
The Efficient and Environmentally Friendly Chlorination of Arene, Alcohol, Halobenzene, and Peroxide Catalyzed by Fe–Ba Binary Oxides Using Hydrochloric Acid as Chlorine Source and Aqueous H2O2 as Oxidant

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
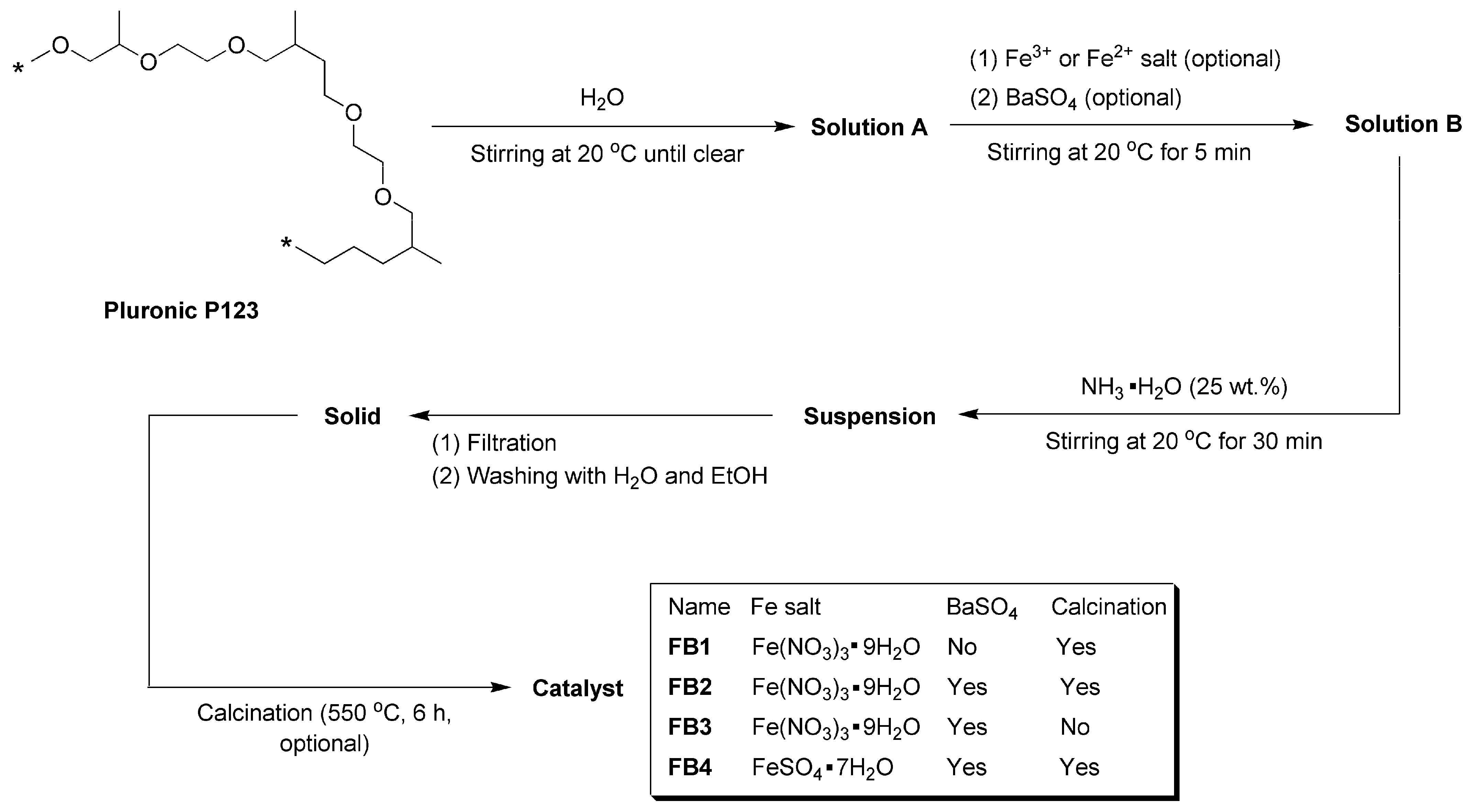
2.1. Synthesis of Catalysts
2.2. Characterization of Synthesized Catalysts
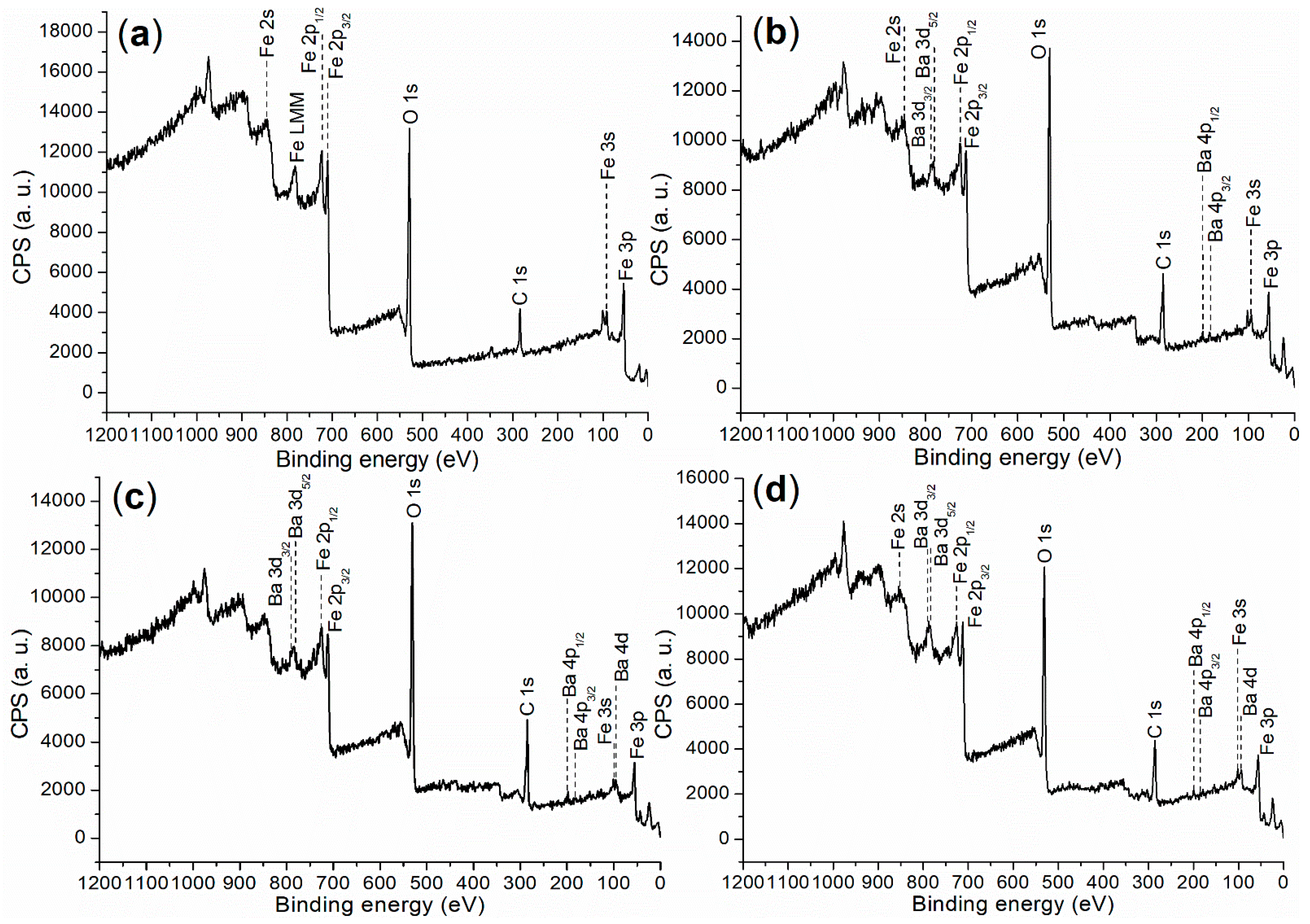
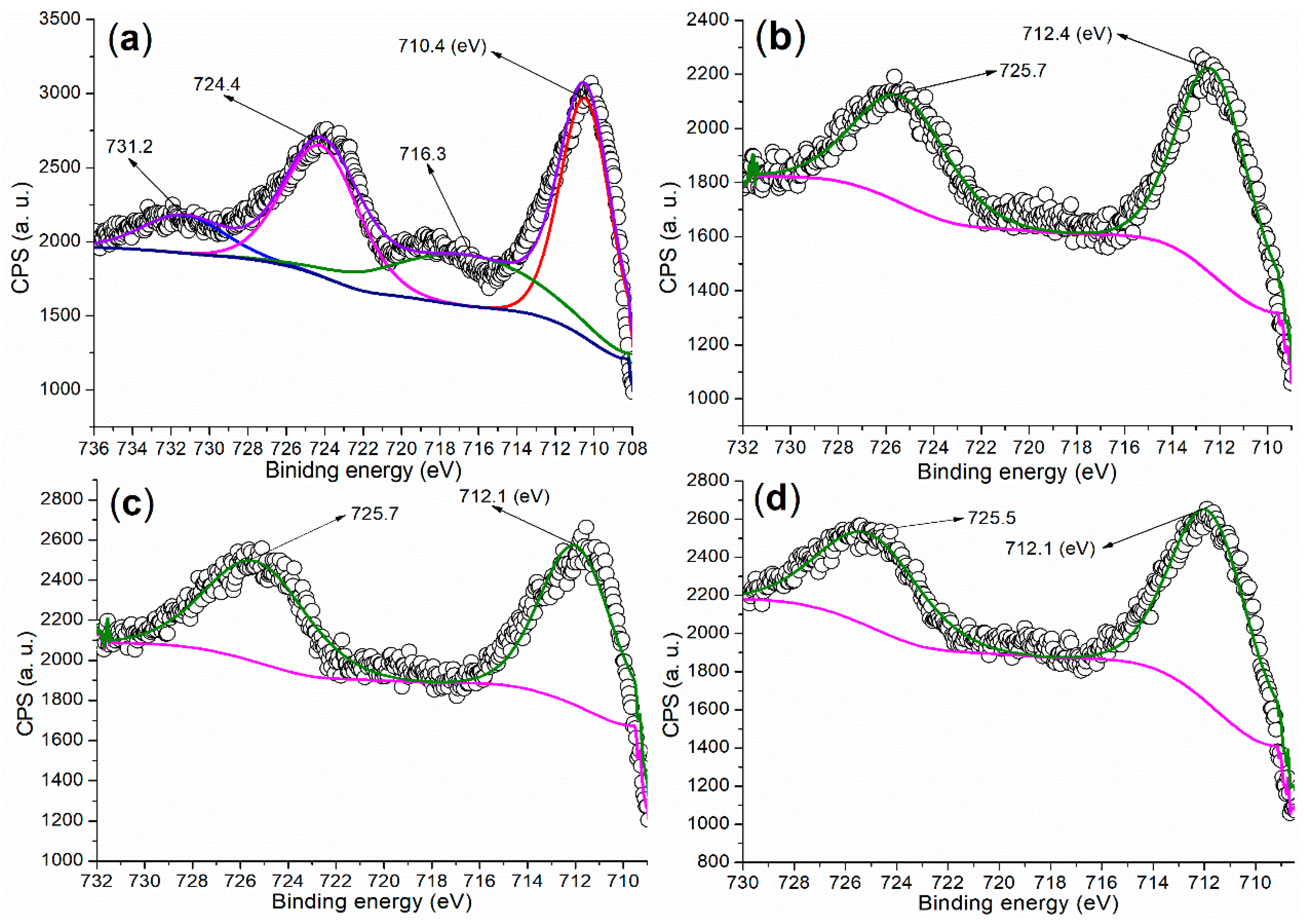
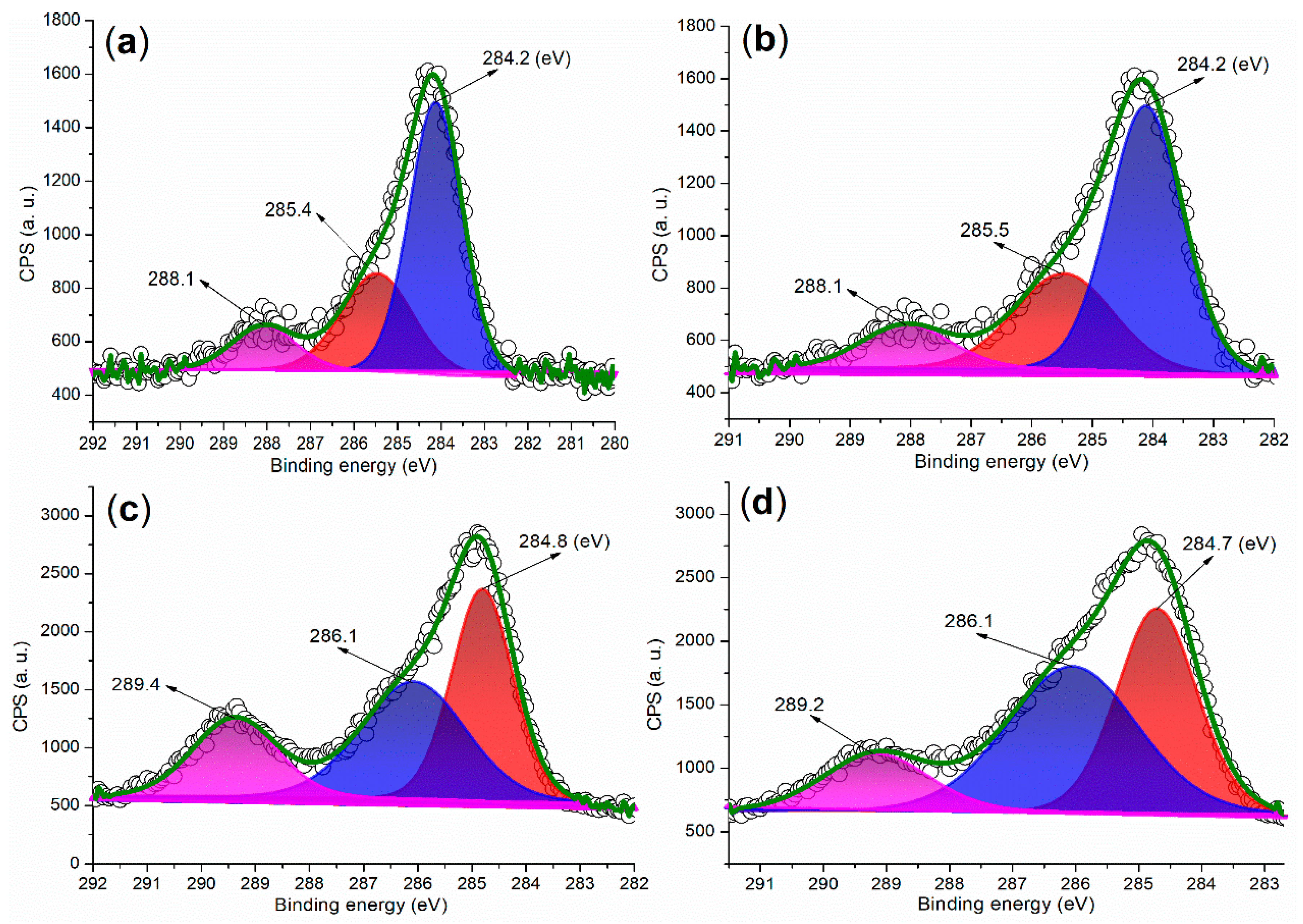
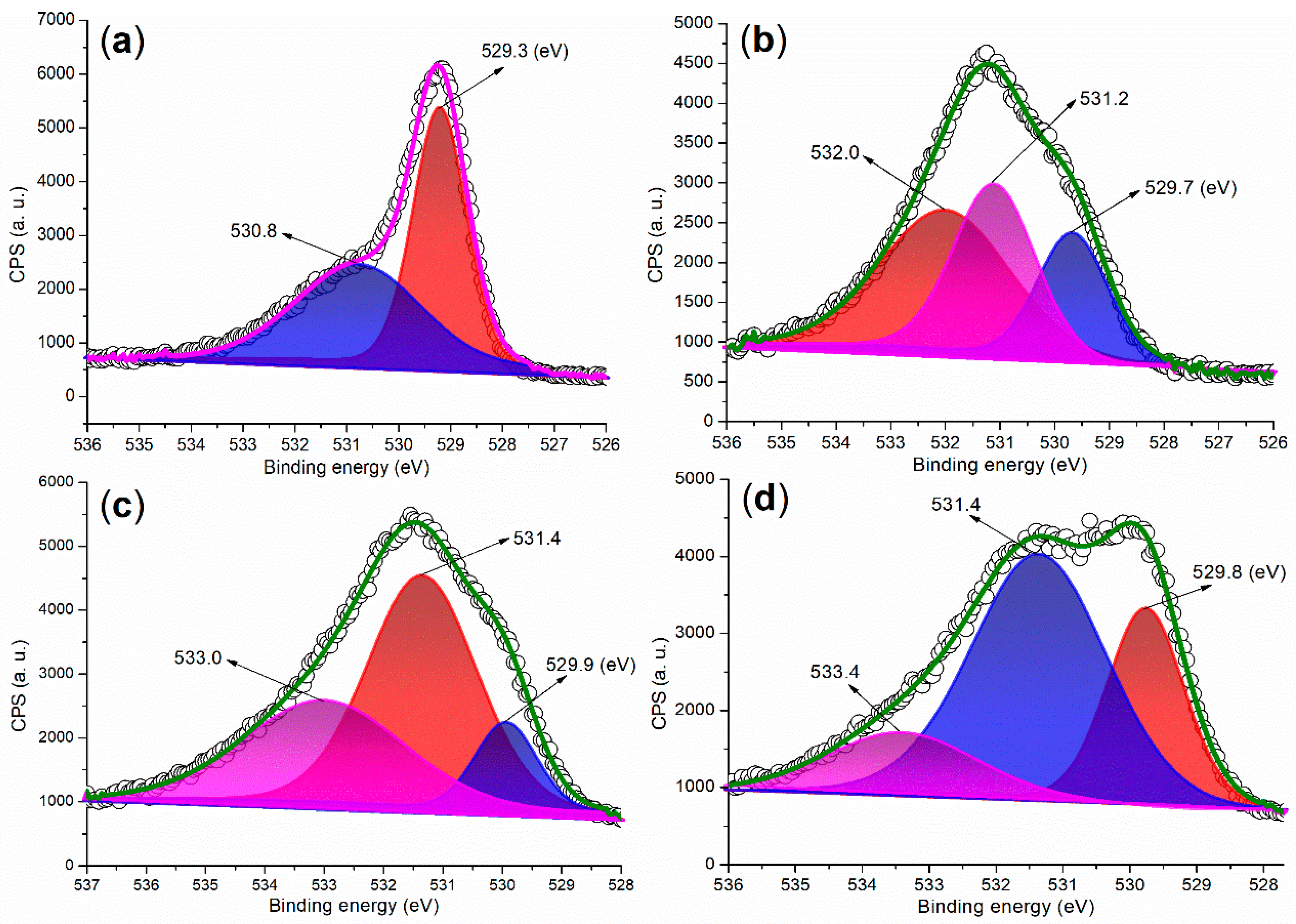
2.2.1. Elemental and Crystalline Analysis
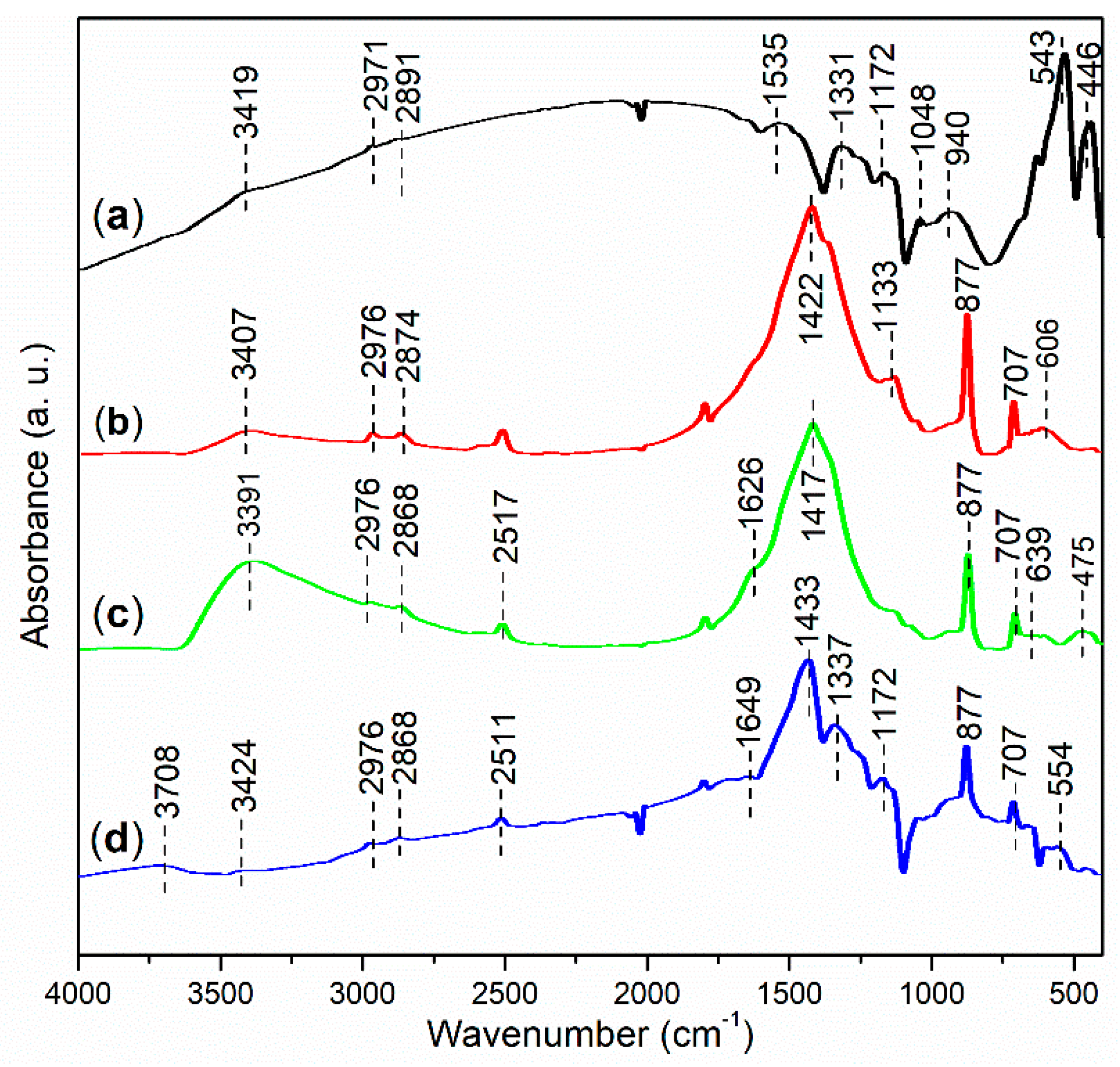
2.2.2. Functional Group
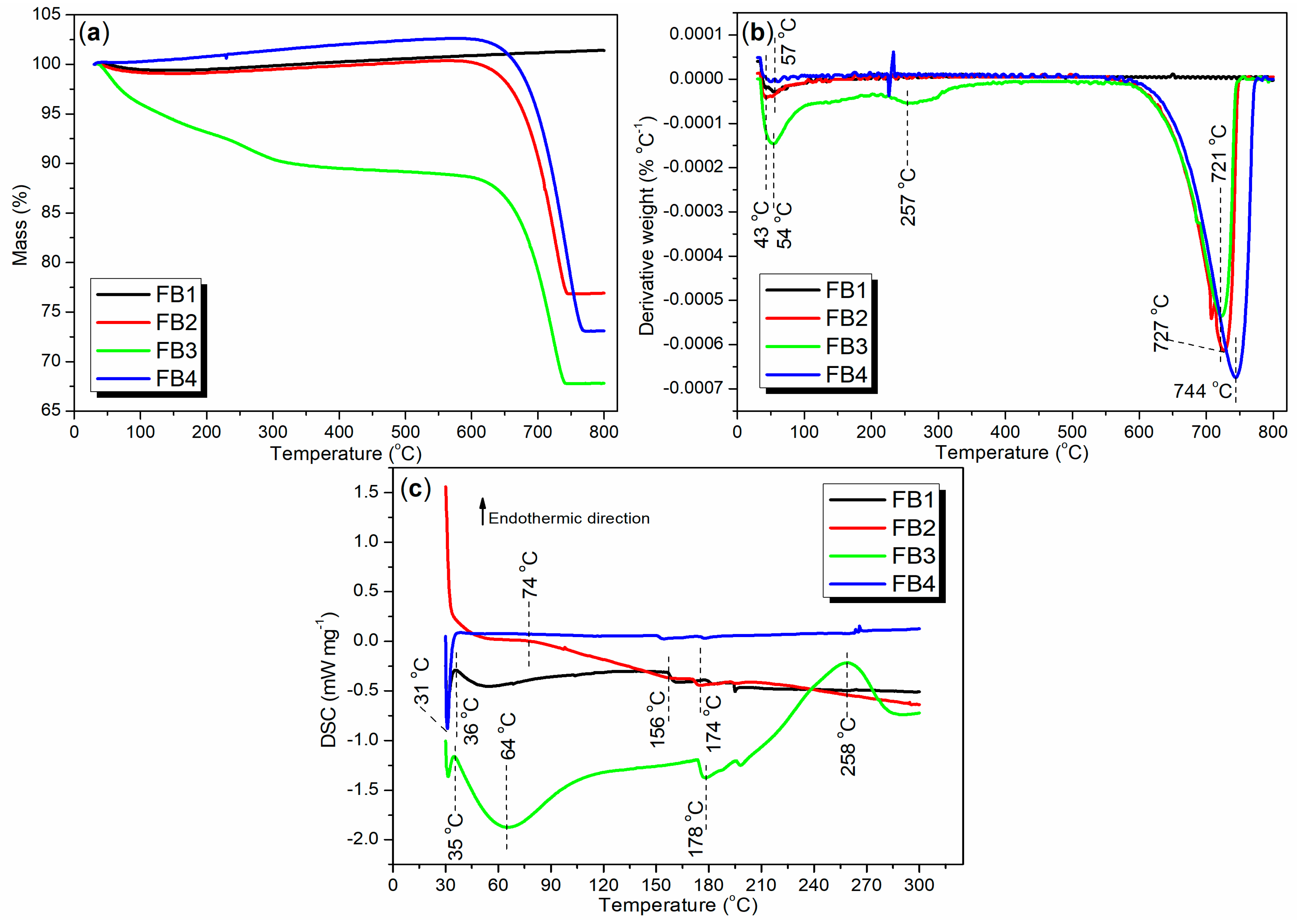
2.2.3. Thermal Stability
2.2.4. Morphology and Energy-Dispersive Spectroscopy
2.3. Catalytic Chlorination
2.3.1. Effects of Catalysts
2.3.2. Effects of Catalyst and Hydrogen Chloride Dosages
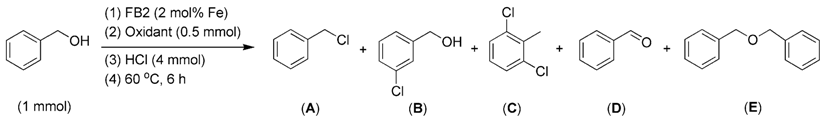
2.3.3. Effects of Oxidants
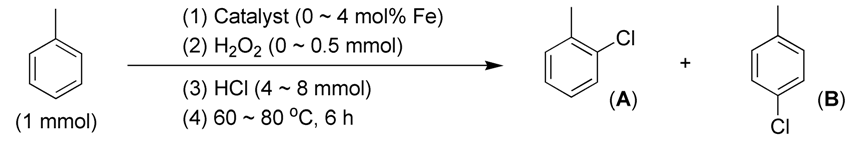
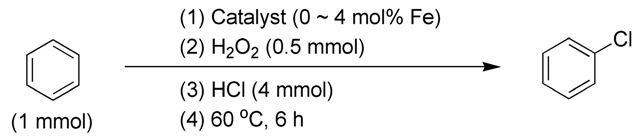
2.3.4. Effects of Substrates
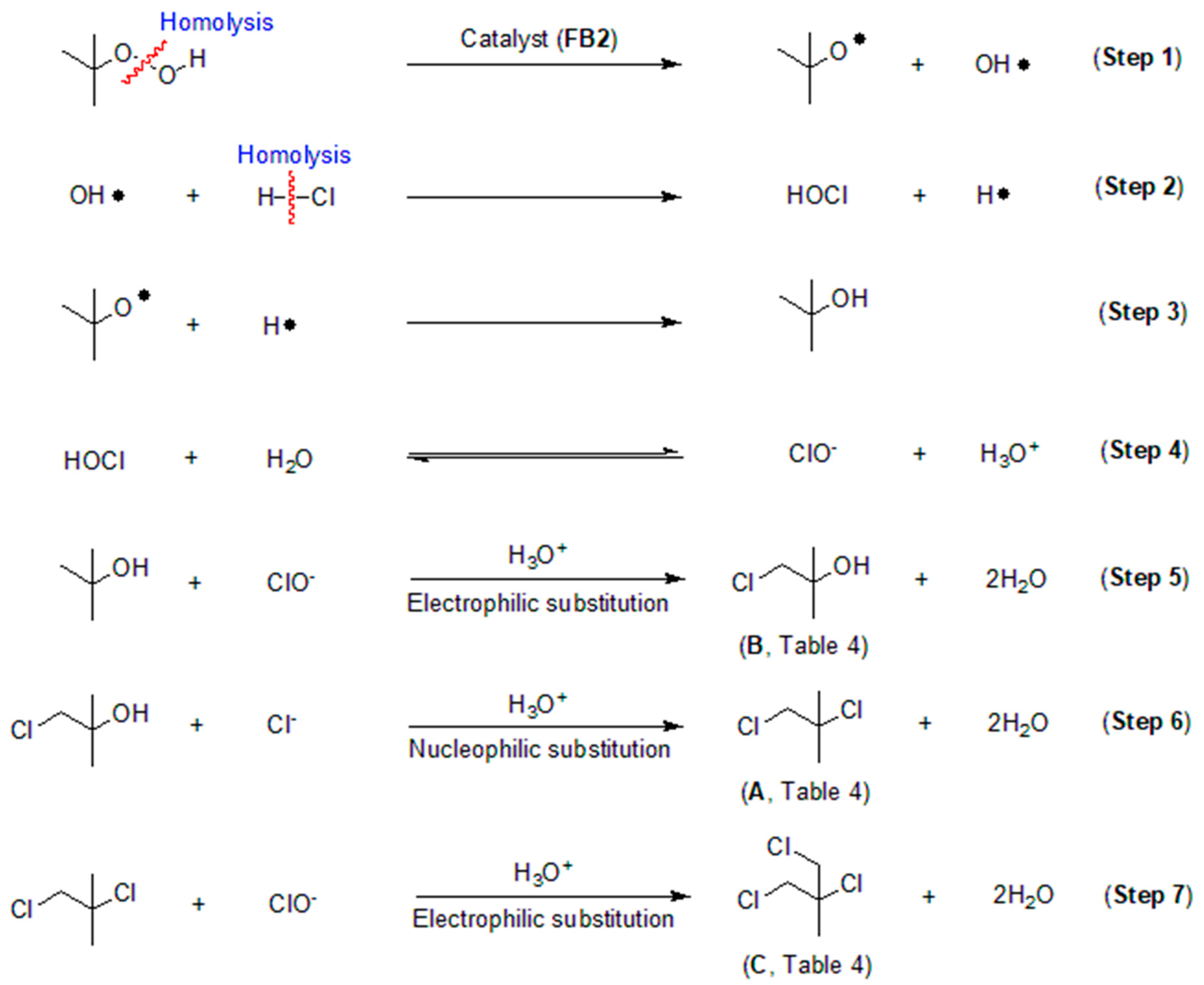
2.4. Proposed Process for Catalytic Chlorination
3. Experimental Section
3.1. Starting Materials
3.2. Analytical Instruments
3.3. Synthesis of Catalysts
3.4. Catalytic Chlorination
4. Conclusions
- (1)
- The synthesized oxide catalyst, composed solely of Fe, primarily consists of Fe2O3, with a small proportion of Fe3O4. This catalyst demonstrates excellent thermal stability when heated from 30 °C to 800 °C and exhibits moderate to high conversions in the chlorination of toluene, benzene, and tert-butyl hydroperoxide. Notably, this catalyst displays the highest ortho-selectivity among the Fe–Ba mixed oxide catalysts in the chlorination of toluene.
- (2)
- The incorporation of Fe3+ salt with BaSO4 in the sol–gel process, followed by calcination at 550 °C, yields a Fe–Ba mixed oxide catalyst composed of BaO2, BaFe4O7, and Fe2O3. This catalyst experiences weight loss when heated to 600 °C or higher, attributed to the thermal reduction of BaO2. It demonstrates significantly higher conversions compared to the pure Fe-containing catalyst in the chlorination of toluene and benzene. Notably, this catalyst produces a tri-chlorinated product compared to the pure Fe-containing catalyst in the chlorination of tert-butyl hydroperoxide (TBHP).
- (3)
- Other Fe–Ba mixed oxide catalysts, including those that do not undergo calcination at 550 °C and those that utilize Fe2+ salt as the raw material in the sol–gel synthesis, exhibit comparatively lower chlorination activity. This reduced performance is likely due to the presence of organic residues in the catalysts, as well as the excessive Fe2+-containing components.
- (4)
- The chlorination of tert-butyl hydroperoxide (TBHP) does not require additional oxidants such as H2O2 or peracetic acid, as TBHP itself serves as both the substrate and the oxidant during the reaction. This process encompasses both electrophilic and nucleophilic substitutions within the catalytic process.
- (5)
- The chlorination of bromobenzene yields chlorobenzene as the sole product, with a yield of 18%. Meanwhile, the chlorination of iodobenzene produces chlorobenzene, 1-chloro-4-iodobenzene, and 1-chloro-2-iodobenzene. To the best of our knowledge, these two reactions have not been previously reported, marking a significant advancement in the field of chlorination and its industrial applications.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
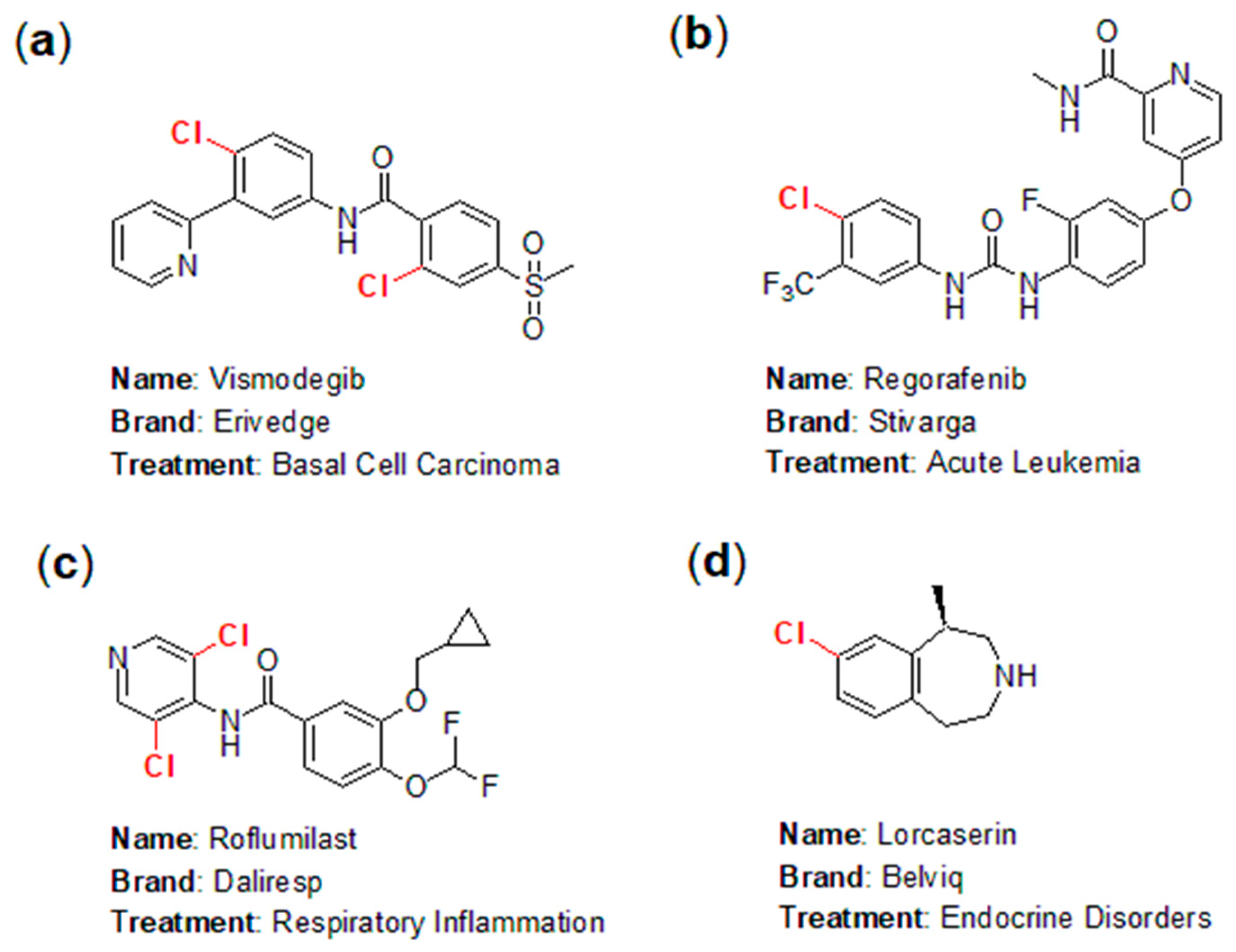
- Fang, W.; Ravindar, L.; Rakesh, K.P.; Manukumar, H.M.; Shantharam, C.S.; Alharbi, N.S.; Qin, H. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. [Google Scholar] [CrossRef] [PubMed]
- Klaus, N. Influence of chlorine substituents on biological activity of chemicals: A review. Pest Manag. Sci. 2000, 56, 3–21. [Google Scholar]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; de Azevedo, D.W.; Leite, A.C.L. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Calienni, M.N.; Febres-Molina, C.; Llovera, R.E.; Zevallos-Delgado, C.; Tuttolomondo, M.E.; Paolino, D.; Fresta, M.; Barazorda-Ccahuana, H.L.; Gómez, B.; Alonso, S.d.V.; et al. Nanoformulation for potential topical delivery of Vismodegib in skin cancer treatment. Int. J. Pharm. 2019, 565, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Wiedey, R.; Hoheisel, W.; Serno, P.; Breitkreutz, J. Impact of co-administered stabilizers on the biopharmaceutical performance of regorafenib amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2021, 169, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Viertelhaus, M.; Holst, H.C.; Volz, J.; Hummel, R. Roflumilast—A reversible single-crystal to single-crystal phase transition at 50 °C. J. Mol. Struct. 2013, 1031, 254–262. [Google Scholar] [CrossRef]
- Jacobs, D.S.; Barkin, C.E.; Kohut, M.R.; Bergman, J.; Kohut, S.J. Effects of lorcaserin (Belviq®) on nicotine- and food-maintained responding in non-human primates. Drug Alcohol. Depend. 2017, 181, 94–101. [Google Scholar] [CrossRef]
- Saito, F.; Zhang, Q.; Kano, J. Mechanochemical dechlorination of waste PVC resin and feedstock recycling. Powder Technol. 2024, 448, 120330. [Google Scholar] [CrossRef]
- Kim, J.; Zhang, G.; Shi, M.; Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 2021, 374, 212–216. [Google Scholar] [CrossRef]
- Jordan, A.; Stoy, P.; Sneddon, H.F. Chlorinated solvents: Their advantages, disadvantages, and alternatives in organic and medicinal chemistry. Chem. Rev. 2021, 121, 1582–1622. [Google Scholar] [CrossRef]
- Liu, M.; Wu, X.; Dyson, P.J. Tandem catalysis enables chlorine-containing waste as chlorination reagents. Nat. Chem. 2024, 16, 700–708. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C−H Activation: Toward sustainability and applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, M.; Sahadev, S. Pollution control of HCl synthesis unit in chloro-alkali industry. Procedia Technol. 2016, 24, 696–703. [Google Scholar] [CrossRef]
- Liu, Y.; Long, Z.; Zeng, J.; Wu, H.; Zhang, W.; Deng, B.; Jiang, T. A DFT study of detection of toxic gases (Cl2, SO2, HCHO, NH3) by SnS2 monolayer modified with Ag2O(1,2) cluster. Surf. Interfaces 2023, 37, 102735. [Google Scholar] [CrossRef]
- Romer, D.R. Synthesis of 2,3-dichloroquinoxalines via Vilsmeier reagent chlorination. J. Heterocycl. Chem. 2009, 46, 317–319. [Google Scholar] [CrossRef]
- Bortoluzzi, M.; Marchetti, F.; Murrali, M.G.; Pampaloni, G. Revisitation of the PCl5-chlorination reaction of a-amino acids: Spectroscopic and DFT insights, and synthesis of the L-proline-derived 2,5-diketopiperazine. Inorg. Chim. Acta 2015, 427, 150–154. [Google Scholar] [CrossRef]
- Chen, J.; Lin, J.-H.; Xiao, J.-C. Halogenation through deoxygenation of alcohols and aldehydes. Org. Lett. 2018, 20, 3061–3064. [Google Scholar] [CrossRef]
- Ben-Daniel, R.; de Visser, S.P.; Shaik, S.; Neumann, R. Electrophilic aromatic chlorination and haloperoxidation of chloride catalyzed by polyfluorinated alcohols: A new manifestation of template catalysis. J. Am. Chem. Soc. 2003, 125, 12116–12117. [Google Scholar] [CrossRef]
- Faxon, C.B.; Allen, D.T. Chlorine chemistry in urban atmospheres: A review. Environ. Chem. 2013, 10, 221–233. [Google Scholar] [CrossRef]
- Galabov, B.; Nalbantova, D.; Schleyer, P.v.R.; Schaefer, H.F., III. Electrophilic aromatic substitution: New insights into an old class of reactions. Acc. Chem. Res. 2016, 49, 1191–1199. [Google Scholar] [CrossRef]
- Konishi, M.; Tsuchida, K.; Sano, K.; Kochi, T.; Kakiuchi, F. Palladium-catalyzed ortho-Selective C−H chlorination of benzamide derivatives under anodic oxidation conditions. J. Org. Chem. 2017, 82, 8716–8724. [Google Scholar] [CrossRef] [PubMed]
- Vinoth, P.; Karuppasamy, M.; Vachan, B.S.; Muthukrishnan, I.; Maheswari, C.U.; Nagarajan, S.; Pace, V.; Roller, A.; Bhuvanesh, N.; Sridharan, V. Palladium-catalyzed regioselective syn-chloropalladation−olefin insertion−oxidative chlorination cascade: Synthesis of dichlorinated tetrahydroquinolines. Org. Lett. 2019, 21, 3465–3469. [Google Scholar] [CrossRef] [PubMed]
- Farshadfar, K.; Tizhoush, S.K.; Ariafard, A. Role of Brønsted acids in promoting Pd(OAc)2-catalyzed chlorination of phenol carbamates using N-chlorosuccinimide. ACS Catal. 2022, 12, 2681–2693. [Google Scholar] [CrossRef]
- Mostafa, M.A.B.; Bowley, R.M.; Racys, D.T.; Henry, M.C.; Sutherland, A. Iron(III)-catalyzed chlorination of activated arenes. J. Org. Chem. 2017, 82, 7529–7537. [Google Scholar] [CrossRef]
- Grell, Y.; Xie, X.; Ivlev, S.I.; Meggers, E. Enantioselective α-fluorination and α-chlorination of N-acyl pyrazoles catalyzed by a non-C2-symmetric chiral-at-rhodium catalyst. ACS Catal. 2021, 11, 11396–11406. [Google Scholar] [CrossRef]
- Dinh, A.N.; Maddox, S.M.; Vaidya, S.D.; Saputra, M.A.; Nalbandian, C.J.; Gustafson, J.L. Catalyst-controlled regioselective chlorination of phenols and anilines through a Lewis basic selenoether catalyst. J. Org. Chem. 2020, 85, 13895–13905. [Google Scholar] [CrossRef]
- Li, D.; Shen, J.; Zhang, J.; Chai, Y.; Xie, Y.; Qiu, C.; Ni, M.; Zheng, Y.; Wang, X.; Zhang, Z. Photocatalytic chlorination of methane using alkali chloride solution. ACS Catal. 2022, 12, 7004–7013. [Google Scholar] [CrossRef]
- Cui, H.; Chen, X. POCl3/sulfoxide and AcCl/sulfoxide mediated chlorination of pyrrolo[2,1-a]isoquinolines. J. Org. Chem. 2023, 88, 11935–11944. [Google Scholar] [CrossRef]
- Dey, A.; Singsardar, M.; Sarkar, R.; Hajra, A. Environment-friendly protocol for the chlorination of imidazoheterocycles by Chloramine-T. ACS Omega 2018, 3, 3513–3521. [Google Scholar] [CrossRef]
- Mahajan, T.; Kumar, L.; Dwivedi, K.; Agarwal, D.D. Efficient and facile chlorination of industrially-important aromatic compounds using NaCl/p-TsOH/NCS in aqueous media. Ind. Eng. Chem. Res. 2012, 51, 3881–3886. [Google Scholar] [CrossRef]
- Brussino, P.; Mehring, E.L.; Ulla, M.A.; Bortolozzi, J.P. Tuning the properties of NiO supported on silicon-aluminum oxides: Influence of the silica amount in the ODH of ethane. Catal. Today 2022, 394–396, 133–142. [Google Scholar] [CrossRef]
- Ali, A.M.; Messaoud, H. Barium sulphate deposits. Energy Procedia 2019, 157, 879–891. [Google Scholar] [CrossRef]
- Hanor, J.S. Barite-celestine geochemistry and environments of formation. Rev. Mineral. Geochem. 2000, 40, 193–275. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.M.; Zhang, L.; Gerritsen, G.; Abbenhuis, H.C.L.; van Santen, R.A.; Li, C. Enantioselective epoxidation of β-methylstyrene catalyzed by immobilized Mn(salen) catalysts in different mesoporous silica supports. J. Catal. 2008, 256, 226–236. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Ramana, C.V.; Bandi, M.; Nair, A.N.; Manciu, F.S.; Sreenivasan, S.; Shutthanandan, V. Electronic structure, chemical bonding, and electrocatalytic activity of Ba(Fe0.7Ta0.3)O3−δ compounds. ACS Appl. Energy Mater. 2021, 4, 1313–1322. [Google Scholar] [CrossRef]
- Kou, Y.; Wang, Y.; Zhang, J.; Guo, K.; Zhang, X.; Yu, Z.; Song, X. Nano BaSO4 prepared by microreactor and its effect on thermal decomposition of some energetics. FirePhysChem 2022, 2, 174–184. [Google Scholar] [CrossRef]
- Haselbach, L.M.; Ma, S. Potential for carbon adsorption on concrete: Surface XPS Analyses. Environ. Sci. Technol. 2008, 42, 5329–5334. [Google Scholar] [CrossRef]
- Kara, Y.S.; Koparal, S.; Tekin, N.; Ömür, N. New approach: Solvent effects of benzaldehyde in aliphatic alcohol solvents with FT-IR spectroscopy and augmented vertex-adjacency matrices. J. Mol. Struct. 2024, 1321, 139815. [Google Scholar] [CrossRef]
- Saravanan, V.; Lakshmanan, P.; Ramalingan, C. Palladium decorated carbon nitride: A pragmatic catalyst for the construction of carbon-carbon double bonds in water. J. Mol. Struct. 2024, 1299, 137097. [Google Scholar] [CrossRef]
- Devipriya, C.P.; Deepa, S.; Udayaseelan, J.; Chandrasekaran, R.; Aravinthraj, M.; Sabari, V. Quantum chemical and MD investigations on molecular structure, vibrational (FT-IR and FT-Raman), electronic, thermal, topological, molecular docking analysis of 1-carboxy-4-ethoxybenzene. Chem. Phys. Impact 2024, 8, 100495. [Google Scholar] [CrossRef]
- Jiang, H.; Bai, L.; Yang, B.; Zeng, S.; Dong, H.; Zhang, X. The effect of protic ionic liquids incorporation on CO2 separation performance of Pebax-based membranes. Chin. J. Chem. Eng. 2022, 43, 169–176. [Google Scholar] [CrossRef]
- Soriano, N.U., Jr.; Migo, V.P.; Matsumura, M. Functional group analysis during ozonation of sunflower oil methyl esters by FT-IR and NMR. Chem. Phys. Lipids 2003, 126, 133–140. [Google Scholar] [CrossRef]
- Li, J.; Hu, J.; Chung, L.; Zhou, J.; Borah, P.; Lin, Z.; Zhong, Y.; Zhou, H.; Yang, X.; Xu, Z.; et al. Insight into stable, concentrated radicals from sulfur-functionalized alkyne-rich crystalline frameworks and application in solar-to-vapor conversion. Chin. J. Struct. Chem. 2024, 43, 100380. [Google Scholar] [CrossRef]
- Kuang, J.; Guo, H.; Si, Q.; Guo, W.; Ma, F. Highly dispersed natural Goethite/Fe3O4 heterogeneous Fenton-like catalyst for efficient tetracycline degradation: Rapid accumulation of hydroxyl radicals by Fe–O–Si bonds. J. Clean. Prod. 2023, 420, 138262. [Google Scholar] [CrossRef]
- Lei, F.; Dyall, A.; AuYeung, N. An in-depth investigation of BaO2/BaO redox oxides for reversible solar thermochemical energy storage. Sol. Energy Mater. Sol. Cells 2021, 223, 110957. [Google Scholar] [CrossRef]
- Amani, V.; Norouzi, F.; Esmaeili, M.; Khavasi, H.R. Effect of substituent position on assembly of compounds bearing ortho/para over meta directing groups. Results Chem. 2024, 10, 101748. [Google Scholar] [CrossRef]
- Solans-Monfort, X.; Fierro, J.L.G.; Hermosilla, L.; Sieiro, C.; Sodupe, M.; Mas-Ballesté, R. O–O Bond activation in H2O2 and (CH3)3C-OOH mediated by [Ni(cyclam)(CH3CN)2](ClO4)2: Different mechanisms to form the same Ni(III) product? Dalton Trans. 2011, 40, 6868–6876. [Google Scholar] [CrossRef]
- Balasubramanian, P.N.; Smith, J.R.L.; Davies, M.J.; Kaaret, T.W.; Bruice, T.C. Dynamics of reaction of (meso-tetrakis(2,6-dimethyl-3-sulfonatophenyl)porphinato)iron(III) hydrate with tert-butyl hydroperoxide in aqueous solution. 2. Establishment of a mechanism that involves homolytic O–O bond breaking and one-electron oxidation of the iron(III) porphyrin. J. Am. Chem. Soc. 1989, 111, 1477–1483. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | C (1s) | O (1s) | Fe (2p) | Ba (3d) | ζ (mV) a |
|---|---|---|---|---|---|
| FB1 | 283.80 (29.49) b | 528.80 (56.38) | 709.80 (14.13) | - c | 0.0531 |
| FB2 | 284.80 (46.23) | 530.80 (51.26) | 710.80 (2.49) | 782.80 (0.02) | −13.3000 |
| FB3 | 284.80 (45.07) | 530.80 (47.28) | 710.80 (7.38) | 782.80 (0.26) | −13.1000 |
| FB4 | 284.80 (42.86) | 531.80 (48.51) | 711.80 (7.90) | 782.80 (0.73) | 0.0070 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry a | Catalyst | Cat. Loading (mol% Fe) c | H2O2 (mmol) | HCl (mmol) | T (°C) | Conversion (%) b | Yield (%) c |
| 1 | blank | 0 | 0.5 | 4 | 60 | 64 | 46 (A), 18 (B) |
| 2 | FB2 | 2 | 0 | 4 | 60 | 0 | 0 |
| 3 | FB2 | 2 | 0 | 4 | 80 | 0 | 0 |
| 4 | FB1 | 2 | 0.5 | 4 | 60 | 79 | 62 (A), 17 (B) |
| 5 | FB2 | 2 | 0.5 | 4 | 60 | 82 | 56 (A), 26 (B) |
| 6 | FB3 | 2 | 0.5 | 4 | 60 | 36 | 19 (A), 17 (B) |
| 7 | FB4 | 2 | 0.5 | 4 | 60 | 63 | 32 (A), 31 (B) |
| 8 | FB2 | 2 | 0.5 | 8 | 60 | 100 | 70 (A), 30 (B) |
| 9 | FB2 | 4 | 0.5 | 4 | 60 | 100 | 73 (A), 27 (B) |
 | ||||
|---|---|---|---|---|
| Entry a | Catalyst | Cat. Loading (mol% Fe) | Conversion (%) b | Yield (%) c |
| 1 | blank | 0 | 83 | 83 |
| 2 | FB1 | 2 | 98 | 98 |
| 3 | FB2 | 2 | 100 | 100 |
| 4 | FB3 | 2 | 90 | 90 |
| 5 | FB4 | 2 | 100 | 100 |
| 6 | FB1 | 4 | 100 | 100 |
| 7 | FB2 | 4 | 100 | 100 |
 | |||
|---|---|---|---|
| Entry a | Catalyst b | Conversion (%) c | Yield (%) d |
| 1 | FB2 | 100 | 62 (A), 27 (B), 11 (C) |
| 2 | FB1 | 100 | 49 (A), 51 (B) |
 | |||
|---|---|---|---|
| Entry a | Initiator | Conversion (%) b | Yield (%) c |
| 1 | H2O2 | 70 | 45 (A), 14 (B), 11 (C) |
| 2 | CH3(C=O)OOH | 59 | 45 (A), 2 (D), 12 (E) |
| Entry a | Substrate | Conversion (%) b | Yield (%) c |
|---|---|---|---|
| 1 | 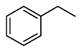 | 4 |  3 3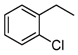 1 1 |
| 2 | 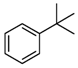 | 21 | 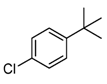 21 21 |
| 3 | 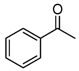 | 7 | 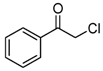 7 7 |
| 4 | 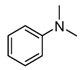 | 34 | 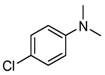 13 13 21 21 |
| 5 | 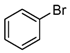 | 18 | 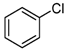 18 18 |
| 6 | 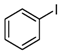 | 5 | 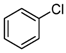 2.5 2.5  1.5 1.5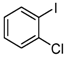 1 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, S.; Pan, Q.; Wu, Y.; Bibi, Z.; Li, X.; Jia, Q.; Sun, Y. The Efficient and Environmentally Friendly Chlorination of Arene, Alcohol, Halobenzene, and Peroxide Catalyzed by Fe–Ba Binary Oxides Using Hydrochloric Acid as Chlorine Source and Aqueous H2O2 as Oxidant. Molecules 2024, 29, 5451. https://doi.org/10.3390/molecules29225451
Chaudhary S, Pan Q, Wu Y, Bibi Z, Li X, Jia Q, Sun Y. The Efficient and Environmentally Friendly Chlorination of Arene, Alcohol, Halobenzene, and Peroxide Catalyzed by Fe–Ba Binary Oxides Using Hydrochloric Acid as Chlorine Source and Aqueous H2O2 as Oxidant. Molecules. 2024; 29(22):5451. https://doi.org/10.3390/molecules29225451
Chicago/Turabian StyleChaudhary, Sidra, Qin Pan, Yong Wu, Zainab Bibi, Xiaoyong Li, Qinxiang Jia, and Yang Sun. 2024. "The Efficient and Environmentally Friendly Chlorination of Arene, Alcohol, Halobenzene, and Peroxide Catalyzed by Fe–Ba Binary Oxides Using Hydrochloric Acid as Chlorine Source and Aqueous H2O2 as Oxidant" Molecules 29, no. 22: 5451. https://doi.org/10.3390/molecules29225451
APA StyleChaudhary, S., Pan, Q., Wu, Y., Bibi, Z., Li, X., Jia, Q., & Sun, Y. (2024). The Efficient and Environmentally Friendly Chlorination of Arene, Alcohol, Halobenzene, and Peroxide Catalyzed by Fe–Ba Binary Oxides Using Hydrochloric Acid as Chlorine Source and Aqueous H2O2 as Oxidant. Molecules, 29(22), 5451. https://doi.org/10.3390/molecules29225451