
Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds


Abstract
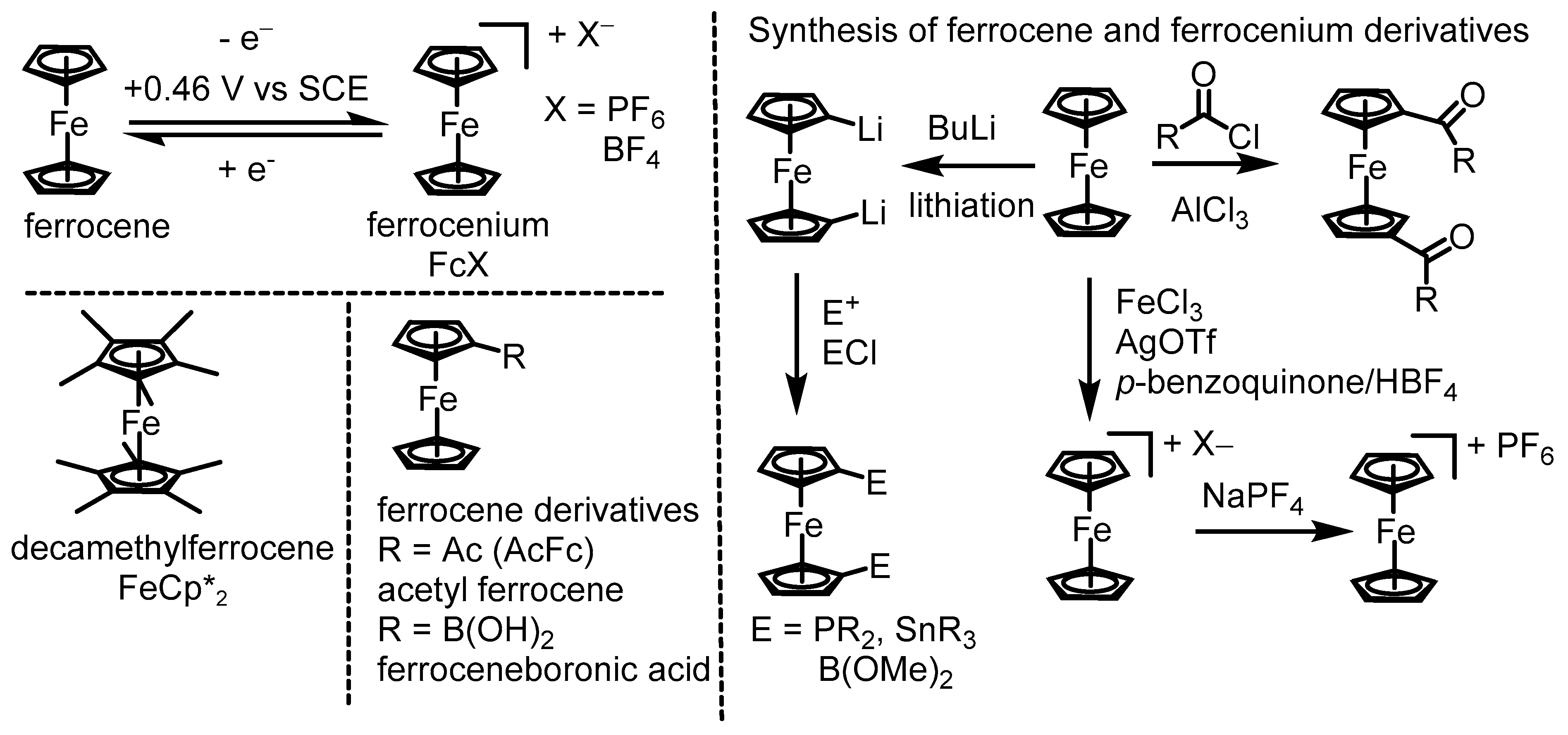
1. Introduction
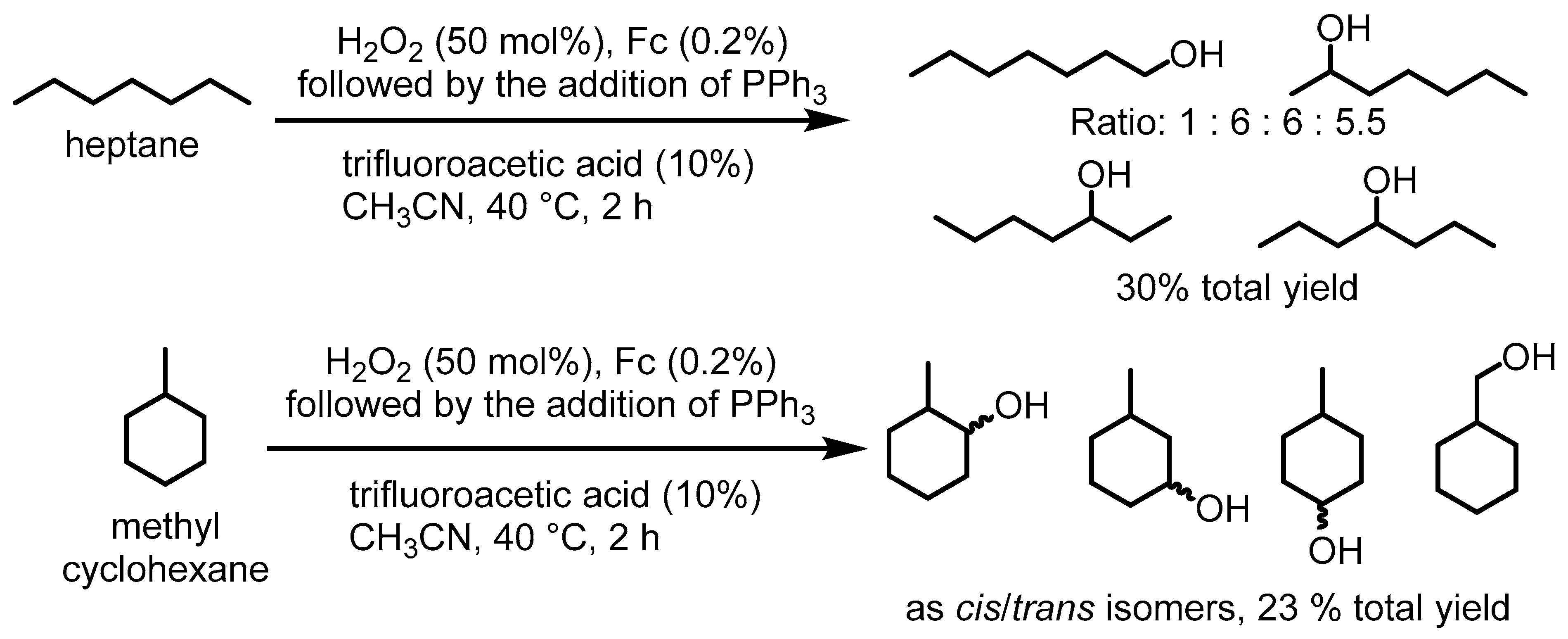
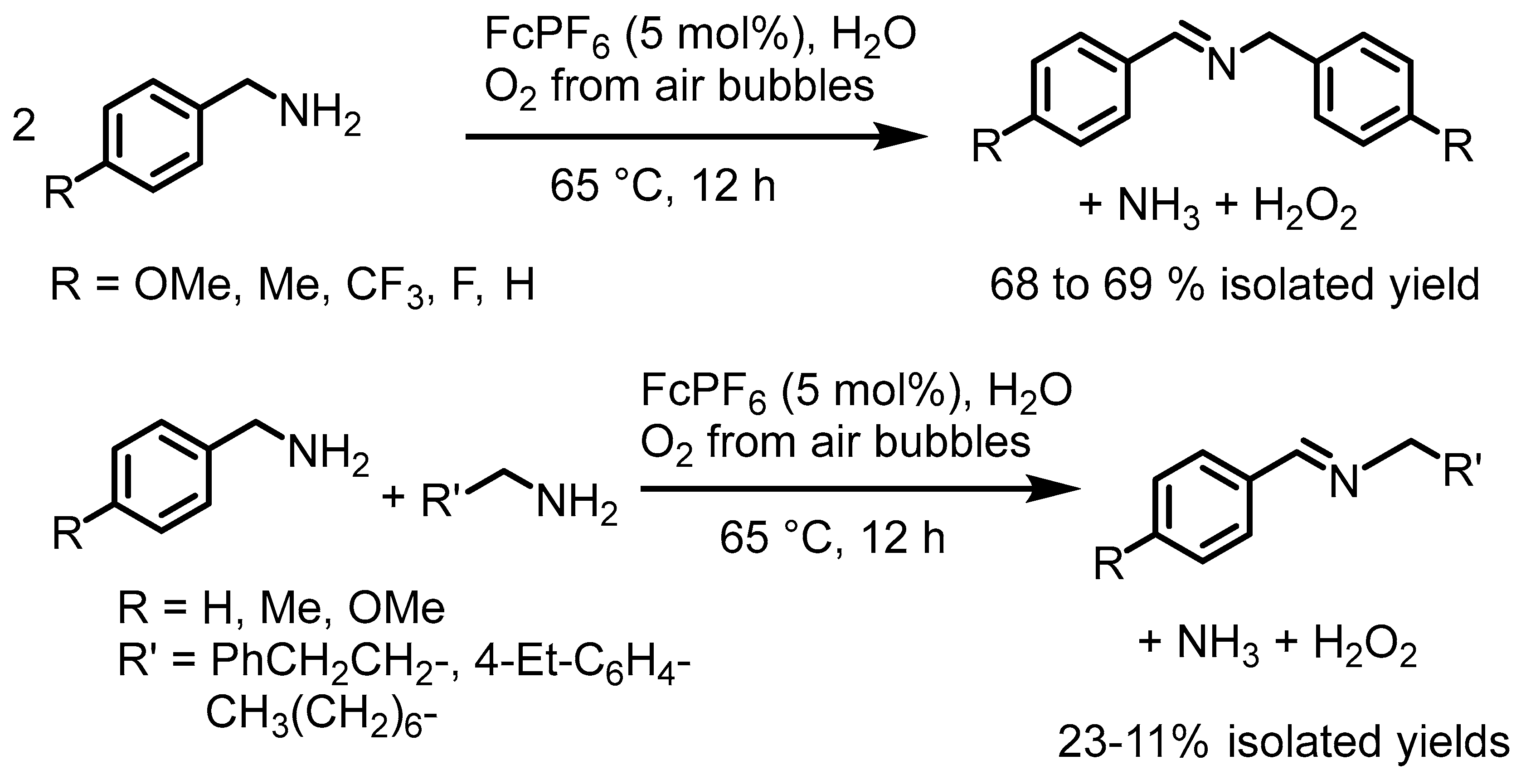
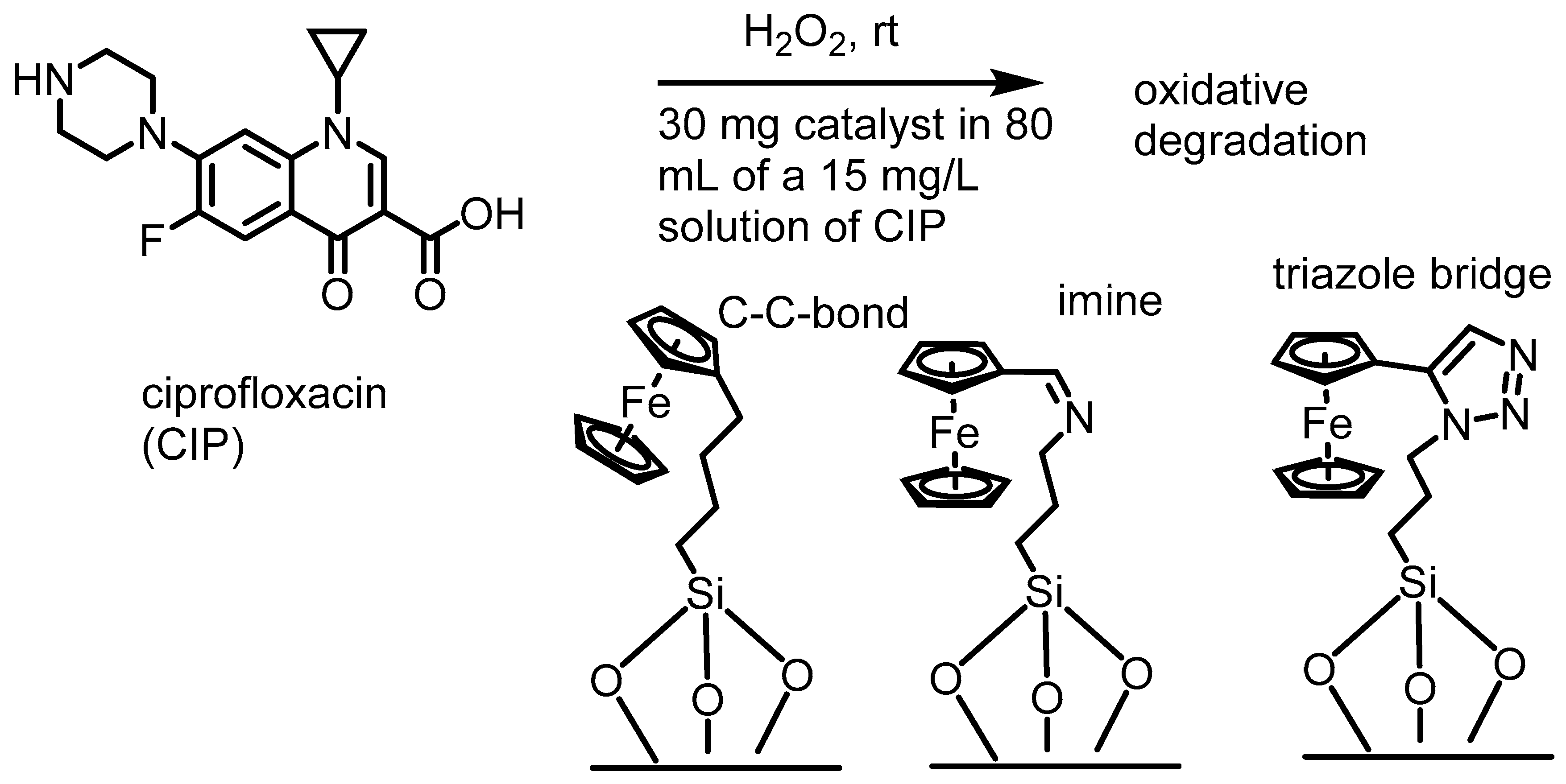
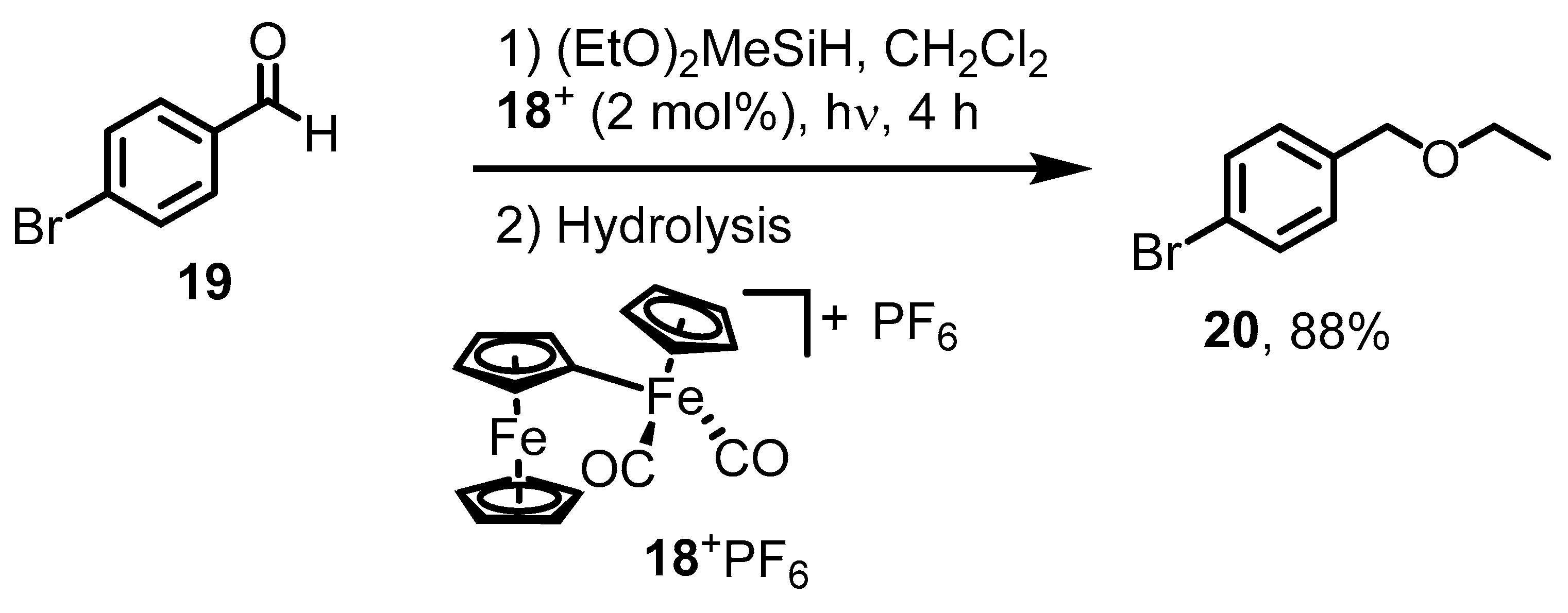
2. Ferrocenium-Catalyzed Oxidation Reactions
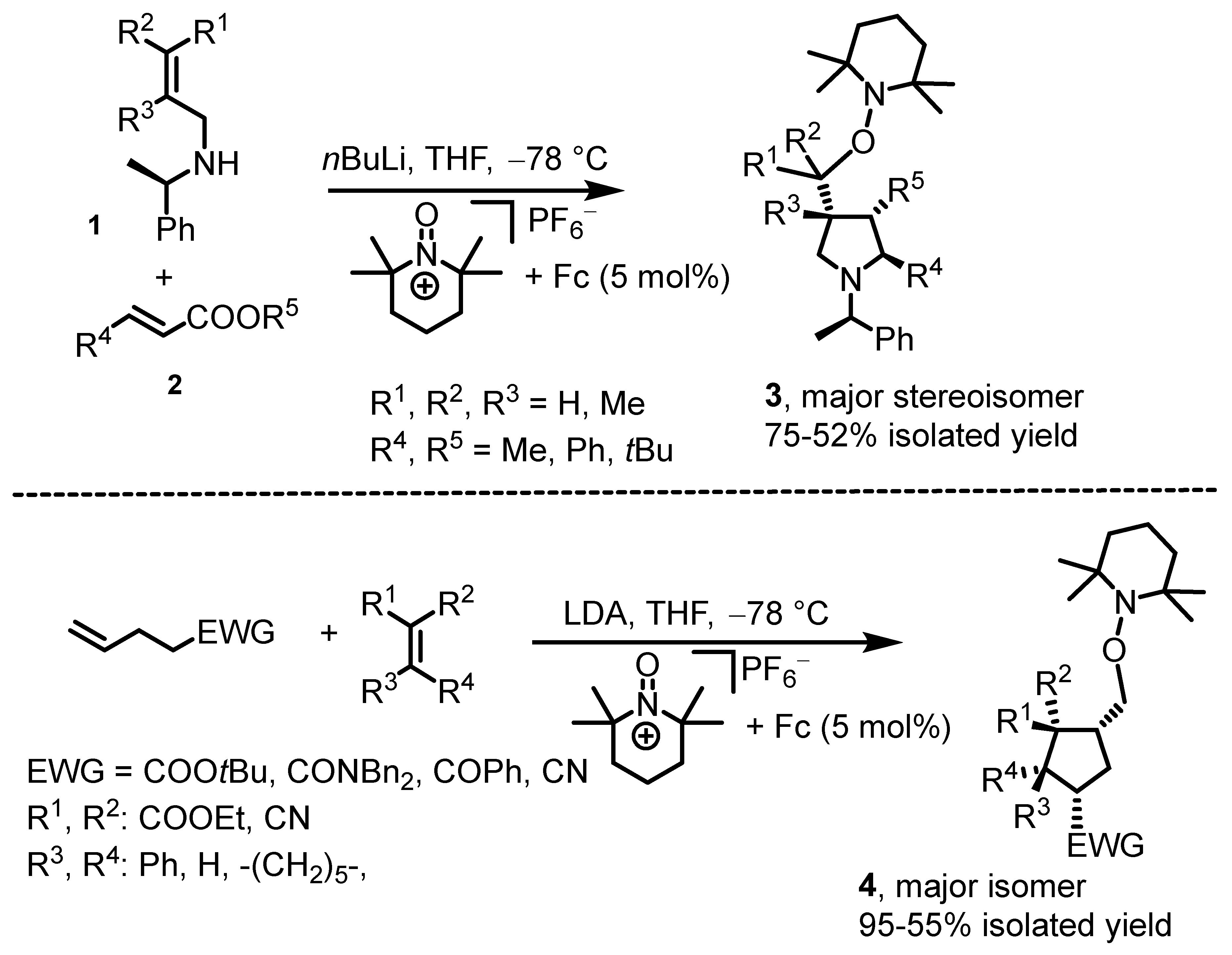
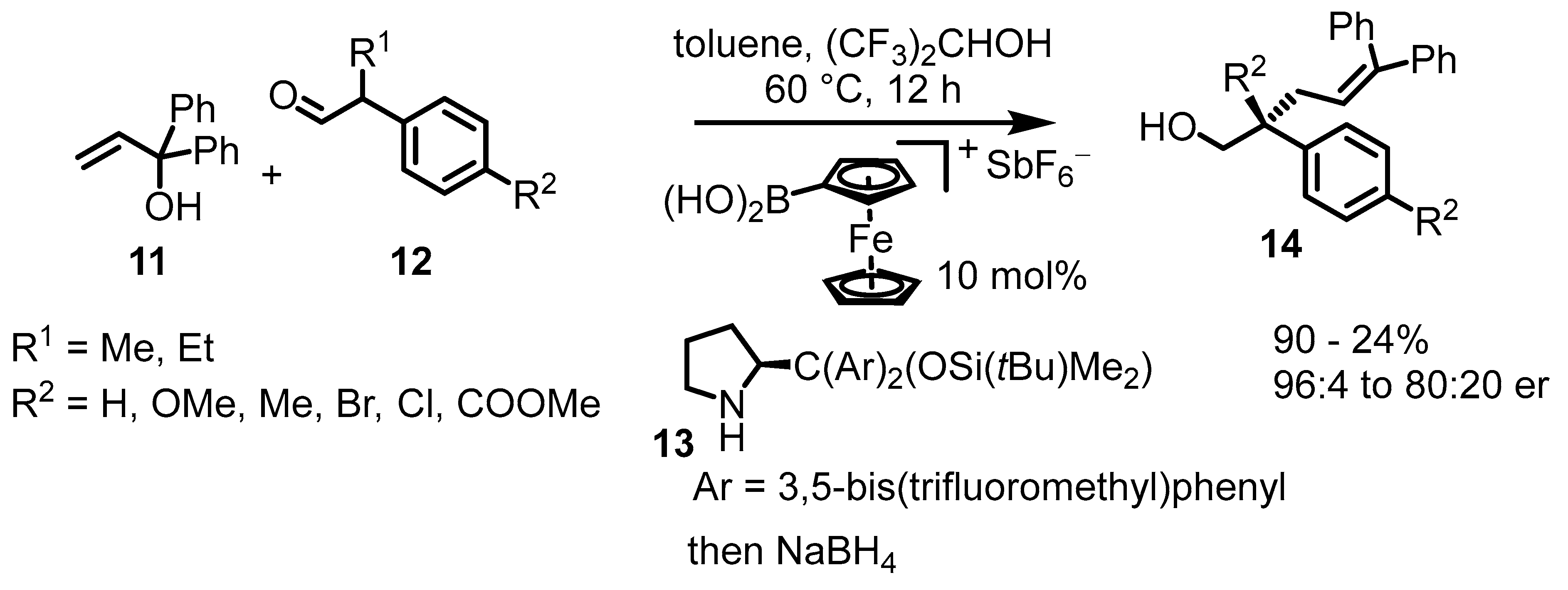
3. Ferrocenium-Catalyzed Coupling Reactions
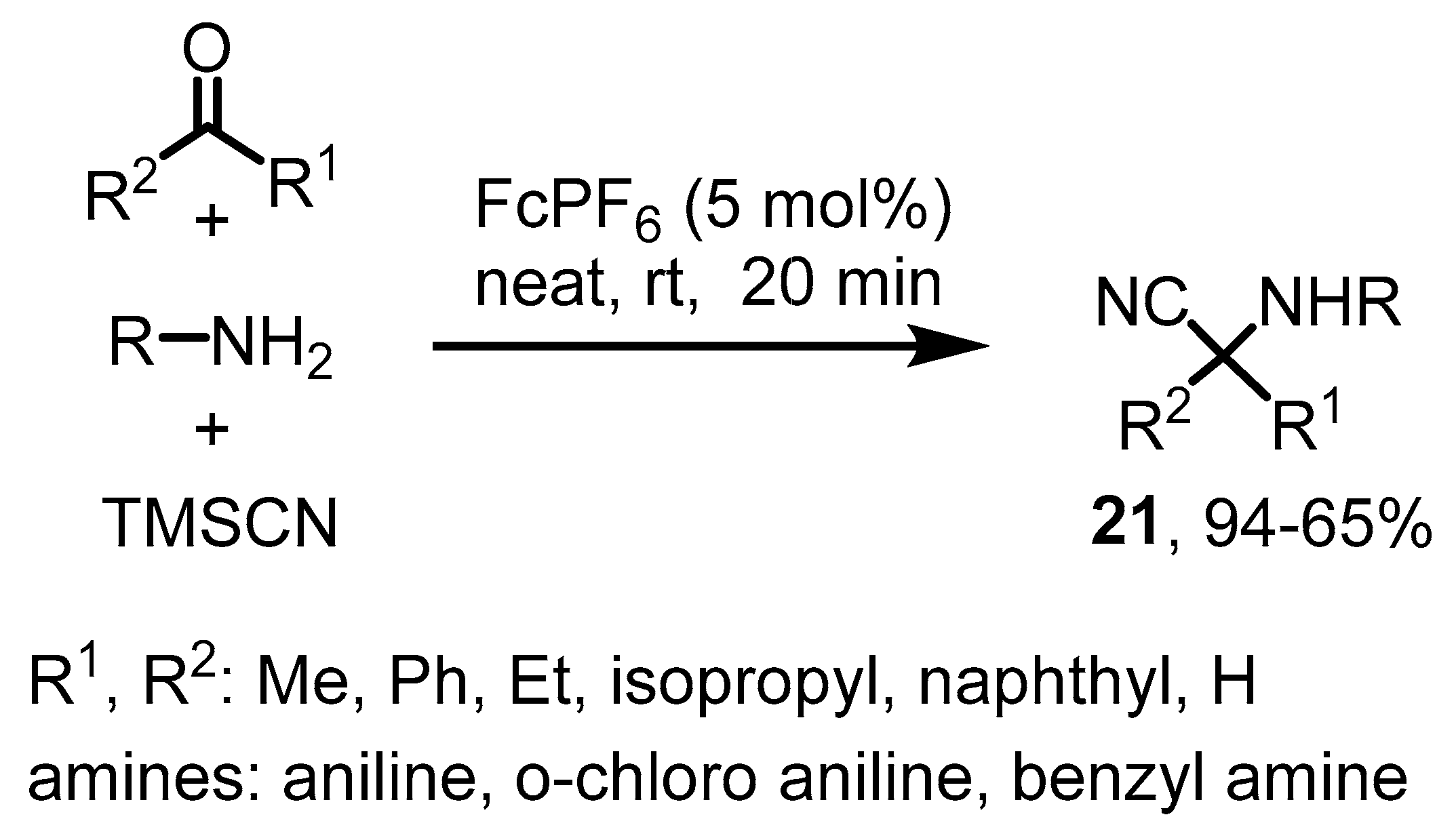
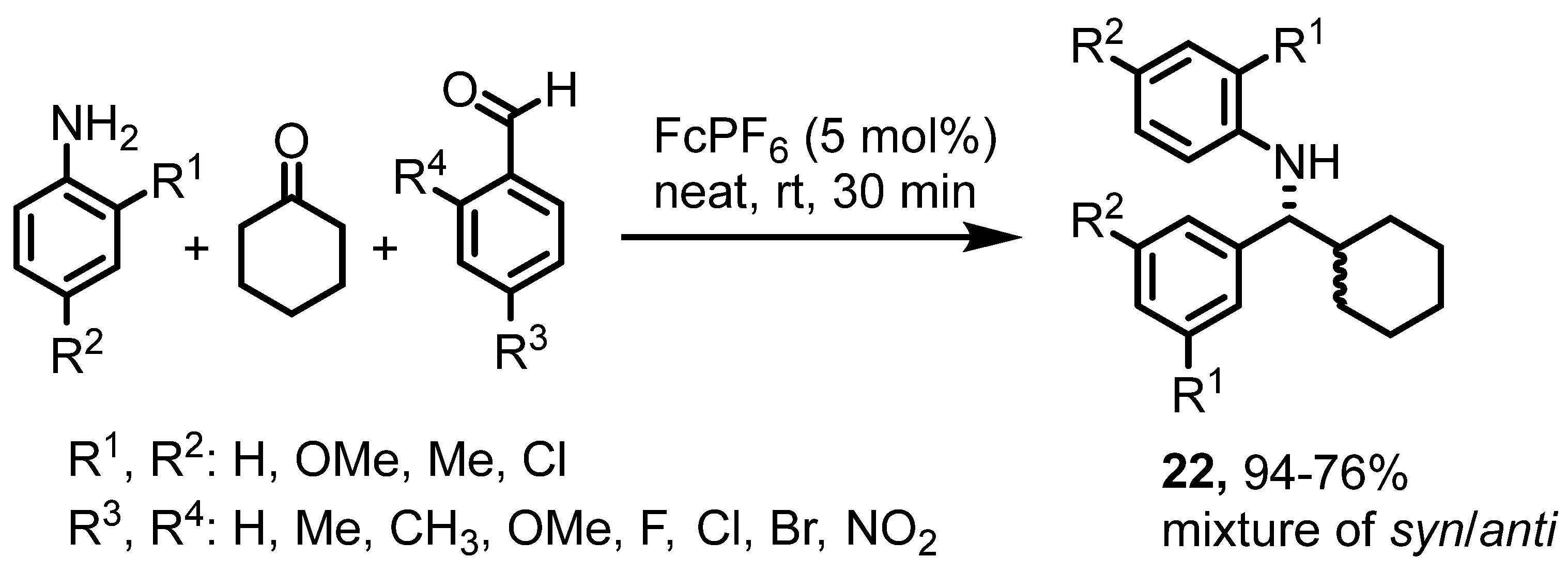
4. Various Reactions Where the Ferrocenium Serves as a Lewis Acid Catalyst
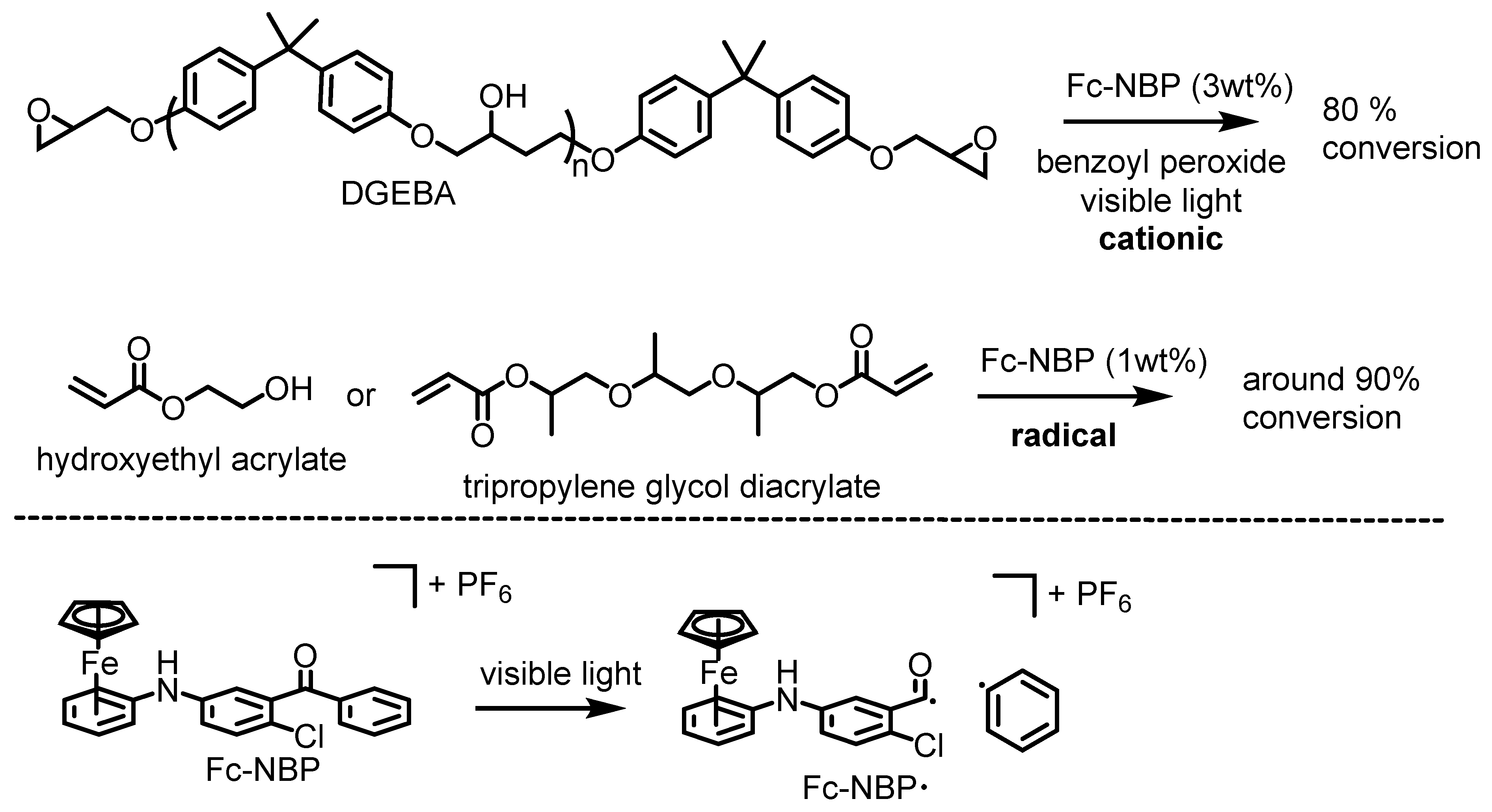
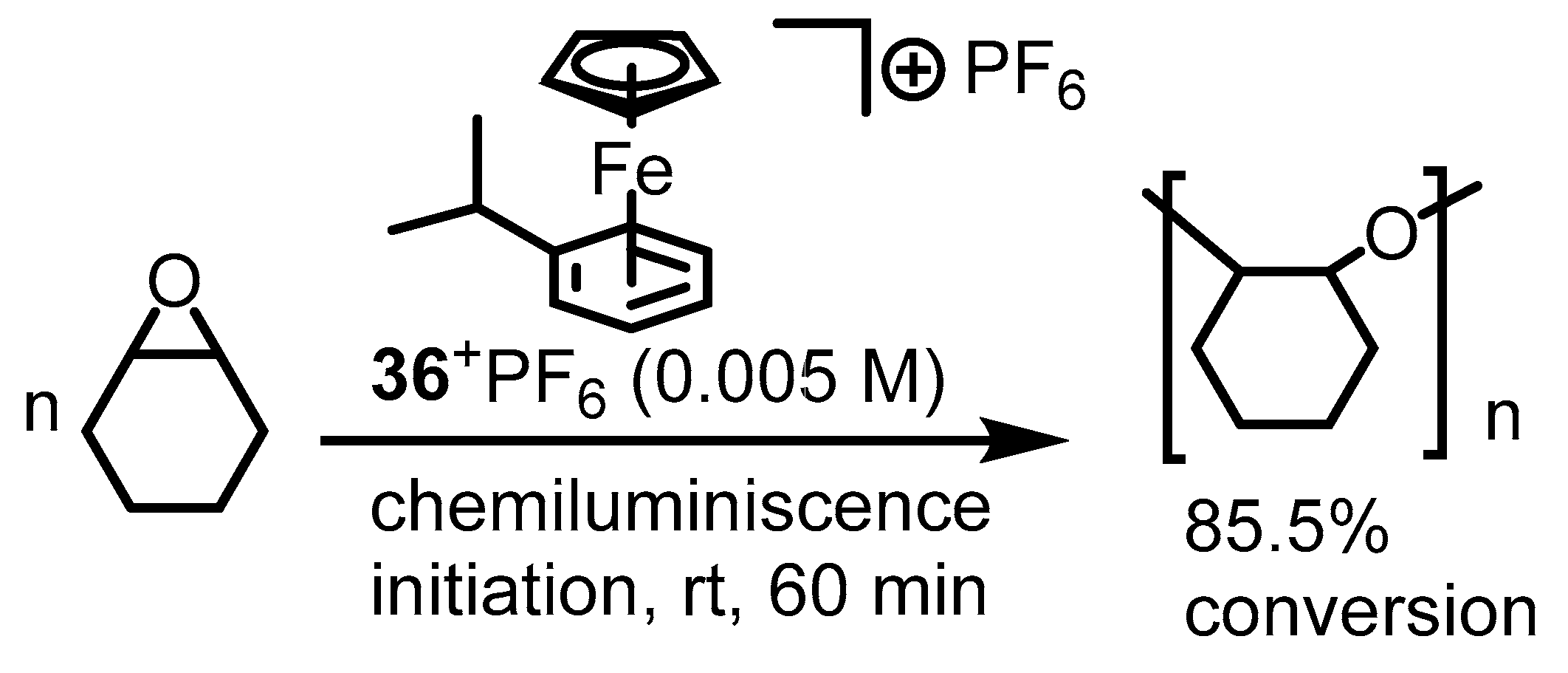
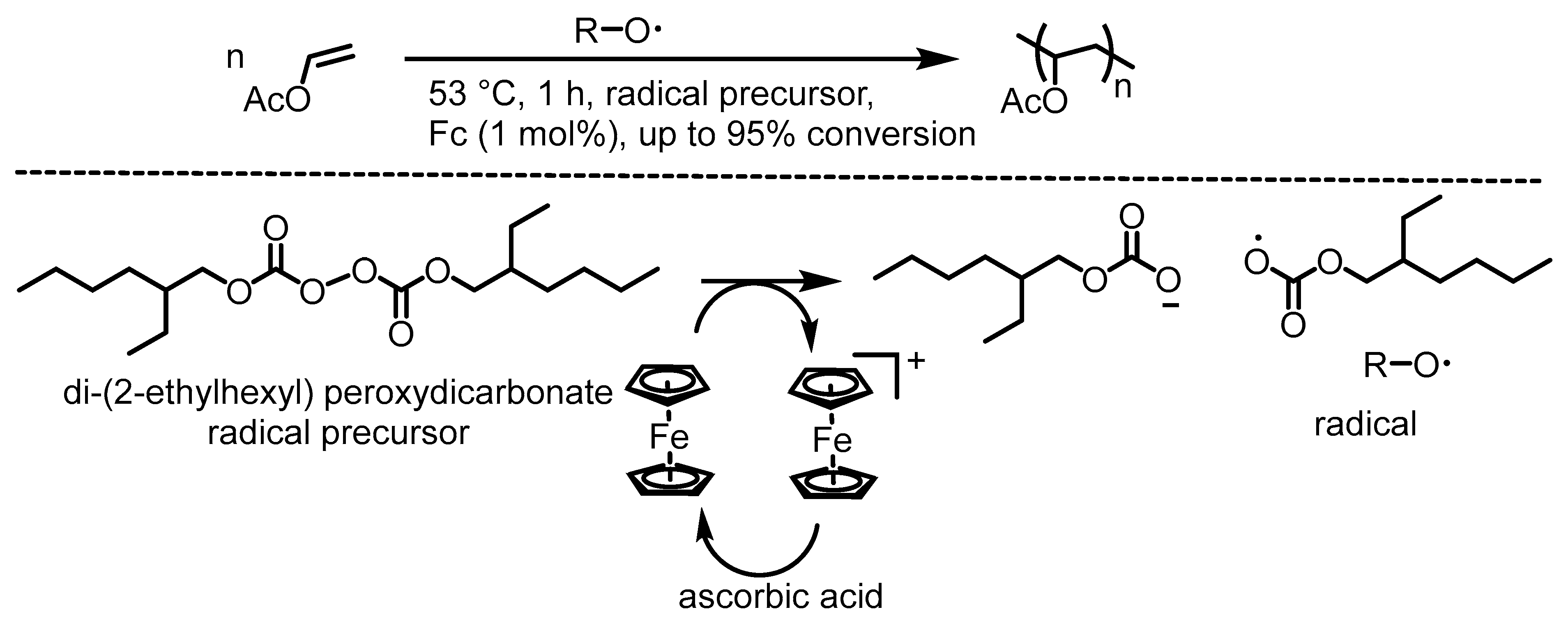
5. Ferrocenium-Catalyzed Oligomerization and Polymerization Reactions
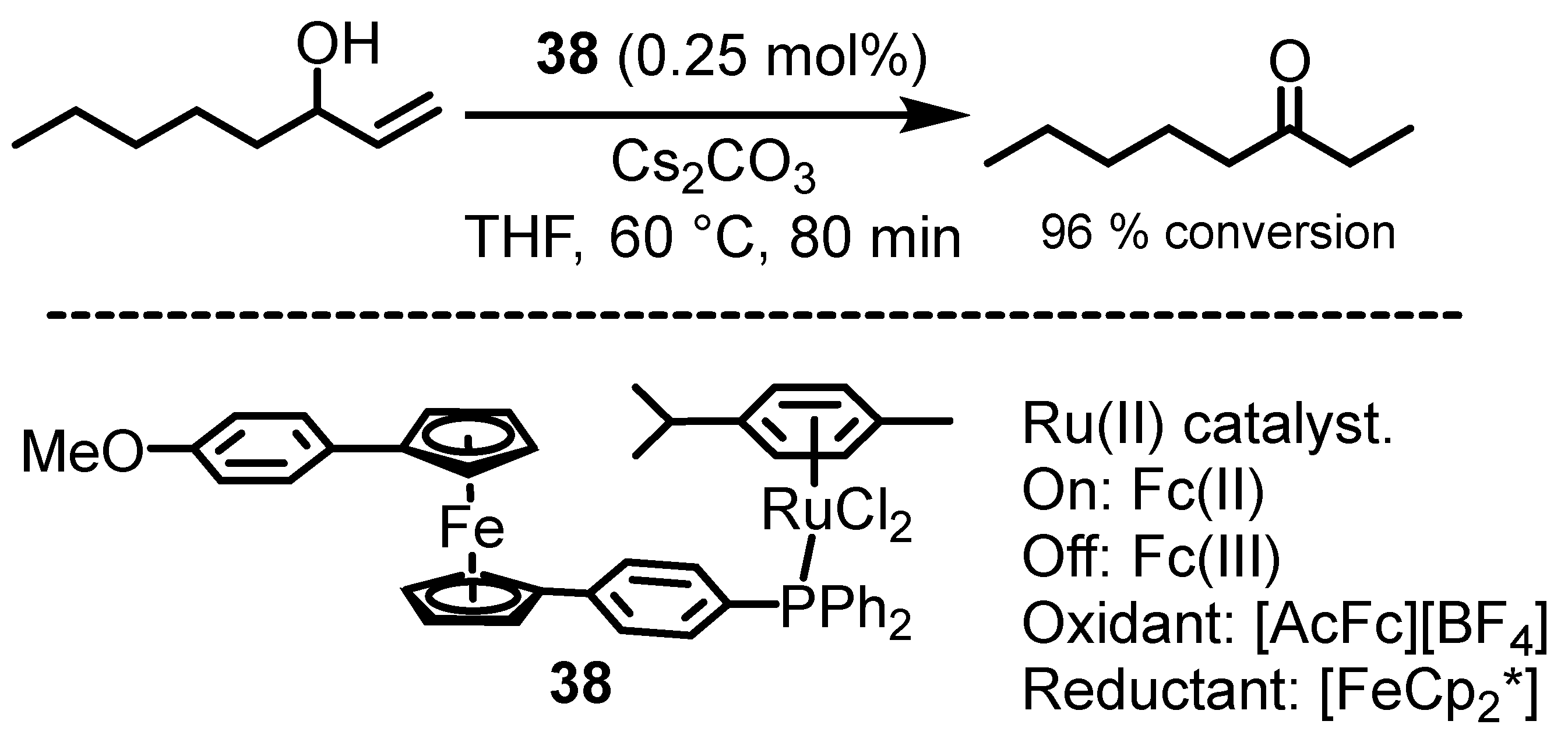
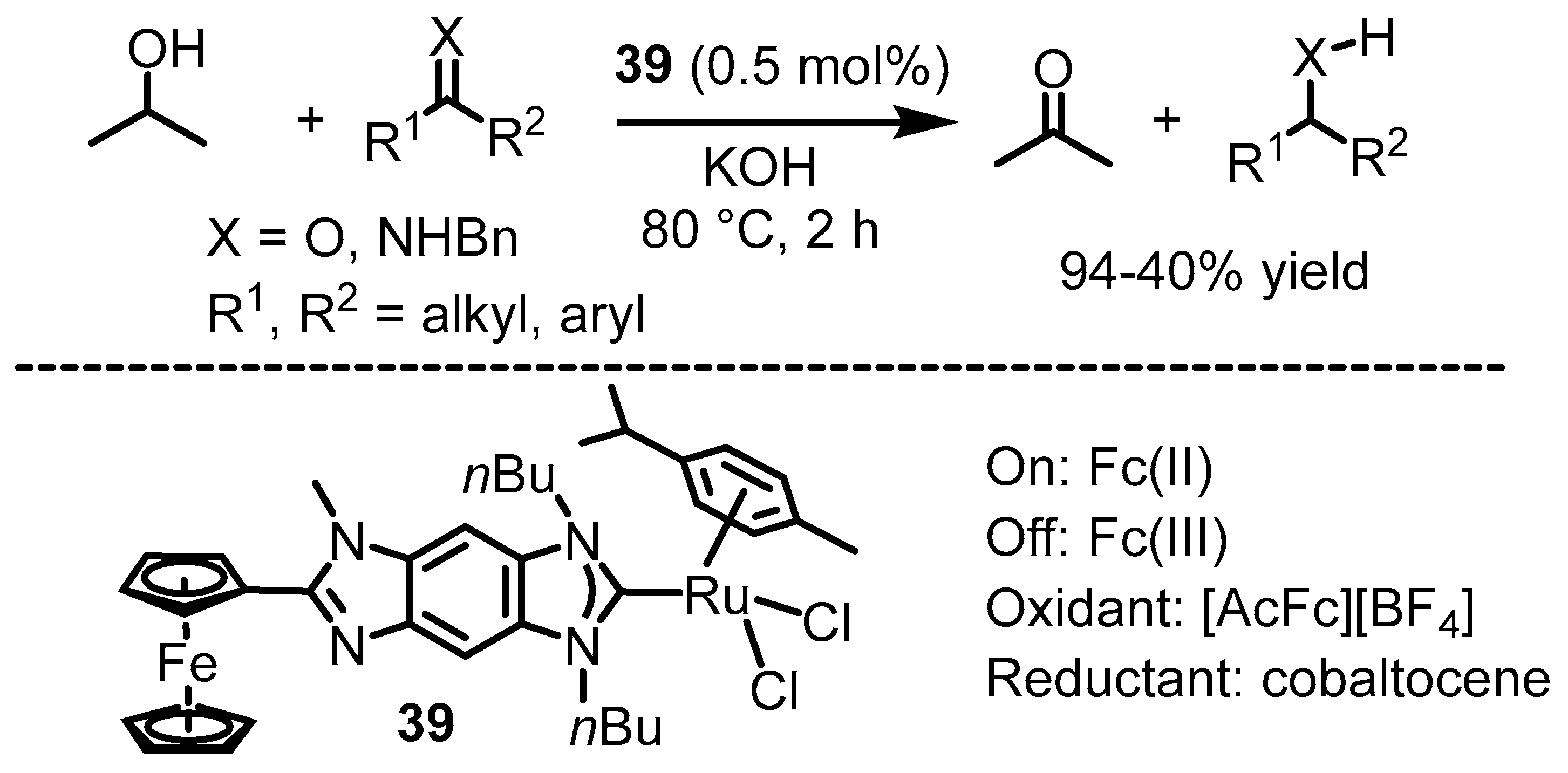
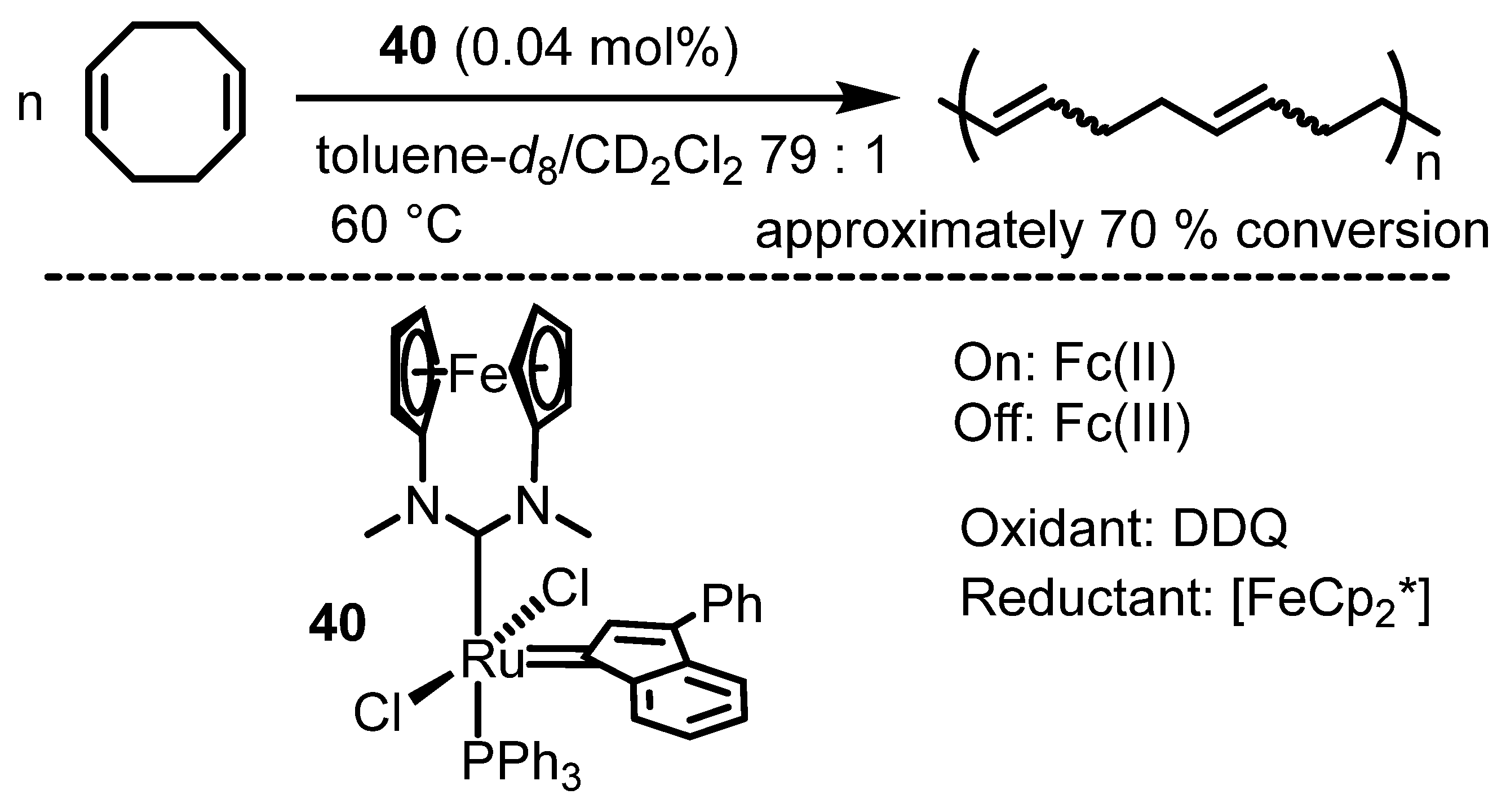
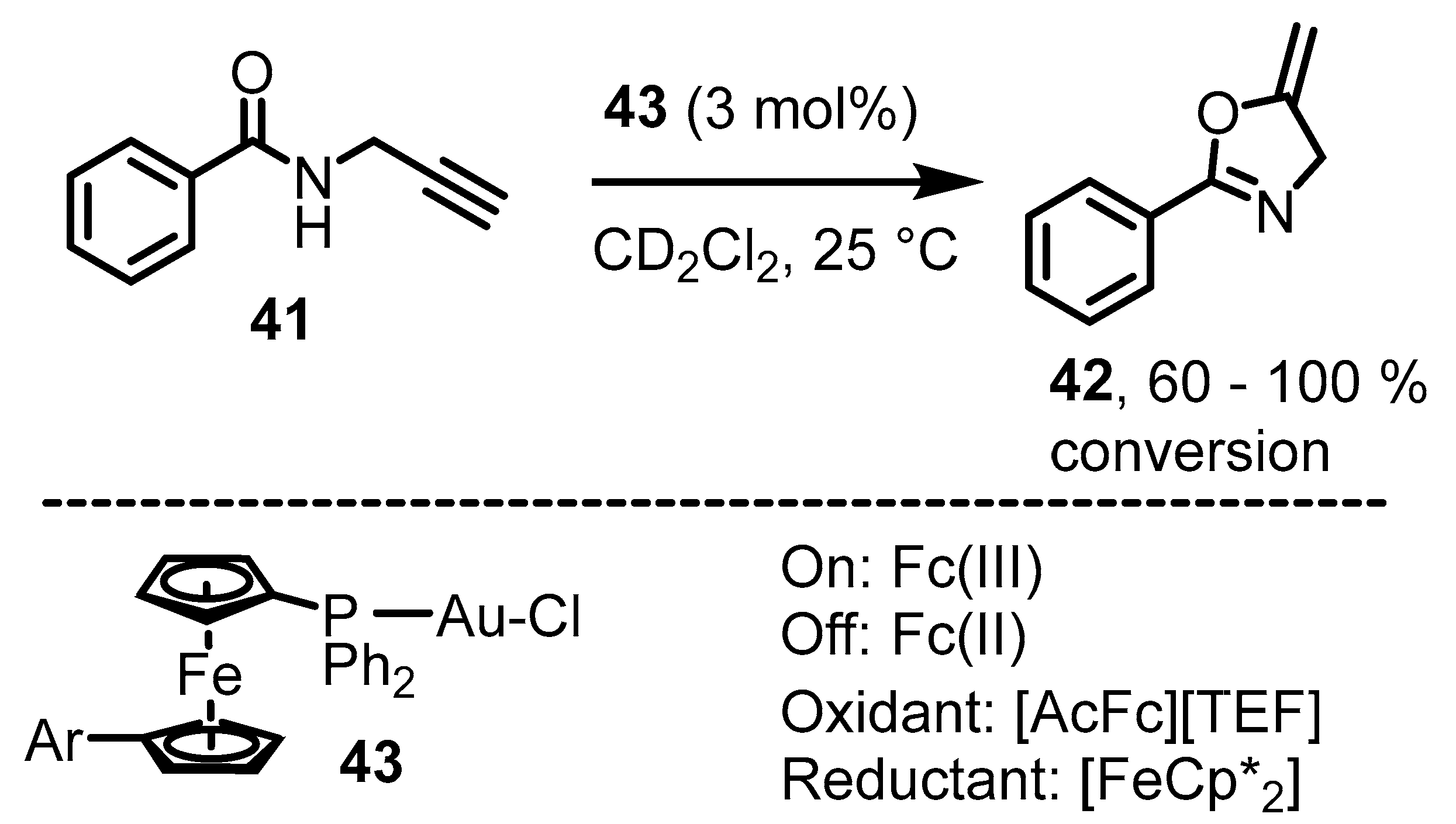
6. Ferrocenium in Redox-Switchable Catalysts
7. Conclusions and Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Štěpnička, P. Forever young: The first seventy years of ferrocene. Dalton Trans. 2017, 51, 8085–8102. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. Why is ferrocene so exceptional? Eur. J. Inorg. Chem. 2017, 2017, 6–29. [Google Scholar] [CrossRef]
- Young, D.J.; Chien, S.W.; Hor, T.A. 1,1′-Bis (diphenylphosphino) ferrocene in functional molecular materials. Dalton Trans. 2012, 41, 12655–12665. [Google Scholar] [CrossRef] [PubMed]
- Miner, N.E.; Nataro, C. Hydroamination and carboxylative cyclization reactions catalyzed by of gold (I) compounds with 1,1′-bis (phosphino) metallocene ligands. J. Organomet. Chem. 2022, 963, 22283. [Google Scholar] [CrossRef]
- Leiby, I.S.; Parparcén, V.; Ding, N.; Kunz, K.J.; Wolfarth, S.A.; Stevens, J.E.; Nataro, C. Cleavage of [Pd2 (PP)2(μ-Cl)2][BArF24]2 (PP= Bis (phosphino) ferrocene, BArF24 = Tetrakis (3, 5-bis (trifluoromethyl) phenyl) borate) with Monodentate Phosphines. Molecules 2024, 29, 2047. [Google Scholar] [CrossRef]
- Weigand, S.; Sünkel, K. 2-Pyridylmetallocenes, Part IX. Sulphur-Substituted 2-Pyridylferrocene: Synthesis and Reactivity towards Pt (II) and Hg (II). Molecules 2024, 29, 4884. [Google Scholar] [CrossRef]
- Atkinson, R.C.; Gibson, V.C.; Long, N.J. The syntheses and catalytic applications of unsymmetrical ferrocene ligands. Chem. Soc. Rev. 2004, 33, 313–328. [Google Scholar] [CrossRef]
- Gómez Arrayás, R.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef]
- Cunningham, L.; Benson, A.; Guiry, P.J. Recent developments in the synthesis and applications of chiral ferrocene ligands and organocatalysts in asymmetric catalysis. Org. Biomol. Chem. 2020, 18, 9329–9370. [Google Scholar] [CrossRef]
- Larik, F.A.; Saeed, A.; Fattah, T.A.; Muqadar, U.; Channar, P.A. Recent advances in the synthesis, biological activities and various applications of ferrocene derivatives. Appl. Organomet. Chem. 2017, 31, 3664. [Google Scholar] [CrossRef]
- Cybulski, M.; Michalak, O.; Buchowicz, W.; Mazur, M. Ansa–Ferrocene Derivatives as Potential Therapeutics. Molecules 2024, 29, 4903. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Correia, J.D.; Kühn, F.E. Ferrocene derivatives as anti-infective agents. Coord. Chem. Rev. 2019, 396, 22–48. [Google Scholar] [CrossRef]
- Khan, A.; Wang, L.; Yu, H.; Haroon, M.; Ullah, R.S.; Nazir, A.; Elshaarani, T.; Usman, M.; Fahad, S.; Haq, F. Research advances in the synthesis and applications of ferrocene-based electro and photo responsive materials. Appl. Organomet. Chem. 2018, 32, 4575. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Toma, Š.; Šebesta, R. Applications of ferrocenium salts in organic synthesis. Synthesis 2015, 47, 1683–1695. [Google Scholar] [CrossRef]
- Lerayer, E.; Radal, L.; Nguyen, T.A.; Dwadnia, N.; Cattey, H.; Amardeil, R.; Pirio, N.; Roger, J.; Hierso, J.C. Highly functionalized ferrocenes. Eur. J. Inorg. Chem. 2020, 419–445. [Google Scholar] [CrossRef]
- Khobragade, D.A.; Mahamulkar, S.G.; Pospíšil, L.; Císařová, I.; Rulíšek, L.; Jahn, U. Acceptor-Substituted Ferrocenium Salts as Strong, Single-Electron Oxidants: Synthesis, Electrochemistry, Theoretical Investigations, and Initial Synthetic Application. Chem. Eur. J. 2012, 18, 12267–12277. [Google Scholar] [CrossRef]
- López, L.A.; Lopez, E. Recent advances in transition metal-catalyzed C–H bond functionalization of ferrocene derivatives. Dalton Trans. 2015, 44, 10128–10135. [Google Scholar] [CrossRef]
- Butler, I.R. The Simple Synthesis of Ferrocene Ligands from a Practitioner’s Perspective. Eur. J. Inorg. Chem. 2012, 4387–4406. [Google Scholar] [CrossRef]
- Nesmeyanov, A.N.; Leonova, E.V.; Kochetkova, N.S.; Malkova, A.I.; Makarovskaya, A.G. 1,3-Diacetylferrocene. J. Organomet. Chem. 1975, 96, 275–278. [Google Scholar] [CrossRef]
- Ravindran, N.A.; Deepak, R.J.; Bhuvanesh, N.; Karvembu, R. Synthesis of substituted quinazolines using Ru (II)-p-cymene catalysts containing ferrocene acylthiourea ligand via acceptorless dehydrogenative coupling. Inorg. Chem. Commun. 2024, 168, 112963. [Google Scholar] [CrossRef]
- Erb, W.; Hurvois, J.P.; Roisnel, T.; Dorcet, V. Ferroceneboronic Acid and Derivatives: Synthesis, Structure, Electronic Properties, and Reactivity in Directed C–H Bond Activation. Organometallics 2018, 37, 3780–3790. [Google Scholar] [CrossRef]
- Bakke, A.A.; Jolly, W.L.; Pinsky, B.L.; Smart, J.C. Unusual Charge Distributions in Some Bis(fulvalene)dimetal complexes As Revealed by X-ray Photoelectron Spectroscopy. Inorg. Chem. 1979, 18, 1343–1345. [Google Scholar] [CrossRef]
- Curley, J.J.; Bergman, R.G.; Tilley, T.D. Preparation and physical properties of early-late heterobimetallic compounds featuring Ir–M bonds (M = Ti, Zr, Hf). Dalton Trans. 2012, 41, 192–200. [Google Scholar] [CrossRef]
- Bradley, S.; Camm, K.D.; Liu, X.; McGowan, P.C.; Mumtaz, R.; Oughton, K.A.; Podesta, T.J.; Thornton-Pett, M. Synthesis and Structural Studies of 1,1′-Bis-Amino-Functionalized Ferrocenes, Ferrocene Salts, and Ferrocenium Salts. Inorg. Chem. 2002, 41, 715–726. [Google Scholar] [CrossRef]
- Martinez, R.; Tiripicchio, A. Structure of ferrocenium hexafluorophosphate. Acta Cryst. 1990, C46, 202–205. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar]
- Rana, S.; Biswas, J.P.; Paul, S.; Paik, A.; Maiti, D. Organic synthesis with the most abundant transition metal–iron: From rust to multitasking catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar]
- Wei, D.; Darcel, C. Iron catalysis in reduction and hydrometalation reactions. Chem. Rev. 2018, 119, 2550–2610. [Google Scholar] [CrossRef]
- Bauer, E.B. Iron Catalysis: Historic Overview and Current Trends. In Iron Catalysis II; Bauer, E., Ed.; Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2015; Volume 50, pp. 1–18. [Google Scholar]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C−O Bond Activation: Opportunity for Sustainable Catalysis. ChemSusChem 2017, 10, 3964–3981. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, N.; Decavoli, C.; Boldrini, C.L.; Coluccini, C.; Abbotto, A. Ferrocene derivatives functionalized with donor/acceptor (Hetero) Aromatic substituents: Tuning of redox properties. Energies 2020, 13, 3937. [Google Scholar] [CrossRef]
- Maity, R.; Birenheide, B.S.; Breher, F.; Sarkar, B. Cooperative effects in multimetallic complexes applied in catalysis. ChemCatChem 2021, 13, 2337–2370. [Google Scholar] [CrossRef]
- Bauer, E.B. Recent advances in iron catalyzed oxidation reactions of organic compounds. Isr. J. Chem. 2017, 57, 1131–1150. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nakajima, K.; Nishibayashi, Y. Phosphine Oxidation with Water and Ferrocenium (III) Cation Induced by Visible-Light Irradiation. Chem. Eur. J. 2018, 24, 18618–18622. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, Z.J.; Xu, H.C. Ferrocene as a Redox Catalyst for Organic Electrosynthesis. Isr. J. Chem. 2024, 64, e202300097. [Google Scholar] [CrossRef]
- Gozzo, F. Radical and non-radical chemistry of the Fenton-like systems in the presence of organic substrates. J. Mol. Catal. A Chem. 2001, 171, 1–22. [Google Scholar] [CrossRef]
- Shul’pina, L.S.; Kudinov, A.R.; Mandelli, D.; Carvalho, W.A.; Kozlov, Y.N.; Vinogradov, M.M.; Ikonnikov, N.S.; Shul’pin, G.B. Oxidation of alkanes and benzene with hydrogen peroxide catalyzed by ferrocene in the presence of acids. J. Organomet. Chem. 2015, 793, 217–231. [Google Scholar] [CrossRef]
- Deb, M.; Hazra, S.; Dolui, P.; Elias, A.J. Ferrocenium promoted oxidation of benzyl amines to imines using water as the solvent and air as the oxidant. ACS Sustainable Chem. Eng. 2018, 7, 479–486. [Google Scholar] [CrossRef]
- Tijani, J.O.; Fatoba, O.O.; Madzivire, G.; Petrik, L.F. A review of combined advanced oxidation technologies for the removal of organic pollutants from water. Water Air Soil. Pollut. 2014, 225, 2102. [Google Scholar] [CrossRef]
- Babuponnusami, A.; Muthukumar, K. A review on Fenton and improvements to the Fenton process for wastewater treatment. J. Environ. Chem. Eng. 2014, 2, 557–572. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, H.; Zhao, X.; Zhai, Q.; Yin, D.; Sun, Y.; Li, J. The marriage of ferrocene and silicotungstate: An ingenious heterogeneous Fenton-like synergistic photocatalyst. Appl. Catal. B 2016, 193, 47–57. [Google Scholar] [CrossRef]
- Walkowiak, A.; Wolski, L.; Ziolek, M. The influence of ferrocene anchoring method on the reactivity and stability of SBA-15-based catalysts in the degradation of ciprofloxacin via photo-Fenton process. RSC Adv. 2023, 13, 8360–8373. [Google Scholar] [CrossRef] [PubMed]
- Amin, B.U.; Yu, H.; Wang, L.; Nazir, A.; Fahad, S.; Haq, F.; Mahmood, S.; Liang, R.; Uddin, M.A.; Lin, T. Recent advances on ferrocene-based compounds and polymers as a burning rate catalysts for propellants. J. Organomet. Chem. 2020, 921, 121368. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, L.; Yu, H.; Tong, R.; Chen, Q.; Wang, J.; Yang, X.; Saleem, M. Progress on the synthesis and catalytic and anti-migration properties of ferrocene-based burning rate catalysts. Appl. Organometal. Chem. 2016, 30, 796–805. [Google Scholar] [CrossRef]
- Tong, R.; Zhao, Y.; Wang, L.; Yu, H.; Ren, F.; Saleem, M.; Amer, W.A. Recent research progress in the synthesis and properties of burning rate catalysts based on ferrocene-containing polymers and derivatives. J. Organomet. Chem. 2014, 755, 16–32. [Google Scholar] [CrossRef]
- Sun, F.; Li, Z.; Xu, H.; Fu, Y.; Li, H.; Yao, Y.; Ren, L.; He, X.; Li, Y.; Yang, R.; et al. Exploring Structural Evolution Behaviors of Ligand-Defect-Rich Ferrocene-Based Metal-Organic Frameworks for Electrochemical Oxygen Evolution via Operando X-Ray Absorption Spectroscopy. Adv. Energy Mater. 2024, 14, 2400875. [Google Scholar] [CrossRef]
- Liang, J.; Gao, X.; Guo, B.; Ding, Y.; Yan, J.; Guo, Z.; Tse, E.C.; Liu, J. Ferrocene-based metal–organic framework nanosheets as a robust oxygen evolution catalyst. Angew. Chem. Int. Ed. 2021, 60, 2770–12774. [Google Scholar] [CrossRef]
- Wertz, S.; Leifert, D.; Studer, A. Cross dehydrogenative coupling via base-promoted homolytic aromatic substitution (BHAS): Synthesis of fluorenones and xanthones. Org. Lett. 2013, 15, 928–931. [Google Scholar] [CrossRef]
- Corbet, J.P.; Mignani, G. Selected patented cross-coupling reaction technologies. Chem. Rev. 2006, 106, 2651–2710. [Google Scholar] [CrossRef]
- Buskes, M.J.; Blanco, M.J. Impact of cross-coupling reactions in drug discovery and development. Molecules 2020, 25, 3493. [Google Scholar] [CrossRef] [PubMed]
- Drusan, M.; Šebesta, R. Enantioselective C–C and C–heteroatom bond forming reactions using chiral ferrocene catalysts. Tetrahedron 2014, 70, 759–786. [Google Scholar] [CrossRef]
- Kafka, F.; Holan, M.; Hidasova, D.; Pohl, R.; Císařová, I.; Klepetářová, B.; Jahn, U. Oxidative Catalysis Using the Stoichiometric Oxidant as a Reagent: An Efficient Strategy for Single-Electron-Transfer-Induced Tandem Anion–Radical Reactions. Angew. Chem. Int. Ed. 2014, 53, 9944–9948. [Google Scholar] [CrossRef] [PubMed]
- Foo, K.; Sella, E.; Thomé, I.; Eastgate, M.D.; Baran, P.S. A mild, ferrocene-catalyzed C–H imidation of (hetero) arenes. J. Am. Chem. Soc. 2014, 136, 5279–5282. [Google Scholar] [CrossRef]
- Mo, X.; Yakiwchuk, J.; Dansereau, J.; McCubbin, J.A.; Hall, D.G. Unsymmetrical diarylmethanes by ferroceniumboronic acid catalyzed direct Friedel–Crafts reactions with deactivated benzylic alcohols: Enhanced reactivity due to ion-pairing effects. J. Am. Chem. Soc. 2015, 137, 9694–9703. [Google Scholar] [CrossRef]
- Ang, H.T.; Rygus, J.P.; Hall, D.G. Two-component boronic acid catalysis for increased reactivity in challenging Friedel–Crafts alkylations with deactivated benzylic alcohols. Org. Biomol. Chem. 2019, 17, 6007–6014. [Google Scholar] [CrossRef]
- Mo, X.; Hall, D.G. Dual catalysis using boronic acid and chiral amine: Acyclic quaternary carbons via enantioselective alkylation of branched aldehydes with allylic alcohols. J. Am. Chem. Soc. 2016, 138, 10762–10765. [Google Scholar] [CrossRef]
- Zhang, Q.; Cui, X.; Zhang, L.; Luo, S.; Wang, H.; Wu, Y. Redox tuning of a direct asymmetric aldol reaction. Angew. Chem. Int. Ed. 2015, 54, 5210–5213. [Google Scholar] [CrossRef]
- Argouarch, G.; Grelaud, G.; Roisnel, T.; Humphrey, M.G.; Paul, F. [Fp*Fc][PF6]: A remarkable non-symmetric dinuclear cation in a very stable mixed-valent state. J. Organomet. Chem. 2017, 847, 218–223. [Google Scholar] [CrossRef]
- Noor-ul, H.K.; Agrawal, S.; Kureshy, R.I.; Abdi, S.H.; Singh, S.; Suresh, E.; Jasra, R.V. Fe(Cp)2PF6 catalyzed efficient Strecker reactions of ketones and aldehydes under solvent-free conditions. Tetrahedron Lett. 2008, 49, 640–644. [Google Scholar]
- Kureshy, R.I.; Agrawal, S.; Saravanan, S.; Noor-ul, H.K.; Shah, A.K.; Abdi, S.H.; Bajaj, H.C.; Suresh, E. Direct Mannich reaction mediated by Fe(Cp)2PF6 under solvent-free conditions. Tetrahedron Lett. 2010, 51, 489–494. [Google Scholar] [CrossRef]
- Yadav, G.D.; Singh, S. Ring opening of epoxides with alcohols using Fe(Cp)2BF4 as catalyst. Tetrahedron Lett. 2014, 55, 3979–3983. [Google Scholar] [CrossRef]
- Yadav, G.D.; Chauhan, M.S.; Singh, S. Fe(Cp)2BF4: An efficient Lewis acid catalyst for the aminolysis of epoxides. Synthesis 2014, 46, 629–634. [Google Scholar]
- Queensen, M.J.; Rabus, J.M.; Bauer, E.B. Ferrocenium hexafluorophosphate as an inexpensive, mild catalyst for the etherification of propargylic alcohols. J. Mol. Catal. A Chem. 2015, 407, 221–229. [Google Scholar] [CrossRef]
- Talasila, D.S.; Queensen, M.J.; Barnes-Flaspoler, M.; Jurkowski, K.; Stephenson, E.; Rabus, J.M.; Bauer, E.B. Ferrocenium Cations as Catalysts for the Etherification of Cyclopropyl-Substituted Propargylic Alcohols: Ene-yne Formation and Mechanistic Insights. Eur. J. Org. Chem. 2019, 7348–7358. [Google Scholar] [CrossRef]
- Burcher, B.; Breuil, P.A.R.; Magna, L.; Olivier-Bourbigou, H. Iron-catalyzed oligomerization and polymerization Reactions. In Iron Catalysis II; Bauer, E., Ed.; Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2015; Volume 50, pp. 217–258. [Google Scholar]
- Wang, T.; Huang, Y.L. Cyclopentadiene—Iron—Naphthalene tetrafluoroborate salt as a cationic photoinitiator for preparing photosensitive materials. Imaging Sci. J. 2003, 51, 247–253. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.H. Cationic photopolymerization of epoxy systems initiated by cyclopentadien-iron-biphenyl hexafluorophosphate ([Cp-Fe-biphenyl]+PF6−). Polym. Bull. 2005, 53, 323–331. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Simon-Masseron, A.; Lalevée, J. Radical photoinitiation with LEDs and applications in the 3D printing of composites. Chem. Soc. Rev. 2021, 50, 3824–3841. [Google Scholar] [CrossRef]
- Dietlin, C.; Schweizer, S.; Xiao, P.; Zhang, J.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P.; Lalevée, J. Photopolymerization upon LEDs: New photoinitiating systems and strategies. Polym. Chem. 2015, 6, 3895–3912. [Google Scholar] [CrossRef]
- Wang, T.; Chen, J.W.; Li, Z.Q.; Wan, P.Y. Several ferrocenium salts as efficient photoinitiators and thermal initiators for cationic epoxy polymerization. J. Photochem. Photobiol. A 2007, 187, 389–394. [Google Scholar] [CrossRef]
- Li, M.; Chen, Y.; Zhang, H.; Wang, T. A novel ferrocenium salt as visible light photoinitiator for cationic and radical photopolymerization. Prog. Org. Coat. 2010, 68, 234–239. [Google Scholar] [CrossRef]
- Wang, T.; Li, B.S.; Zhang, L.X. Carbazole-bound ferrocenium salt as an efficient cationic photoinitiator for epoxy polymerization. Polym. Int. 2005, 54, 1251–1255. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, D.; Dumur, F.; Xiao, P.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. The carbazole-bound ferrocenium salt as a specific cationic photoinitiator upon near-UV and visible LEDs (365–405 nm). Polym. Bull. 2016, 73, 493–507. [Google Scholar] [CrossRef]
- Liu, X.; Luo, Y.; Chen, Y.; Wu, S.; Li, Z. From Glow to Growth: Chemiluminescence Induced Cationic Polymerization Using Ferrocenium Salt. Macromol. Chem. Phys. 2023, 224, 2300048. [Google Scholar] [CrossRef]
- Shan, J.; Yang, Z.; Chen, G.; Hu, Y.; Luo, Y.; Dong, X.; Zheng, W.; Zhou, W. Design and synthesis of free-radical/cationic photosensitive resin applied for 3D printer with liquid crystal display (LCD) irradiation. Polymers 2020, 12, 1346. [Google Scholar] [CrossRef]
- Zouhri, Y.; Berthé, J.M.M.; Bonnet, F.; Stasik, B.; Colemonts, C.; Lasuye, T.; Mortreux, A.; Champouret, Y.; Visseaux, M. Ferrocene/ferrocenium redox shuttling: Peroxide decomposition catalysis applied to vinyl monomers polymerization under suspension conditions. Polymer 2022, 260, 125368. [Google Scholar] [CrossRef]
- Fujimura, K.; Ouchi, M.; Sawamoto, M. Ferrocene cocatalysis in metal-catalyzed living radical polymerization: Concerted redox for highly active catalysis. ACS Macro Lett. 2012, 1, 321–323. [Google Scholar] [CrossRef]
- Fujimura, K.; Ouchi, M.; Sawamoto, M. Ferrocene cocatalysis for iron-catalyzed living radical polymerization: Active, robust, and sustainable system under concerted catalysis by two iron complexes. Macromolecules 2015, 48, 4294–4300. [Google Scholar] [CrossRef]
- Ghorbani-Choghamarani, A.; Taherinia, Z. Recent advances utilized in artificial switchable catalysis. RSC Advances 2022, 12, 23595–23617. [Google Scholar] [CrossRef]
- Blanco, V.; Leigh, D.A.; Marcos, V. Artificial switchable catalysts. Chem. Soc. Rev. 2015, 44, 5341–5370. [Google Scholar] [CrossRef]
- Luca, O.R.; Crabtree, R.H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikova, S.V.; Poddel’sky, A.I. Heteroligand Metal Complexes with Extended Redox Properties Based on Redox-Active Chelating Ligands of o-Quinone Type and Ferrocene. Molecules 2022, 27, 3928. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Calle, S.; Miller, A.J. Tunable and switchable catalysis enabled by cation-controlled gating with crown ether ligands. Acc. Chem. Res. 2023, 56, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizzi, L. The ferrocenium/ferrocene couple: A versatile redox switch. ChemTexts 2020, 6, 22. [Google Scholar] [CrossRef]
- Wei, J.; Diaconescu, P.L. Redox-switchable ring-opening polymerization with ferrocene derivatives. Acc. Chem. Res. 2019, 52, 415–424. [Google Scholar] [CrossRef]
- Lorkovic, I.M.; Duff Jr, R.R.; Wrighton, M.S. Use of the redox-active ligand 1,1′-bis (diphenylphosphino) cobaltocene to reversibly alter the rate of the rhodium (I)-catalyzed reduction and isomerization of ketones and alkenes. J. Am. Chem. Soc. 1995, 117, 3617–3618. [Google Scholar] [CrossRef]
- Slone, C.S.; Mirkin, C.A.; Yap, G.P.; Guzei, I.A.; Rheingold, A.L. Oxidation-state-dependent reactivity and catalytic properties of a Rh (I) complex formed from a redox-switchable hemilabile ligand. J. Am. Chem. Soc. 1997, 119, 10743–10753. [Google Scholar] [CrossRef]
- Neumann, P.; Dib, H.; Caminade, A.M.; Hey-Hawkins, E. Redox control of a dendritic ferrocenyl-based homogeneous catalyst. Angew. Chem. Int. Ed. 2015, 54, 311–314. [Google Scholar] [CrossRef]
- Ibáñez, S.; Poyatos, M.; Peris, E. A Ferrocenyl-Benzo-Fused Imidazolylidene Complex of Ruthenium as Redox-Switchable Catalyst for the Transfer Hydrogenation of Ketones and Imines. ChemCatChem 2016, 8, 3790–3795. [Google Scholar] [CrossRef]
- Varnado, C.D.; Rosen, E.L.; Collins, M.S.; Lynch, V.M.; Bielawski, C.W. Synthesis and study of olefin metathesis catalysts supported by redox-switchable diaminocarbene [3]-ferrocenophanes. Dalton Trans. 2013, 42, 13251–13264. [Google Scholar] [CrossRef]
- Straube, A.; Coburger, P.; Dütsch, L.; Hey-Hawkins, E. Triple the fun: Tris (ferrocenyl) arene-based gold (I) complexes for redox-switchable catalysis. Chem. Sci. 2020, 11, 10657–10668. [Google Scholar] [CrossRef] [PubMed]
- Broderick, E.M.; Guo, N.; Vogel, C.S.; Xu, C.; Sutter, J.; Miller, J.T.; Meyer, K.; Mehrkhodavandi, P.; Diaconescu, P.L. Redox control of a ring-opening polymerization catalyst. J. Am. Chem. Soc. 2011, 133, 9278–9281. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Diaconescu, P.L. Reactivity and Properties of Metal Complexes Enabled by Flexible and Redox-Active Ligands with a Ferrocene Backbone. Inorg. Chem. 2016, 55, 10013–10023. [Google Scholar] [CrossRef] [PubMed]
- Shepard, S.M.; Diaconescu, P.L. Redox-switchable hydroelementation of a cobalt complex supported by a ferrocene-based ligand. Organometallics 2016, 35, 2446–2453. [Google Scholar] [CrossRef]
- Bora, D.; Ali, A.A.; Saha, B. A redox switchable ferrocene decorated n-heterocyclic carbene (NHC) palladium catalyst for cross coupling of arylboronic acid and acetic anhydride in phosphine, base and additive free conditions. New J. Chem. 2024, 48, 615–620. [Google Scholar] [CrossRef]
- Jurkowski, K.; Bauer, E.B. Ferrocenium Ions as Catalysts: Decomposition Studies and Counteranion Influence on Catalytic Activity. Synthesis 2021, 53, 2007–2014. [Google Scholar]
- Hurvois, J.P.; Moinet, C. Reactivity of ferrocenium cations with molecular oxygen in polar organic solvents: Decomposition, redox reactions and stabilization. J. Organomet. Chem. 2005, 690, 1829–1839. [Google Scholar] [CrossRef]



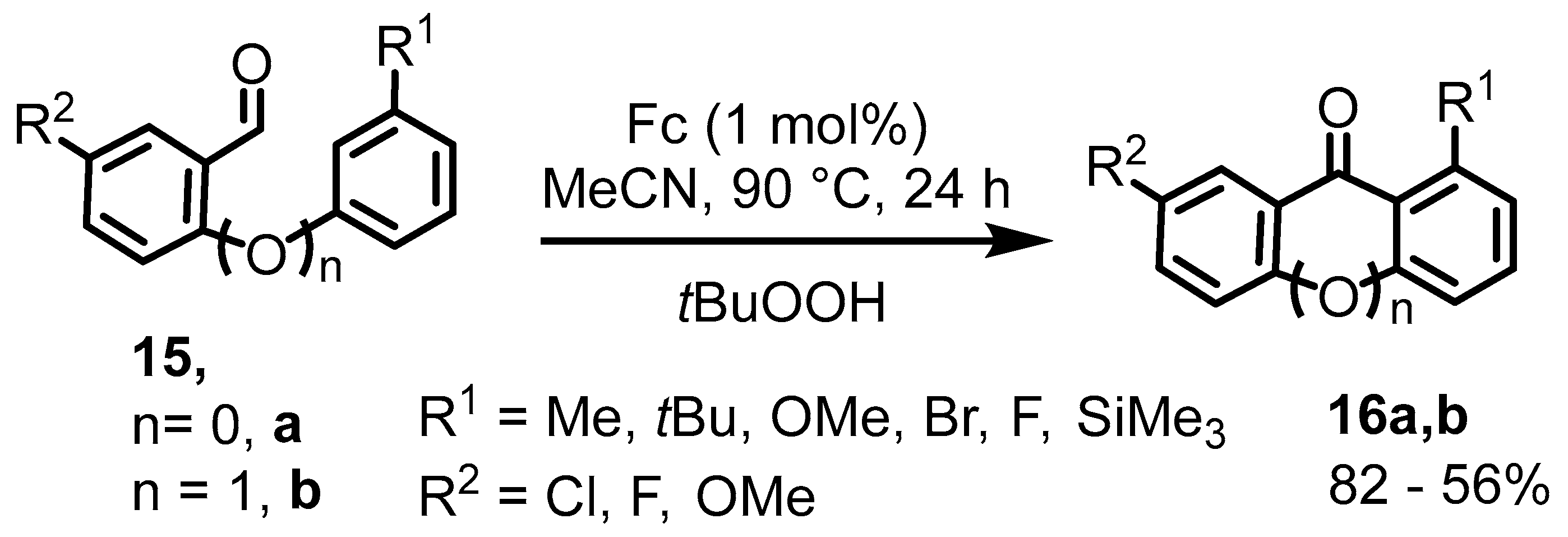







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, E.B. Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds. Molecules 2024, 29, 5544. https://doi.org/10.3390/molecules29235544
Bauer EB. Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds. Molecules. 2024; 29(23):5544. https://doi.org/10.3390/molecules29235544
Chicago/Turabian StyleBauer, Eike B. 2024. "Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds" Molecules 29, no. 23: 5544. https://doi.org/10.3390/molecules29235544
APA StyleBauer, E. B. (2024). Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds. Molecules, 29(23), 5544. https://doi.org/10.3390/molecules29235544