
Electrochemical Water Oxidation and CO2 Reduction with a Nickel Molecular Catalyst

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Description of Crystal Structures
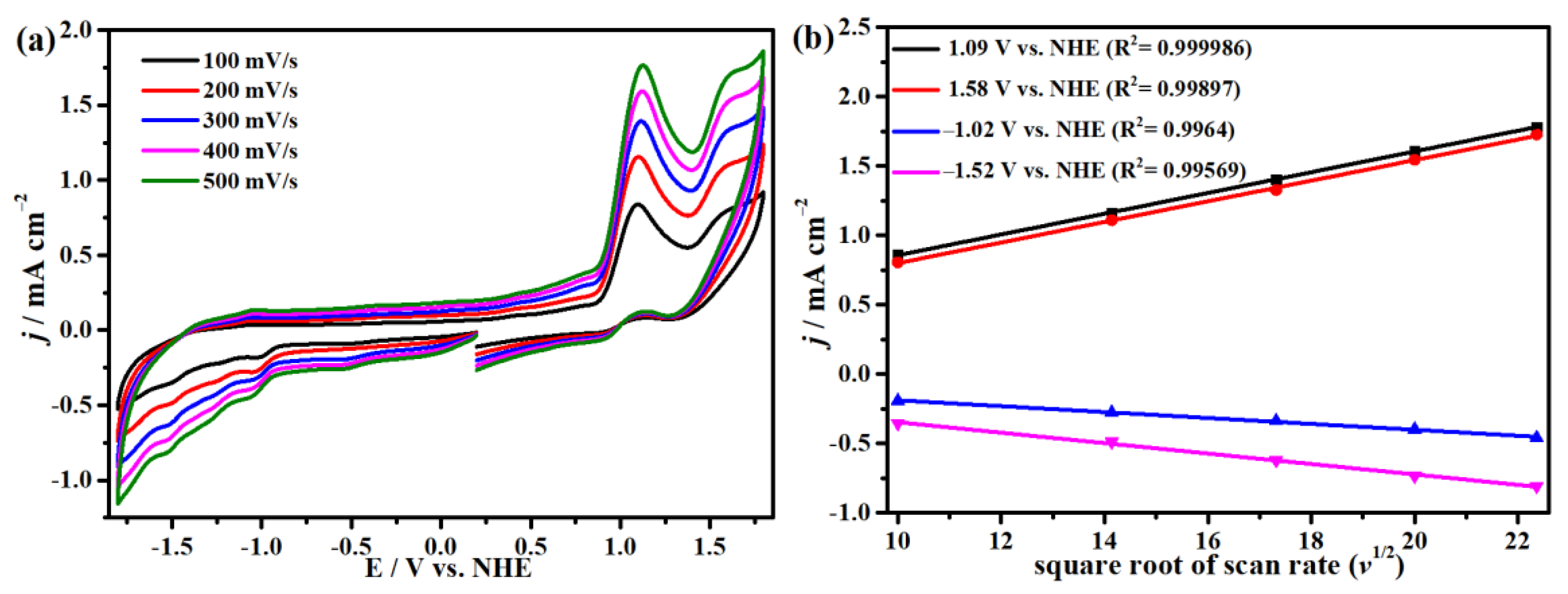
2.2. Electrochemistry under Argon Atmosphere
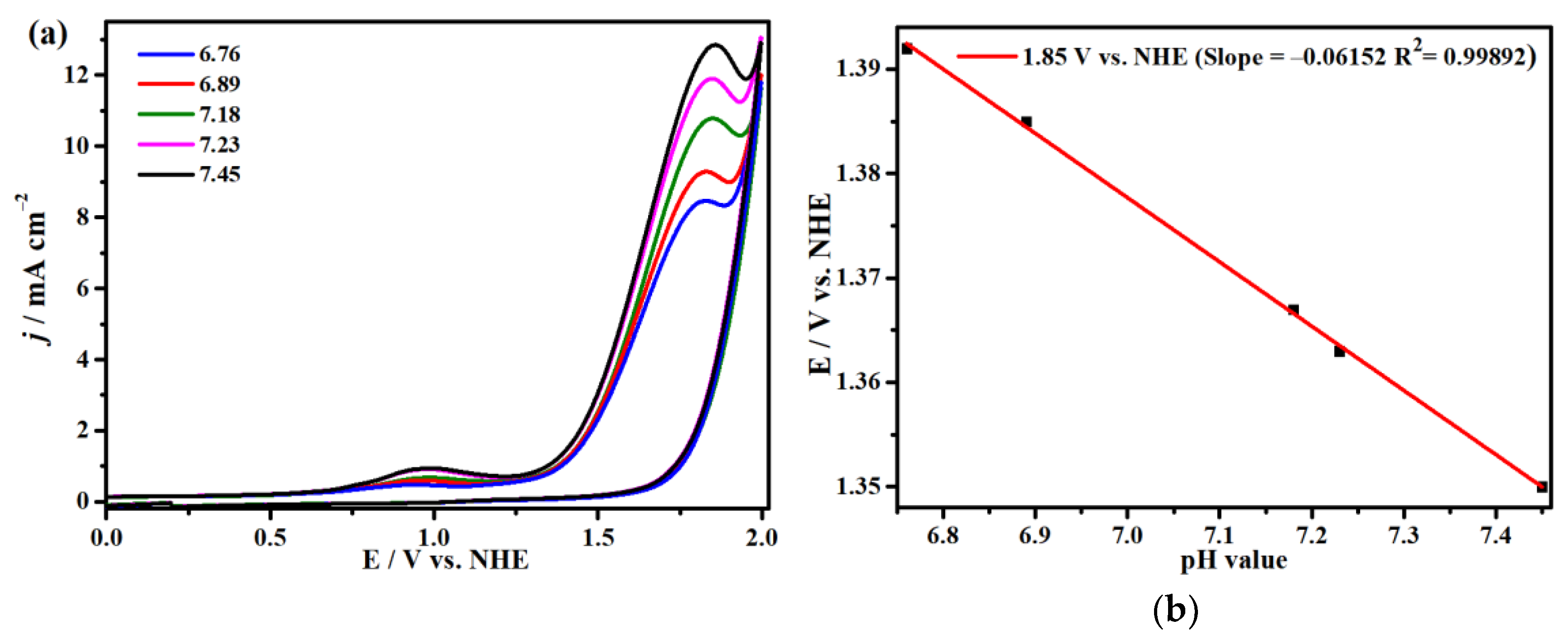
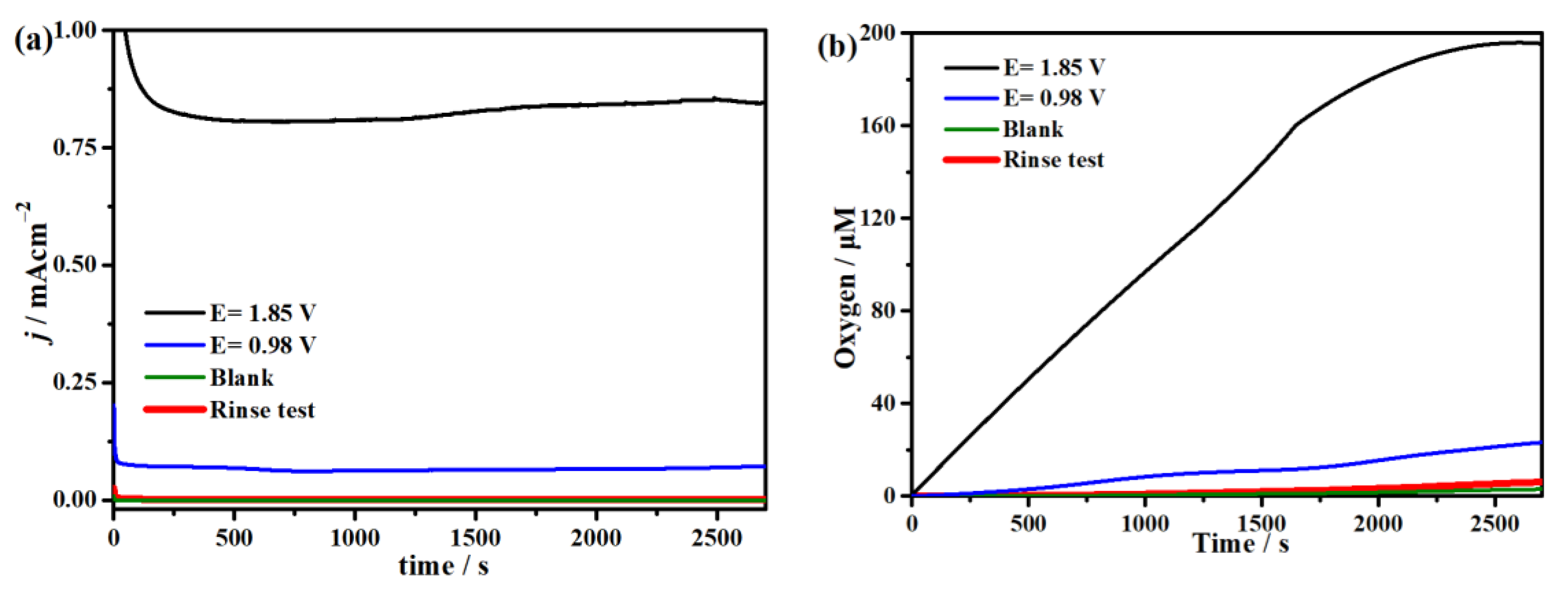
2.3. Electrocatalytic Water Oxidation
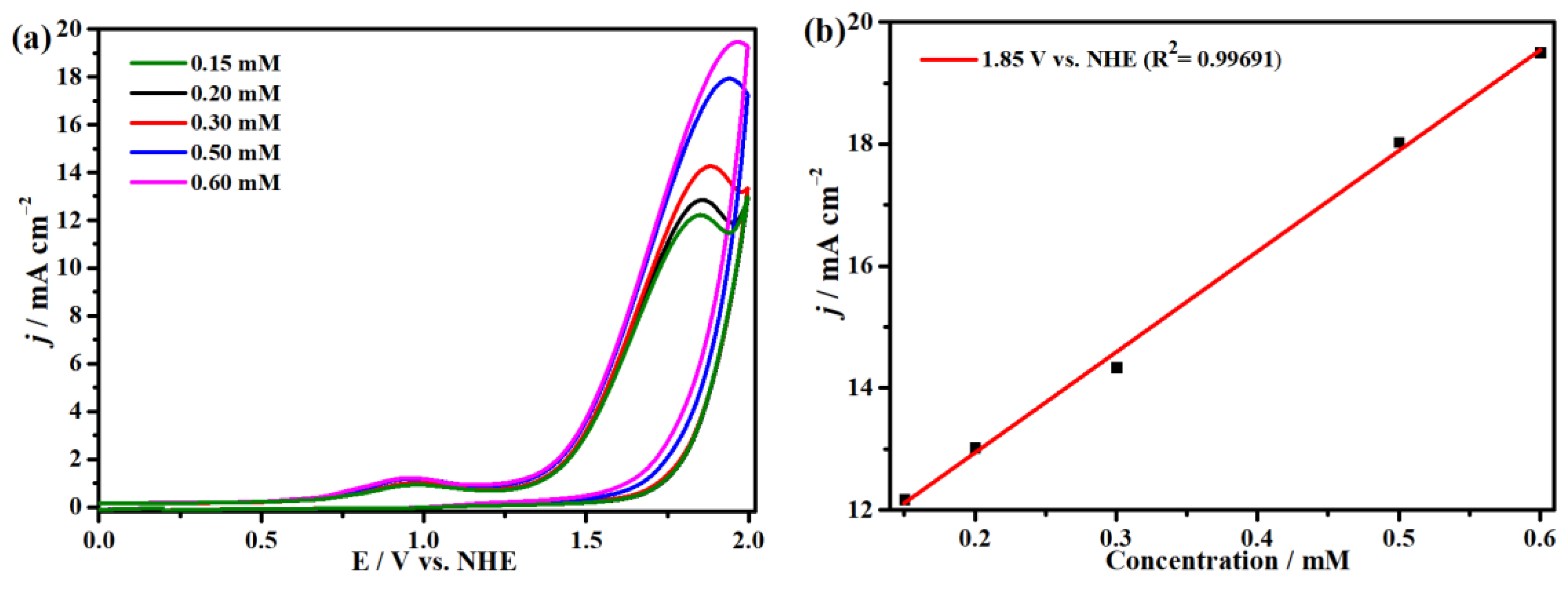
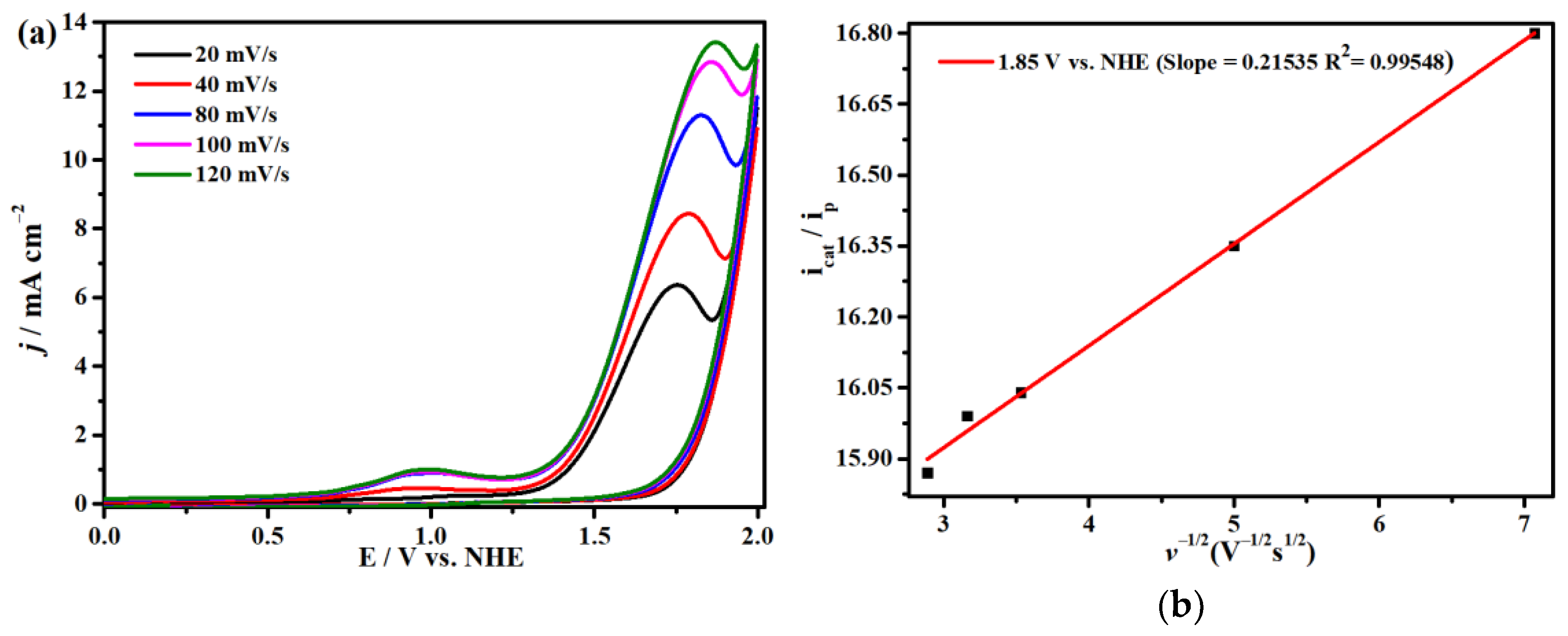
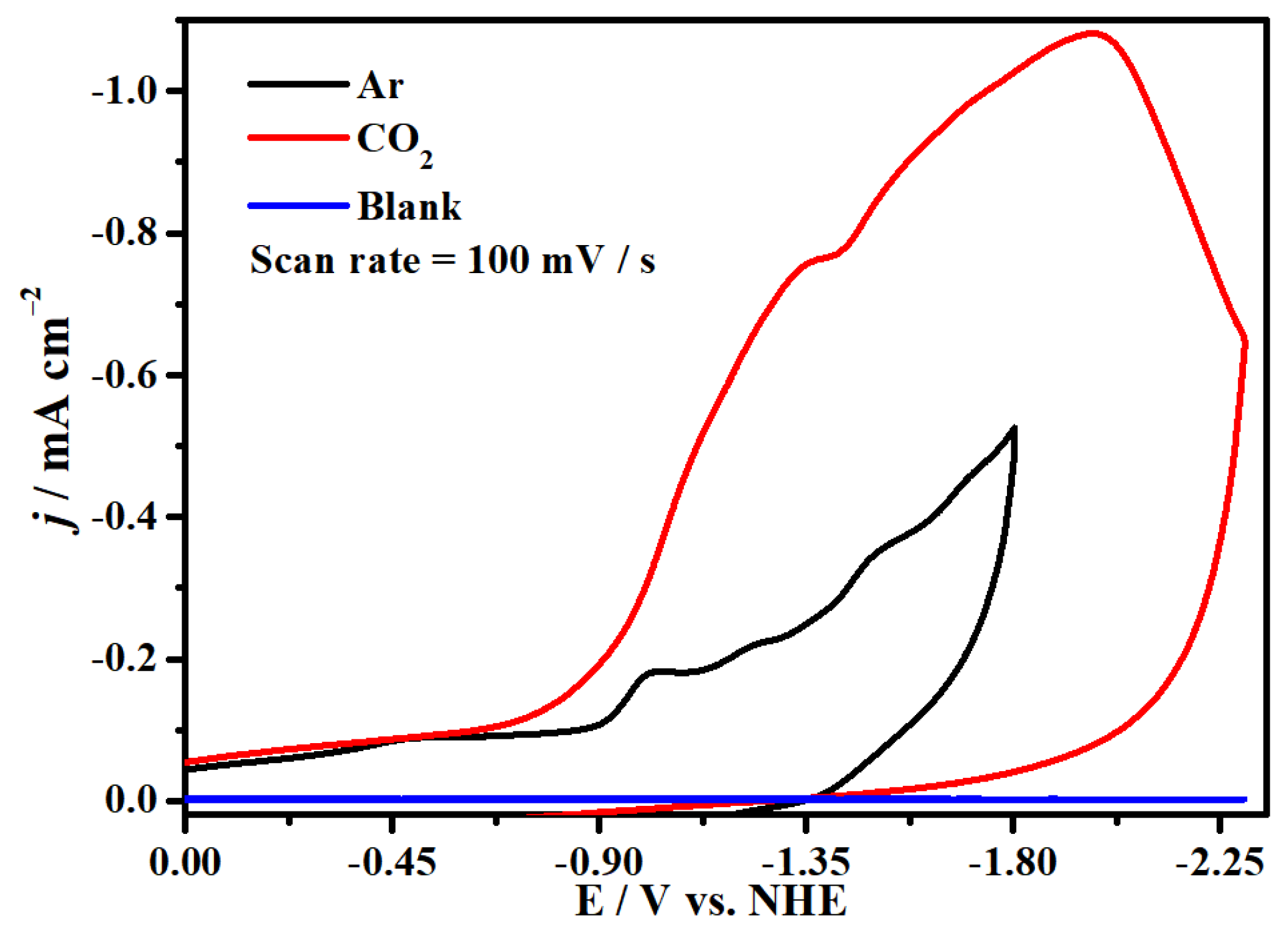
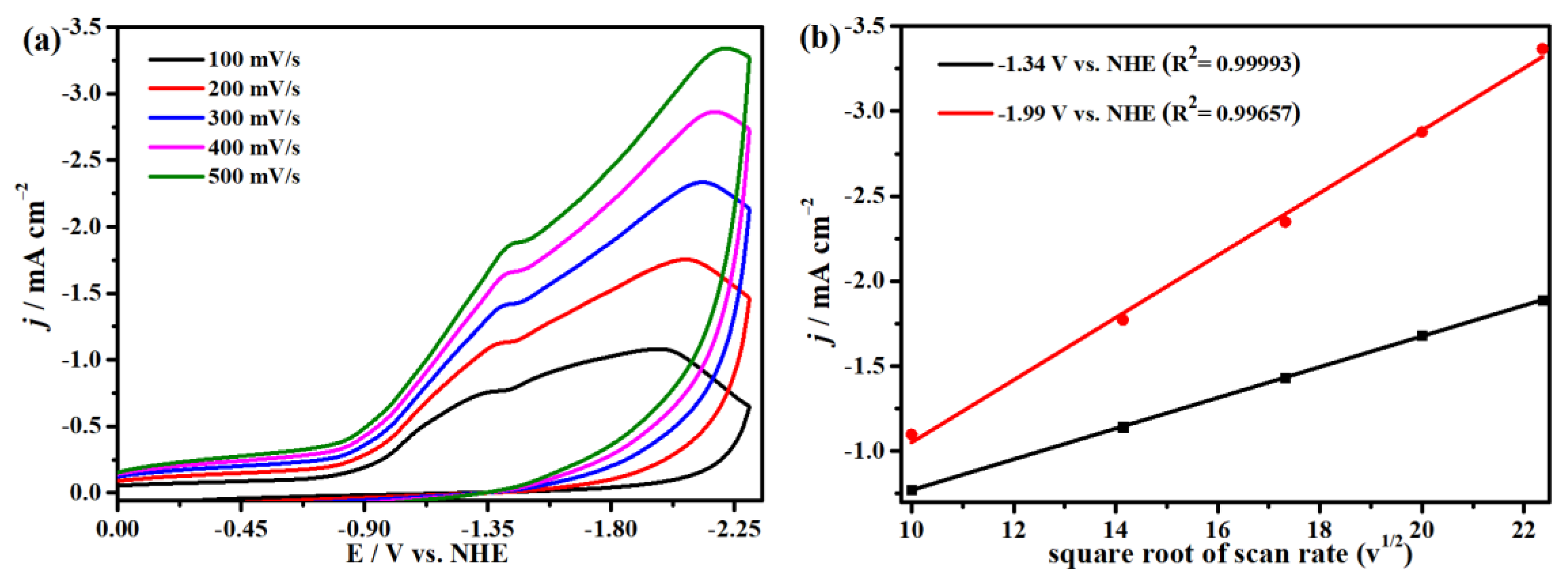
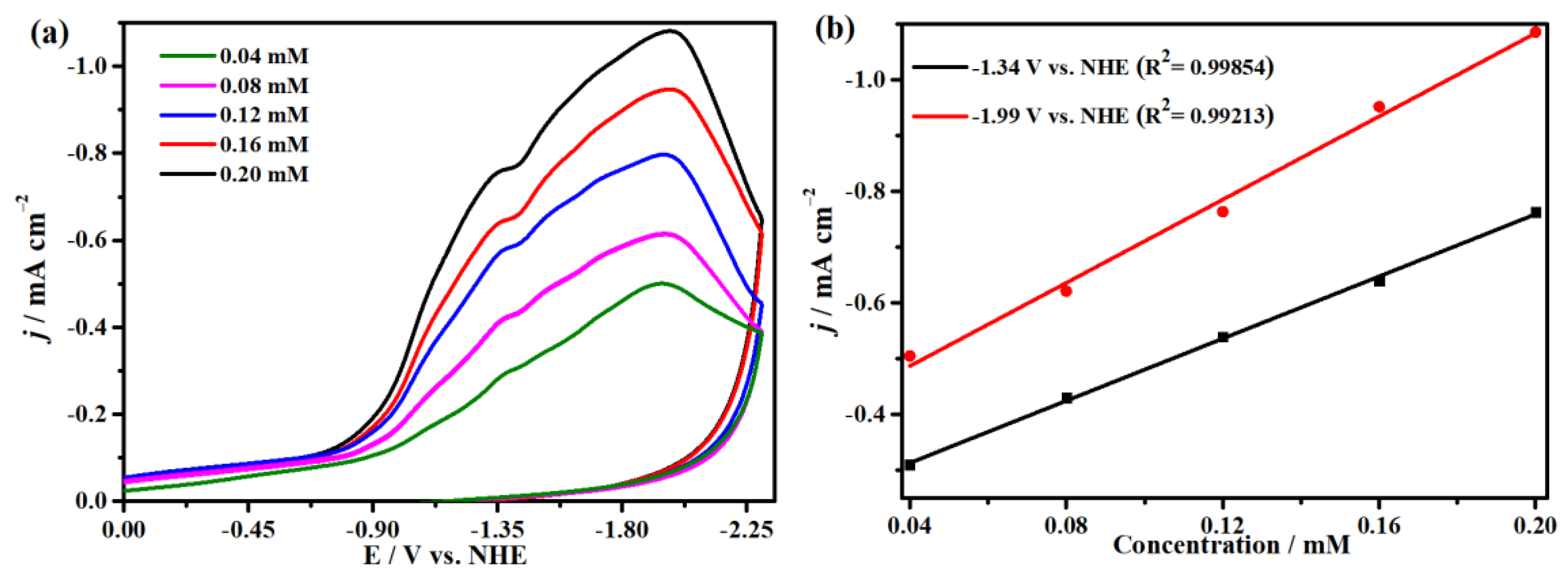
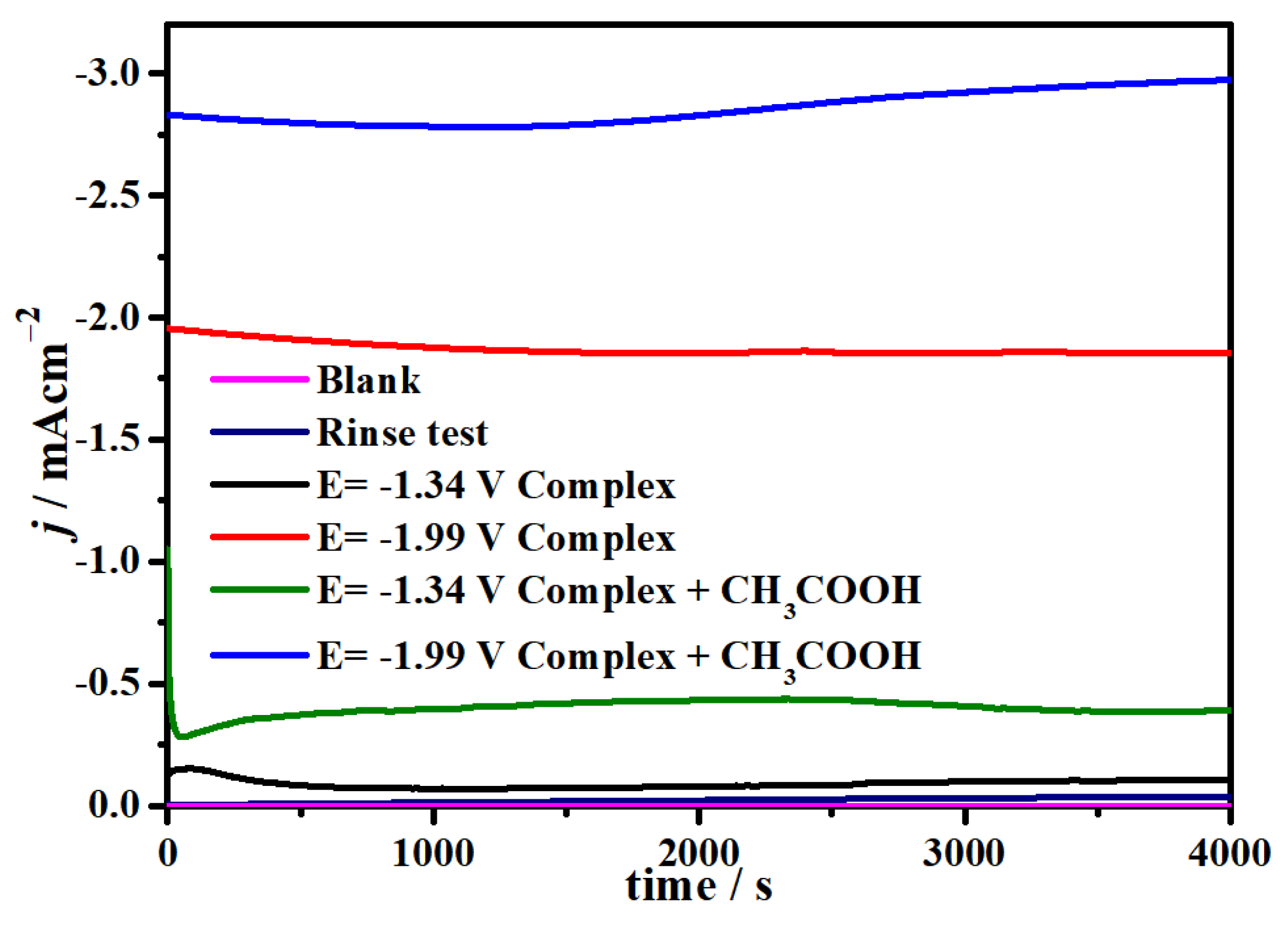
2.4. Electrocatalytic CO2 Reduction
3. Experimental Sections
3.1. Chemical Materials and Methods
3.1.1. Chemical Materials
3.1.2. Physical Measurements
3.1.3. X-ray Crystallographic Data Collection and Refinement of the Structures
3.1.4. Electrochemical Measurements and Electrolysis Product Analysis
3.2. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chang, X.; Wang, T.; Yang, P.; Zhang, G.; Gong, J. The Development of Cocatalysts for Photoelectrochemical CO2 Reduction. Adv. Mater. 2019, 31, 1804710. [Google Scholar]
- Raciti, D.; Wang, C. Recent Advances in CO2 Reduction Electrocatalysis on Copper. ACS Energy Lett. 2018, 3, 1545–1556. [Google Scholar]
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.-T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2018, 31, 1807166. [Google Scholar]
- Rahman, F.A.; Aziz, M.M.A.; Saidur, R.; Abu Bakar, W.A.W.; Hainin, M.R.; Putrajaya, R.; Hassan, N.A. Pollution to solution: Capture and sequestration of carbon dioxide (CO2) and its utilization as a renewable energy source for a sustainable future. Renew. Sustain. Energy Rev. 2017, 71, 112–126. [Google Scholar]
- Vermaas, D.A.; Smith, W.A. Synergistic Electrochemical CO2 Reduction and Water Oxidation with a Bipolar Membrane. ACS Energy Lett. 2016, 1, 1143–1148. [Google Scholar]
- Nema, P.; Nema, S.; Roy, P. An overview of global climate changing in current scenario and mitigation action. Renew. Sustain. Energy Rev. 2012, 16, 2329–2336. [Google Scholar]
- Möller, D. SONNE: Solar-Based Man-Made Carbon Cycle and the Carbon Dioxide Economy. Ambio 2012, 41, 413–419. [Google Scholar] [PubMed]
- Chen, Y.; Xia, M.; Zhou, C.; Zhang, Y.; Zhou, C.; Xu, F.; Feng, B.; Wang, X.; Yang, L.; Hu, Z.; et al. Hierarchical Dual Single-Atom Catalysts with Coupled CoN4 and NiN4 Moieties for Industrial-Level CO2 Electroreduction to Syngas. ACS Nano 2023, 17, 22095–22105. [Google Scholar]
- Wang, C.; Liu, L.; Li, H.; Wang, L.; Xiao, F.-S. Hydrophobic catalysts for syngas conversion. Matter 2023, 6, 2697–2710. [Google Scholar]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accom plishment, Challenges, and Prospects. Adv. Mater. 2014, 26, 4607–4626. [Google Scholar]
- Dau, H.; Zaharieva, I.; Haumann, M. Recent developments in research on water oxidation by photosystem II. Curr. Opin. Chem. Biol. 2012, 16, 3–10. [Google Scholar] [PubMed]
- Concepcion, J.J.; Jurss, J.W.; Templeton, J.L.; Meyer, T.J. Mediator-assisted water oxidation by the ruthenium “blue dimer” cis,cis-[(bpy)2(H2O)RuORu(OH2)(bpy)2]4+. Proc. Natl. Acad. Sci. USA 2008, 105, 17632–17635. [Google Scholar] [PubMed]
- Dau, H.; Zaharieva, I. Principles, Efficiency, and Blueprint Character of Solar-Energy Conversion in Photosynthetic Water Oxidation. Acc. Chem. Res. 2009, 42, 1861–1870. [Google Scholar] [PubMed]
- Duan, L.; Wang, L.; Li, F.; Li, F.; Sun, L. Highly Efficient Bioinspired Molecular Ru Water Oxidation Catalysts with Neg atively Charged Backbone Ligands. Acc. Chem. Res. 2015, 48, 2084–2096. [Google Scholar] [PubMed]
- Zhang, X.; Ma, H.; Zhang, M.; Ma, Y. Interfacial Charge-Transfer Excitons Help the Photoreduction of CO2 on TiO2. ACS Catal. 2022, 12, 11024–11035. [Google Scholar]
- Guan, D.; Xu, H.; Zhang, Q.; Huang, Y.-C.; Shi, C.; Chang, Y.-C.; Xu, X.; Tang, J.; Gu, Y.; Pao, C.-W.; et al. Identifying a Universal Activity Descriptor and a Unifying Mechanism Concept on Perovskite Oxides for Green Hydrogen Production. Adv. Mater. 2023, 35, 2305074. [Google Scholar]
- Cho, S.; Noh, W.; Lee, I. Hybrid systems design for blue and green hydrogen co-production: Integration of autothermal re forming with solid oxide electrolysis. Energy Convers. Manag. 2024, 300, 117969. [Google Scholar]
- Barber, J. Photosynthetic water splitting by the Mn4Ca2+ OX catalyst of photosystem II: Its structure, robustness and mechanism. Q. Rev. Biophys. 2017, 50, e13. [Google Scholar]
- Najafpour, A.M.M.; Wiechen, M. Chem Soc Rev—Growing stronger. Energy Environ.-UK 2014, 7, 2203–2212. [Google Scholar] [CrossRef]
- Yagi, M.; Kaneko, M. Molecular Catalysts for Water Oxidation. Chem. Rev. 2001, 101, 21–36. [Google Scholar]
- Meyer, T.J.; Huynh, M.H.V.; Thorp, H.H. The Possible Role of Proton-Coupled Electron Transfer (PCET) in Water Oxidation by Photosystem II. Angew. Chem. Int. Ed. 2007, 46, 5284–5304. [Google Scholar]
- Zhang, L.-H.; Mathew, S.; Hessels, J.; Reek, J.N.H.; Yu, F. Homogeneous Catalysts Based on First-Row Transition-Metals for Electrochemical Water Oxidation. ChemSusChem 2021, 14, 234–250. [Google Scholar] [PubMed]
- Nocera, D.G. Proton-Coupled Electron Transfer: The Engine of Energy Conversion and Storage. J. Am. Chem. Soc. 2022, 144, 1069–1081. [Google Scholar] [PubMed]
- Wang, Y.; He, T. Recent advances in and comprehensive consideration of the oxidation half reaction in photocatalytic CO2 conversion. J. Mater. Chem. A 2021, 9, 87–110. [Google Scholar]
- Kamdar, J.M.; Grotjahn, D.B. An Overview of Significant Achievements in Ruthenium-Based Molecular Water Oxidation Ca talysis. Molecules 2019, 24, 494. [Google Scholar]
- Jiang, N.; Zhu, Z.; Xue, W.; Xia, B.Y.; You, B. Emerging Electrocatalysts for Water Oxidation under Near-Neutral CO2 Reduction Conditions. Adv. Mater. 2022, 34, 2105852. [Google Scholar]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 Reduction: From Homogeneous to Heterogeneous Electrocatalysis. Acc. Chem. Res. 2020, 53, 255–264. [Google Scholar]
- Wang, J.-W.; Zhong, D.-C.; Lu, T.-B. Artificial photosynthesis: Catalytic water oxidation and CO2 reduction by dinuclear non-noble-metal molecular catalysts. Coord. Chem. Rev. 2018, 377, 225–236. [Google Scholar]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar]
- Boer, D.D.; Hetterscheid, D.G.H. Correlations between the Electronic Structure and Energetics of the Catalytic Steps in Homogeneous Water Oxidation Catalysis. J. Am. Chem. Soc. 2023, 145, 23057–23067. [Google Scholar]
- Thomas, A.; Ohsaki, Y.; Nakazato, R.; Kuttassery, F.; Mathew, S.; Remello, S.N.; Tachibana, H.; Inoue, H. Molecular Character istics of Water-Insoluble Tin-Porphyrins for Designing the One-Photon-Induced Two-Electron Oxidation of Water in Artificial Photosynthesis. Molecules 2023, 28, 1882. [Google Scholar]
- Wang, J.-W.; Zhang, X.-Q.; Huang, H.-H.; Lu, T.-B. A Nickel(II) Complex as a Homogeneous Electrocatalyst for Water Oxidation at Neutral pH: Dual Role of HPO42− in Catalysis. ChemCatChem 2016, 8, 3287–3293. [Google Scholar]
- Han, X.-B.; Li, Y.-G.; Zhang, Z.-M.; Tan, H.-Q.; Lu, Y.; Wang, E.-B. Polyoxometalate-Based Nickel Clusters as Visible Light-Driven Water Oxidation Catalysts. J. Am. Chem. Soc. 2015, 137, 5486–5493. [Google Scholar] [PubMed]
- Zhang, M.; Zhang, M.-T.; Hou, C.; Ke, Z.-F.; Lu, T.-B. Homogeneous Electrocatalytic Water Oxidation at Neutral pH by a Robust Macrocyclic Nickel(II) Complex. Angew. Chem. Int. Ed. 2014, 53, 13042–13048. [Google Scholar]
- Wang, L.; Duan, L.; Ambre, R.B.; Daniel, Q.; Chen, H.; Sun, J.; Das, B.; Thapper, A.; Uhlig, J.; Dinér, P.; et al. A nickel (II) PY5 complex as an electrocatalyst for water oxidation. J. Catal. 2016, 335, 72–78. [Google Scholar]
- Wang, J.; Meng, X.; Xie, W.; Zhang, X.; Fan, Y.; Wang, M. Two biologically inspired tetranuclear nickel(II) catalysts: Effect of the geometry of Ni4 core on electrocatalytic water oxidation. J. Biol. Inorg. 2021, 26, 205–216. [Google Scholar]
- Chen, Q.-F.; Xiao, Y.; Liao, R.-Z.; Zhang, M.-T. Trinuclear Nickel Catalyst for Water Oxidation: Intramolecular Proton-Cou pled Electron Transfer Triggered Trimetallic Cooperative O–O Bond Formation. CCS Chem. 2023, 5, 245–256. [Google Scholar]
- Cope, J.D.; Liyanage, N.P.; Kelley, P.J.; Denny, J.A.; Valente, E.J.; Webster, C.E.; Delcamp, J.H.; Hollis, T.K. Electrocatalytic reduction of CO2 with CCC-NHC pincer nickel complexes. Chem. Commun. 2017, 53, 9442–9445. [Google Scholar]
- Qiu, T.; Yap, G.P.A.; Rosenthal, J. Nickel(II) Cyclen Complexes Bearing Ancillary Amide Appendages for the Electrocata lytic Reduction of CO2. ACS Appl. Energy Mater. 2019, 2, 8560–8569. [Google Scholar]
- Du, J.; Cheng, B.; Yuan, H.; Tao, Y.; Chen, Y.; Ming, M.; Han, Z.; Eisenberg, R. Molecular Nickel Thiolate Complexes for Electrochemical Reduction of CO2 to C1–3 Hydrocarbons. Angew. Chem. Int. Ed. 2023, 62, e202211804. [Google Scholar]
- Bíró, L.; Tóth, B.; Lihi, N.; Farkas, E.; Buglyó, P. Interaction between [(η6-p-cym)M(H2O)3]2+ (MII = Ru, Os) or [(η5-Cp*)M(H2O)3]2+ (MIII = Rh, Ir) and Phosphonate Derivatives of Iminodiacetic Acid: A Solution Equilibrium and DFT Study. Molecules 2023, 28, 1477. [Google Scholar]
- Bárta, J.; Hermann, P.; Kotek, J. Coordination Behavior of 1,4-Disubstituted Cyclen Endowed with Phosphonate, Phosphonate Monoethylester, and H-Phosphinate Pendant Arms. Molecules 2019, 24, 3324. [Google Scholar]
- Wang, M.; Ma, C.; Wen, H.; Chen, C. Synthesis and characterization of a series of manganese phosphonate complexes with various valences and nuclearity. Dalton Trans. 2009, 14, 994–1003. [Google Scholar]
- Sheikh, J.A.; Jena, H.S.; Clearfield, A.; Konar, S. Phosphonate Based High Nuclearity Magnetic Cages. Acc. Chem. Res. 2016, 49, 1093–1103. [Google Scholar] [PubMed]
- Yao, H.-C.; Li, Y.-Z.; Zheng, L.-M.; Xin, X.-Q. Syntheses, structures and magnetic properties of tetra- and heptanuclear iron(III) clusters incorporating phosphonate ligands. Inorg. Chem. 2005, 358, 2523–2529. [Google Scholar]
- Ma, Y.-S.; Song, Y.; Li, Y.-Z.; Zheng, L.-M. Tridecanuclear and Docosanuclear Manganese Phosphonate Clusters with Slow Magnetic Relaxation. Inorg. Chem. 2007, 46, 5459–5461. [Google Scholar]
- Bortoluzzi, M.; Castro, J.; Di Vera, A.; Palù, A.; Ferraro, V. Manganese(ii) bromo- and iodo-complexes with phospho ramidate and phosphonate ligands: Synthesis, characterization and photoluminescence. New J. Chem. 2021, 45, 12871–12878. [Google Scholar]
- Henderson, W.; Alley, S.R. Platinum(II) complexes containing ferrocene-derived phosphonate ligands; synthesis, structural characterisation and antitumour activity. Inorg. Chem. 2001, 322, 106–112. [Google Scholar]
- Wang, J.-M.; Liu, Y.-R.; Mao, X.-Y.; Shi, N.-N.; Zhang, X.; Wang, H.-S.; Fan, Y.-H.; Wang, M. Two Trinuclear CuII Complexes: Effect of Phosphonate Ligand on the Magnetic Property and Electrocatalytic Reactivity for Water Oxidation. Chem. Asian J. 2019, 14, 2685–2693. [Google Scholar]
- Vereshchuk, N.; Matheu, R.; Benet-Buchholz, J.; Pipelier, M.; Lebreton, J.; Dubreuil, D.; Tessier, A.; Gimbert-Suriñach, C.; Ertem, M.Z.; Llobet, A. Second Coordination Sphere Effects in an Evolved Ru Complex Based on Highly Adaptable Ligand Results in Rapid Water Oxidation Catalysis. J. Am. Chem. Soc. 2020, 142, 5068–5077. [Google Scholar]
- Stamatatos, T.C.; Escuer, A.; Abboud, K.A.; Raptopoulou, C.P.; Perlepes, S.P.; Christou, G. Unusual Structural Types in Nickel Cluster Chemistry from the Use of Pyridyl Oximes: Ni5, Ni12Na2, and Ni14 Clusters. Inorg. Chem. 2008, 47, 11825–11838. [Google Scholar] [PubMed]
- Polyzou, C.D.; Lada, Z.G.; Terzis, A.; Raptopoulou, C.P.; Psycharis, V.; Perlepes, S.P. The fac diastereoisomer of tris(2-pyridinealdoximato)cobalt(III) and a cationic cobalt(III) complex containing both the neutral and anionic forms of the ligand: Synthetic, structural and spectroscopic studies. Polyhedron 2014, 79, 29–36. [Google Scholar]
- Papatriantafyllopoulou, C.; Stamatatos, T.C.; Efthymiou, C.G.; Cunha-Silva, L.; Paz, F.A.A.; Perlepes, S.P.; Christou, G. A High-Nuclearity 3d/4f Metal Oxime Cluster: An Unusual Ni8Dy8 “Core−Shell” Complex from the Use of 2-Pyridinealdoxime. Inorg. Chem. 2010, 49, 9743–9745. [Google Scholar] [PubMed]
- Polyzou, C.D.; Koumousi, E.S.; Lada, Z.G.; Raptopoulou, C.P.; Psycharis, V.; Rouzières, M.; Tsipis, A.C.; Mathonière, C.; Clérac, R.; Perlepes, S.P. “Switching on” the single-molecule magnet properties within a series of dinuclear cobalt(iii)–dysprosium(iii) 2-pyridyloximate complexes. Dalton Trans. 2017, 46, 14812–14825. [Google Scholar] [PubMed]
- Stamatatos, T.C.; Abboud, K.A.; Perlepes, S.P.; Christou, G. The highest nuclearity metal oxime clusters: Ni14 and Ni12Na2 complexes from the use of 2-pyridinealdoximate and azide ligands. Dalton Trans. 2007, 35, 3861–3863. [Google Scholar]
- Esteban, J.; Alcázar, L.; Torres-Molina, M.; Monfort, M.; Font-Bardia, M.; Escuer, A. High Nuclearity in Azido/Oximate Chemistry: Ni14 and Ni13 Clusters with S = 6 and 9 Ground States. Inorg. Chem. 2012, 51, 5503–5505. [Google Scholar]
- Hossain, S.; Das, S.; Chakraborty, A.; Lloret, F.; Cano, J.; Pardo, E.; Chandrasekhar, V. S-shaped decanuclear hetero metallic [Ni8Ln2] complexes [Ln(iii) = Gd, Tb, Dy and Ho]: Theoretical modeling of the magnetic properties of the gadolinium analogue. Dalton Trans. 2014, 43, 10164–10174. [Google Scholar]
- Escuer, A.; Vlahopoulou, G.; Mautner, F.A. Use of 6-methylpyridine-2-carbaldehydeoxime in nickel(ii) carboxylate chemis try: Synthetic, structural and magnetic properties of penta and hexanuclear complexes. Dalton Trans. 2011, 40, 10109–10116. [Google Scholar]
- Chen, H.; Ma, C.-B.; Yuan, D.-Q.; Hu, M.-Q.; Wen, H.-M.; Liu, Q.-T.; Chen, C.-N. Syntheses, Structures, and Magnetic Properties of a Family of Tetra-, Hexa-, and Nonanuclear Mn/Ni Heterometallic Clusters. Inorg. Chem. 2011, 50, 10342–10352. [Google Scholar]
- Escuer, A.; Mayans, J.; Font-Bardia, M. Linked Nickel Metallacrowns from a Phosphonate/2-Pyridyloximate Blend of Lig ands: Structure and Magnetic Properties. Inorg. Chem. 2016, 55, 3161–3168. [Google Scholar] [PubMed]
- Lada, Z.G.; Polyzou, C.D.; Nika, V.; Stamatatos, T.C.; Konidaris, K.F.; Perlepes, S.P. Adventures in the coordination chemistry of 2-pyridyl oximes: On the way to 3d/4f-metal coordination clusters. Inorg. Chem. 2022, 539, 120954. [Google Scholar]
- Liu, W.; Thorp, H.H. Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2. Refined distances and other enzymes. Inorg. Chem. Commun. 1993, 32, 4102–4105. [Google Scholar]
- Zou, D.; Yi, Y.; Song, Y.; Guan, D.; Xu, M.; Ran, R.; Wang, W.; Zhou, W.; Shao, Z. The BaCe0.16Y0.04Fe0.8O3−δ nanocomposite: A new high-performance cobalt-free triple-conducting cathode for protonic ceramic fuel cells operating at reduced temperatures. J. Mater. Chem. A 2022, 10, 5381–5390. [Google Scholar]
- Shen, J.; Wang, M.; He, T.; Jiang, J.; Hu, M. Influence of the backbone of N5-pentadentate ligands on the catalytic perfor mance of Ni(ii) complexes for electrochemical water oxidation in neutral aqueous solutions. Chem. Commun. 2018, 54, 9019–9022. [Google Scholar]
- Shaffer, D.W.; Xie, Y.; Concepcion, J.J. O-O bond formation in ruthenium-catalyzed water oxidation: Single-site nucleophilic attack vs. O-O radical coupling. Chem. Soc. Rev. 2017, 46, 6170–6193. [Google Scholar]
- Lin, J.; Kang, P.; Liang, X.; Ma, B.; Ding, Y. Homogeneous electrocatalytic water oxidation catalyzed by a mononuclear nickel complex. Electrochim. Acta 2017, 258, 353–359. [Google Scholar]
- Hussain, N.; Yang, W.; Dou, J.; Chen, Y.; Qian, Y.; Xu, L. Ultrathin mesoporous F-doped α- Ni(OH)2 nanosheets as an efficient electrode material for water splitting and supercapacitors. J. Mater. Chem. A 2019, 7, 9656–9664. [Google Scholar]
- Li, Q.-J.; Ren, Y.-J.; Xie, Q.; Wu, M.; Feng, H.-X.; Zheng, L.-M.; Zhang, H.-X.; Long, J.-Q.; Wang, T.-S. Nickel (II) tetra pyridyl complexes as electrocatalysts and precatalysts for water oxidation. Appl. Organomet. Chem. 2020, 34, e5813. [Google Scholar]
- Lee, H.; Wu, X.; Sun, L. Copper-based homogeneous and heterogeneous catalysts for electrochemical water oxidation. Nanoscale 2020, 12, 4187–4218. [Google Scholar]
- Zhang, L.-H.; Yu, F.; Shi, Y.; Li, F.; Li, H. Base-enhanced electrochemical water oxidation by a nickel complex in neutralaqueous solution. Chem. Commun. 2019, 55, 6122–6125. [Google Scholar]
- Luo, G.-Y.; Huang, H.-H.; Wang, J.-W.; Lu, T.-B. Further Investigation of a Nickel-Based Homogeneous Water Oxidation Catalyst with Two cis Labile Sites. ChemSusChem 2016, 9, 485–491. [Google Scholar] [PubMed]
- Shevchenko, D.; Anderlund, M.F.; Styring, S.; Dau, H.; Zaharieva, I.; Thapper, A. Water oxidation by manganese ox ides formed from tetranuclear precursor complexes: The influence of phosphate on structure and activity. Phys. Chem. Chem. Phys. 2014, 16, 11965. [Google Scholar]
- Jin, K.; Park, J.; Lee, J.; Yang, K.D.; Pradhan, G.K.; Sim, U.; Jeong, D.; Jang, H.L.; Park, S.; Kim, D.; et al. Hydrated Manganese(II) Phosphate (Mn3(PO4)2·3H2O) as a Water Oxidation Catalyst. J. Am. Chem. Soc. 2014, 136, 7435–7443. [Google Scholar] [PubMed]
- De, R.; Gonglach, S.; Paul, S.; Haas, M.; Sreejith, S.S.; Gerschel, P.; Apfel, U.P.; Vuong, T.H.; Rabeah, J.; Roy, S.; et al. Electrocatalytic reduction of CO2 to acetic acid by a molecular manganese corrole complex. Angew. Chem. Int. Ed. 2020, 59, 10527–10534. [Google Scholar]
- Reid, A.G.; Hooe, S.L.; Moreno, J.J.; Dickie, D.A.; Machan, C.W. Homogeneous Electrocatalytic Reduction of CO2 by a CrN3O Complex: Electronic Coupling with a Redox-Active Terpyridine Fragment Favors Selectivity for CO. Inorg. Chem. 2022, 61, 16963–16970. [Google Scholar]
- Göttle, A.J.; Koper, M.T.M. Proton-coupled electron transfer in the electrocatalysis of CO2 reduction: Prediction of sequential vs. concerted pathways using DFT. Chem. Sci. 2017, 8, 458–465. [Google Scholar]
- Ahsan, H.; Breedlove, B.K.; Cosquer, G.; Yamashita, M. Enhancement of electrocatalytic abilities toward CO2 reduction by tethering redox-active metal complexes to the active site. Dalton Trans. 2021, 50, 13368–13373. [Google Scholar]
- Huang, H.-H.; Zhang, J.-H.; Dai, M.; Liu, L.; Ye, Z.; Liu, J.; Zhong, D.-C.; Wang, J.-W.; Zhao, C.; Ke, Z. Dual electronic effects achieving a high-performance Ni(II) pincer catalyst for CO2 photoreduction in a noble-metal-free system. Proc. Natl. Acad. Sci. USA 2022, 119, e2119267119. [Google Scholar]
- Wang, J.-W.; Huang, H.-H.; Sun, J.-K.; Zhong, D.-C.; Lu, T.-B. Syngas Production with a Highly-Robust Nickel(II) Homogeneous Electrocatalyst in a Water-Containing System. ACS Catal. 2018, 8, 7612–7620. [Google Scholar]
- Reed, B.R.; Yousif, M.; Lord, R.L.; McKinnon, M.; Rochford, J.; Groysman, S. Coordination Chemistry and Reactivity of Bis(al dimino)pyridine Nickel Complexes in Four Different Oxidation States. Organometallics 2017, 36, 582–593. [Google Scholar]
- Rodriguez-Olguin, M.A.; Flox, C.; Ponce-Pérez, R.; Lipin, R.; Ruiz-Zepeda, F.; Winczewski, J.P.; Kallio, T.; Vandichel, M.; Guerrero-Sánchez, J.; Gardeniers, J.G.E.; et al. Chlorine in NiO promotes electroreduction of CO2 to formate. Appl. Mater. 2022, 28, 101528. [Google Scholar]
- Löwe, A.; Schmidt, M.; Bienen, F.; Kopljar, D.; Wagner, N.; Klemm, E. Optimizing reaction conditions and gas diffusion electrodes applied in the CO2 reduction reaction to formate to reach current densities up to 1.8 A cm−2. ACS Sustain. Chem. Eng. 2021, 9, 4213–4223. [Google Scholar]
- Fan, T.; Ma, W.; Xie, M.; Liu, H.; Zhang, J.; Yang, S.; Huang, P.; Dong, Y.; Chen, Z.; Yi, X. Achieving high current density for electrocatalytic reduction of CO2 to formate on bismuth-based catalysts. Cell Rep. Phys. Sci. 2021, 2, 100353. [Google Scholar]
- Díaz-Sainz, G.; Fernández-Caso, K.; Lagarteira, T.; Delgado, S.; Alvarez-Guerra, M.; Mendes, A.; Irabien, A. Coupling continuous CO2 electroreduction to formate with efficient Ni-based anodes. J. Environ. Chem. Eng. 2023, 11, 109171. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C48H46Cl2N12Ni3O20P2 |
| Formula weight | 1419.94 |
| Crystal system | monoclinic |
| Space group | P2/n |
| a [Å] | 12.9435(7) |
| b [Å] | 19.4313(11) |
| c [Å] | 13.9832(8) |
| α [°] | 90 |
| β [°] | 103.797(5) |
| γ [°] | 90 |
| V [Å3] | 3415.4(3) |
| Z | 2 |
| ρcalcd [g m−3] | 1.381 |
| μ [mm−1] | 2.753 mm−1 |
| Reflections collected | 13,086 |
| F(000) | 1452 |
| Rint | 0.05 |
| T [K] | 293(2)K |
| Final R indices | R1 = 0.0525 |
| [I > 2sigma(I)] | wR1 = 0.1723 |
| R indices (all data) | R2 = 0.0677 |
| wR2 = 0.1957 | |
| Gof | 0.85 |
| [a] R1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2− − Fc2)2]/[∑w(Fo2)2]1/2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jian, H.; Lu, M.; Zheng, H.; Yan, S.; Wang, M. Electrochemical Water Oxidation and CO2 Reduction with a Nickel Molecular Catalyst. Molecules 2024, 29, 578. https://doi.org/10.3390/molecules29030578
Jian H, Lu M, Zheng H, Yan S, Wang M. Electrochemical Water Oxidation and CO2 Reduction with a Nickel Molecular Catalyst. Molecules. 2024; 29(3):578. https://doi.org/10.3390/molecules29030578
Chicago/Turabian StyleJian, Hengxin, Mengyu Lu, Haowen Zheng, Shengrui Yan, and Mei Wang. 2024. "Electrochemical Water Oxidation and CO2 Reduction with a Nickel Molecular Catalyst" Molecules 29, no. 3: 578. https://doi.org/10.3390/molecules29030578