



Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target



Abstract
:1. Introduction
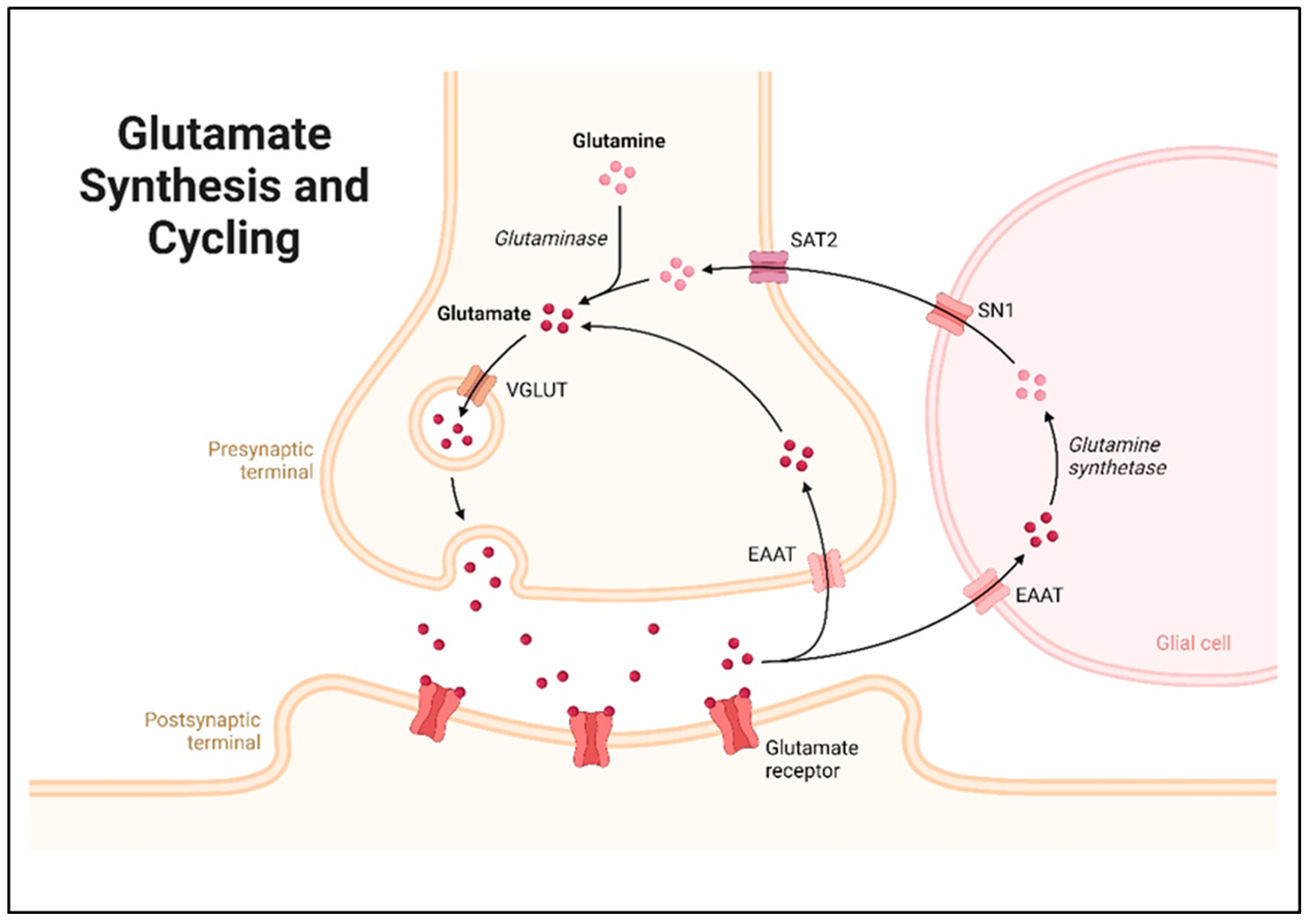
2. Glutamate Synthesis and Metabolism
3. Glutamate Signaling and Receptor Activation
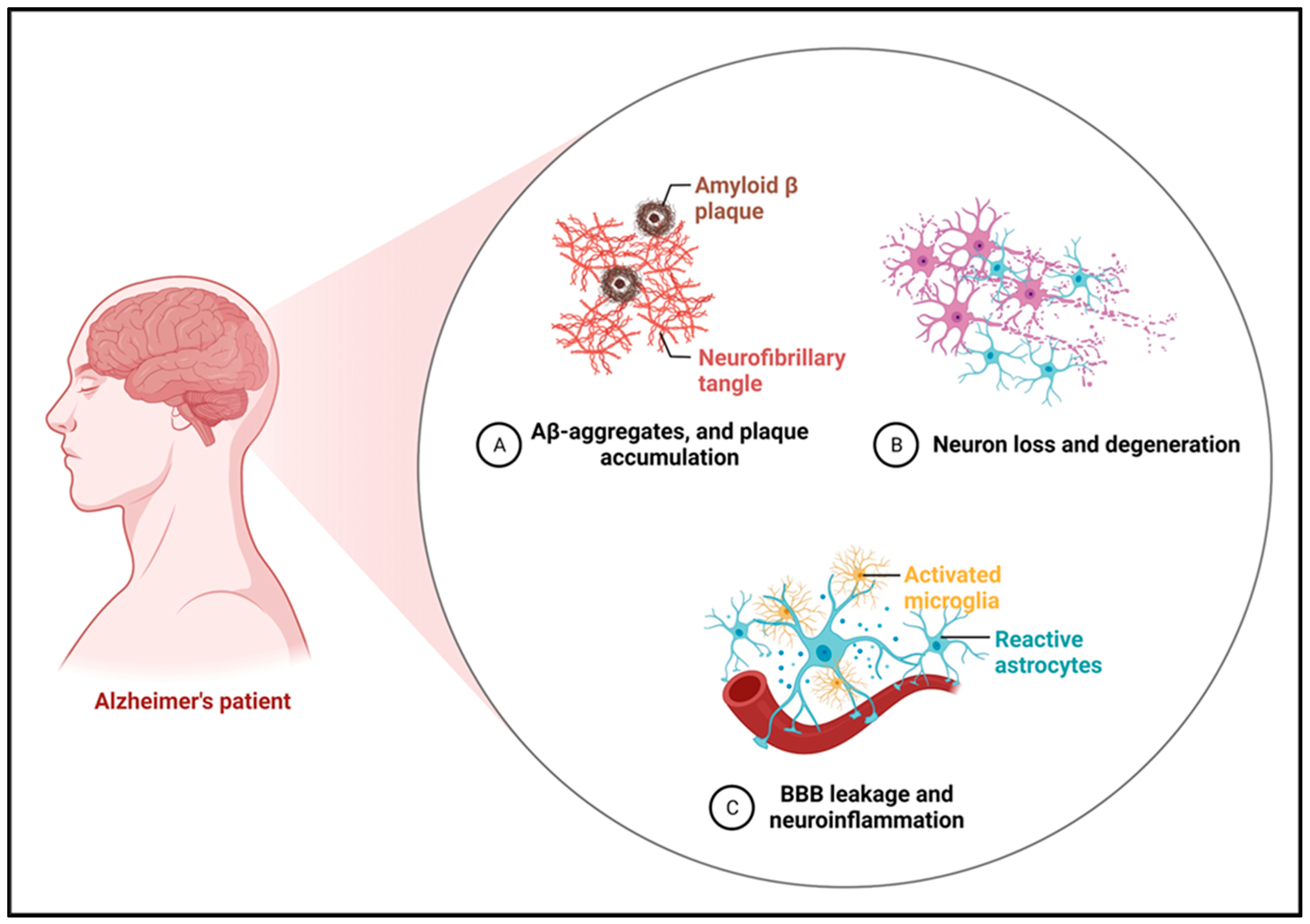
4. Glutamate Dysregulation in Alzheimer’s Disease
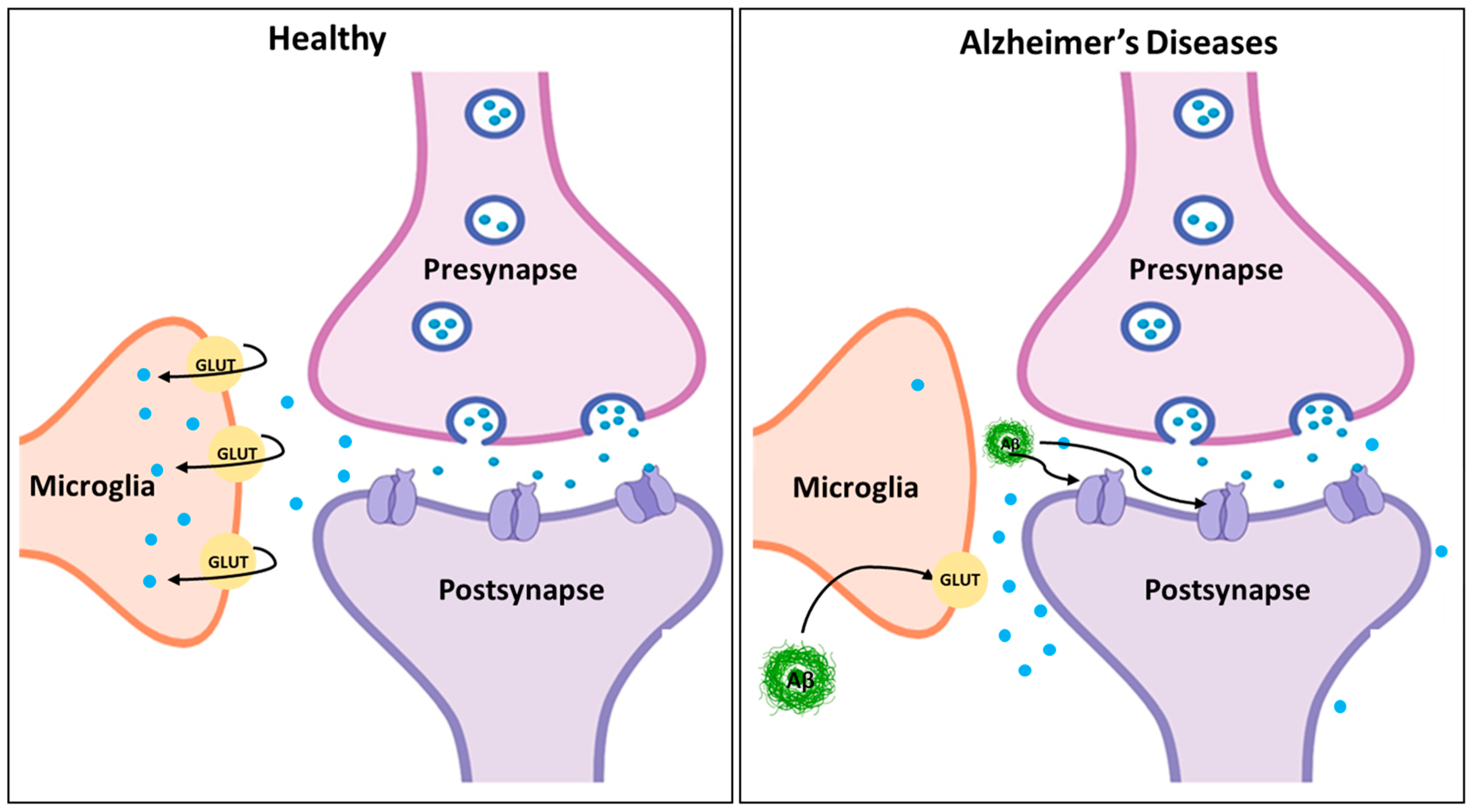
4.1. Excitotoxicity and Neurodegeneration
4.2. Amyloid-β and Tau Interaction with Glutamate Signaling
4.3. Impairment of Synaptic Plasticity and Memory
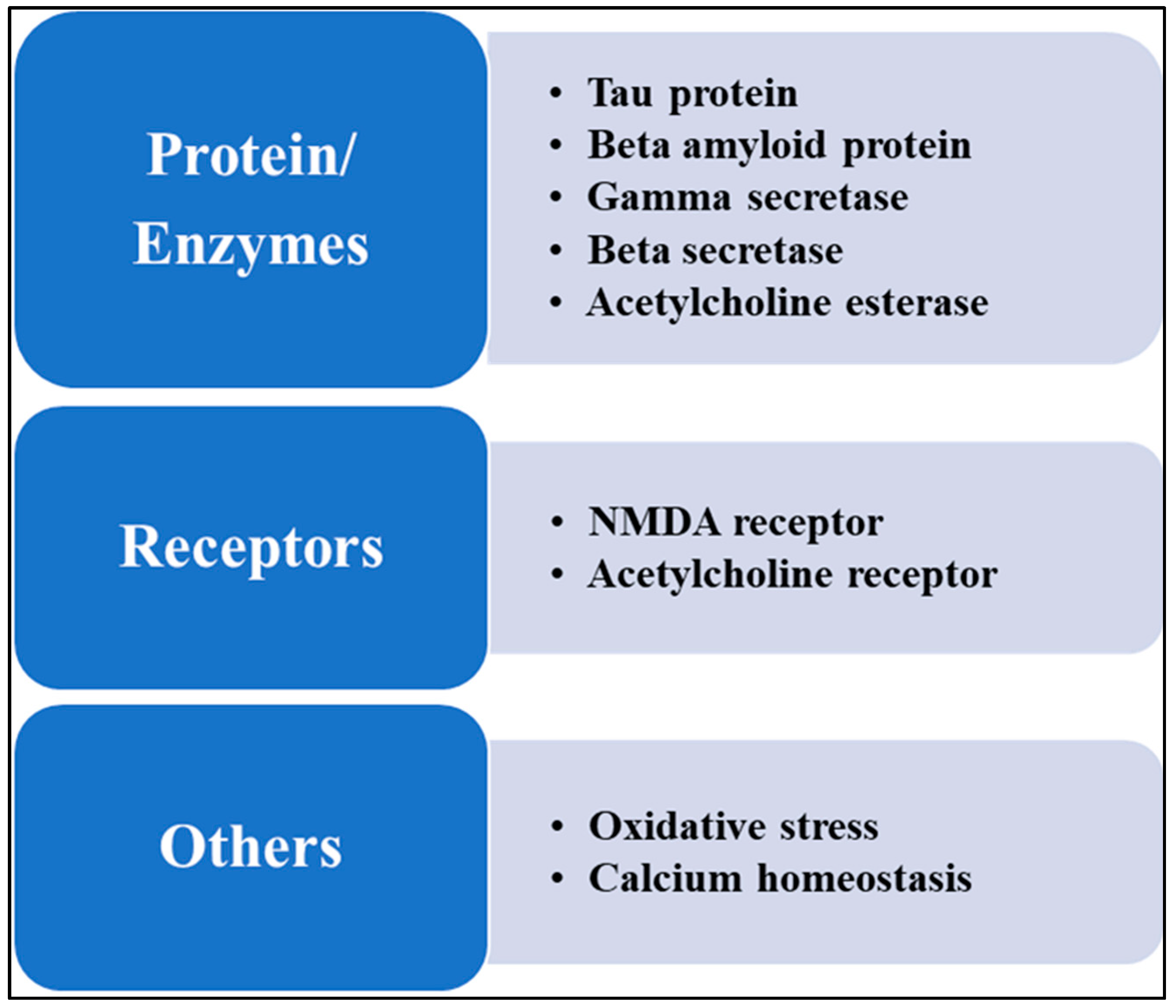
5. Therapeutic Targeting of Glutamate Signaling in Alzheimer’s Disease
5.1. NMDA Receptor Modulators
5.1.1. Memantine
5.1.2. Potential Novel NMDA Antagonists
5.2. Modulation of mGluRs
5.2.1. mGluR2/3 Agonists
5.2.2. mGluR5 Antagonists
6. Glutamate Signaling in the Diagnosis of AD
7. Clinical Drugs
8. Limitations and Challenges
9. Future Directions and Prospects
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. A Blueprint for Dementia Research; WHO: Geneva, Switzerland, 2022; 72p. [Google Scholar]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.; Yadav, D.; Song, M. Advancements in the Application of Nanomedicine in Alzheimer’s Disease: A Therapeutic Perspective. Int. J. Mol. Sci. 2023, 24, 14044. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Singh, D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J. Neuroinflammation 2022, 19, 206. [Google Scholar] [CrossRef]
- Cummings, J.L.; Osse, A.M.L.; Kinney, J.W. Alzheimer’s Disease: Novel Targets and Investigational Drugs for Disease Modification. Drugs 2023, 83, 1387–1408. [Google Scholar] [CrossRef]
- Gabr, M.T.; Ibrahim, M.M. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Castillo-Vazquez, S.K.; Massieu, L.; Rincón-Heredia, R.; la Torre, P.G.-D.; Quiroz-Baez, R.; Gomez-Verjan, J.C.; Rivero-Segura, N.A. Glutamatergic Neurotransmission in Aging and Neurodegenerative Diseases: A Potential Target to Improve Cognitive Impairment in Aging. Arch. Med Res. 2024, 55, 103039. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef]
- Du, X.; Li, J.; Li, M.; Yang, X.; Qi, Z.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. Research progress on the role of type I vesicular glutamate transporter (VGLUT1) in nervous system diseases. Cell Biosci. 2020, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- 2024 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2024, 20, 3708–3821. [CrossRef] [PubMed]
- Abdelnour, C.; Agosta, F.; Bozzali, M.; Fougère, B.; Iwata, A.; Nilforooshan, R.; Takada, L.T.; Viñuela, F.; Traber, M. Perspectives and challenges in patient stratification in Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 112. [Google Scholar] [CrossRef]
- Crabbé, M.; Dirkx, N.; Casteels, C.; Van Laere, K. Excitotoxic neurodegeneration is associated with a focal decrease in metabotropic glutamate receptor type 5 availability: An in vivo PET imaging study. Sci. Rep. 2019, 9, 12916. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Gupta, K.; Hardingham, G.E.; Chandran, S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013, 543, 95–100. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Anastacio, H.T.D.; Matosin, N.; Ooi, L. Neuronal hyperexcitability in Alzheimer’s disease: What are the drivers behind this aberrant phenotype? Transl. Psychiatry 2022, 12, 257. [Google Scholar] [CrossRef]
- Cummings, J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 2. [Google Scholar] [CrossRef]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet. Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowska, A.; Wydrych, M.; Drapich, P.; Zadrozny, M.; Steczkowska, M.; Niewiadomski, W.; Niewiadomska, G. The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front. Aging Neurosci. 2021, 13, 654931. [Google Scholar] [CrossRef]
- Falgàs, N.; Walsh, C.M.; Neylan, T.C.; Grinberg, L.T. Deepen into sleep and wake patterns across Alzheimer’s disease phenotypes. Alzheimer’s Dement. 2021, 17, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Al Mamun, A.; Kabir, T.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Multi-Target Drug Candidates for Multifactorial Alzheimer’s Disease: AChE and NMDAR as Molecular Targets. Mol. Neurobiol. 2021, 58, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Devanand, D.P.; Fremont, R. Cognitive Enhancers and Treatments for Alzheimer’s Disease. In Tasman’s Psychiatry; Springer: Cham, Switzerland, 2024; pp. 4345–4386. [Google Scholar] [CrossRef]
- Cheong, S.L.; Tiew, J.K.; Fong, Y.H.; Leong, H.W.; Chan, Y.M.; Chan, Z.L.; Kong, E.W.J. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals 2022, 15, 1560. [Google Scholar] [CrossRef]
- Kabir, T.; Uddin, S.; Al Mamun, A.; Jeandet, P.; Aleya, L.; Mansouri, R.A.; Ashraf, G.M.; Mathew, B.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Combination Drug Therapy for the Management of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3272. [Google Scholar] [CrossRef]
- Hung, S.-Y.; Fu, W.-M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef]
- Abdallah, A.E. Review on anti-alzheimer drug development: Approaches, challenges and perspectives. RSC Adv. 2024, 14, 11057–11088. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Maciejewska, K.; Czarnecka, K.; Szymański, P. A review of the mechanisms underlying selected comorbidities in Alzheimer’s disease. Pharmacol. Rep. 2021, 73, 1565–1581. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate Metabolism in the Brain Focusing on Astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef] [PubMed]
- Cuellar-Santoyo, A.O.; Ruiz-Rodríguez, V.M.; Mares-Barbosa, T.B.; Patrón-Soberano, A.; Howe, A.G.; Portales-Pérez, D.P.; Graf, A.M.; Estrada-Sánchez, A.M. Revealing the contribution of astrocytes to glutamatergic neuronal transmission. Front. Cell. Neurosci. 2023, 16, 1037641. [Google Scholar] [CrossRef]
- Ding, L.; Xu, X.; Li, C.; Wang, Y.; Xia, X.; Zheng, J.C. Glutaminase in microglia: A novel regulator of neuroinflammation. Brain, Behav. Immun. 2020, 92, 139–156. [Google Scholar] [CrossRef]
- Katabathula, S.; Davis, P.B.; Xu, R. Comorbidity-driven multi-modal subtype analysis in mild cognitive impairment of Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 1428–1439. [Google Scholar] [CrossRef]
- Zhang, D.; Hua, Z.; Li, Z. The role of glutamate and glutamine metabolism and related transporters in nerve cells. CNS Neurosci. Ther. 2024, 30, e14617. [Google Scholar] [CrossRef]
- Gong, X.; Guo, R.; Li, X.; Yang, Y.; Lin, W. A red-emitting mitochondria targetable fluorescent probe for detecting viscosity in HeLa, zebrafish, and mice. Anal. Methods 2023, 16, 293–300. [Google Scholar] [CrossRef]
- Wang, S.; Wang, L.; Qin, X.; Turdi, S.; Sun, D.; Culver, B.; Reiter, R.J.; Wang, X.; Zhou, H.; Ren, J. ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal Transduct. Target. Ther. 2020, 5, 119. [Google Scholar] [CrossRef]
- Salasova, A.; Monti, G.; Andersen, O.M.; Nykjaer, A. Finding memo: Versatile interactions of the VPS10p-Domain receptors in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Schousboe, A. Milestone Review: Metabolic dynamics of glutamate and GABA mediated neurotransmission—The essential roles of astrocytes. J. Neurochem. 2023, 166, 109–137. [Google Scholar] [CrossRef] [PubMed]
- Marde, V.S.; Atkare, U.A.; Gawali, S.V.; Tiwari, P.L.; Badole, S.P.; Wankhede, N.L.; Taksande, B.G.; Upaganlawar, A.B.; Umekar, M.J.; Kale, M.B. Alzheimer’s disease and sleep disorders: Insights into the possible disease connections and the potential therapeutic targets. Asian J. Psychiatry 2021, 68, 102961. [Google Scholar] [CrossRef]
- Fehsel, K.; Christl, J. Comorbidity of osteoporosis and Alzheimer’s disease: Is ‘AKT’-ing on cellular glucose uptake the missing link? Ageing Res. Rev. 2022, 76, 101592. [Google Scholar] [CrossRef] [PubMed]
- Errasti-Murugarren, E.; Palacín, M. Heteromeric Amino Acid Transporters in Brain: From Physiology to Pathology. Neurochem. Res. 2021, 47, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Anderson, C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 38. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Ullah, G. The role of transporters and synaptic cleft morphology in glutamate and GABA homeostasis and their effect on neuronal function. bioRxiv 2019. [Google Scholar] [CrossRef]
- Gunes, S.; Aizawa, Y.; Sugashi, T.; Sugimoto, M.; Rodrigues, P.P. Biomarkers for Alzheimer’s Disease in the Current State: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 4962. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Brown, P.M.G.E.; Dawe, G.B.; Feltz, A.; Bowie, D. Structural and Functional Properties of Ionotropic and Metabotropic Glutamate Receptors. In Physiology of Neurons; Garland Science: New York, NY, USA, 2020; pp. 231–243. [Google Scholar] [CrossRef]
- Yadav, P.; Podia, M.; Kumari, S.P.; Mani, I. Glutamate receptor endocytosis and signaling in neurological conditions. Prog. Mol. Biol. Transl. Sci. 2023, 196, 167–207. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef] [PubMed]
- Ragnarsson, L.; Dodd, P.R.; Latif, M.R. Role of Ionotropic Glutamate Receptors in Neurodegenerative and Other Disorders. In Handbook of Neurotoxicity, 2nd ed.; Springer: Cham, Switzerland, 2022; Volume 3, pp. 1969–1997. [Google Scholar] [CrossRef]
- Roman, J.Y.M.; González, C.C. Glutamate and excitotoxicity in central nervous system disorders: Ionotropic glutamate receptors as a target for neuroprotection. Neuroprotection 2024, 2, 137–150. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Lloret, M.-A.; Cervera-Ferri, A.; Lopez, B.; Nepomuceno, M.; Monllor, P. When Does Alzheimer′s Disease Really Start? The Role of Biomarkers. Int. J. Mol. Sci. 2019, 20, 5536. [Google Scholar] [CrossRef] [PubMed]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimer’s Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef]
- Nedelec, T.; Couvy-Duchesne, B.; Monnet, F.; Daly, T.; Ansart, M.; Gantzer, L.; Lekens, B.; Epelbaum, S.; Dufouil, C.; Durrleman, S. Identifying health conditions associated with Alzheimer’s disease up to 15 years before diagnosis: An agnostic study of French and British health records. Lancet Digit. Health 2022, 4, e169–e178. [Google Scholar] [CrossRef]
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimer’s Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Logroscino, G. Prevention of Alzheimer’s disease and dementia: The evidence is out there, but new high-quality studies and implementation are needed. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1140–1141. [Google Scholar] [CrossRef]
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From Risk factors to early intervention. Alzheimer’s Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Omura, J.D.; McGuire, L.C.; Patel, R.; Baumgart, M.; Lamb, R.; Jeffers, E.M.; Olivari, B.S.; Croft, J.B.; Thomas, C.W.; Hacker, K. Modifiable Risk Factors for Alzheimer Disease and Related Dementias Among Adults Aged ≥45 Years—United States, 2019. Mmwr-Morbidity Mortal. Wkly. Rep. 2022, 71, 680–685. [Google Scholar] [CrossRef]
- Zhang, D.-F.; Li, M. Toward a Full Understanding of Causal and Modifiable Risk Factors for Alzheimer’s Disease by Integrative Phenome-wide Association Studies. Biol. Psychiatry 2023, 93, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.V.F.; Loures, C.d.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.d.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [PubMed]
- Beata, B.-K.; Wojciech, J.; Johannes, K.; Piotr, L.; Barbara, M. Alzheimer’s Disease—Biochemical and Psychological Background for Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 1059. [Google Scholar] [CrossRef] [PubMed]
- Thakral, S.; Yadav, A.; Singh, V.; Kumar, M.; Kumar, P.; Narang, R.; Sudhakar, K.; Verma, A.; Khalilullah, H.; Jaremko, M.; et al. Alzheimer’s disease: Molecular aspects and treatment opportunities using herbal drugs. Ageing Res. Rev. 2023, 88, 101960. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Chiriac, S.I.B.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2020, 10, 40. [Google Scholar] [CrossRef]
- Hampel, H.; Lista, S.; Khachaturian, Z.S. Development of biomarkers to chart all Alzheimer’s disease stages: The royal road to cutting the therapeutic Gordian Knot. Alzheimer’s Dement. 2012, 8, 312–336. [Google Scholar] [CrossRef]
- Sutphen, C.L.; Fagan, A.M.; Holtzman, D.M. Progress Update: Fluid and Imaging Biomarkers in Alzheimer’s Disease. Biol. Psychiatry 2014, 75, 520–526. [Google Scholar] [CrossRef]
- Lista, S.; Garaci, F.G.; Ewers, M.; Teipel, S.; Zetterberg, H.; Blennow, K.; Hampel, H. CSF Aβ1-42 combined with neuroimaging biomarkers in the early detection, diagnosis and prediction of Alzheimer’s disease. Alzheimer’s Dement. 2013, 10, 381–392. [Google Scholar] [CrossRef]
- Reiman, E.M. Putting AD treatments and biomarkers to the test. Nat. Rev. Neurol. 2017, 13, 74–76. [Google Scholar] [CrossRef]
- Nair, J.D.; Wilkinson, K.A.; Henley, J.M.; Mellor, J.R. Kainate receptors and synaptic plasticity. Neuropharmacology 2021, 196, 108540. [Google Scholar] [CrossRef]
- Qizilbash, N.; Whitehead, A.; Higgins, J.; Wilcock, G.; Schneider, L.; Farlow, M. Cholinesterase Inhibition for Alzheimer Disease: A Meta-analysis of the Tacrine Trials. JAMA 1998, 280, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Bodzęta, A.; Berger, F.; MacGillavry, H.D. Subsynaptic mobility of presynaptic mGluR types is differentially regulated by intra- and extracellular interactions. Mol. Biol. Cell 2022, 33, ar66. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xiao, W.; Wang, Y.; Li, J.; Gong, J.; Tu, E.; Long, L.; Xiao, B.; Yan, X.; Wan, L. Metabotropic glutamate receptors (mGluRs) in epileptogenesis: An update on abnormal mGluRs signaling and its therapeutic implications. Neural Regen. Res. 2023, 19, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Jarrott, B. Tacrine: In vivo veritas. Pharmacol. Res. 2017, 116, 29–31. [Google Scholar] [CrossRef]
- Mango, D.; Ledonne, A. Updates on the Physiopathology of Group I Metabotropic Glutamate Receptors (mGluRI)-Dependent Long-Term Depression. Cells 2023, 12, 1588. [Google Scholar] [CrossRef]
- Su, L.-D.; Wang, N.; Han, J.; Shen, Y. Group 1 Metabotropic Glutamate Receptors in Neurological and Psychiatric Diseases: Mechanisms and Prospective. Neuroscientist 2021, 28, 453–468. [Google Scholar] [CrossRef]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients with Alzheimer’s Disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef]
- Ríos, C.d.L.; Marco-Contelles, J. Tacrines for Alzheimer’s disease therapy. III. The PyridoTacrines. Eur. J. Med. Chem. 2019, 166, 381–389. [Google Scholar] [CrossRef]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 2018. [Google Scholar] [CrossRef]
- Bocchio, M.; Lukacs, I.P.; Stacey, R.; Plaha, P.; Apostolopoulos, V.; Livermore, L.; Sen, A.; Ansorge, O.; Gillies, M.J.; Somogyi, P.; et al. Group II metabotropic glutamate receptors mediate presynaptic inhibition of excitatory transmission in pyramidal neurons of the human cerebral cortex. Front. Cell. Neurosci. 2019, 12, 508. [Google Scholar] [CrossRef]
- Trepanier, C.; Lei, G.; Xie, Y.-F.; MacDonald, J.F. Group II metabotropic glutamate receptors modify N-methyl-D-aspartate receptors via Src kinase. Sci. Rep. 2013, 3, 926. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Lim, Y.J.; Kumar, K.; Baby, N.; Pang, K.L.K.; Benoy, A.; Behnisch, T.; Sajikumar, S.; Neurobiology, S.K.L.O.M. China Group III metabotropic glutamate receptors gate long-term potentiation and synaptic tagging/capture in rat hippocampal area CA2. eLife 2020, 9, e55344. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Guo, Y.-E.; Fang, J.-H.; Shi, C.-J.; Suo, N.; Zhang, R.; Xie, X. Donepezil, a drug for Alzheimer’s disease, promotes oligodendrocyte generation and remyelination. Acta Pharmacol. Sin. 2019, 40, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Brewster, J.T.; Dell’Acqua, S.; Thach, D.Q.; Sessler, J.L. Classics in Chemical Neuroscience: Donepezil. ACS Chem. Neurosci. 2019, 10, 155–167. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Feldman, H.H.; Lane, R. Rivastigmine: A placebo controlled trial of twice daily and three times daily regimens in patients with Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1056–1063. [Google Scholar] [CrossRef]
- Rösler, M.; Anand, R.; Cicin-Sain, A.; Gauthier, S.; Agid, Y.; Dal-Bianco, P.; Stähelin, H.B.; Hartman, R.; Gharabawi, M.; Bayer, T. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: International randomised controlled trial Commentary: Another piece of the Alzheimer’s jigsaw. BMJ 1999, 318, 633–640. [Google Scholar] [CrossRef]
- Coyle, J.; Kershaw, P. Galantamine, a cholinesterase inhibitor that allosterically modulates nicotinic receptors: Effects on the course of Alzheimer’s disease. Biol. Psychiatry 2001, 49, 289–299. [Google Scholar] [CrossRef]
- Scott, L.J.; Goa, K.L. Galantamine: A review of its use in Alzheimer’s disease. Drugs 2000, 60, 1095–1122. [Google Scholar] [CrossRef]
- Marco-Contelles, J.; do Carmo Carreiras, M.; Rodríguez, C.; Villarroya, M.; García, A.G. Synthesis and pharmacology of Galantamine. Chem. Rev. 2006, 106, 116–133. [Google Scholar] [CrossRef]
- Robinson, D.M.; Keating, G.M. Memantine: A review of its use in Alzheimer’s disease. Drugs 2006, 66, 1515–1534. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.J.; Badraoui, R.; Jahan, S.; Alshahrani, M.M.; Siddiqui, M.A.; Khan, A.; Adnan, M. Targeting NMDA receptor in Alzheimer’s disease: Identifying novel inhibitors using computational approaches. Front. Pharmacol. 2023, 14, 1208968. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Babaei, P. NMDA and AMPA receptors dysregulation in Alzheimer’s disease. Eur. J. Pharmacol. 2021, 908, 174310. [Google Scholar] [CrossRef]
- Ning, L.; Shen, R.; Xie, B.; Jiang, Y.; Geng, X.; Dong, W. AMPA receptors in Alzheimer disease: Pathological changes and potential therapeutic targets. J. Neuropathol. Exp. Neurol. 2024, 83, 895–906. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.; Bennett, M.; Zukin, R. Ampa/kainate receptor gene expression in normal and alzheimer’s disease hippocampus. Neuroscience 1994, 61, 41–49. [Google Scholar] [CrossRef]
- Srivastava, A.; Das, B.; Yao, A.Y.; Yan, R. Metabotropic Glutamate Receptors in Alzheimer’s Disease Synaptic Dysfunction: Therapeutic Opportunities and Hope for the Future. J. Alzheimer’s Dis. 2020, 78, 1345–1361. [Google Scholar] [CrossRef]
- Li, S.H.; Abd-Elrahman, K.S.; Ferguson, S.S. Targeting mGluR2/3 for treatment of neurodegenerative and neuropsychiatric diseases. Pharmacol. Ther. 2022, 239, 108275. [Google Scholar] [CrossRef]
- Abd-Elrahman, K.S.; Sarasija, S.; Ferguson, S.S. The Role of Neuroglial Metabotropic Glutamate Receptors in Alzheimer’s Disease. Curr. Neuropharmacol. 2023, 21, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Wood, O.W.G.; Yeung, J.H.Y.; Faull, R.L.M.; Kwakowsky, A. EAAT2 as a therapeutic research target in Alzheimer’s disease: A systematic review. Front. Neurosci. 2022, 16, 952096. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Serviddio, G.; Gaetani, S.; Romano, A.; Dipasquale, P.; Cianci, S.; Bellanti, F.; Laconca, L.; Romano, A.D.; Padalino, I.; et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol. Aging 2011, 33, 1121.e1–1121.e12. [Google Scholar] [CrossRef]
- Rupsingh, R.; Borrie, M.; Smith, M.; Wells, J.; Bartha, R. Reduced hippocampal glutamate in Alzheimer disease. Neurobiol. Aging 2011, 32, 802–810. [Google Scholar] [CrossRef]
- Zott, B.; Konnerth, A. Impairments of glutamatergic synaptic transmission in Alzheimer’s disease. Semin. Cell Dev. Biol. 2023, 139, 24–34. [Google Scholar] [CrossRef]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in Moderate-to-Severe Alzheimer’s Disease. New Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Greig, S.L. Memantine ER/Donepezil: A Review in Alzheimer’s Disease. CNS Drugs 2015, 29, 963–970. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front Neurosci 2015, 9, 170294. [Google Scholar] [CrossRef]
- Maestú, F.; de Haan, W.; Busche, M.A.; DeFelipe, J. Neuronal excitation/inhibition imbalance: Core element of a translational perspective on Alzheimer pathophysiology. Ageing Res. Rev. 2021, 69, 101372. [Google Scholar] [CrossRef]
- Deardorff, W.J.; Grossberg, G.T. A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer’s disease. Drug Des. Dev. Ther. 2016, 10, 3267–3279. [Google Scholar] [CrossRef] [PubMed]
- Rudy, C.C.; Hunsberger, H.C.; Weitzner, D.S.; Reed, M.N. The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology of Alzheimer’s Disease. Aging Dis. 2015, 6, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine-Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Benarroch, E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef]
- Fairless, R.; Bading, H.; Diem, R. Pathophysiological Ionotropic Glutamate Signalling in Neuroinflammatory Disease as a Therapeutic Target. Front. Neurosci. 2021, 15, 741280. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Wang, T.; Kuang, W.; Chen, W.; Xu, W.; Zhang, L.; Li, Y.; Li, H.; Peng, Y.; Chen, Y.; Wang, B.; et al. A phase II randomized trial of sodium oligomannate in Alzheimer’s dementia. Alzheimer’s Res. Ther. 2020, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Luboeinski, J.; Tetzlaff, C. Memory consolidation and improvement by synaptic tagging and capture in recurrent neural networks. Commun. Biol. 2021, 4, 110. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-X.; Wang, Y.; Qin, Z.-H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Bellone, C.; Lüscher, C.; Mameli, M. Mechanisms of synaptic depression triggered by metabotropic glutamate receptors. Cell. Mol. Life Sci. 2008, 65, 2913–2923. [Google Scholar] [CrossRef]
- Xiao, S.; Chan, P.; Wang, T.; Hong, Z.; Wang, S.; Kuang, W.; He, J.; Pan, X.; Zhou, Y.; Ji, Y.; et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimer’s Res. Ther. 2021, 13, 62. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA receptors in the central nervous system. Methods Mol. Biol. 2017, 1677, 1. [Google Scholar] [CrossRef]
- Hanson, J.E.; Yuan, H.; Perszyk, R.E.; Banke, T.G.; Xing, H.; Tsai, M.-C.; Menniti, F.S.; Traynelis, S.F. Therapeutic potential of N-methyl-D-aspartate receptor modulators in psychiatry. Neuropsychopharmacology 2023, 49, 51–66. [Google Scholar] [CrossRef]
- Chen, H.; Dong, Y.; Wu, Y.; Yi, F. Targeting NMDA receptor signaling for therapeutic intervention in brain disorders. Prog. Neurobiol. 2023, 34, 635–647. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Monaghan, D.T.; Irvine, M.W.; Costa, B.M.; Fang, G.; Jane, D.E. Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochem. Int. 2012, 61, 581–592. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate Use Recommendations. J. Prev. Alzheimer’s Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.C.; Wang, Y.T.; Ren, J. Basic information about memantine and its treatment of Alzheimer’s disease and other clinical applications. Ibrain 2023, 9, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-Methyl D-Aspartate (NMDA) Receptor Antagonists and Memantine Treatment for Alzheimer’s Disease, Vascular Dementia and Parkinson’s Disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef]
- Tampi, R.R.; van Dyck, C.H. Memantine: Efficacy and safety in mild-to-severe Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 245–258. [Google Scholar] [CrossRef]
- Tamm, L.N.; Miller, R. NMDA Receptors in Stroke: Pathways and Potential Treatments. J. Undergrad. Res. 2024, 22. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Sharma, N.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Felemban, S.G.; Alsubayiel, A.M.; et al. “Aducanumab” making a comeback in Alzheimer’s disease: An old wine in a new bottle. Biomedicine & Pharmacotherapy 2022, 148, 112746. [Google Scholar] [CrossRef]
- Larkin, H.D. Lecanemab Gains FDA Approval for Early Alzheimer Disease. JAMA 2023, 329, 363. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimer’s Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef]
- Tari, P.K.; Parsons, C.G.; Collingridge, G.L.; Rammes, G. Memantine: Updating a rare success story in pro-cognitive therapeutics. Neuropharmacology 2023, 244, 109737. [Google Scholar] [CrossRef]
- Dominguez, E.; Chin, T.-Y.; Chen, C.-P.; Wu, T.-Y. Management of moderate to severe Alzheimer’s disease: Focus on memantine. Taiwan. J. Obstet. Gynecol. 2011, 50, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Harris, E. Alzheimer Drug Lecanemab Gains Traditional FDA Approval. JAMA 2023, 330, 495. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.-S.H.; Tseng, P.-T.; Liang, C.-S. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 1630–1632. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Nery, E.S.M.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Hippius, H.; Neundörfer, G. The discovery of Alzheimer’s disease. Dialog-Clin. Neurosci. 2003, 5, 101–108. [Google Scholar] [CrossRef]
- Hajjo, R.; Sabbah, D.A.; Abusara, O.H.; Al Bawab, A.Q. A Review of the Recent Advances in Alzheimer’s Disease Research and the Utilization of Network Biology Approaches for Prioritizing Diagnostics and Therapeutics. Diagnostics 2022, 12, 2975. [Google Scholar] [CrossRef]
- Burns, S.; Selman, A.; Sehar, U.; Rawat, P.; Reddy, A.P.; Reddy, P.H. Therapeutics of Alzheimer’s Disease: Recent Developments. Antioxidants 2022, 11, 2402. [Google Scholar] [CrossRef]
- Jin, L.E.; Wang, M.; Galvin, V.C.; Lightbourne, T.C.; Conn, P.J.; Arnsten, A.F.; Paspalas, C.D. mGluR2 versus mGluR3 Metabotropic Glutamate Receptors in Primate Dorsolateral Prefrontal Cortex: Postsynaptic mGluR3 Strengthen Working Memory Networks. Cereb. Cortex 2017, 28, 974–987. [Google Scholar] [CrossRef]
- Kumar, A.; Dhull, D.K.; Mishra, P.S. Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front. Neurosci. 2015, 9, 215. [Google Scholar] [CrossRef]
- Thal, D.R. Excitatory amino acid transporter EAAT-2 in tangle-bearing neurons in Alzheimer’s disease. Brain Pathol. 2002, 12, 405–411. [Google Scholar] [CrossRef]
- Manisha, C.; Selvaraj, A.; Jubie, S.; Nanjan, C.M.J.; Joghee, N.M.; Clement, J.P.; Justin, A. Positive allosteric activation of glial EAAT-2 transporter protein: A novel strategy for Alzheimer’s disease. Med. Hypotheses 2020, 142, 109794. [Google Scholar] [CrossRef] [PubMed]
- O’donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. Npj Schizophr. 2017, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Foster, J.B.; Lin, C.-L.G. Glutamate transporter EAAT2: Regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell. Mol. Life Sci. 2015, 72, 3489–3506. [Google Scholar] [CrossRef] [PubMed]
- Kabir, T.; Sufian, M.A.; Uddin, S.; Begum, M.M.; Akhter, S.; Islam, A.; Mathew, B.; Islam, S.; Amran, S.; Ashraf, G.M. NMDA Receptor Antagonists: Repositioning of Memantine as a Multitargeting Agent for Alzheimer’s Therapy. Curr. Pharm. Des. 2019, 25, 3506–3518. [Google Scholar] [CrossRef]
- Hu, N.-W.; Klyubin, I.; Anwyl, R.; Rowan, M.J. GluN2B subunit-containing NMDA receptor antagonists prevent Aβ-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20504–20509. [Google Scholar] [CrossRef]
- Bayraktar, A.; Li, X.; Kim, W.; Zhang, C.; Turkez, H.; Shoaie, S.; Mardinoglu, A. Drug repositioning targeting glutaminase reveals drug candidates for the treatment of Alzheimer’s disease patients. J. Transl. Med. 2023, 21, 332. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Piras, A.; Ismail, M.A.M.; Manchanda, S.; Eyjolfsdottir, H.; Saido, T.C.; Johansson, J.; Eriksdotter, M.; Winblad, B.; Nilsson, P. Targeting Alzheimer’s disease with gene and cell therapies. J. Intern. Med. 2018, 284, 2–36. [Google Scholar] [CrossRef]
- Takahashi, K.; Kong, Q.; Lin, Y.; Stouffer, N.; Schulte, D.A.; Lai, L.; Liu, Q.; Chang, L.-C.; Dominguez, S.; Xing, X.; et al. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J. Exp. Med. 2015, 212, 319–332. [Google Scholar] [CrossRef]
- Bhole, R.P.; Chikhale, R.V.; Rathi, K.M. Current biomarkers and treatment strategies in Alzheimer disease: An overview and future perspectives. IBRO Neurosci. Rep. 2024, 16, 8–42. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Márquez, F.; Yassa, M.A. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Young, P.N.E.; Estarellas, M.; Coomans, E.; Srikrishna, M.; Beaumont, H.; Maass, A.; Venkataraman, A.V.; Lissaman, R.; Jiménez, D.; Betts, M.J.; et al. Imaging biomarkers in neurodegeneration: Current and future practices. Alzheimer’s Res. Ther. 2020, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- van Oostveen, W.M.; de Lange, E.C.M. Imaging Techniques in Alzheimer’s Disease: A Review of Applications in Early Diagnosis and Longitudinal Monitoring. Int. J. Mol. Sci. 2021, 22, 2110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Huber, H.; Huber, H.; Blennow, K.; Blennow, K.; Zetterberg, H.; Zetterberg, H.; Boada, M.; Boada, M.; Jeromin, A.; Jeromin, A.; et al. Biomarkers of Alzheimer’s disease and neurodegeneration in dried blood spots—A new collection method for remote settings. Alzheimer’s Dement. 2024, 20, 2340–2352. [Google Scholar] [CrossRef]
- Gautam, D.; Naik, U.P.; Naik, M.U.; Yadav, S.K.; Chaurasia, R.N.; Dash, D. Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications. Biomolecules 2023, 13, 1609. [Google Scholar] [CrossRef]
- De Ninno, G.; Giuffrè, G.M.; Urbani, A.; Baroni, S. Current perspectives on Alzheimer’s disease fluid biomarkers and future challenges: A narrative review. J. Lab. Precis. Med. 2024, 9, 25. [Google Scholar] [CrossRef]
- Khoury, R.; Ghossoub, E. Diagnostic biomarkers of Alzheimer’s disease: A state-of-the-art review. Biomark. Neuropsychiatry 2019, 1, 100005. [Google Scholar] [CrossRef]
- Qiu, S.; Cai, Y.; Yao, H.; Lin, C.; Xie, Y.; Tang, S.; Zhang, A. Small molecule metabolites: Discovery of biomarkers and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 132. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef]
- Singh, B.; Day, C.M.; Abdella, S.; Garg, S. Alzheimer’s disease current therapies, novel drug delivery systems and future directions for better disease management. J. Control. Release 2024, 367, 402–424. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Dinh, L.; Lee, S.; Abuzar, S.M.; Park, H.; Hwang, S.-J. Formulation, Preparation, Characterization, and Evaluation of Dicarboxylic Ionic Liquid Donepezil Transdermal Patches. Pharmaceutics 2022, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.; Rezaei, K.; Quazi, A.; Goldmeier, G.; Silverglate, B.; Grossberg, G.T. The donepezil transdermal system for the treatment of patients with mild, moderate, or severe Alzheimer’s disease: A critical review. Expert Rev. Neurother. 2024, 24, 607–614. [Google Scholar] [CrossRef]
- Siddique, Y.H.; Naz, F.; Rahul; Varshney, H. Comparative study of rivastigmine and galantamine on the transgenic Drosophila model of Alzheimer’s disease. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100120. [Google Scholar] [CrossRef]
- Miculas, D.C.; Negru, P.A.; Bungau, S.G.; Behl, T.; Hassan, S.S.U.; Tit, D.M. Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions. Cells 2022, 12, 131. [Google Scholar] [CrossRef]
- Dighe, S.N.; De la Mora, E.; Chan, S.; Kantham, S.; McColl, G.; Miles, J.A.; Veliyath, S.K.; Sreenivas, B.Y.; Nassar, Z.D.; Silman, I.; et al. Rivastigmine and metabolite analogues with putative Alzheimer’s disease-modifying properties in a Caenorhabditis elegans model. Commun. Chem. 2019, 2, 35. [Google Scholar] [CrossRef]
- Guo, J.; Wang, Z.; Liu, R.; Huang, Y.; Zhang, N.; Zhang, R. Memantine, Donepezil, or Combination Therapy—What is the best therapy for Alzheimer’s Disease? A Network Meta-Analysis. Brain Behav. 2020, 10, e01831. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Bosch, M.E.; Dodiya, H.B.; Michalkiewicz, J.; Lee, C.; Shaik, S.M.; Weigle, I.Q.; Zhang, C.; Osborn, J.; Nambiar, A.; Patel, P.; et al. Sodium oligomannate alters gut microbiota, reduces cerebral amyloidosis and reactive microglia in a sex-specific manner. Mol. Neurodegener. 2024, 19, 18. [Google Scholar] [CrossRef]
- Huang, L.-K.; Kuan, Y.-C.; Lin, H.-W.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease: A 2020–2023 update. J. Biomed. Sci. 2023, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- Haddad, H.W.; Malone, G.W.; Comardelle, N.J.; Degueure, A.E.; Kaye, A.M.; Kaye, A.D. Aducanumab, a Novel Anti-Amyloid Monoclonal Antibody, for the Treatment of Alzheimer’s Disease: A Comprehensive Review. Health Psychol. Res. 2022, 10, 31925. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chu, F.; Zhu, F.; Zhu, J. Impact of Anti-amyloid-beta Monoclonal Antibodies on the Pathology and Clinical Profile of Alzheimer’s Disease: A Focus on Aducanumab and Lecanemab. Front Aging Neurosci 2022, 14, 870517. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Osse, A.M.L.; Cammann, D.; Powell, J.; Chen, J. Anti-Amyloid Monoclonal Antibodies for the Treatment of Alzheimer’s Disease. BioDrugs 2023, 38, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Slomkowski, M.; Hefting, N.; Chen, D.; Larsen, K.G.; Kohegyi, E.; Hobart, M.; Cummings, J.L.; Grossberg, G.T. Brexpiprazole for the Treatment of Agitation in Alzheimer Dementia. JAMA Neurol. 2023, 80, 1307–1316. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, F.; Tang, J.; Zhou, Y.; Fu, Z.; Zhang, P.; Haines, J.L.; Leverenz, J.B.; Gan, L.; Hu, J.; et al. Artificial intelligence and open science in discovery of disease-modifying medicines for Alzheimer’s disease. Cell Rep. Med. 2024, 5, 101379. [Google Scholar] [CrossRef]
- Wu, T.; Lin, R.; Cui, P.; Yong, J.; Yu, H.; Li, Z. Deep learning-based drug screening for the discovery of potential therapeutic agents for Alzheimer’s disease. J. Pharm. Anal. 2024, 14, 101022. [Google Scholar] [CrossRef]
- Patwekar, M.; Patwekar, F.; Shaikh, D.; Fatema, S.R.; Aher, S.J.; Sharma, R. Receptor-based approaches and therapeutic targets in Alzheimer’s disease along with role of AI in drug designing: Unraveling pathologies and advancing treatment strategies. Appl. Chem. Eng. 2023, 6. [Google Scholar] [CrossRef]
- Lukiw, W.J. Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer’s disease (AD). Expert Opin. Emerg. Drugs 2012, 17, 43–60. [Google Scholar] [CrossRef]
- Xiao, D.; Zhang, C. Current therapeutics for Alzheimer’s disease and clinical trials. Open Explor. 2024, 3, 255–271. [Google Scholar] [CrossRef]
- Hoffmann, T.; Rahfeld, J.-U.; Schenk, M.; Ponath, F.; Makioka, K.; Hutter-Paier, B.; Lues, I.; Lemere, C.A.; Schilling, S. Combination of the Glutaminyl Cyclase Inhibitor PQ912 (Varoglutamstat) and the Murine Monoclonal Antibody PBD-C06 (m6) Shows Additive Effects on Brain Aβ Pathology in Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 11791. [Google Scholar] [CrossRef] [PubMed]
- Vijverberg, E.G.B.; Axelsen, T.M.; Bihlet, A.R.; Henriksen, K.; Weber, F.; Fuchs, K.; Harrison, J.E.; Kühn-Wache, K.; Alexandersen, P.; Prins, N.D.; et al. Rationale and study design of a randomized, placebo-controlled, double-blind phase 2b trial to evaluate efficacy, safety, and tolerability of an oral glutaminyl cyclase inhibitor varoglutamstat (PQ912) in study participants with MCI and mild AD—VIVIAD. Alzheimer’s Res. Ther. 2021, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Seripa, D.; Solfrizzi, V.; Imbimbo, B.P.; Santamato, A.; Lozupone, M.; Capozzo, R.; Prete, C.; Pilotto, A.; Greco, A.; et al. Tau aggregation inhibitors: The future of Alzheimer’s pharmacotherapy? Expert Opin. Pharmacother. 2015, 17, 457–461. [Google Scholar] [CrossRef]
- Cummings, J.L.; Gonzalez, M.I.; Pritchard, M.C.; May, P.C.; Toledo-Sherman, L.M.; Harris, G.A. The therapeutic landscape of tauopathies: Challenges and prospects. Alzheimer’s Res. Ther. 2023, 15, 168. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Daniele, A.; Lozupone, M. Passive tau-based immunotherapy for tauopathies. Handb. Clin. Neurol. 2023, 196, 611–619. [Google Scholar] [CrossRef]
- Chen, T.-S.; Huang, T.-H.; Lai, M.-C.; Huang, C.-W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- Pal, M.M. Glutamate: The Master Neurotransmitter and Its Implications in Chronic Stress and Mood Disorders. Front. Hum. Neurosci. 2021, 15, 722323. [Google Scholar] [CrossRef]
- Hoffmann, J.; Charles, A. Glutamate and Its Receptors as Therapeutic Targets for Migraine. Neurotherapeutics 2018, 15, 361–370. [Google Scholar] [CrossRef]
- Conway, M.E. Alzheimer’s disease: Targeting the glutamatergic system. Biogerontology 2020, 21, 257–274. [Google Scholar] [CrossRef]
- Pinky, P.D.; Pfitzer, J.C.; Senfeld, J.; Hong, H.; Bhattacharya, S.; Suppiramaniam, V.; Qureshi, I.; Reed, M.N. Recent Insights on Glutamatergic Dysfunction in Alzheimer’s Disease and Therapeutic Implications. Neuroscientist 2022, 29, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, P.; Du, T.; Jiang, W.; Peng, L.; Butterworth, R.F. Pathogenesis of hepatic encephalopathy and brain edema in acute liver failure: Role of glutamine redefined. Neurochem. Int. 2012, 60, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Schneider, M.; Müller, C.E.; Vázquez, S.; Griñán-Ferré, C.; Pallàs, M. NMDA receptor antagonists reduce amyloid-β deposition by modulating calpain-1 signaling and autophagy, rescuing cognitive impairment in 5XFAD mice. Cell. Mol. Life Sci. 2022, 79, 408. [Google Scholar] [CrossRef]
- Ebrahimi, Z.; Talaei, S.; Aghamiri, S.; Goradel, N.H.; Jafarpour, A.; Negahdari, B. Overcoming the blood–brain barrier in neurodegenerative disorders and brain tumours. IET Nanobiotechnology 2020, 14, 441–448. [Google Scholar] [CrossRef]
- Aragón-González, A.; Shaw, P.J.; Ferraiuolo, L. Blood–Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 15271. [Google Scholar] [CrossRef]
- Song, Q.; Li, J.; Li, T.; Li, H. Nanomaterials that Aid in the Diagnosis and Treatment of Alzheimer’s Disease, Resolving Blood–Brain Barrier Crossing Ability. Adv. Sci. 2024, 11, 2403473. [Google Scholar] [CrossRef]
- Xie, J.; Shen, Z.; Anraku, Y.; Kataoka, K.; Chen, X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials 2019, 224, 119491. [Google Scholar] [CrossRef]
- Wu, D.-D.; Salah, Y.A.; Ngowi, E.E.; Zhang, Y.-X.; Khattak, S.; Khan, N.H.; Li, T.; Guo, Z.-H.; Wang, Y.-M.; Ji, X.-Y. Nanotechnology prospects in brain therapeutics concerning gene-targeting and nose-to-brain administration. iScience 2023, 26, 107321. [Google Scholar] [CrossRef]
- Nunes, D.; Loureiro, J.A.; Pereira, M.C. Drug Delivery Systems as a Strategy to Improve the Efficacy of FDA-Approved Alzheimer’s Drugs. Pharmaceutics 2022, 14, 2296. [Google Scholar] [CrossRef]
- Peng, Y.; Jin, H.; Xue, Y.-H.; Chen, Q.; Yao, S.-Y.; Du, M.-Q.; Liu, S. Current and future therapeutic strategies for Alzheimer’s disease: An overview of drug development bottlenecks. Front. Aging Neurosci. 2023, 15, 1206572. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Type | Dysregulation in Alzheimer’s Disease (AD) | Therapeutic Targeting | Reference |
|---|---|---|---|
| NMDA Receptors |
|
| [96,97,98,99] |
| AMPA Receptors |
|
| [100,101] |
| Kainate Receptors |
|
| [102] |
| mGluR1/5 (Group I) |
|
| [103] |
| mGluR2/3 (Group II) |
|
| [104,105] |
| EAAT Transporters |
|
| [106] |
| Therapeutic Strategy | Target/Mechanism | Clinical Status | Challenges/Limitations | References |
|---|---|---|---|---|
| NMDA Receptor Antagonists | Blocks pathological overactivation of NMDA receptors, reducing excitotoxicity without disrupting normal signaling | Memantine (FDA-approved for moderate-to-severe AD) | Limited efficacy in advanced stages of AD; potential off-target effects on normal neurotransmission | [96,97,158] |
| Selective NMDA Subunit Antagonists | Targets specific NMDA receptor subunits (e.g., NR2B) to enhance selectivity and reduce side effectsGluN2B subunit-containing NMDARs preventing cognitive deficits in early AD | In preclinical and early clinical trials | High specificity is required to avoid interference with essential NMDA receptor functions | [159] |
| mGluR5 Antagonists | Reduces Aβ-induced excitotoxicity by inhibiting Group I mGLURs (mGluR5) | In early-stage clinical trials for AD | Potential for disrupting normal mGluR5-mediated plasticity and learning | [153] |
| mGluR2/3 Agonists | Activates Group II mGLURs to inhibit GLU release, reducing excitotoxicity and neuronal damage | In clinical trials for neuroprotection in AD | Limited understanding of long-term effects on synaptic transmission | [104] |
| EAAT Enhancers | Enhances the function of EAAT transporters to clear excess GLU from the synaptic cleft | Experimental phase, preclinical research | Difficulty in delivering EAAT-enhancing compounds across the blood–brain barrier | [106] |
| Glutaminase Inhibitors | Reduces GLU synthesis by inhibiting the conversion of GLN to GLU | Investigational, in preclinical trials for neurodegenerative diseases | Potential disruption of essential GLU-dependent brain functions | [160] |
| Gene Therapy Approaches | Targets genes involved in the GLU metabolism or receptor regulation (e.g., increasing EAAT expression) | Experimental, early-stage research | Ethical and technical challenges in gene therapy; long-term safety not yet established | [161,162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puranik, N.; Song, M. Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target. Molecules 2024, 29, 5744. https://doi.org/10.3390/molecules29235744
Puranik N, Song M. Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target. Molecules. 2024; 29(23):5744. https://doi.org/10.3390/molecules29235744
Chicago/Turabian StylePuranik, Nidhi, and Minseok Song. 2024. "Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target" Molecules 29, no. 23: 5744. https://doi.org/10.3390/molecules29235744
APA StylePuranik, N., & Song, M. (2024). Glutamate: Molecular Mechanisms and Signaling Pathway in Alzheimer’s Disease, a Potential Therapeutic Target. Molecules, 29(23), 5744. https://doi.org/10.3390/molecules29235744