
Targeting EZH2 in Cancer: Mechanisms, Pathways, and Therapeutic Potential

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
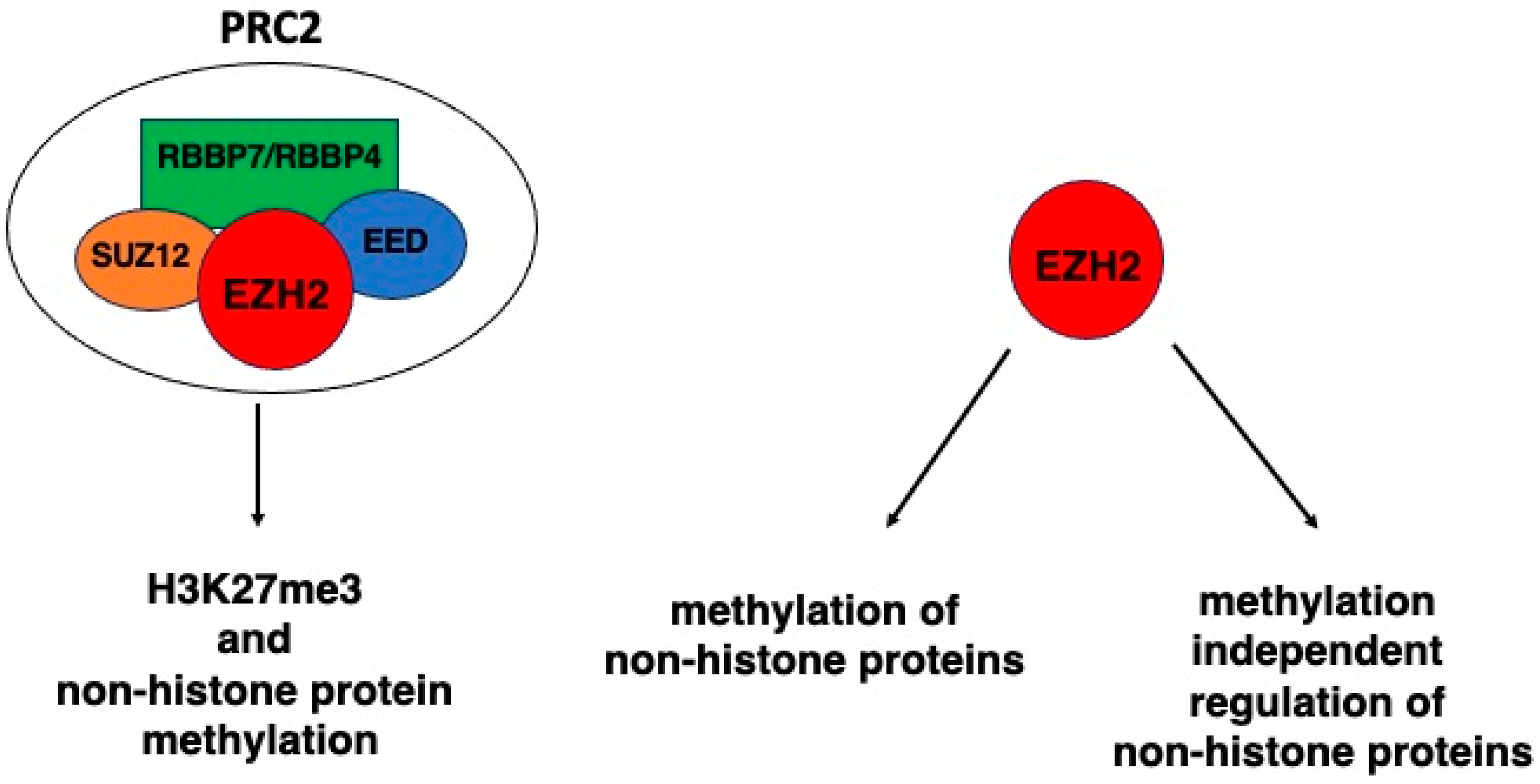
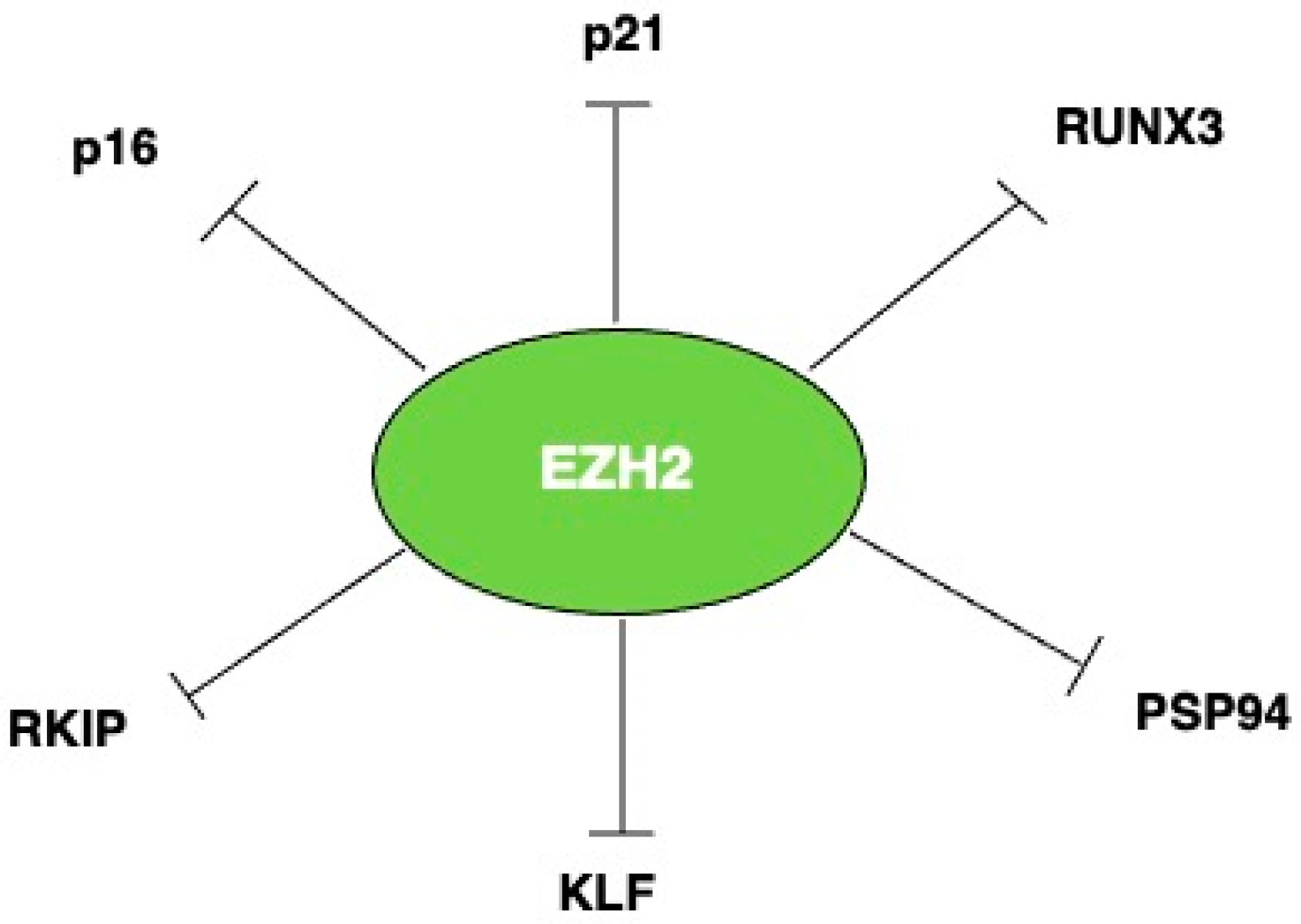
2. EZH2 and H3K27 and Tumor Suppressor Genes
3. EZH2 and Non-Histone Protein Methylation: The Case of GATA4, RORα, and PLZF
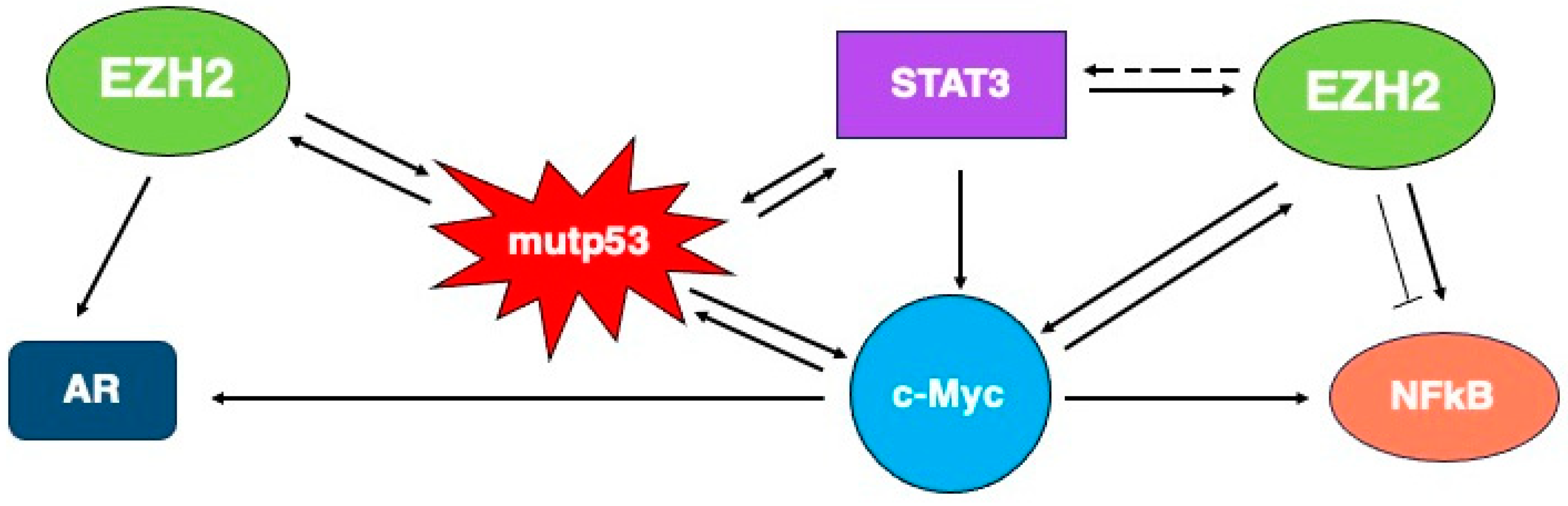
EZH2 and STAT3
4. EZH2 and Non-Histone Protein Regulation Independent on Methylation
4.1. EZH2 and p53
4.2. EZH2 and c-Myc
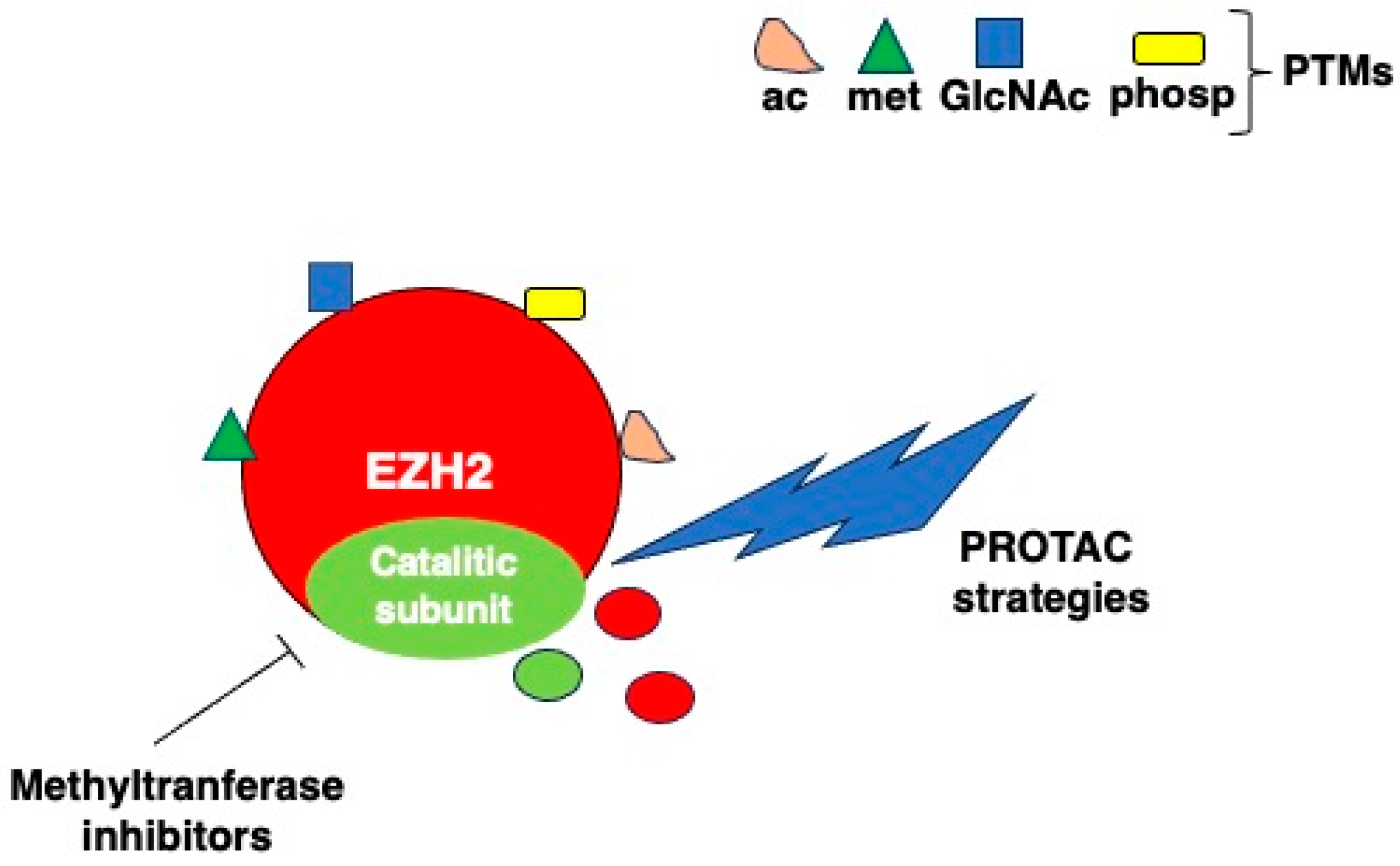
5. EZH2 Posttranslational Modifications
6. EZH2 and Tumor Immune Escape
7. Targeting EZH2 in Cancer Therapy
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.; Mignon, C.; Hetet, G.; Grandchamps, B.; Fontes, M.; Colleaux, L. The human EZH2 gene: Genomic organisation and revised mapping in 7q35 within the critical region for malignant myeloid disorders. Eur. J. Hum. Genet. 2000, 8, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Laible, G.; Dorn, R.; Reuter, G. SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci. 1998, 54, 80–93. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef]
- Muller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; de la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Curry, E.; Green, I.; Chapman-Rothe, N.; Shamsaei, E.; Kandil, S.; Cherblanc, F.L.; Payne, L.; Bell, E.; Ganesh, T.; Srimongkolpithak, N.; et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenetics 2015, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Sun, S.; Yu, F.; Xu, D.; Zheng, H.; Li, M. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188700. [Google Scholar] [CrossRef]
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2016, 7, 2284–2296. [Google Scholar] [CrossRef]
- Karanikolas, B.D.; Figueiredo, M.L.; Wu, L. Polycomb group protein enhancer of zeste 2 is an oncogene that promotes the neoplastic transformation of a benign prostatic epithelial cell line. Mol. Cancer Res. 2009, 7, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Chen, J.; Ma, M.; Cai, M.; Xu, F.; Wang, G.; Tao, K.; Shuai, X. Inhibiting enhancer of zeste homolog 2 promotes cellular senescence in gastric cancer cells SGC-7901 by activation of p21 and p16. DNA Cell Biol. 2014, 33, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Beke, L.; Nuytten, M.; Van Eynde, A.; Beullens, M.; Bollen, M. The gene encoding the prostatic tumor suppressor PSP94 is a target for repression by the Polycomb group protein EZH2. Oncogene 2007, 26, 4590–4595. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef]
- Fujii, S.; Ito, K.; Ito, Y.; Ochiai, A. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J. Biol. Chem. 2008, 283, 17324–17332. [Google Scholar] [CrossRef]
- Taniguchi, H.; Jacinto, F.V.; Villanueva, A.; Fernandez, A.F.; Yamamoto, H.; Carmona, F.J.; Puertas, S.; Marquez, V.E.; Shinomura, Y.; Imai, K.; et al. Silencing of Kruppel-like factor 2 by the histone methyltransferase EZH2 in human cancer. Oncogene 2012, 31, 1988–1994. [Google Scholar] [CrossRef]
- Ren, G.; Baritaki, S.; Marathe, H.; Feng, J.; Park, S.; Beach, S.; Bazeley, P.S.; Beshir, A.B.; Fenteany, G.; Mehra, R.; et al. Polycomb protein EZH2 regulates tumor invasion via the transcriptional repression of the metastasis suppressor RKIP in breast and prostate cancer. Cancer Res. 2012, 72, 3091–3104. [Google Scholar] [CrossRef]
- Deb, G.; Singh, A.K.; Gupta, S. EZH2: Not EZHY (easy) to deal. Mol. Cancer Res. 2014, 12, 639–653. [Google Scholar] [CrossRef]
- He, A.; Shen, X.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Wang, G.; Marquez, V.E.; Orkin, S.H.; Pu, W.T. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes. Dev. 2012, 26, 37–42. [Google Scholar] [CrossRef]
- Moretti, R.M.; Montagnani Marelli, M.; Motta, M.; Limonta, P. Role of the orphan nuclear receptor ROR alpha in the control of the metastatic behavior of androgen-independent prostate cancer cells. Oncol. Rep. 2002, 9, 1139–1143. [Google Scholar] [PubMed]
- Lee, J.M.; Kim, I.S.; Kim, H.; Lee, J.S.; Kim, K.; Yim, H.Y.; Jeong, J.; Kim, J.H.; Kim, J.Y.; Lee, H.; et al. RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol. Cell 2010, 37, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Xu, D.; Lun, A.T.; Kueh, A.J.; van Gisbergen, K.P.; Iannarella, N.; Li, X.; Yu, L.; Wang, D.; Williams, B.R.; et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep. 2017, 18, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Savage, A.K.; Constantinides, M.G.; Han, J.; Picard, D.; Martin, E.; Li, B.; Lantz, O.; Bendelac, A. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 2008, 29, 391–403. [Google Scholar] [CrossRef]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef]
- Hu, Y.; Dong, Z.; Liu, K. Unraveling the complexity of STAT3 in cancer: Molecular understanding and drug discovery. J. Exp. Clin. Cancer Res. 2024, 43, 23. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314 e297. [Google Scholar] [CrossRef]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Santarelli, R.; D’Orazi, G.; Cirone, M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front. Oncol. 2020, 10, 1102. [Google Scholar] [CrossRef]
- Pan, Y.M.; Wang, C.G.; Zhu, M.; Xing, R.; Cui, J.T.; Li, W.M.; Yu, D.D.; Wang, S.B.; Zhu, W.; Ye, Y.J.; et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol. Cancer 2016, 15, 79. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, Z.; Li, J.; Hu, T. EZH2 Exacerbates Breast Cancer by Methylating and Activating STAT3 Directly. J. Cancer 2021, 12, 5220–5230. [Google Scholar] [CrossRef] [PubMed]
- Ozes, A.R.; Pulliam, N.; Ertosun, M.G.; Yilmaz, O.; Tang, J.; Copuroglu, E.; Matei, D.; Ozes, O.N.; Nephew, K.P. Protein kinase A-mediated phosphorylation regulates STAT3 activation and oncogenic EZH2 activity. Oncogene 2018, 37, 3589–3600. [Google Scholar] [CrossRef]
- Di Crosta, M.; Arena, A.; Benedetti, R.; Gilardini Montani, M.S.; Cirone, M. 5-AZA Upregulates SOCS3 and PTPN6/SHP1, Inhibiting STAT3 and Potentiating the Effects of AG490 against Primary Effusion Lymphoma Cells. Curr. Issues Mol. Biol. 2024, 46, 2468–2479. [Google Scholar] [CrossRef]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820 e2804. [Google Scholar] [CrossRef]
- Yan, J.; Ng, S.B.; Tay, J.L.; Lin, B.; Koh, T.L.; Tan, J.; Selvarajan, V.; Liu, S.C.; Bi, C.; Wang, S.; et al. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 2013, 121, 4512–4520. [Google Scholar] [CrossRef]
- Benedetti, R.; Di Crosta, M.; D’Orazi, G.; Cirone, M. Post-Translational Modifications (PTMs) of mutp53 and Epigenetic Changes Induced by mutp53. Biology 2024, 13, 508. [Google Scholar] [CrossRef]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerod, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; van de Vijver, M.J.; van Lohuizen, M. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. 2008, 10, R109. [Google Scholar] [CrossRef]
- Tang, X.; Milyavsky, M.; Shats, I.; Erez, N.; Goldfinger, N.; Rotter, V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 2004, 23, 5759–5769. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.Z.; He, Y.Y.; Wang, H.H.; Zhang, H.L.; Zhang, J.; Yan, X.F.; Wang, X.J.; Che, Q.; Ke, J.Q.; Chen, Z.; et al. Mutant p53 induces EZH2 expression and promotes epithelial-mesenchymal transition by disrupting p68-Drosha complex assembly and attenuating miR-26a processing. Oncotarget 2015, 6, 44660–44674. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ding, L.; Wang, D.; Ye, Z.; He, Y.; Ma, L.; Zhu, R.; Pan, Y.; Wu, Q.; Pang, K.; et al. EZH2 cooperates with gain-of-function p53 mutants to promote cancer growth and metastasis. EMBO J. 2019, 38, e99599. [Google Scholar] [CrossRef] [PubMed]
- Versemann, L.; Patil, S.; Steuber, B.; Zhang, Z.; Kopp, W.; Krawczyk, H.E.; Kaulfuss, S.; Wollnik, B.; Strobel, P.; Neesse, A.; et al. TP53-Status-Dependent Oncogenic EZH2 Activity in Pancreatic Cancer. Cancers 2022, 14, 3541. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, R.; Collura, F.; Corrado, V.; Di Crosta, M.; Santarelli, R.; Cirone, M. EZH2 Inhibition by DS3201 Triggers the Kaposi’s Sarcoma-Associated Herpesvirus Lytic Cycle and Potentiates the Effects Induced by SAHA in Primary Effusion Lymphoma Cells. Viruses 2024, 16, 1490. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Gong, L.; Yan, J.; Liu, Q.; Zeng, W.; Williamson, A.; Lim, C.B.; Molloy, M.E.; Little, J.B.; Huang, L.; et al. An EZH2-mediated epigenetic mechanism behind p53-dependent tissue sensitivity to DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 3452–3457. [Google Scholar] [CrossRef]
- Sander, S.; Bullinger, L.; Klapproth, K.; Fiedler, K.; Kestler, H.A.; Barth, T.F.; Moller, P.; Stilgenbauer, S.; Pollack, J.R.; Wirth, T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 2008, 112, 4202–4212. [Google Scholar] [CrossRef]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef]
- Wang, L.; Chen, C.; Song, Z.; Wang, H.; Ye, M.; Wang, D.; Kang, W.; Liu, H.; Qing, G. EZH2 depletion potentiates MYC degradation inhibiting neuroblastoma and small cell carcinoma tumor formation. Nat. Commun. 2022, 13, 12. [Google Scholar] [CrossRef]
- Li, Z.; Li, M.; Wang, D.; Hou, P.; Chen, X.; Chu, S.; Chai, D.; Zheng, J.; Bai, J. Post-translational modifications of EZH2 in cancer. Cell Biosci. 2020, 10, 143. [Google Scholar] [CrossRef]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Chen, S.; Huang, H. Phosphorylation of EZH2 by CDK1 and CDK2: A possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle 2011, 10, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabuddhe, A.A.; Chen, X.; Chung, F.; Velusamy, T.; Lim, M.S.; Elenitoba-Johnson, K.S. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene 2015, 34, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.W.; Lee, H.H.; Huo, L.; Xia, W.; Yang, C.C.; Hsu, J.L.; Li, L.Y.; Lai, C.C.; Chan, L.C.; Cheng, C.C.; et al. GSK3beta inactivation promotes the oncogenic functions of EZH2 and enhances methylation of H3K27 in human breast cancers. Oncotarget 2016, 7, 57131–57144. [Google Scholar] [CrossRef]
- Wan, L.; Xu, K.; Wei, Y.; Zhang, J.; Han, T.; Fry, C.; Zhang, Z.; Wang, Y.V.; Huang, L.; Yuan, M.; et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol. Cell 2018, 69, 279–291 e275. [Google Scholar] [CrossRef]
- Chu, C.S.; Lo, P.W.; Yeh, Y.H.; Hsu, P.H.; Peng, S.H.; Teng, Y.C.; Kang, M.L.; Wong, C.H.; Juan, L.J. O-GlcNAcylation regulates EZH2 protein stability and function. Proc. Natl. Acad. Sci. USA 2014, 111, 1355–1360. [Google Scholar] [CrossRef]
- Lo, P.W.; Shie, J.J.; Chen, C.H.; Wu, C.Y.; Hsu, T.L.; Wong, C.H. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2018, 115, 7302–7307. [Google Scholar] [CrossRef]
- Zeng, Y.; Qiu, R.; Yang, Y.; Gao, T.; Zheng, Y.; Huang, W.; Gao, J.; Zhang, K.; Liu, R.; Wang, S.; et al. Regulation of EZH2 by SMYD2-Mediated Lysine Methylation Is Implicated in Tumorigenesis. Cell Rep. 2019, 29, 1482–1498 e1484. [Google Scholar] [CrossRef]
- Yuan, H.; Han, Y.; Wang, X.; Li, N.; Liu, Q.; Yin, Y.; Wang, H.; Pan, L.; Li, L.; Song, K.; et al. SETD2 Restricts Prostate Cancer Metastasis by Integrating EZH2 and AMPK Signaling Pathways. Cancer Cell 2020, 38, 350–365 e357. [Google Scholar] [CrossRef]
- Wang, X.; Long, Y.; Paucek, R.D.; Gooding, A.R.; Lee, T.; Burdorf, R.M.; Cech, T.R. Regulation of histone methylation by automethylation of PRC2. Genes Dev. 2019, 33, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Sanulli, S.; Justin, N.; Teissandier, A.; Ancelin, K.; Portoso, M.; Caron, M.; Michaud, A.; Lombard, B.; da Rocha, S.T.; Offer, J.; et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol. Cell 2015, 57, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Lu, J.; Huang, B.; Wang, Y.; Dong, M.; Fan, D.; Li, H.; Gao, Y.; Hou, P.; et al. Methylation of EZH2 by PRMT1 regulates its stability and promotes breast cancer metastasis. Cell Death Differ. 2020, 27, 3226–3242. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Eccleston, M.; Clermont, P.L.; Latarani, M.; Male, D.K.; Wang, Y.; Crea, F. EZH2 inhibition: A promising strategy to prevent cancer immune editing. Epigenomics 2020, 12, 1457–1476. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Takada, K.; Tagawa, T.; Hamamoto, R.; Yamada, Y.; Shimokawa, M.; Oda, Y.; Maehara, Y. A Positive Correlation Between the EZH2 and PD-L1 Expression in Resected Lung Adenocarcinomas. Ann. Thorac. Surg. 2019, 107, 393–400. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401 e388. [Google Scholar] [CrossRef]
- Huang, B.; Huang, M.; Li, Q. Cancer-Associated Fibroblasts Promote Angiogenesis of Hepatocellular Carcinoma by VEGF-Mediated EZH2/VASH1 Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819879905. [Google Scholar] [CrossRef]
- Bugide, S.; Green, M.R.; Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 2018, 115, E3509–E3518. [Google Scholar] [CrossRef]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef]
- Pang, B.; Zheng, X.R.; Tian, J.X.; Gao, T.H.; Gu, G.Y.; Zhang, R.; Fu, Y.B.; Pang, Q.; Li, X.G.; Liu, Q. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1alpha signaling. Oncotarget 2016, 7, 45134–45143. [Google Scholar] [CrossRef]
- Xia, L.; Zhu, X.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. 2020, 67, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, J.; Sun, Y.; Wang, J.; Ren, C.; Banerjee, S.; Ouyang, L.; Wang, Y. Targeting EZH2 for cancer therapy: From current progress to novel strategies. Eur. J. Med. Chem. 2022, 238, 114419. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef]
- Dou, F.; Tian, Z.; Yang, X.; Li, J.; Wang, R.; Gao, J. Valemetostat: First approval as a dual inhibitor of EZH1/2 to treat adult T-cell leukemia/lymphoma. Drug Discov. Ther. 2022, 16, 297–299. [Google Scholar] [CrossRef]
- Eich, M.L.; Athar, M.; Ferguson, J.E., 3rd; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef]
- Wang, X.; Cao, W.; Zhang, J.; Yan, M.; Xu, Q.; Wu, X.; Wan, L.; Zhang, Z.; Zhang, C.; Qin, X.; et al. A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. EMBO J. 2017, 36, 1243–1260. [Google Scholar] [CrossRef]
- Lee, S.W.; Oh, Y.M.; Lu, Y.L.; Kim, W.K.; Yoo, A.S. MicroRNAs Overcome Cell Fate Barrier by Reducing EZH2-Controlled REST Stability during Neuronal Conversion of Human Adult Fibroblasts. Dev. Cell 2018, 46, 73–84 e77. [Google Scholar] [CrossRef]
- Corbin, J.; Yu, X.; Jin, J.; Cai, L.; Wang, G.G. EZH2 PROTACs target EZH2- and FOXM1-associated oncogenic nodes, suppressing breast cancer cell growth. Oncogene 2024, 43, 2722–2736. [Google Scholar] [CrossRef]
- Shi, M.X.; Ding, X.; Tang, L.; Cao, W.J.; Su, B.; Zhang, J. PROTAC EZH2 degrader-1 overcomes the resistance of podophyllotoxin derivatives in refractory small cell lung cancer with leptomeningeal metastasis. BMC Cancer 2024, 24, 504. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilardini Montani, M.S.; Benedetti, R.; Cirone, M. Targeting EZH2 in Cancer: Mechanisms, Pathways, and Therapeutic Potential. Molecules 2024, 29, 5817. https://doi.org/10.3390/molecules29245817
Gilardini Montani MS, Benedetti R, Cirone M. Targeting EZH2 in Cancer: Mechanisms, Pathways, and Therapeutic Potential. Molecules. 2024; 29(24):5817. https://doi.org/10.3390/molecules29245817
Chicago/Turabian StyleGilardini Montani, Maria Saveria, Rossella Benedetti, and Mara Cirone. 2024. "Targeting EZH2 in Cancer: Mechanisms, Pathways, and Therapeutic Potential" Molecules 29, no. 24: 5817. https://doi.org/10.3390/molecules29245817
APA StyleGilardini Montani, M. S., Benedetti, R., & Cirone, M. (2024). Targeting EZH2 in Cancer: Mechanisms, Pathways, and Therapeutic Potential. Molecules, 29(24), 5817. https://doi.org/10.3390/molecules29245817