
Iridium-Catalyzed Highly Selective 1,4-Reduction of α,β-Unsaturated Carbonyl Compounds



Abstract
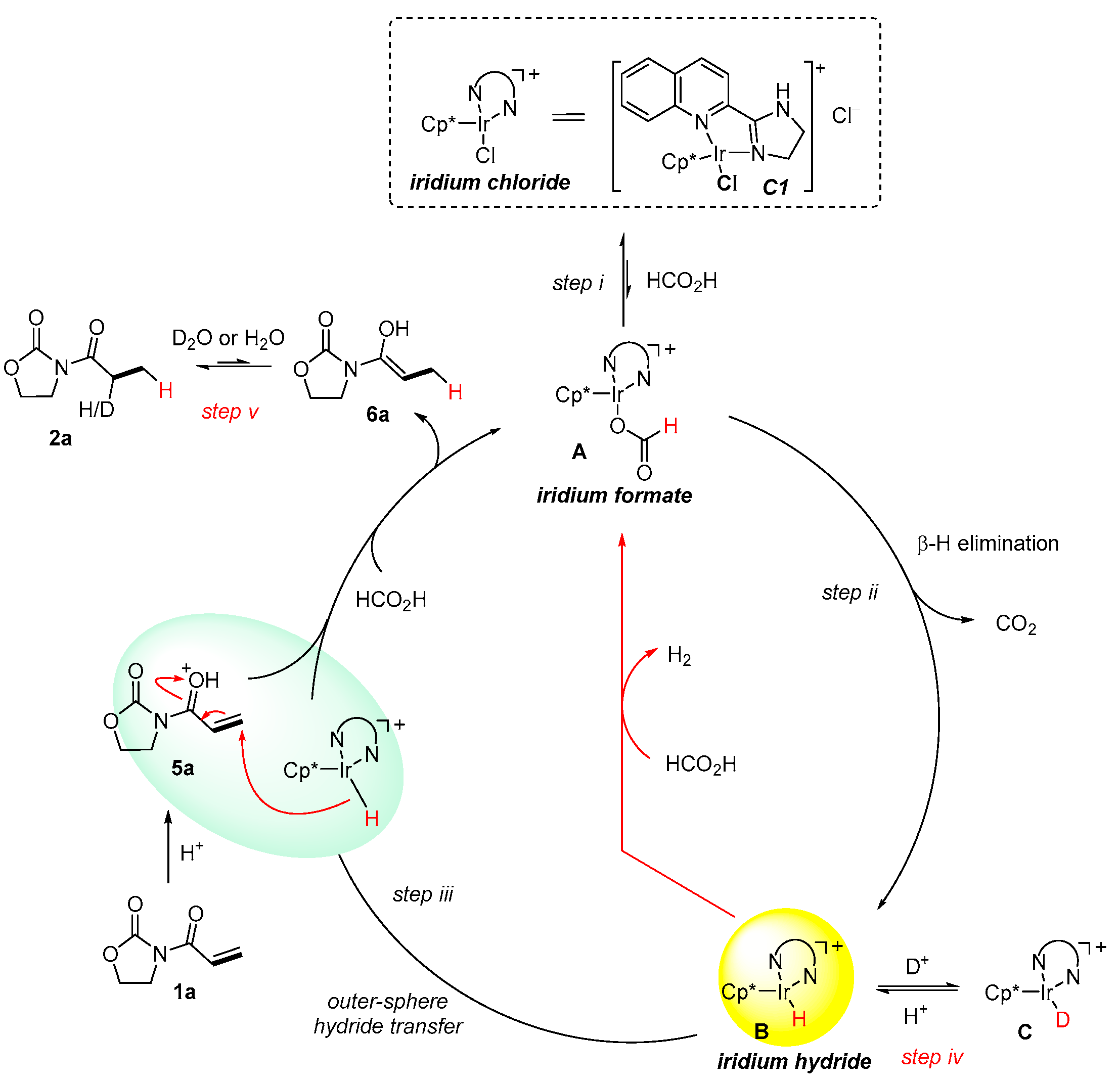
:1. Introduction
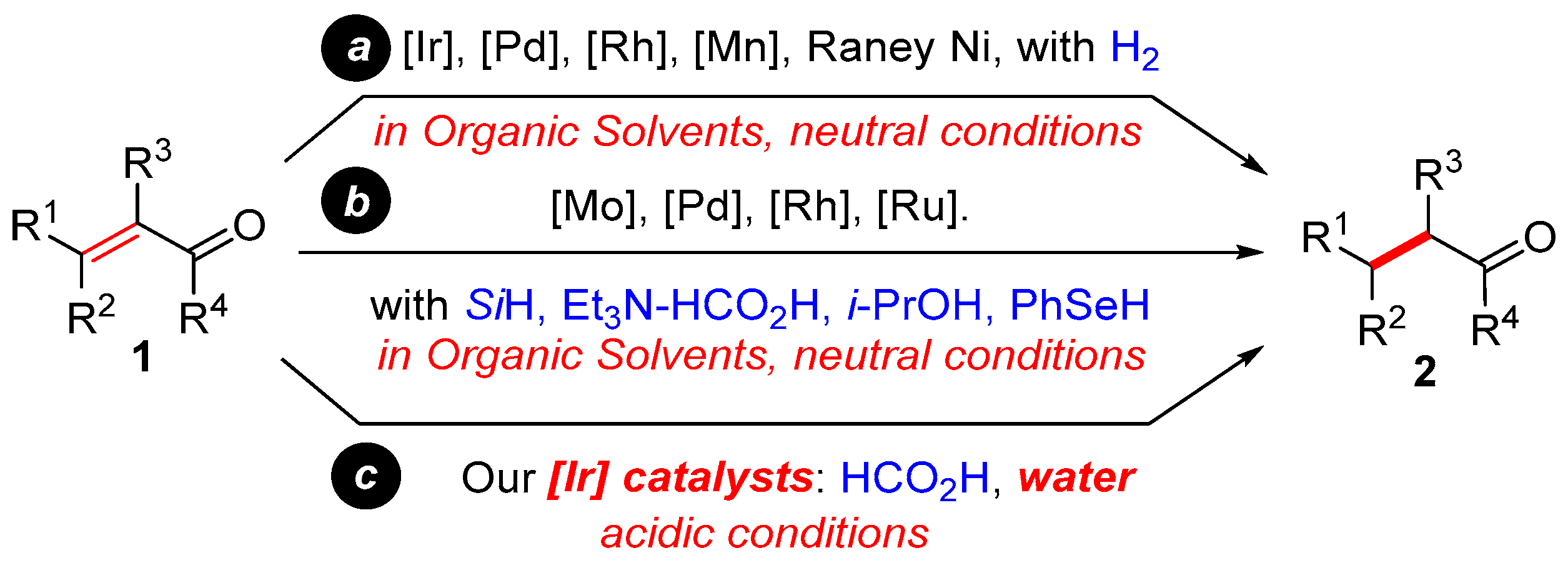
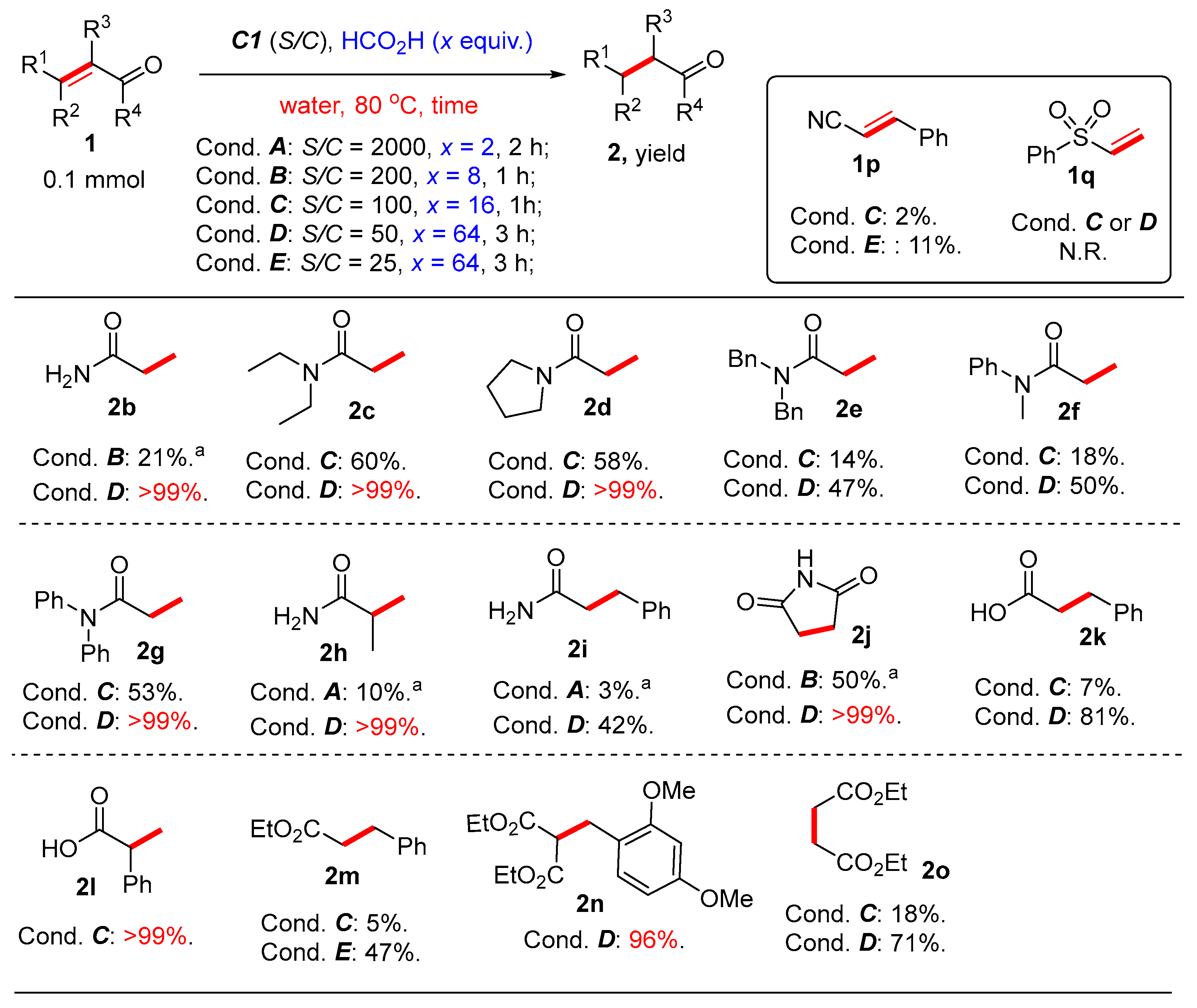
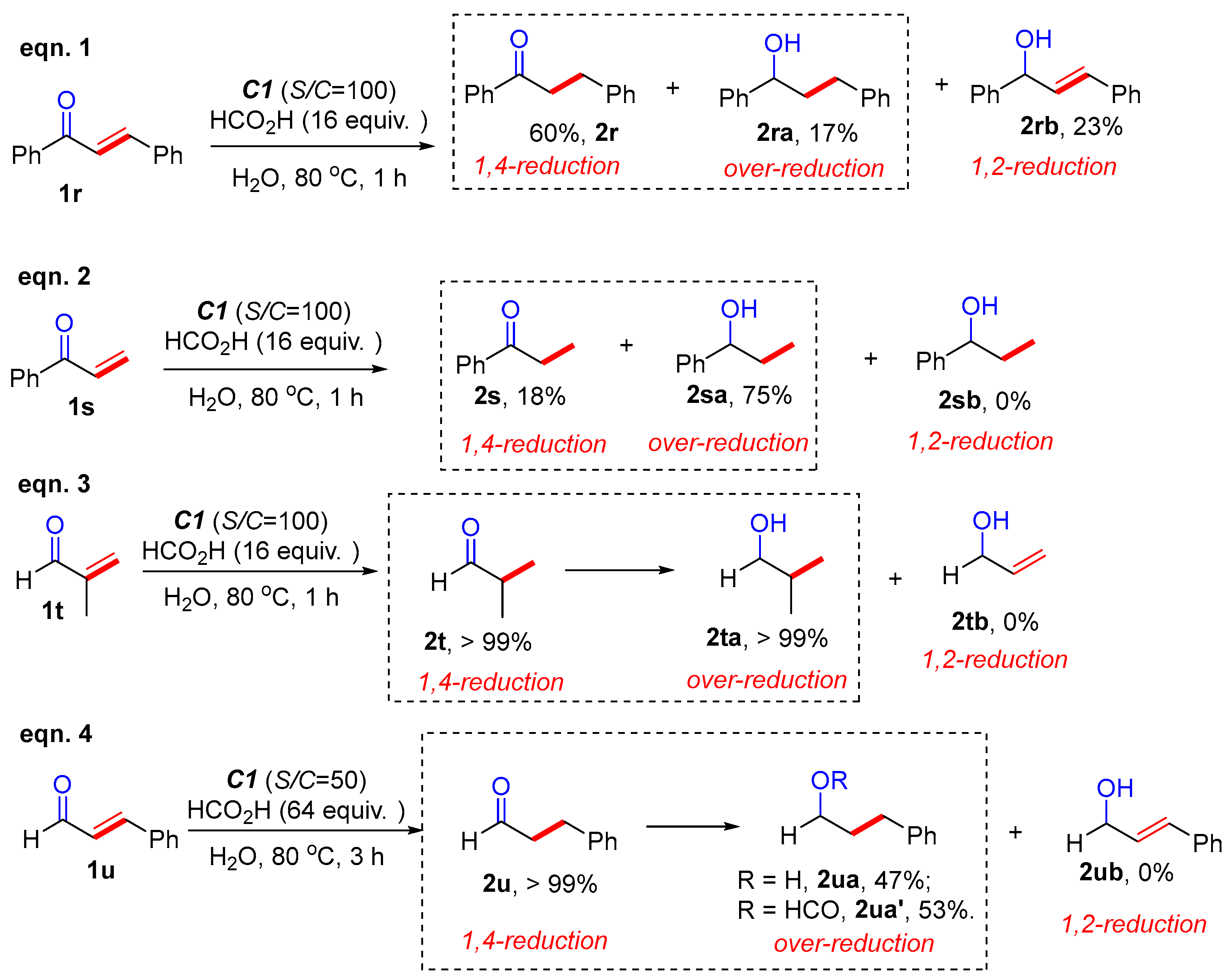
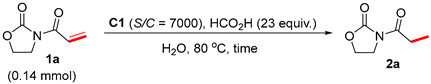
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Instruments
3.2. Preparation of the Catalyst C1
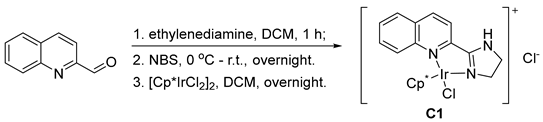
3.3. Optimization of Reaction Conditions
3.3.1. Optimization of Catalysts in Entries 1–11, Table 1
3.3.2. Optimization of Catalyst Type in Entries 12–16, Table 1
3.3.3. Optimization of Catalyst Loading in Entries 17–18, Table 1
3.3.4. Optimization of Equivalents of Formic Acid in Entries 19–22, Table 1
3.4. Procedure for Measuring TOF Values
3.5. General Procedure for Synthesis of Acrylamide [49]
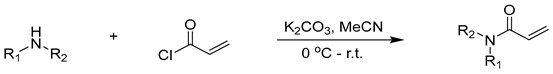
3.5.1. N,N-Diethylacrylamide (1c)
3.5.2. N,N-Diethylacrylamide (1d)
3.5.3. N,N-Dibenzylacrylamide (1e)
3.5.4. N-Methyl-N-Phenylacrylamide (1f)
3.5.5. N,N-Diphenylacrylamide (1g)
3.6. Synthesis of (Vinylsulfonyl)benzene (1q) [50]
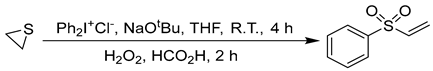
3.7. General Procedure
3.7.1. N-Propanoyl-2-Oxazolidinone (2a)
3.7.2. Propionamide (2b)
3.7.3. N,N-Diethylpropionamide (2c)
3.7.4. N-Propionylpyrrolidine (2d)
3.7.5. N,N-Dibenzylpropionamide (2e)
3.7.6. N-Methylpropionanilide (2f)
3.7.7. N,N-Diphenylpropionamide (2g)
3.7.8. Isobutyramide (2h)
3.7.9. 3-Phenylpropanamide (2i)
3.7.10. Pyrrolidine-2,5-Dione (2j)
3.7.11. 3-Phenylpropanoic Acid (2k)
3.7.12. 2-Phenylpropionic Acid (2l)
3.7.13. Ethyl 3-Phenylpropanoate (2m)
3.7.14. Diethyl 2-(2,4-Dimethoxybenzyl)malonate (2n)
3.7.15. Dimethyl Succinate (2o)
3.7.16. 3-Phenylpropanenitrile (2p)
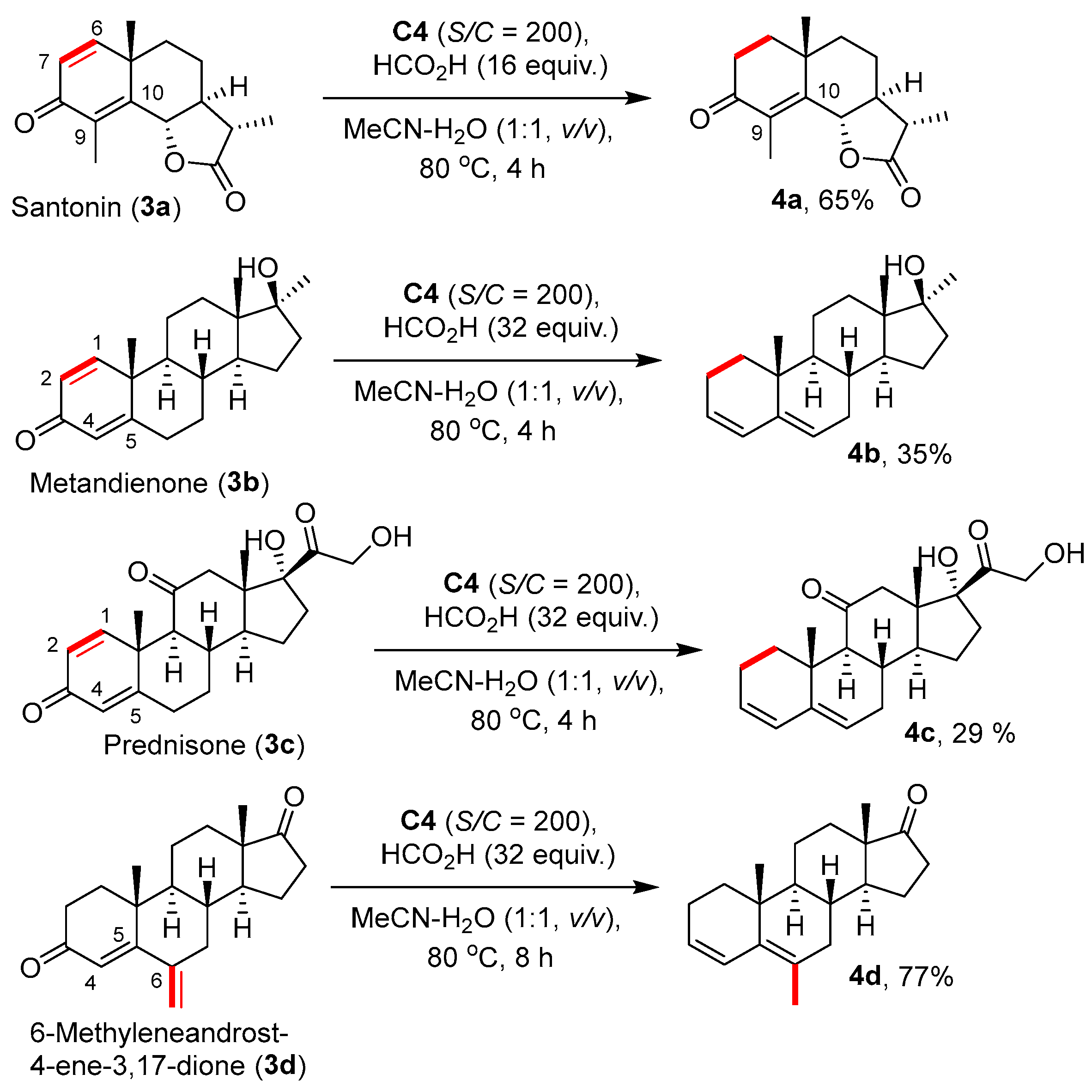
3.8. Applications in Structural Modifications of Medicinal Molecules [35]
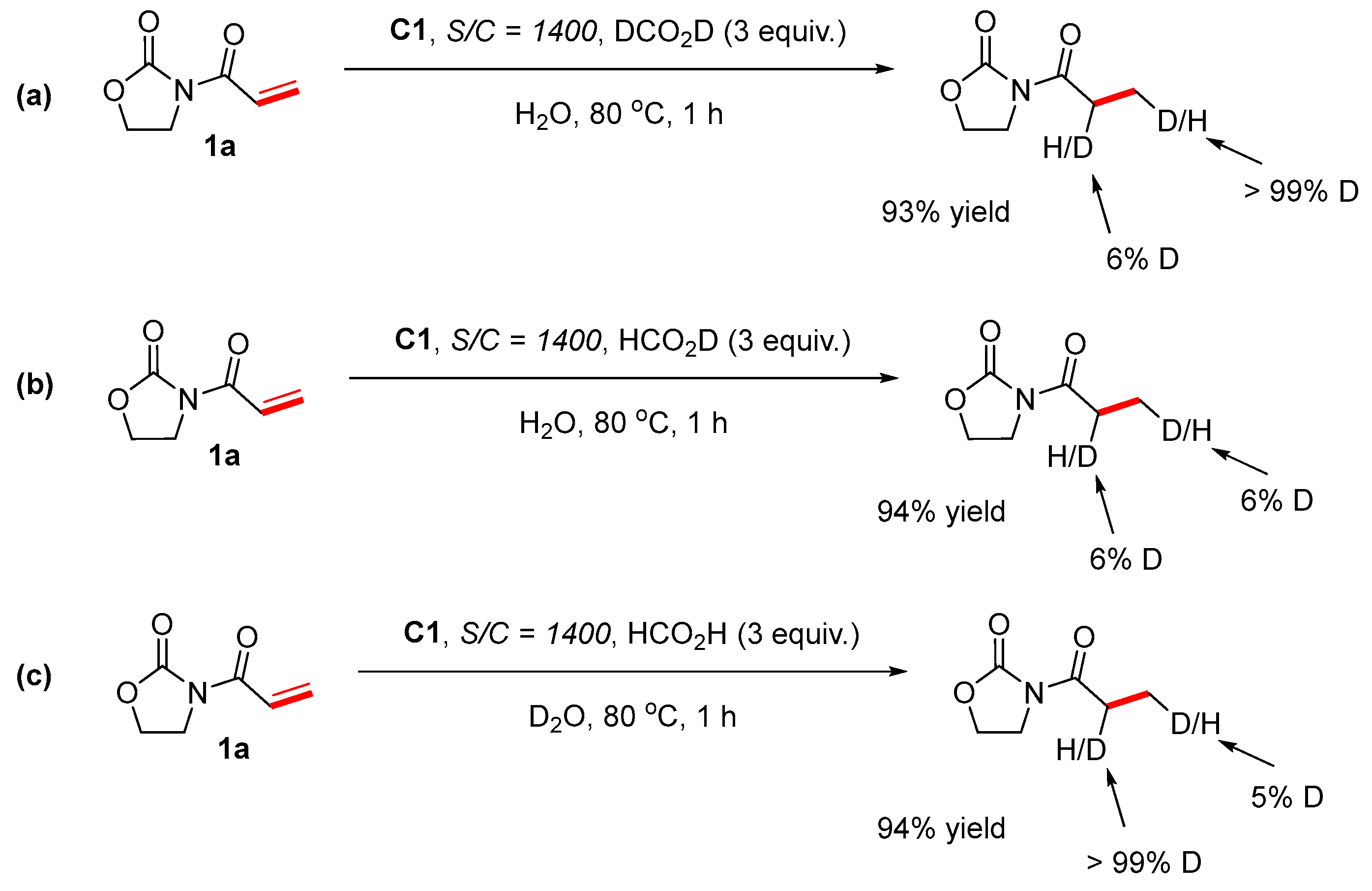
3.9. Deuterium Substitution Experiments
3.10. Gram-Scale Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keinan, E.; Greenspoon, N. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1991; Volume 8, Chapter 3.5. [Google Scholar]
- Paquette, L.A.; Tae, J.; Arrington, M.P.; Sadoun, A.H. Enantioselective Double Michael Addition/Cyclization with an Oxygen-Centered Nucleophile as the First Step in a Concise Synthesis of Natural (+)-Asteriscanolide. J. Am. Chem. Soc. 2000, 122, 2742–2748. [Google Scholar] [CrossRef]
- Tietze, L.F.; Stecker, F.; Zinngrebe, J.; Sommer, K.M. Enantioselective Palladium-Catalyzed Total Synthesis of Vitamin E by Employing a Domino Wacker–Heck Reaction. Chem. Eur. J. 2006, 12, 8770–8776. [Google Scholar] [CrossRef]
- Shibata, I.; Suwa, T.; Ryu, K.; Baba, A. Conjugate Hydrostannation of Unsaturated Esters by Iodotin Hydride Ate Complex. J. Org. Chem. 2001, 66, 8690–8692. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, J.M.; Ganem, B. Lithium and potassium trialkylborohydrides. Reagents for direct reduction of.alpha.,.beta.-unsaturated carbonyl compounds to synthetically versatile enolate anions. J. Org. Chem. 1976, 41, 2194–2200. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Stauffer, R.D.; Yamashita, A. Reductions of conjugated carbonyl compounds with copper hydride—Preparative and mechanistic aspects. J. Org. Chem. 1977, 42, 3180–3188. [Google Scholar] [CrossRef]
- Masamune, S.; Bates, G.S.; Georghiou, P.E. Reactions of lithium alkyl and alkynyl cuprates. Selective removal of halo and mesyloxy groups and reduction of.alpha.,.beta.-unsaturated ketones. J. Am. Chem. Soc. 1974, 96, 3686–3688. [Google Scholar] [CrossRef]
- Ashby, E.C.; Lin, J.-J.; Goel, A.B. Reactions of complex metal hydrides of copper with alkyl halides, enones, and cyclic ketones. J. Org. Chem. 1978, 43, 183–188. [Google Scholar] [CrossRef]
- Mahoney, W.S.; Brestensky, D.M.; Stryker, J.M. Selective hydride-mediated conjugate reduction of.alpha.,.beta.-unsaturated carbonyl compounds using [(Ph3P)CuH]6. J. Am. Chem. Soc. 1988, 110, 291–293. [Google Scholar] [CrossRef]
- Russo, A.T.; Amezcua, K.L.; Huynh, V.A.; Rousslang, Z.M.; Cordes, D.B. A simple borohydride-based method for selective 1,4-conjugate reduction of α,β-unsaturated carbonyl compounds. Tetrahedron Lett. 2011, 52, 6823–6826. [Google Scholar] [CrossRef]
- Sheng, Y.; You, Y.; Cao, Z.; Liu, L.; Wu, H.C. Rapid and selective DNA-based detection of melamine using α-hemolysin nanopores. Analyst 2018, 143, 2411–2415. [Google Scholar] [CrossRef]
- Hudlicky, T.; Sinai-Zingde, G.; Natchus, M.G. Selective reduction of α,β-unsaturated esters in the presence of olefins. Tetrohedron Lett. 1987, 28, 5287–5290. [Google Scholar] [CrossRef]
- Johns, W.F. Synthesis of 3-Methoxy-17-acetyl-18-norestra-1,3,5(10),16-tetraene. J. Org. Chem. 1963, 28, 1856–1861. [Google Scholar] [CrossRef]
- Yanto, Y.; Winkler, C.K.; Lohr, S.; Hall, M.; Faber, K.; Bommarius, A.S. Asymmetric Bioreduction of Alkenes Using Ene–Reductases YersER and KYE1 and Effects of Organic Solvents. Org. Lett. 2011, 13, 2540–2543. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Fujii, M.; Ohno, A.; Oka, S. NAD(P)+-NAD(P)H model. 52. Reduction of olefins by hantzsch ester on silica gel. Tetrahedron Lett. 1984, 25, 3983–3986. [Google Scholar] [CrossRef]
- Chikashita, H.; Miyazaki, M.; Itoh, K. Lewis acid-promoted conjugate reduction of α,β-unsaturated carbonyl compounds by 2-phenylbenzothiazoline (2-phenyl-2,3-dihydrobenzothiazole). J. Chem. Soc. Perkin Trans. I 1987, 4, 699–706. [Google Scholar] [CrossRef]
- Temperini, A.; Ballarotto, M.; Siciliano, C. Chemoselective and metal-free reduction of α,β-unsaturated ketones by in situ produced benzeneselenol from O-(tert-butyl) Se-phenyl selenocarbonate. RSC Adv. 2020, 10, 33706–33717. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.J.; Mudd, R.J.; Brown, J.A. Iridium(I) N-Heterocyclic Carbene (NHC)/Phosphine Catalysts for Mild and Chemoselective Hydrogenation Processes. Chem. Eur. J. 2016, 22, 4738–4742. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; Meneses, R. Raney Nickel: An Efficient Reagent to Achieve the Chemoselective Hydrogenation of α,β-Unsaturated Carbonyl Compounds. Synlett 1999, 1999, 1663–1666. [Google Scholar] [CrossRef]
- Oyamada, H.; Naito, T.; Miyamoto, S.; Akiyama, R.; Hagio, H.; Kobayashi, S. Development of polysilane-supported palladium/alumina hybrid catalysts and their application to hydrogenation. Org. Biomol. Chem. 2008, 6, 61–65. [Google Scholar] [CrossRef]
- Hagiwara, H.; Nakamura, T.; Hoshi, T.; Suzuki, T. Palladium-supported ionic liquid catalyst (Pd-SH-SILC) immobilized on mercaptopropyl silica gel as a chemoselective, reusable and heterogeneous catalyst for catalytic hydrogenation. Green Chem. 2011, 13, 1133–1137. [Google Scholar] [CrossRef]
- Dey, K.; de Ruiter, G. Chemoselective Hydrogenation of α,β-Unsaturated Ketones Catalyzed by a Manganese(I) Hydride Complex. Org. Lett. 2024, 26, 4173–4177. [Google Scholar] [CrossRef] [PubMed]
- Keinan, E.; Perez, D. Silicon hydrides and molybdenum(0) catalyst: A novel approach for conjugate reduction of .alpha.,.beta.-unsaturated carbonyl compounds. J. Org. Chem. 1987, 52, 2576–2580. [Google Scholar] [CrossRef]
- Mai, V.H.; Nikonov, G.I. Transfer Hydrogenation of Nitriles, Olefins, and N-Heterocycles Catalyzed by an N-Heterocyclic Carbene-Supported Half-Sandwich Complex of Ruthenium. Organometallics 2016, 35, 943–949. [Google Scholar] [CrossRef]
- Xue, D.; Chen, Y.-C.; Cui, X.; Wang, Q.-W.; Zhu, J.; Deng, J.-G. Transfer Hydrogenation of Activated C=C Bonds Catalyzed by Ruthenium Amido Complexes: Reaction Scope, Limitation, and Enantioselectivity. J. Org. Chem. 2005, 70, 3584–3591. [Google Scholar] [CrossRef]
- Keinan, E.; Greenspoon, N. Highly chemoselective palladium-catalyzed conjugate reduction of α.,.β-unsaturated carbonyl compounds with silicon hydrides and zinc chloride cocatalyst. J. Am. Chem. Soc. 1986, 108, 7314–7325. [Google Scholar] [CrossRef]
- Zhou, Y.; Bandar, J.S.; Liu, R.Y.; Buchwald, S.L. CuH-Catalyzed Asymmetric Reduction of α,β-Unsaturated Carboxylic Acids to β-Chiral Aldehydes. J. Am. Chem. Soc. 2018, 140, 606–609. [Google Scholar] [CrossRef]
- Yu, X.; Wan, S.; Zhou, W. 1,4-Reduction of α,β-Unsaturated Ketones through Rhodium(III)-Catalyzed Transfer Hydrogenation. Synlett 2022, 34, 1399–1402. [Google Scholar] [CrossRef]
- Wang, C.; Xiao, J. Iridacycles for hydrogenation and dehydrogenation reactions. Chem. Commun. 2017, 53, 3399–3411. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wu, X.; Wang, C.; Xiao, J. Transfer hydrogenation in aqueous media. Catal. Today 2015, 247, 104–116. [Google Scholar] [CrossRef]
- Yang, Z.; Luo, R.; Zhu, Z.; Yang, X.; Tang, W. Harnessing the Reactivity of Iridium Hydrides by Air: Iridium-Catalyzed Oxidation of Aldehydes to Acids in Water. Organometallics 2017, 36, 4095–4098. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Z.; Luo, R.; Qiu, X.; Liu, J.-T.; Yang, J.-K.; Tang, W. Iridium-catalyzed highly efficient chemoselective reduction of aldehydes in water using formic acid as the hydrogen source. Green Chem. 2017, 19, 3296–3301. [Google Scholar] [CrossRef]
- Liu, J.T.; Yang, S.; Tang, W.; Yang, Z.; Xu, J. Iridium-catalyzed efficient reduction of ketones in water with formic acid as a hydride donor at low catalyst loading. Green Chem. 2018, 20, 2118–2124. [Google Scholar] [CrossRef]
- Yang, S.; Tang, W.; Yang, Z.; Xu, J. Iridium-Catalyzed Highly Efficient and Site-Selective Deoxygenation of Alcohols. ACS Catal. 2018, 8, 9320–9326. [Google Scholar] [CrossRef]
- Li, J.; Tang, W.; Ren, D.; Xu, J.; Yang, Z. Iridium-Catalysed Highly Selective Reduction–Elimination of Steroidal 4-En-3-ones to 3,5-Dienes in Water. Green Chem. 2019, 21, 2088–2094. [Google Scholar] [CrossRef]
- Xu, D.; Chen, Y.; Liu, C.; Xu, J.; Yang, Z. Iridium-Catalyzed Highly Chemoselective and Efficient Reduction of Nitroalkenes to Nitroalkanes in Water. Green Chem. 2021, 23, 6050–6058. [Google Scholar] [CrossRef]
- Wang, T.; Liu, C.; Xu, D.; Xu, J.; Yang, Z. Iridium-Catalyzed and pH-Dependent Reductions of Nitroalkenes to Ketones. Molecules 2022, 27, 7822. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Y.; Yang, Z. Iridium-Catalyzed Stereoselective Transfer Hydrogenation of 1,5-Benzodiazepines. J. Org. Chem. 2022, 87, 12001–12018. [Google Scholar] [CrossRef]
- Wang, T.; Chen, Y.; Chen, N.; Xu, J.; Yang, Z. Iridium-Catalyzed Highly Stereoselective Deoxygenation of Tertiary Cycloalkanols: Stereoelectronic Insights and Synthetic Applications. Org. Biomol. Chem. 2021, 19, 9004–9011. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, T.; Du, H.; Chen, N.; Xu, J.; Yang, Z. Accessing para-Alkylphenols via Iridium-Catalyzed Site-Specific Deoxygenation of Alcohols. J. Org. Chem. 2023, 88, 12572–12584. [Google Scholar] [CrossRef]
- Wang, T.; Miao, R.; Luo, R.; Xu, J.; Yang, Z. Furan-2-yl Anions as γ-Oxo/Hydroxyl Acyl Anion Equivalents Enabled by Iridium-Catalyzed Chemoselective Reduction. Org. Lett. 2023, 25, 4705–4710. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Du, H.; Luo, R.; Xu, J.; Yang, Z. Iridium-Catalyzed Reductive γ-Lactonization of ortho-Acylbenzoic Acids in Water: Sustainable Access to Phthalides. Org. Chem. Front. 2024, 11, 2220–2230. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Wang, Y.; Du, H.; Xu, J.; Yang, Z. En Route to Diastereopure Polycyclic γ-Lactones by Iridium-Catalyzed Hydride Transfer. Chin. J. Chem. 2024, 42, 3047–3055. [Google Scholar] [CrossRef]
- Li, W.; Yin, G.; Huang, L.; Xiao, Y.; Fu, Z.; Xin, X.; Liu, F.; Li, Z.; He, W. Regioselective and stereoselective sulfonylation of alkynylcarbonyl compounds in water. Green Chem. 2016, 18, 4879–4883. [Google Scholar] [CrossRef]
- Wu, C.; Xin, X.; Fu, Z.-M.; Xie, L.-Y.; Liu, K.-J.; Wang, Z.; Li, W.; Yuan, Z.-H.; He, W.-M. Water-controlled selective preparation of α-mono or α,α′-dihalo ketones via catalytic cascade reaction of unactivated alkynes with 1,3-dihalo-5,5-dimethylhydantoin. Green Chem. 2017, 19, 1983–1989. [Google Scholar] [CrossRef]
- Fu, Z.; Yuan, W.; Chen, N.; Yang, Z.; Xu, J. Na2S2O8-mediated efficient synthesis of isothiocyanates from primary amines in water. Green Chem. 2018, 20, 4484–4491. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, S.-M.; Li, J.; Wang, J.; Li, C. Unprecedentedly High Formic Acid Dehydrogenation Activity on an Iridium Complex with an N, N’-Diimine Ligand in Water. Green Chem. 2015, 21, 12592–12595. [Google Scholar] [CrossRef]
- Lu, S.M.; Wang, Z.; Wang, J.; Li, J.; Li, C. Hydrogen generation from formic acid decomposition on a highly efficient iridium catalyst bearing a diaminoglyoxime ligand. Green Chem. 2018, 20, 1835–1840. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, F.-P.; Jin, X.-L.; Wang, X.-J.; Chen, H.; Wu, L.-Z. Visible-Light-Driven Intermolecular [2+2] Cycloadditions between Coumarin-3-Carboxylates and Acrylamide Analogs. Chem. Eur. J. 2015, 21, 10326–10329. [Google Scholar] [CrossRef]
- Dong, J.; Xu, J. Safe and Metal-Free Synthesis of 1-Alkenyl Aryl Sulfides and Their Sulfones from Thiiranes and Diaryliodonium Salts. Synthesis 2018, 50, 2407–2415. [Google Scholar]
- Kang, T.; Kim, Y.; Lee, D.; Wang, Z.; Chang, S. Iridium-Catalyzed Intermolecular Amidation of sp3 C–H Bonds: Late-Stage Functionalization of an Unactivated Methyl Group. J. Am. Chem. Soc. 2014, 136, 4141–4144. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Entry | Cat. | S/C | Conv. (%) b | Entry | Cat. | S/C | Conv. (%) b |
|---|---|---|---|---|---|---|---|
| 1 | C1 | 700 | 100 | 12 | C1 | 1400 | 100 |
| 2 | C2 | 700 | 100 | 13 | C2 | 1400 | 72 |
| 3 | C3 | 700 | 38 | 14 | C5 | 1400 | 63 |
| 4 | C4 | 700 | 88 | 15 | C10 | 1400 | 60 |
| 5 | C5 | 700 | 100 | 16 | C11 | 1400 | 59 |
| 6 | C6 | 700 | 45 | 17 | C1 | 3500 | 87 |
| 7 | C7 | 700 | 22 | 18 | C1 | 7000 | 55 |
| 8 | C8 | 700 | 12 | 19 c | C1 | 1400 | >99 |
| 9 | C9 | 700 | 49 | 20 d | C1 | 1400 | >99 |
| 10 | C10 | 700 | 100 | 21 e | C1 | 1400 | >99 |
| 11 | C11 | 700 | 100 | 22 f | C1 | 1400 | >99 |
| Time (min) | 5 | 10 | 30 | 60 | 120 |
| Conv. (%) a | 22 | 27 | 42 | 55 | 61 |
| TOF (h−1) | 18,480 | 11,340 | 5880 | 3850 | 2135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Li, J.; Xu, J.; Yang, Z. Iridium-Catalyzed Highly Selective 1,4-Reduction of α,β-Unsaturated Carbonyl Compounds. Molecules 2024, 29, 5912. https://doi.org/10.3390/molecules29245912
Chen Y, Li J, Xu J, Yang Z. Iridium-Catalyzed Highly Selective 1,4-Reduction of α,β-Unsaturated Carbonyl Compounds. Molecules. 2024; 29(24):5912. https://doi.org/10.3390/molecules29245912
Chicago/Turabian StyleChen, Youwei, Jide Li, Jiaxi Xu, and Zhanhui Yang. 2024. "Iridium-Catalyzed Highly Selective 1,4-Reduction of α,β-Unsaturated Carbonyl Compounds" Molecules 29, no. 24: 5912. https://doi.org/10.3390/molecules29245912
APA StyleChen, Y., Li, J., Xu, J., & Yang, Z. (2024). Iridium-Catalyzed Highly Selective 1,4-Reduction of α,β-Unsaturated Carbonyl Compounds. Molecules, 29(24), 5912. https://doi.org/10.3390/molecules29245912