003, 38, 684–689. [Google Scholar]
- Patrício, M.I.; Barnard, A.R.; Cox, C.I.; Blue, C.; MacLaren, R.E. The Biological Activity of AAV Vectors for Choroideremia Gene Therapy Can Be Measured by In Vitro Prenylation of RAB6A. Mol. Ther. Methods Clin. Dev. 2018, 9, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.; Pili, L.; Buffa, V.; Caroff, M.; Bigot, A.; Gicquel, E.; Rouby, G.; Richard, I.; Fragnoud, R. CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy. Cells 2023, 12, 2444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cho, J.-H.; Arnaoutova, I.; Mansfield, B.C.; Chou, J.Y. An Evolutionary Approach to Optimizing Glucose-6-Phosphatase-α Enzymatic Activity for Gene Therapy of Glycogen Storage Disease Type Ia. J. Inherit. Metab. Dis. 2019, 42, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Clarner, P.; Lau, S.K.; Chowdhury, T.; Guilmette, E.; Trapa, P.; Lo, S.-C.; Shen, S. Development of a One-Step RT-ddPCR Method to Determine the Expression and Potency of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2021, 23, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Vesely, C.; Jantsch, M.F. An I for an A: Dynamic Regulation of Adenosine Deamination-Mediated RNA Editing. Genes 2021, 12, 1026. [Google Scholar] [CrossRef]
- Qu, L.; Yi, Z.; Zhu, S.; Wang, C.; Cao, Z.; Zhou, Z.; Yuan, P.; Yu, Y.; Tian, F.; Liu, Z.; et al. Author Correction: Programmable RNA Editing by Recruiting Endogenous ADAR Using Engineered RNAs. Nat. Biotechnol. 2019, 37, 1380. [Google Scholar] [CrossRef]
- Montiel-Gonzalez, M.F.; Diaz Quiroz, J.F.; Rosenthal, J.J.C. Current Strategies for Site-Directed RNA Editing Using ADARs. Methods 2019, 156, 16–24. [Google Scholar] [CrossRef]
- Li, M.; Yan, C.; Jiao, Y.; Xu, Y.; Bai, C.; Miao, R.; Jiang, J.; Liu, J. Site-Directed RNA Editing by Harnessing ADARs: Advances and Challenges. Funct. Integr. Genom. 2022, 22, 1089–1103. [Google Scholar] [CrossRef]
- Park, M.J.; Jeong, E.; Lee, E.J.; Choi, H.J.; Moon, B.H.; Kang, K.; Chang, S. RNA Editing Enzyme ADAR1 Suppresses the Mobility of Cancer Cells via ARPIN. Mol. Cells 2023, 46, 351–359. [Google Scholar] [CrossRef]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A Third Member of the RNA-Specific Adenosine Deaminase Gene Family, ADAR3, Contains Both Single- and Double-Stranded RNA Binding Domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.-D. ADAR1 and ZBP1 in Innate Immunity, Cell Death, and Disease. Trends Immunol. 2023, 44, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Kaseniit, K.E.; Katz, N.; Kolber, N.S.; Call, C.C.; Wengier, D.L.; Cody, W.B.; Sattely, E.S.; Gao, X.J. Modular, Programmable RNA Sensing Using ADAR Editing in Living Cells. Nat. Biotechnol. 2023, 41, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Koob, J.; Chen, X.D.; Krajeski, R.N.; Zhang, Y.; Volf, V.; Zhou, W.; Sgrizzi, S.R.; Villiger, L.; Gootenberg, J.S.; et al. Programmable Eukaryotic Protein Synthesis with RNA Sensors by Harnessing ADAR. Nat. Biotechnol. 2023, 41, 698–707. [Google Scholar] [CrossRef]
- Chng, J.; Wang, T.; Nian, R.; Lau, A.; Hoi, K.M.; Ho, S.C.L.; Gagnon, P.; Bi, X.; Yang, Y. Cleavage Efficient 2A Peptides for High Level Monoclonal Antibody Expression in CHO Cells. MAbs 2015, 7, 403–412. [Google Scholar] [CrossRef]
- Zhu, X.; Ricci-Tam, C.; Hager, E.R.; Sgro, A.E. Self-Cleaving Peptides for Expression of Multiple Genes in Dictyostelium Discoideum. PLoS ONE 2023, 18, e0281211. [Google Scholar] [CrossRef]
- Burns, C.J.; Silva, M.M.C.G.; Gray, E.; Robinson, C.J. Quantitative RT-PCR as an Alternative to Late-Stage Bioassays for Vascular Endothelial Growth Factor. J. Pharm. Biomed. Anal. 2008, 47, 460–468. [Google Scholar] [CrossRef]
- Mahajan, R.; Feher, B.; Jones, B.; Jones, D.; Marjerison, L.; Sam, M.; Hartikka, J.; Wloch, M.; Lalor, P.; Kaslow, D.; et al. A TaqMan Reverse Transcription Polymerase Chain Reaction (RT-PCR) in Vitro Potency Assay for Plasmid-Based Vaccine Products. Mol. Biotechnol. 2008, 40, 47–57. [Google Scholar] [CrossRef]
- Ranheim, T.; Mathis, P.K.; Joelsson, D.B.; Smith, M.E.; Campbell, K.M.; Lucas, G.; Barmat, S.; Melissen, E.; Benz, R.; Lewis, J.A.; et al. Development and Application of a Quantitative RT-PCR Potency Assay for a Pentavalent Rotavirus Vaccine (RotaTeq). J. Virol. Methods 2006, 131, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational Readthrough Potential of Natural Termination Codons in Eucaryotes—The Impact of RNA Sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef]
- Floquet, C.; Hatin, I.; Rousset, J.-P.; Bidou, L. Statistical Analysis of Readthrough Levels for Nonsense Mutations in Mammalian Cells Reveals a Major Determinant of Response to Gentamicin. PLoS Genet. 2012, 8, e1002608. [Google Scholar] [CrossRef]
- Galipon, J.; Ishii, R.; Suzuki, Y.; Tomita, M.; Ui-Tei, K. Differential Binding of Three Major Human ADAR Isoforms to Coding and Long Non-Coding Transcripts. Genes 2017, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Katrekar, D.; Xiang, Y.; Palmer, N.; Saha, A.; Meluzzi, D.; Mali, P. Comprehensive Interrogation of the ADAR2 Deaminase Domain for Engineering Enhanced RNA Editing Activity and Specificity. eLife 2022, 11, e75555. [Google Scholar] [CrossRef] [PubMed]
- Doherty, E.E.; Wilcox, X.E.; van Sint Fiet, L.; Kemmel, C.; Turunen, J.J.; Klein, B.; Tantillo, D.J.; Fisher, A.J.; Beal, P.A. Rational Design of RNA Editing Guide Strands: Cytidine Analogs at the Orphan Position. J. Am. Chem. Soc. 2021, 143, 6865–6876. [Google Scholar] [CrossRef] [PubMed]
- ‹1034› Analysis of Biological Assays. Available online: https://doi.usp.org/USPNF/USPNF_M5677_02_01.html (accessed on 20 October 2024).
- ‹1032› Design and Development of Biological Assays. Available online: https://doi.usp.org/USPNF/USPNF_M1354_01_01.html (accessed on 20 October 2024).
- Li, H.; Witkos, T.M.; Umlauf, S.; Thompson, C. Potency Assay Variability Estimation in Practice. Pharm. Stat. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Yu, X.; Yu, C.; Wang, K.; Liu, C.; Wang, L.; Wang, J. A Robust Reporter Assay for the Determination of the Bioactivity of IL-4R-Targeted Therapeutic Antibodies. J. Pharm. Biomed. Anal. 2021, 199, 114033. [Google Scholar] [CrossRef]
- Lei, Y.; Yong, Z.; Junzhi, W. Development and Application of Potency Assays Based on Genetically Modified Cells for Biological Products. J. Pharm. Biomed. Anal. 2023, 230, 115397. [Google Scholar] [CrossRef]
- Yu, L.; Jia, C.; Yao, W.; Pei, D.; Qin, X.; Rao, C.; Wang, J. Development and Validation of a Reporter-Cell-Line-Based Bioassay for Therapeutic Soluble Gp130-Fc. Molecules 2019, 24, 3845. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Yu, C.; Wang, J. Development of a Robust Reporter Gene Assay for Measuring the Bioactivity of OX40-Targeted Therapeutic Antibodies. Luminescence 2021, 36, 885–893. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Genomic Titer (vg/mL) | Sensor | Test 1 (%) | Test 2 (%) | Test 3 (%) | Test 4 (%) | Test 5 (%) | Mean (%) | RSD (%) |
|---|---|---|---|---|---|---|---|---|---|
| AAV2-GFP-WPRE reference | 4.80 × 1012 | GFP sensor | / | / | / | / | / | / | / |
| AAV2-GFP-WPRE sample 1 | 1.61 × 1011 | GFP sensor | 3.35 | 3.19 | 2.50 | 2.46 | 2.78 | 2.86 | 14.0 |
| AAV2-GFP-WPRE sample 2 | 2.86 × 1012 | GFP sensor | 76.9 | 68.3 | 60.7 | 70.4 | 70.0 | 69.3 | 8.4 |
| AAV2-SMN-GFP-WPRE reference | 5.21 × 1012 | SMN sensor 1 | / | / | / | / | / | / | / |
| AAV2-SMN-GFP-WPRE sample 1 | 4.17 × 1012 | SMN sensor 1 | 20.4 | 19.3 | 20.8 | 19.7 | 18.3 | 19.7 | 4.9 |
| Expected Value (%) | AAV2-GFP | AAV2-SMN-GFP | ||
|---|---|---|---|---|
| Measured Value (%) | Recovery Rate (%) | Measured Value (%) | Recovery Rate (%) | |
| 100 | 100.0 | / | 100.0 | / |
| 80 | 74.2 | 92.7 | 74.0 | 92.5 |
| 60 | 56.6 | 94.3 | 57.7 | 96.2 |
| 40 | 41.3 | 103.4 | 42.5 | 106.3 |
| 20 | 22.5 | 112.3 | 22.5 | 112.3 |
| Linear equation * | y = 0.974x | y = 0.9788x | ||
| R2 | 0.9981 | 0.9979 | ||
| Sensor | Target Site | Sequence |
|---|---|---|
| GFP sensor | +587nt~+685nt of GFP | 5′-cggcggcggtcacgaactccagcaggaccatgtgatcgcgcttctcgtTAG*ggtctttgctcagggcggactgggtgctcaggtagtggttgtcgggca-3′ |
| WEPR sensor | +58nt~+156nt of WPRE | 5′-gatttatacaaggaggagaaaatgaaagccatacgggaagcaatagcaTAAtacaaaggcattaaagcagcgtatccacatagcgtaaaaggagcaaca-3′ |
| SMN sensor | +30nt~+128nt of SMN | 5′-tatgcttttatcagtgctgtatcatcccaaatgtcagaatcatcgctcTAGcctgtgccgcgccggaacagcacggaatcctcctgctccgggacgccg-3′ |
| SMN dsensor 1 | +691nt~789nt of SMN | 5′-catacttcccaaagcatcagcatcatcaagagaatctggacatatgggaggtggTAGgggaattattggtggtccagaaggaaatggaggcagccagca-3′ |
| SMN dsensor 1 + 2 | +691nt~789nt of SMN | 5′-catacttcccaaagcatcagcatcatcaagagaatctggacatatgggaggTAGTAGgggaattattggtggtccagaaggaaatggaggcagccagca-3′ |
| SMN dsensor 1 + 3 | +691nt~789nt of SMN | 5′-catacttcccaaagcatcagcatcatcaagagaatcTAGacatatgggaggtggTAGgggaattattggtggtccagaaggaaatggaggcagccagca-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Zhou, Y.; Wang, G.; Fu, J.; Fu, Z.; Liang, C.; Wang, J. An In Vitro RNA Editing-Based Reporter Assay for Transcriptional Activity of Therapeutic Gene in Gene Therapy Products. Molecules 2024, 29, 5312. https://doi.org/10.3390/molecules29225312
Yu L, Zhou Y, Wang G, Fu J, Fu Z, Liang C, Wang J. An In Vitro RNA Editing-Based Reporter Assay for Transcriptional Activity of Therapeutic Gene in Gene Therapy Products. Molecules. 2024; 29(22):5312. https://doi.org/10.3390/molecules29225312
Chicago/Turabian StyleYu, Lei, Yong Zhou, Guangyu Wang, Jianning Fu, Zhihao Fu, Chenggang Liang, and Junzhi Wang. 2024. "An In Vitro RNA Editing-Based Reporter Assay for Transcriptional Activity of Therapeutic Gene in Gene Therapy Products" Molecules 29, no. 22: 5312. https://doi.org/10.3390/molecules29225312
APA StyleYu, L., Zhou, Y., Wang, G., Fu, J., Fu, Z., Liang, C., & Wang, J. (2024). An In Vitro RNA Editing-Based Reporter Assay for Transcriptional Activity of Therapeutic Gene in Gene Therapy Products. Molecules, 29(22), 5312. https://doi.org/10.3390/molecules29225312