
Development of a Detailed Chemical Kinetic Model for 1-Methylnaphthalene

 ,
,
Abstract
1. Introduction
2. Results and Discussion
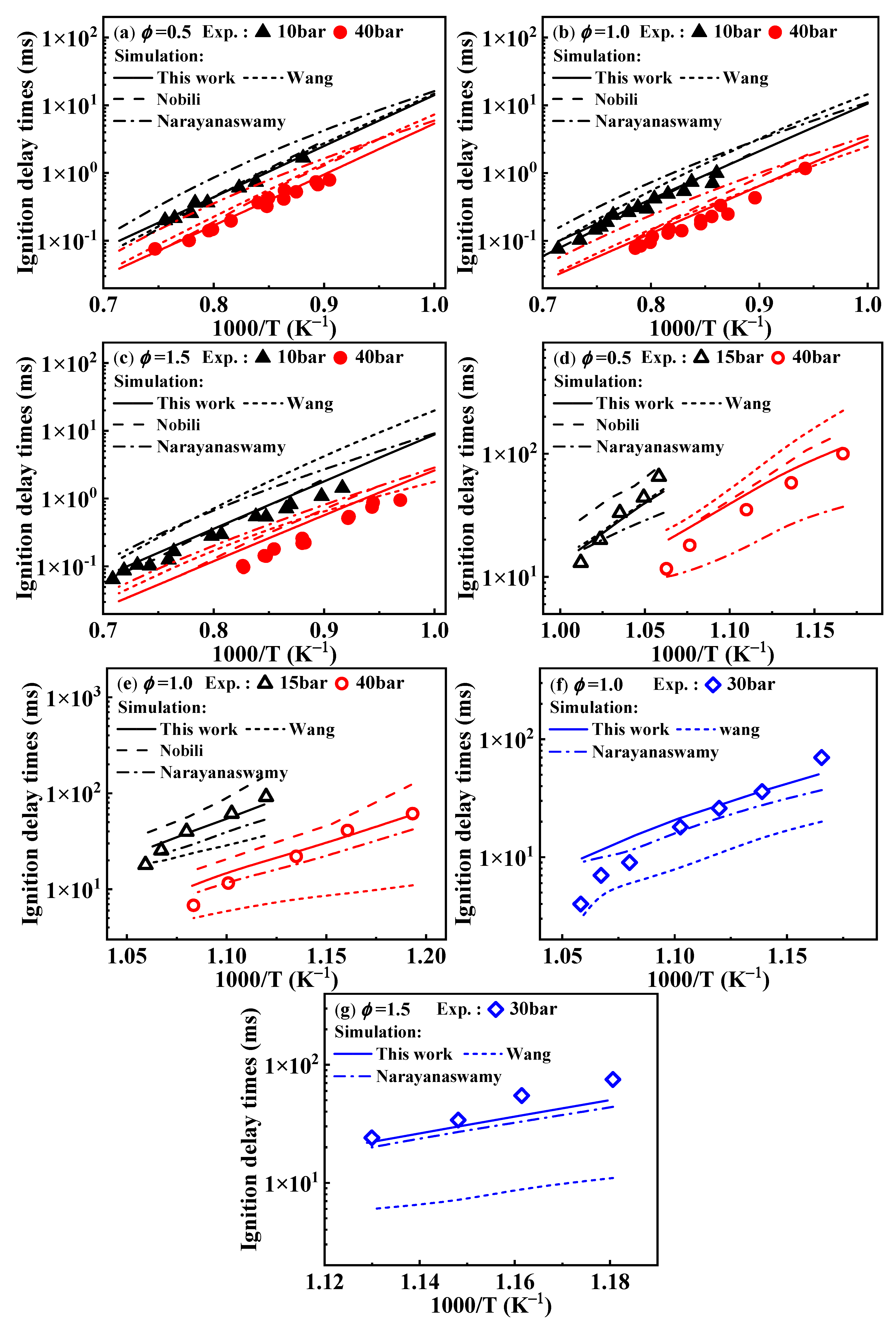
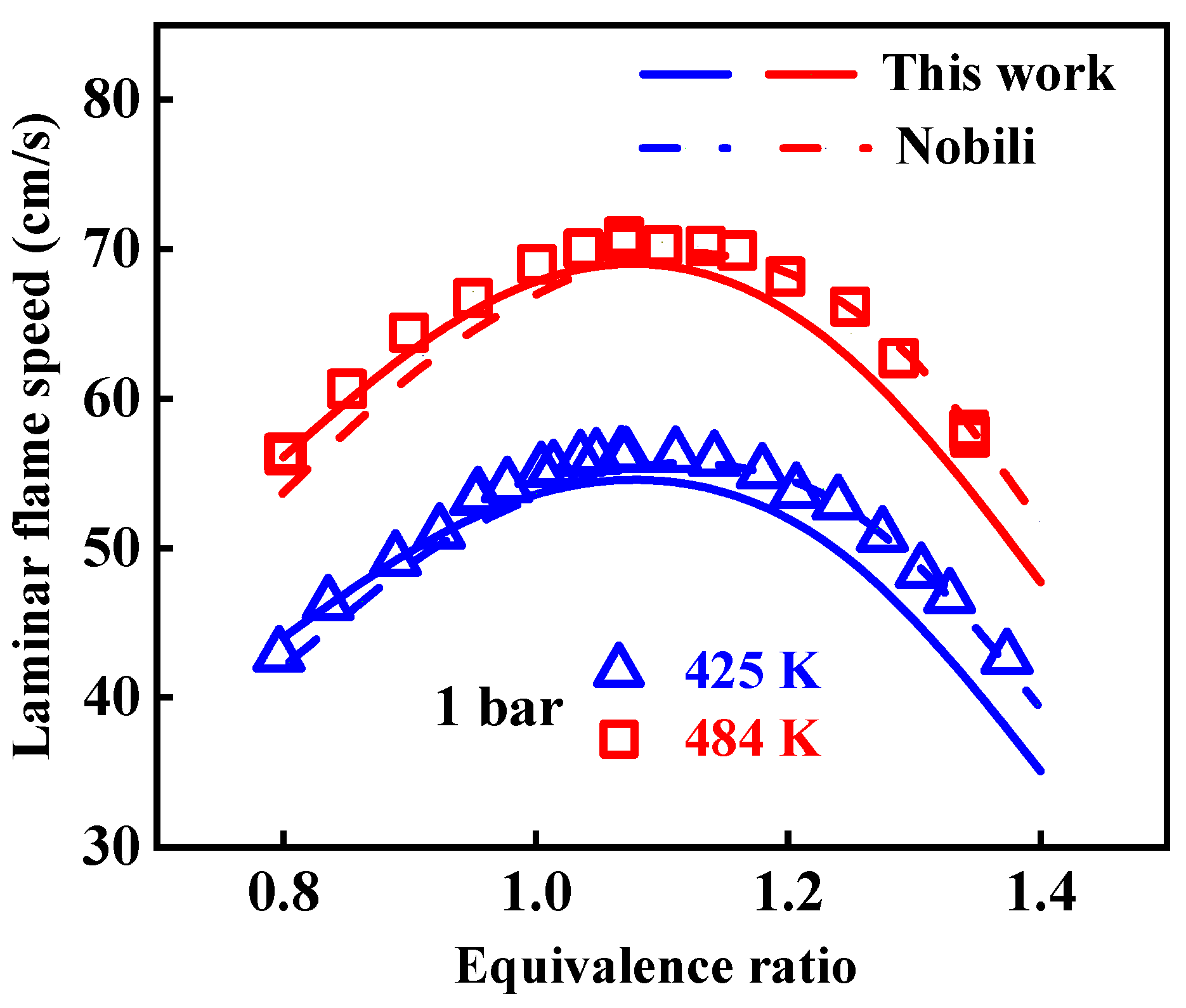
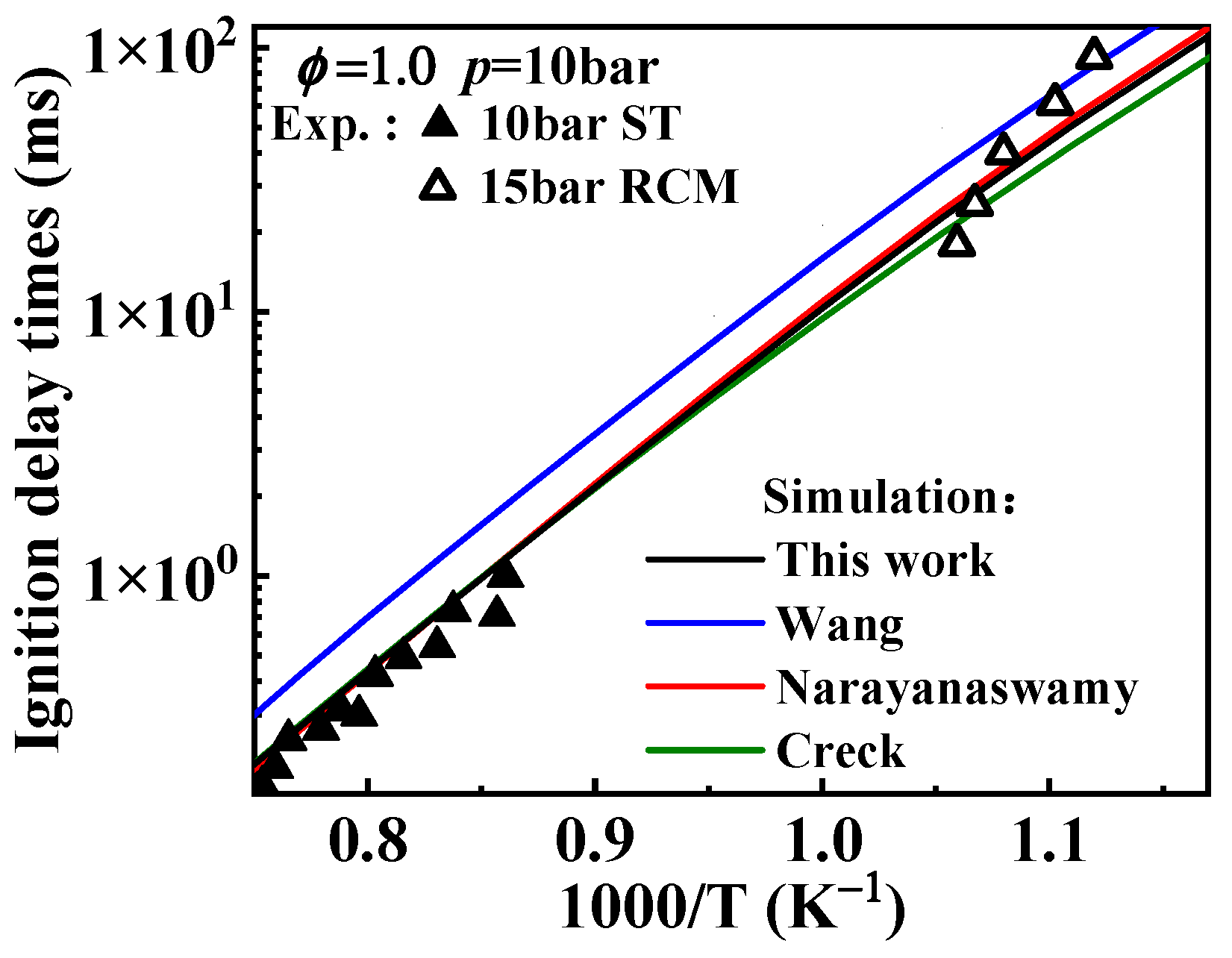
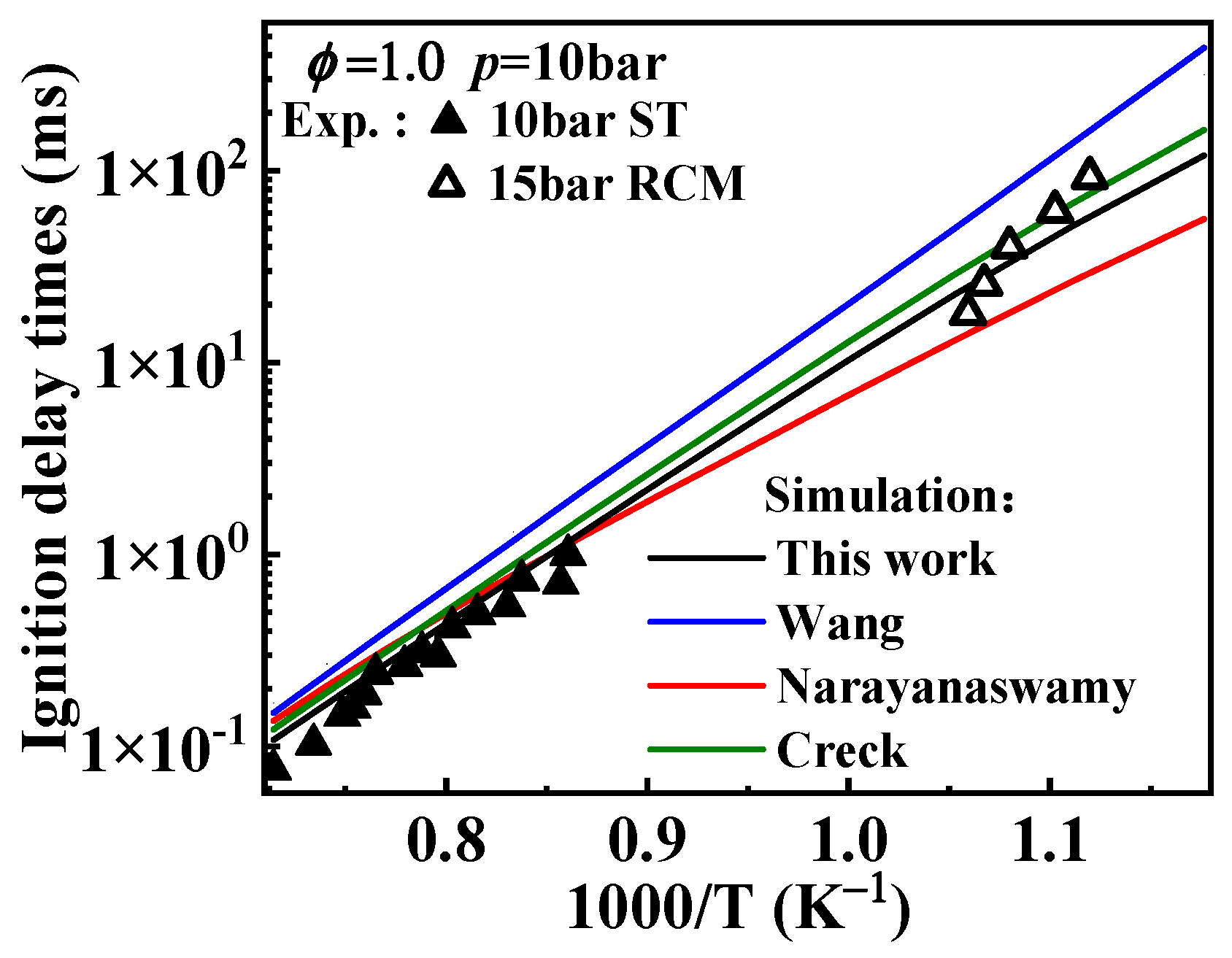
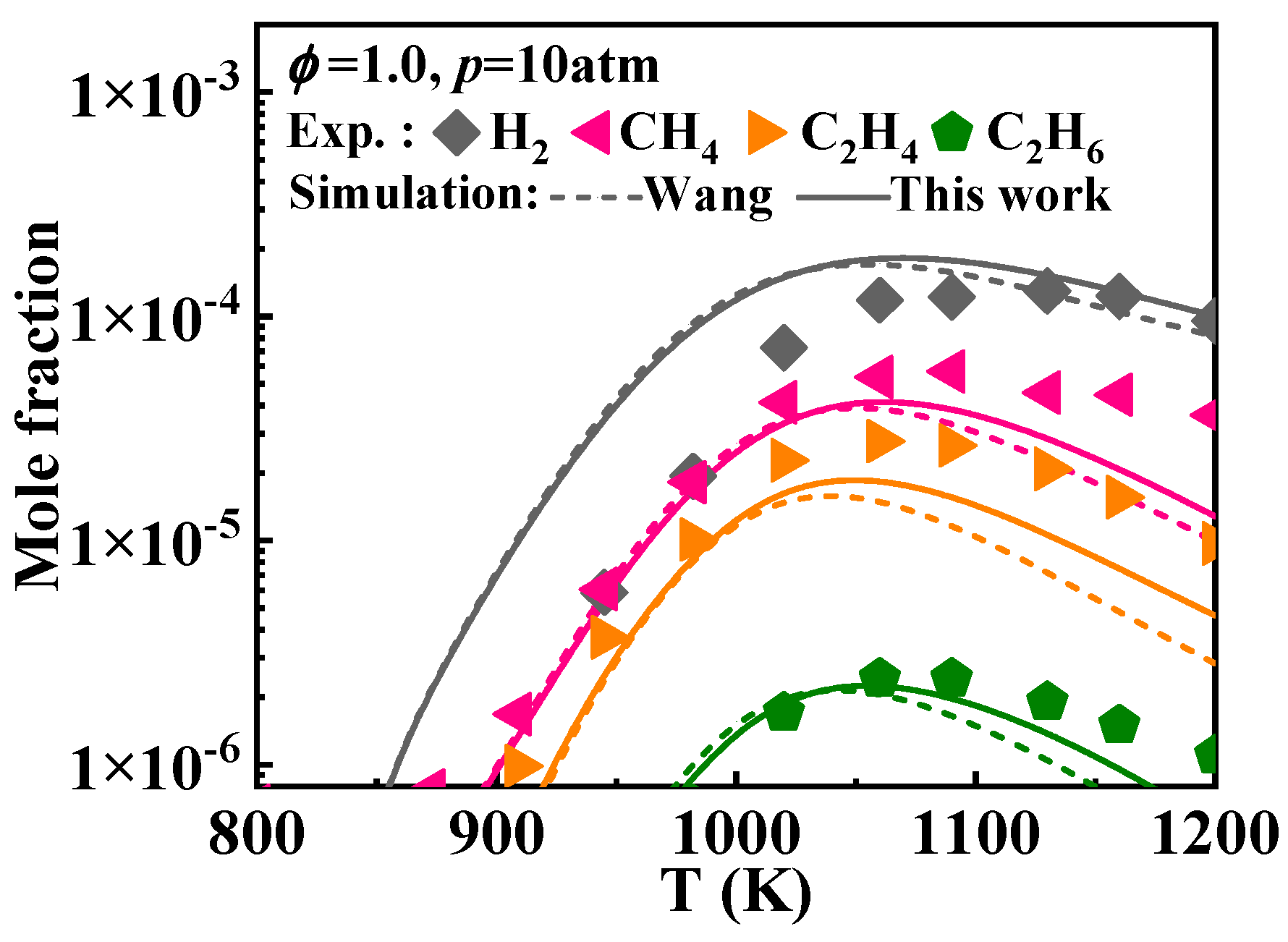
2.1. Validation of Chemical Kinetic Model
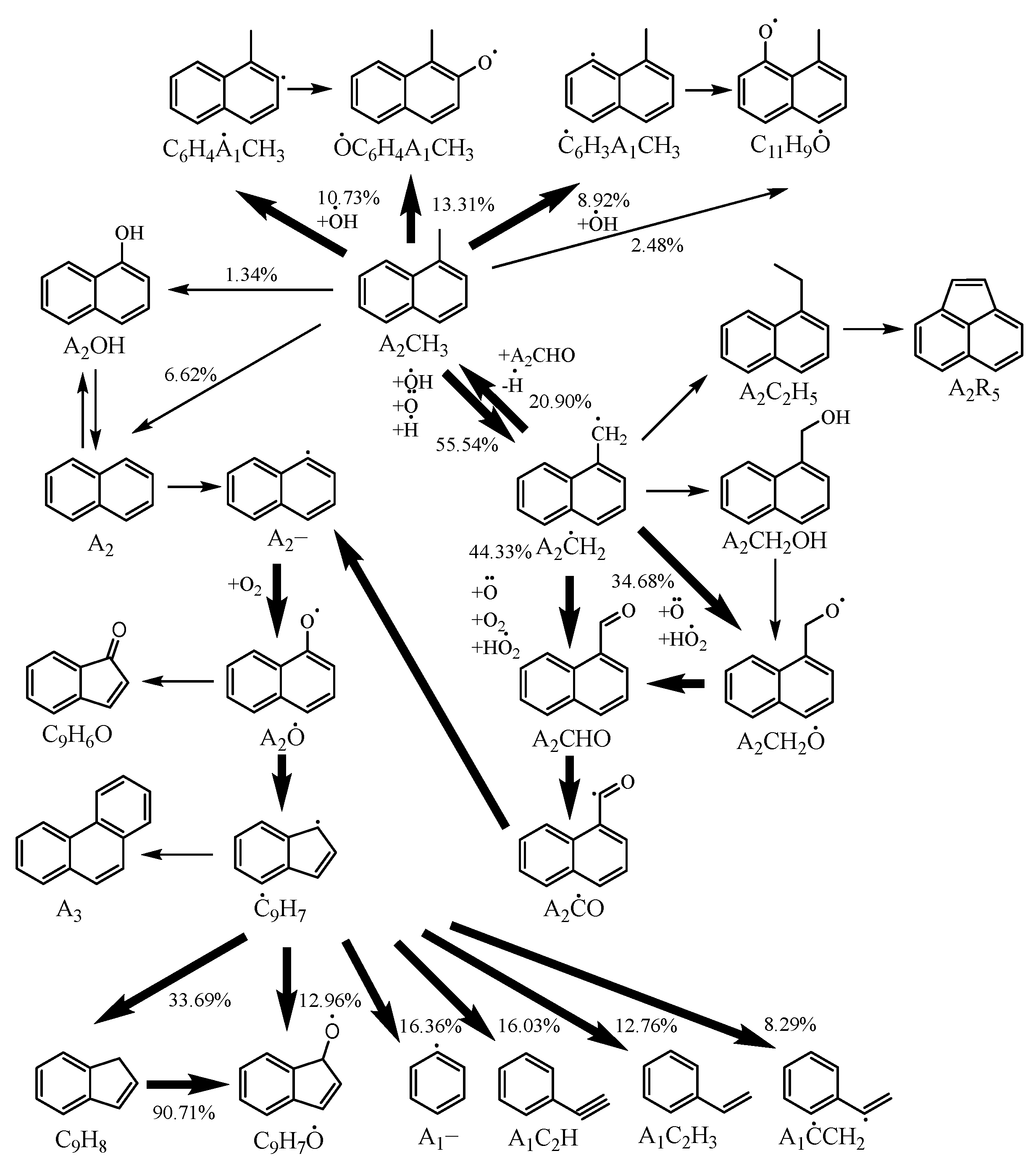
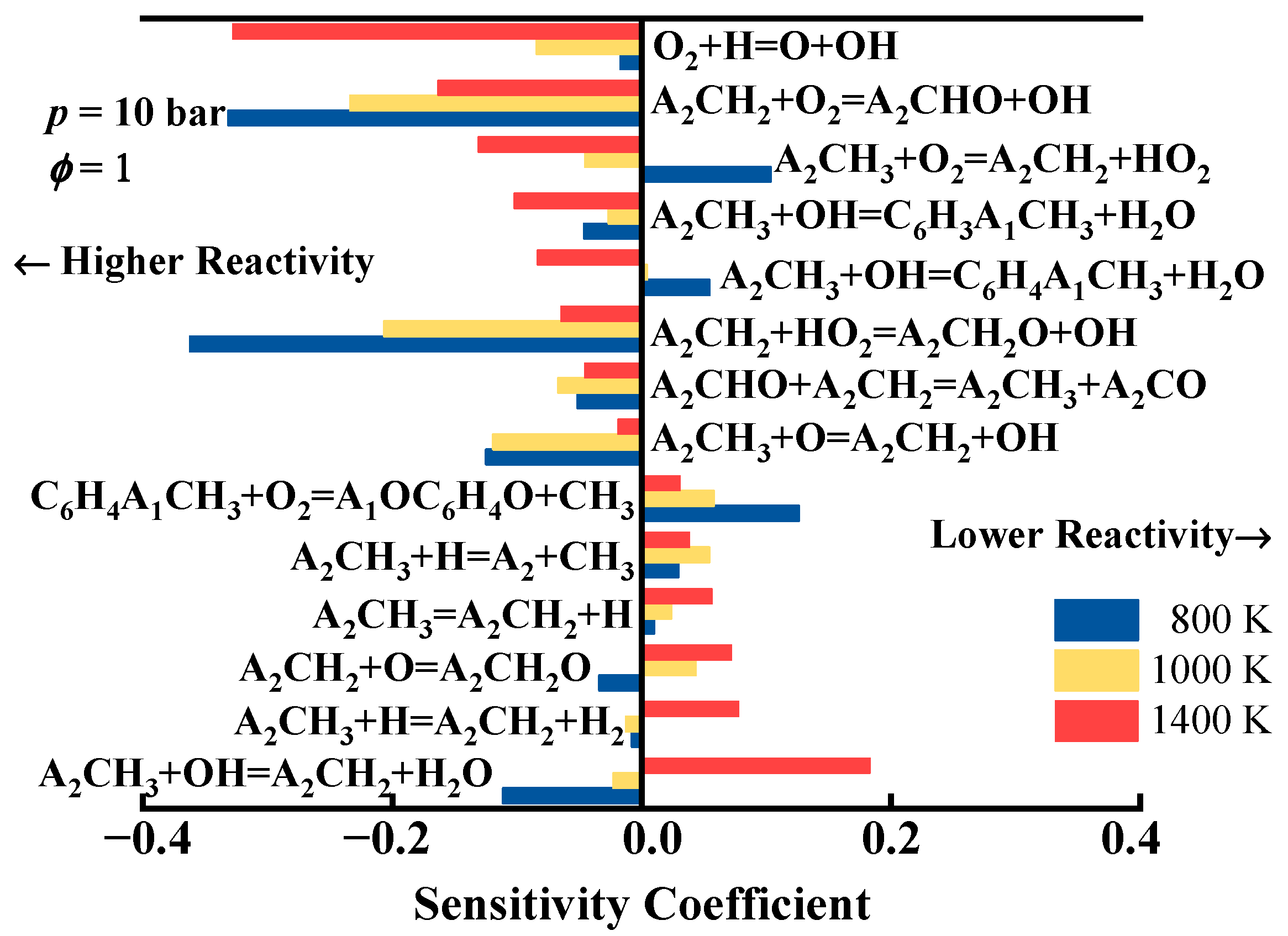
2.2. Chemical Kinetic Analysis of the Ignition of 1-Methylnaphthalene
3. Materials and Methods
3.1. Chemical Kinetic Model
3.2. Rate Constants of Reactions of 1-Methylnaphthalene
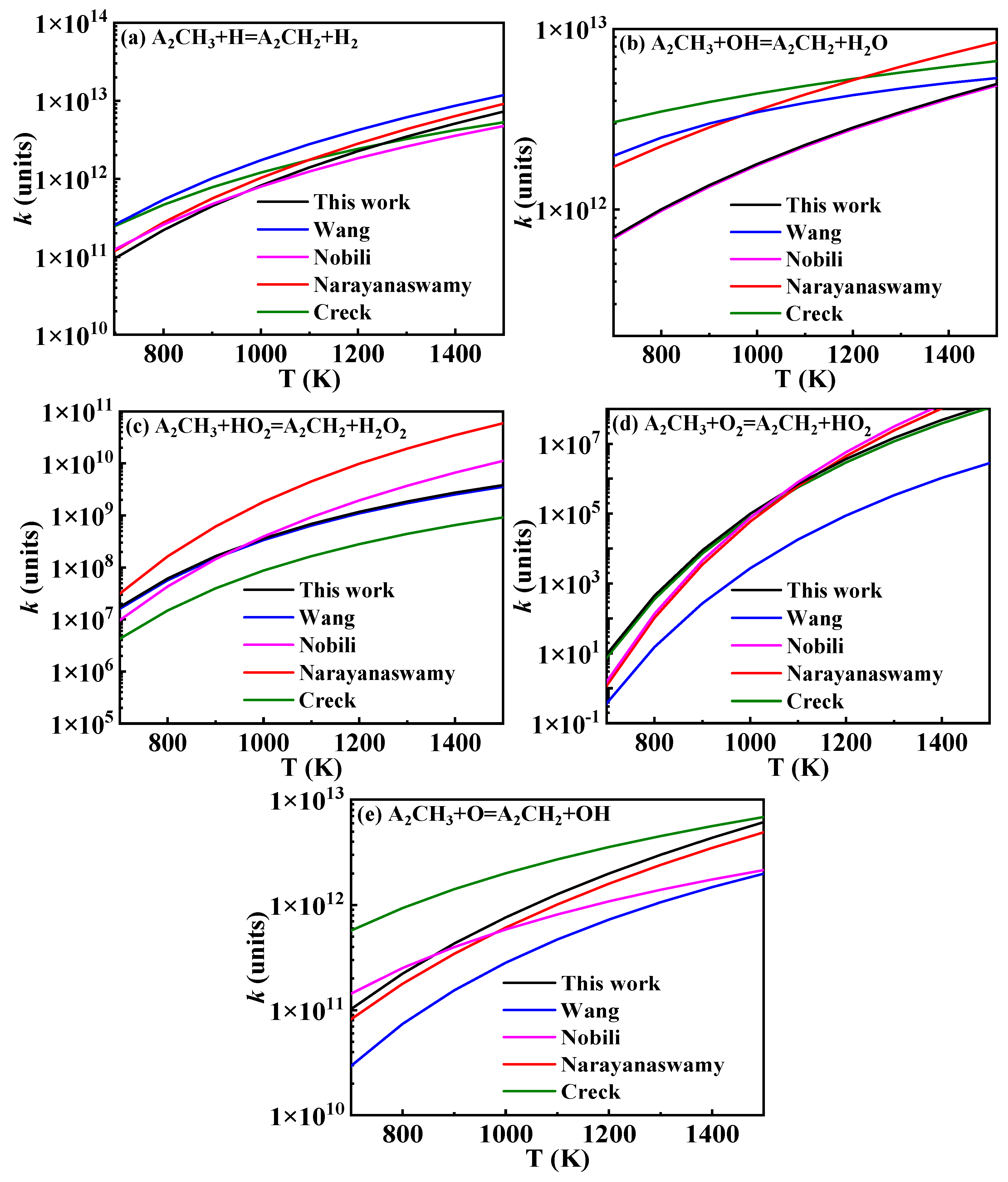
3.2.1. Hydrogen Abstraction of 1-Methylnaphthalene
 ), , A2−, Ċ9H7, A1CHĊH, , and A1−.
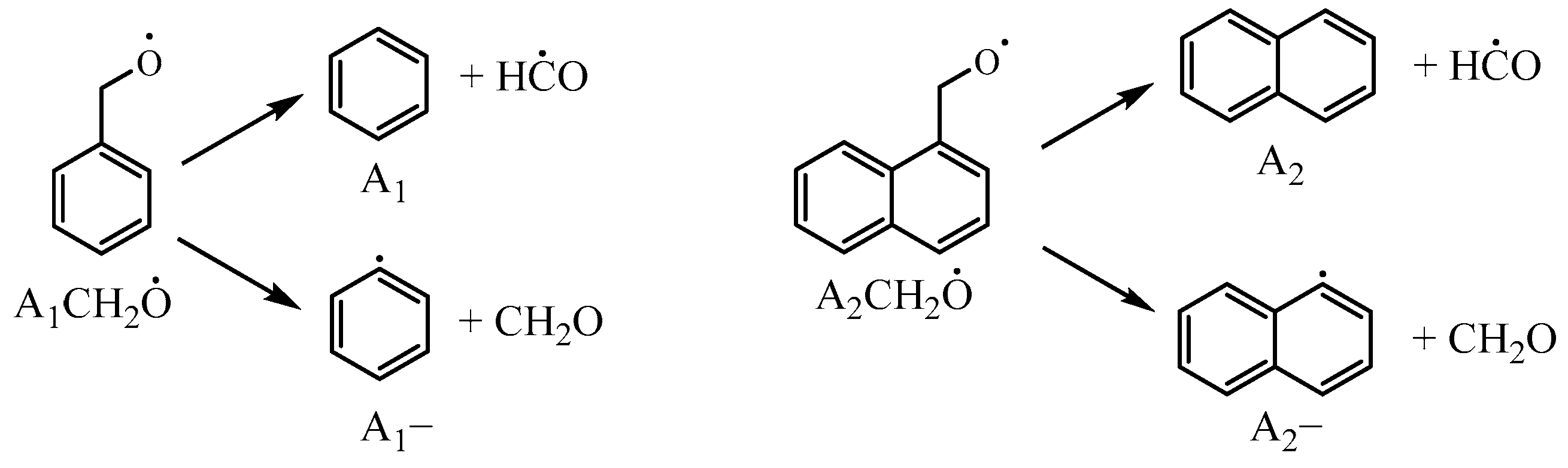
), , A2−, Ċ9H7, A1CHĊH, , and A1−.3.2.2. Other Reaction Classes of 1-Methylnaphthalene
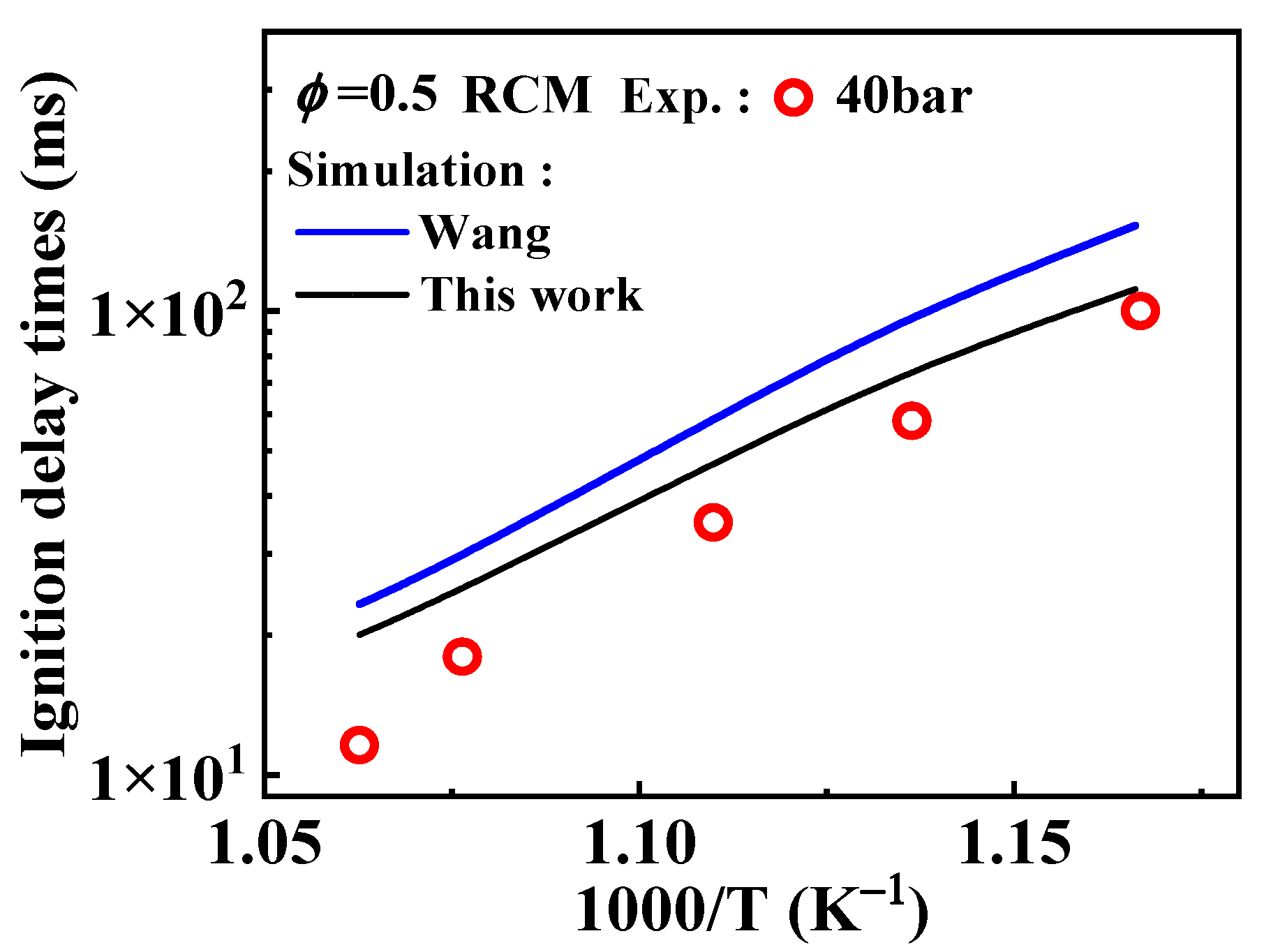
 ). Additionally, the substitution of a Ḣ atom with an radical at the same positions produces HOC6H4A1CH3 and C11H9OH. The formation and consumption of and significantly influence the simulations under conditions of ϕ = 0.5 and p = 40 bar with low temperatures. In this study, the rate constants of the following reactions from the Wang mechanism were adjusted: A2CH3 + Ö = + Ḣ, A2CH3 + Ö = + Ḣ, and = Ċ10H9 (
). Additionally, the substitution of a Ḣ atom with an radical at the same positions produces HOC6H4A1CH3 and C11H9OH. The formation and consumption of and significantly influence the simulations under conditions of ϕ = 0.5 and p = 40 bar with low temperatures. In this study, the rate constants of the following reactions from the Wang mechanism were adjusted: A2CH3 + Ö = + Ḣ, A2CH3 + Ö = + Ḣ, and = Ċ10H9 ( ) + CO, as shown in Table 2. From Figure 9, it can be observed that after the adjustments, the simulated ignition delay times for 1-methylnaphthalene under ϕ = 0.5 and p = 40 bar significantly decrease, bringing them closer to the experimental values. Additionally, the methyl group of A2CH3 can undergo substitution reactions with and Ḣ to form A2OH and A2, respectively. However, these two reaction classes contribute only a small fraction to the overall consumption of 1-methylnaphthalene.
) + CO, as shown in Table 2. From Figure 9, it can be observed that after the adjustments, the simulated ignition delay times for 1-methylnaphthalene under ϕ = 0.5 and p = 40 bar significantly decrease, bringing them closer to the experimental values. Additionally, the methyl group of A2CH3 can undergo substitution reactions with and Ḣ to form A2OH and A2, respectively. However, these two reaction classes contribute only a small fraction to the overall consumption of 1-methylnaphthalene.3.3. Rate Constants of Reactions of 1-Naphthylmethyl
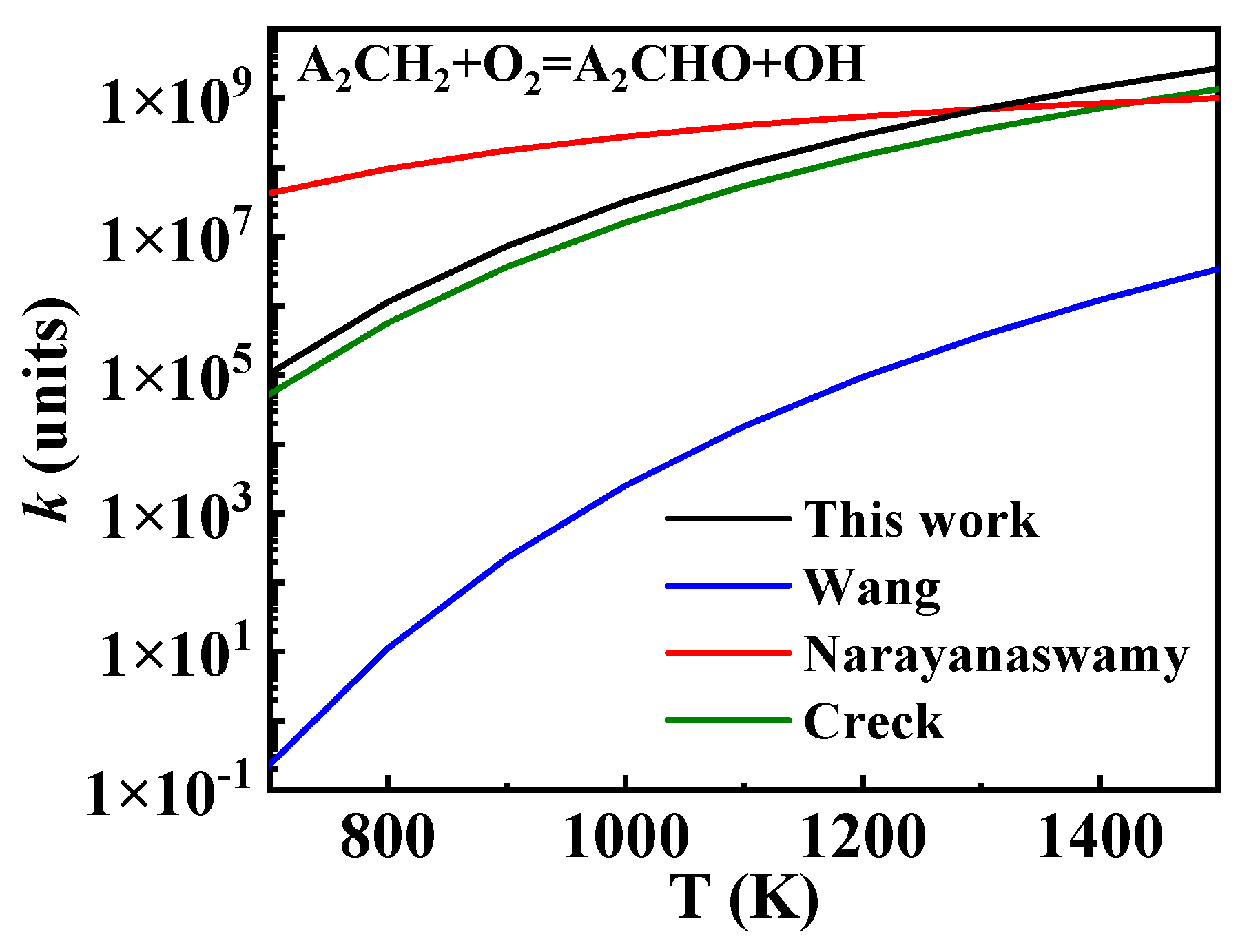
3.3.1. Oxidation of 1-Naphthylmethyl
3.3.2. Other Reaction Classes of 1-Naphthylmethyl
3.4. Rate Constants of Reactions of Indene and Indenyl
3.5. Rate Constants of Reactions of Naphthalene
4. Conclusions
- (1)
- Reactions on the ring of 1-methylnaphthalene, isomerization of 1-naphthylmethyl, formation and consumption of polycyclic aromatic hydrocarbons such as acenaphthene, phenanthrene, and pyrene, as well as formation and consumption of 1-naphthylmethyl-oxy, indenyl-oxy, and indanone were all taken into account in the reaction pathway of 1-methylnaphthalene.
- (2)
- The rate constants of reactions involving 1-methylnaphthalene and its intermediate species were discussed, including hydrogen abstraction, decomposition, and substitution reactions of 1-methylnaphthalene, followed by post-substitution decomposition. Additionally, the oxidation, decomposition, and addition reactions of 1-naphthylmethyl were examined, along with the oxidation of the resulting products. Further reactions discussed include the oxidation of naphthalene, hydrogen abstraction, oxidation, and decomposition of indene, and the oxidation of the indenyl radical. Specific recommended reaction rate constant values were provided for these reactions.
- (3)
- The newly developed chemical kinetic model of 1-methylnaphthalene was comprised of 1389 species and 7185 reactions. This kinetic model was validated against the ignition delay times of 1-methylnaphthalene under equivalence ratios of 0.5, 1.0, and 1.5, pressures ranging from 10 to 40 bar, and temperatures between 800 and 1400 K and the concentration profiles at an equivalence ratio of 1.0, pressure of 10 atm, and temperatures between 800 and 1200 K. Additionally, the present mechanism was also validated against the laminar flame speeds at pressure of 1 bar, initial temperature from 425 to 484 K, and equivalence ratio between 0.8 and 1.35, and the result shows a good agreement was achieved.
- (4)
- The reaction pathway and sensitivity analyses of the ignition process of 1-methylnaphthalene at an equivalence ratio of 1 and a pressure of 10 bar revealed that the conversion of 1-methylnaphthalene to 1-naphthaldehyde through processes such as hydrogen abstraction and oxidation plays an important role in the ignition of 1-methylnaphthalene.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Knecht, W. Diesel engine development in view of reduced emission standards. Energy 2008, 33, 264–271. [Google Scholar] [CrossRef]
- Song, C.; Liang, J.; Zhang, Z.; Li, G.; Zhang, C. Interpretation of role of methane in low-temperature oxidation processes of me-thane/n-heptane mixtures. Fuel 2022, 328, 125373. [Google Scholar] [CrossRef]
- Farrell, J.T.; Cernansky, N.P.; Dryer, F.L.; Law, C.K.; Friend, D.G.; Hergart, C.A.; McDavid, R.M.; Patel, A.K.; Mueller, C.J.; Pitsch, H. Development of an Experimental Database and Kinetic Models for Surrogate Diesel Fuels; SAE Technical Paper: Warrendale, PA, USA, 2007. [Google Scholar]
- Bai, Y.; Wang, Y.; Wang, X.; Wang, P. Development of a skeletal mechanism for tri-component diesel surrogate fuel: N-hexadecane/iso-cetane/1-methylnaphthalene. Fuel 2020, 259, 116217. [Google Scholar] [CrossRef]
- İçıngür, Y.; Altiparmak, D. Effect of fuel cetane number and injection pressure on a DI Diesel engine performance and emissions. Energy Convers. Manag. 2003, 44, 389–397. [Google Scholar] [CrossRef]
- Mueller, C.J.; Cannella, W.J.; Bruno, T.J.; Bunting, B.; Dettman, H.D.; Franz, J.A.; Huber, M.L.; Natarajan, M.; Pitz, W.J.; Ratcliff, M.A.; et al. Methodology for formulating diesel surrogate fuels with accurate compositional, ignition-quality, and volatility characteristics. Energy Fuels 2012, 26, 3284–3303. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, D.; Yu, L.; Qian, Y.; Lu, X. Construction of a skeletal multi-component diesel surrogate model by integrating chemical lumping and genetic algorithm. Fuel 2022, 313, 122711. [Google Scholar] [CrossRef]
- Mensch, A.; Santoro, R.J.; Litzinger, T.A.; Lee, S.-Y. Sooting characteristics of surrogates for jet fuels. Combust. Flame 2010, 157, 1097–1105. [Google Scholar] [CrossRef]
- Pfahl, U.; Fieweger, K.; Adomeit, G. Self-ignition of diesel-relevant hydrocarbon-air mixtures under engine conditions. Proc. Combust. Inst. 1996, 26, 781–789. [Google Scholar] [CrossRef]
- Kukkadapu, G.; Sung, C.J. Autoignition study of 1-methylnaphthalene in a rapid compression machine. Energy Fuels 2017, 31, 854–866. [Google Scholar] [CrossRef]
- Wang, H.; Warner, S.J.; Oehlschlaeger, M.A.; Bounaceur, R.; Biet, J.; Glaude, P.-A.; Battin-Leclerc, F. An experimental and kinetic modeling study of the autoignition of α-methylnaphthalene/air and α-methylnaphthalene/n-decane/air mixtures at elevated pressures. Combust. Flame 2010, 157, 1976–1988. [Google Scholar] [CrossRef]
- Mati, K.; Ristori, A.; Pengloan, G.; Dagaut, P. Oxidation of 1-methylnaphthalene at 1–13 atm: Experimental study in a JSR and detailed chemical kinetic modeling. Combust. Sci. Technol. 2007, 179, 1261–1285. [Google Scholar] [CrossRef]
- Nobili, A.; Maffei, L.P.; Pelucchi, M.; Mehl, M.; Frassoldati, A.; Comandini, A.; Chaumeix, N. Experimental and kinetic modeling study of α-methylnaphthalene laminar flame speeds. Proc. Combust. Inst. 2023, 39, 243–251. [Google Scholar] [CrossRef]
- Narayanaswamy, K.; Blanquart, G.; Pitsch, H. A consistent chemical mechanism for oxidation of substituted aromatic species. Combust. Flame 2010, 157, 1879–1898. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Stagni, A.; Pelucchi, M.; Cuoci, A.; Faravelli, T. Reduced kinetic schemes of complex reaction systems: Fossil and biomass-derived transportation fuels. Int. J. Chem. Kinet. 2014, 46, 512–542. [Google Scholar] [CrossRef]
- Jin, H.; Farooq, A. C7 reaction mechanism and its self-imitation in the kinetic modeling of PAH formation. Combust. Flame 2023, 253, 112816. [Google Scholar] [CrossRef]
- Bounaceur, R.; Glaude, P.A.; Fournet, R.; Battin-Leclerc, F.; Jay, S.; Cruz, A. Kinetic modelling of a surrogate diesel fuel applied to 3D auto-ignition in HCCI engines. Int. J. Veh. Des. 2007, 44, 124–142. [Google Scholar] [CrossRef]
- Blanquart, G.; Pepiot-Desjardins, P.; Pitsch, H. Chemical mechanism for high temperature combustion of engine relevant fuels with emphasis on soot precursors. Combust. Flame 2009, 156, 588–607. [Google Scholar] [CrossRef]
- Sun, S.; Yu, L.; Wang, S.; Mao, Y.; Lu, X. Experimental and kinetic modeling study on self-ignition of α-methylnaphthalene in a heated rapid compression machine. Energy Fuels 2017, 31, 11304–11314. [Google Scholar] [CrossRef]
- Weber, B.W.; Kumar, K.; Zhang, Y.; Sung, C.-J. Autoignition of n-butanol at elevated pressure and low-to-intermediate temperature. Combust. Flame 2011, 158, 809–819. [Google Scholar] [CrossRef]
- Weber, B.W.; Sung, C.-J.; Renfro, M.W. On the uncertainty of temperature estimation in a rapid compression machine. Combust. Flame 2015, 162, 2518–2528. [Google Scholar] [CrossRef]
- Kee, R.J.; Grcar, J.F.; Smooke, M.D.; Miller, J.A.; Meeks, E. PREMIX: A Fortran Program for Modeling Steady Laminar One-Dimensional Premixed Flames, Report No. SAND85-8240; Sandia National Laboratories: Albuquerque, New Mexico/Livermore, CA, USA, 1985; p. 72. [Google Scholar]
- Westbrook, C.K.; Pitz, W.J.; Herbinet, O.; Curran, H.J.; Silke, E.J. A comprehensive detailed chemical kinetic reaction mechanism for combustion of n-alkane hydrocarbons from n-octane to n-hexadecane. Combust. Flame 2009, 156, 181–199. [Google Scholar] [CrossRef]
- Panigrahy, S.; Mohamed, A.A.E.-S.; Wang, P.; Bourque, G.; Curran, H.J. When hydrogen is slower than methane to ignite. Proc. Combust. Inst. 2023, 39, 253–263. [Google Scholar] [CrossRef]
- Goos, E.; Burcat, A.; Ruscic, B. Extended third millennium ideal gas and condensed phase thermochemical database for com-bustion with updates from active thermochemical tables. Elke Goos Remchingen Ger. Accessed Sept 2010, 19, 2016. [Google Scholar]
- Emdee, J.L.; Brezinsky, K.; Glassman, I. A kinetic model for the oxidation of toluene near 1200 K. J. Phys. Chem. 1992, 96, 2151–2161. [Google Scholar] [CrossRef]
- Westmoreland, P.R.; Howard, J.B.; Longwell, J.P.; Dean, A.M. Prediction of rate constants for combustion and pyrolysis reactions by bimolecular QRRK. AIChE J. 1986, 32, 1971–1979. [Google Scholar] [CrossRef]
- Seta, T.; Nakajima, M.; Miyoshi, A. High-temperature reactions of oh radicals with benzene and toluene. J. Phys. Chem. A 2006, 110, 5081–5090. [Google Scholar] [CrossRef]
- Jasper, A.W.; Hansen, N. Hydrogen-assisted isomerizations of fulvene to benzene and of larger cyclic aromatic hydrocarbons. Proc. Combust. Inst. 2013, 34, 279–287. [Google Scholar] [CrossRef]
- Brand, U.; Hippler, H.; Lindemann, L.; Troe, J. Carbon-carbon and carbon-hydrogen bond splits of laser-excited aromatic molecules. 1. Specific and thermally averaged rate constants. J. Phys. Chem. 1990, 94, 6305–6316. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Reaction Class | Key Reactions |
|---|---|---|
| 1-Methylnaphthalene | Hydrogen abstraction | A2−CH3 + RH |
| Substitution | A2CH3/HOA2CH3 + Ḣ | |
| Decomposition | A2CH3→A2ĊH2 + Ḣ; A2CH3→A2− + ĊH3 | |
| 1-Naphthylmethyl | Oxidation | |
| reverse reactions | ||
| Addition | →A2CH2R | |
| Isomerization | A2ĊH2 = cĊ11H9 = Ċ9H6C2H3 | |
| Indenyl | conversion of indene to indenyl | C9H8 + →Ċ9H7 + RH |
| Naphthalene | Oxidation | A2 + Ö = A2OH |
| Reaction Classes | Key Reactions | A | A0× | n | E | Source |
|---|---|---|---|---|---|---|
| Hydrogen abstraction of 1-methylnaphthalene | 2.40 × 1014 | 1.2 | 0 | 43,000 | Creck [15] | |
| 1.48 | 1.2 | 4.09 | 2545 | Narayanaswamy [14] | ||
| = C6H4A1CH3 + H2O | 9.60 × 107 | 1.2 | 1.42 | 1450 | Wang [11] | |
| = A2ĊH2 + HOC6H4A1CH3 | 1.92 × 1011 | 1.2 | 0 | 15,100 | Wang [11] | |
| Decomposition of 1-methylnaphthalene | A2CH3 = A2ĊH2 + Ḣ | 6.25 × 1017 | 0.5 | −0.6 | 94,787 | Narayanaswamy [14] |
| Substitution reactions of 1-methylnaphthalene and post-substitution decomposition | + Ḣ | 1.73 × 1013 | 2.0 | 0 | 3600 | Wang [11] |
| Substitution reactions of 1-methylnaphthalene and post-substitution decomposition Oxidation of 1-naphthylmethyl | + Ḣ | 4.33 × 1012 | 0.5 | 0 | 3600 | Wang [11] |
| = Ċ10H9 + CO | 6.00 × 1011 | 2.0 | 0 | 43,800 | Wang [11] | |
| 2.38 × 109 | 2.0 | 1.03 | −2249 | Narayanaswamy [14] | ||
| Oxidation of 1-naphthylmethyl Addition reactions of 1-naphthylmethyl with radicals and oxidation of the products | + Ḣ | 1.00 × 1013 | 0.5 | 0 | 0 | Narayanaswamy [14] |
| 1.14 × 1014 | 0.5 | 0 | 0 | Narayanaswamy [14] | ||
| 2.00 × 1013 | 2.0 | 0 | 40,000 | Creck [15] | ||
| = A2CH2OH | 1.00 × 1013 | 0.5 | 0 | 0 | Wang [11] | |
| Addition reactions of 1-naphthylmethyl with radicals and oxidation of the products Decomposition of 1-naphthylmethyl-oxy | A2ĊH2 + ĊH3 = A2C2H5 | 5.95 × 1012 | 0.5 | 0 | 221 | Wang [11] |
| + Ḣ | 6.00 × 1014 | 2.0 | 0 | 41,348 | Wang [11] | |
| 1.00 × 1014 | 0.5 | 0 | 41,400 | Wang [11] | ||
| = A2CHO + Ḣ | 2.63 × 1028 | 2.0 | −5.08 | 22,249 | Narayanaswamy [14] | |
| Oxidation of naphthalene | A2 + Ö = A2OH | 3.66 × 1013 | 2.0 | 0 | 4529 | Narayanaswamy [14] |
| Decomposition of indene | Ċ9H7 + Ḣ = C9H8 | 6.32 × 1013 | 0.5 | 0.281 | 179 | NUIG-mech [24] |
| Oxidation of indenyl | 7.50 × 1012 | 0.5 | 0 | 0 | Wang [11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, J.; Zhang, Q.; Heng, Y.; Li, G.; Yang, K.; Wang, R.; Dong, F.; Zhu, N. Development of a Detailed Chemical Kinetic Model for 1-Methylnaphthalene. Molecules 2024, 29, 5660. https://doi.org/10.3390/molecules29235660
Liang J, Zhang Q, Heng Y, Li G, Yang K, Wang R, Dong F, Zhu N. Development of a Detailed Chemical Kinetic Model for 1-Methylnaphthalene. Molecules. 2024; 29(23):5660. https://doi.org/10.3390/molecules29235660
Chicago/Turabian StyleLiang, Junjie, Qianlong Zhang, Yijun Heng, Gesheng Li, Ke Yang, Ruiyang Wang, Fan Dong, and Neng Zhu. 2024. "Development of a Detailed Chemical Kinetic Model for 1-Methylnaphthalene" Molecules 29, no. 23: 5660. https://doi.org/10.3390/molecules29235660
APA StyleLiang, J., Zhang, Q., Heng, Y., Li, G., Yang, K., Wang, R., Dong, F., & Zhu, N. (2024). Development of a Detailed Chemical Kinetic Model for 1-Methylnaphthalene. Molecules, 29(23), 5660. https://doi.org/10.3390/molecules29235660