Theoretical Investigation of Iridium Complex with Aggregation-Induced Emission Properties



Abstract
:
1. Introduction
2. Results
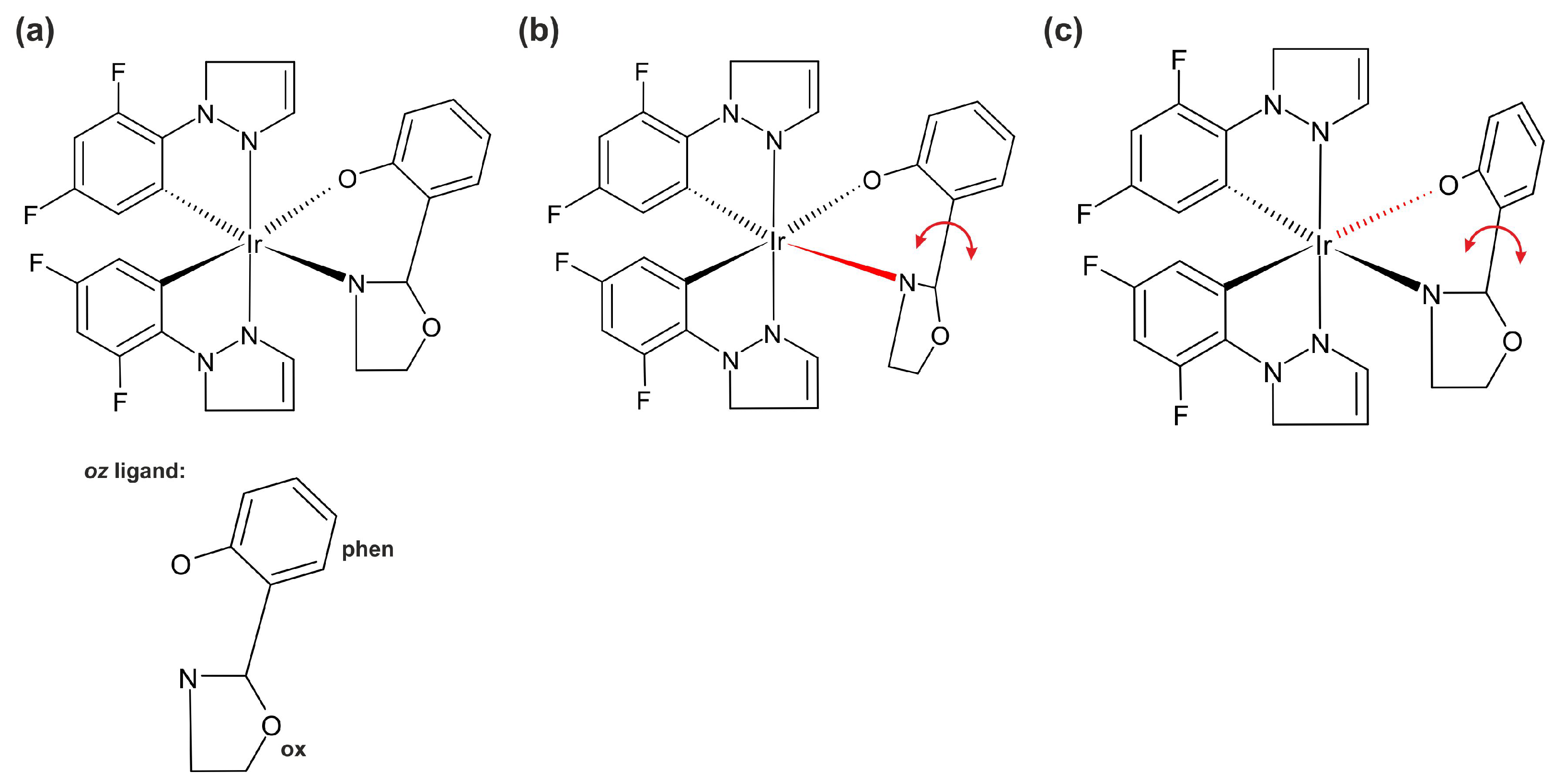
2.1. Geometry
2.2. Electronic Spectrum of 1
2.3. Properties of Monomer Complex 1
2.3.1. Rotation of the Oxazoline Moiety
2.3.2. Rotation of the Phenolate Moiety
2.4. Dimer of Ir(dfppz)2(oz)
3. Discussion
3.1. Monomer Complex 1
3.2. Dimer of Complex 1
3.3. Mechanism of AIE in Complex 1
4. Conclusions
5. Calculation Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIE | Aggregation Induced Emission |
| PEC | Potentia Energy Curve |
| RASTC | Restricted Acess to Singlet-Triplet Crossing |
| ISC | Intersystem Crossing |
| IC | Internal Conversion |
References
- Behera, S.K.; Park, S.Y.; Gierschner, J. Dual Emission: Classes, Mechanisms, and Conditions. Angew. Chem.—Int. Ed. 2021, 60, 22624–22638. [Google Scholar] [CrossRef] [PubMed]
- Girish, Y.; Prashantha, K.; Byrappa, K. Recent advances in aggregation-induced emission of mechanochromic luminescent organic materials. Emergent Mater. 2021, 4, 673–724. [Google Scholar] [CrossRef]
- Gong, Q.; Li, Y.; Wang, H.; Wang, G.; Feng, Q.; Zhong, Y.; Liu, F. Unveiling the Aggregation-Induced Emission (AIE) Mechanism and the Effect of Substituents on Luminescence Properties for Salicylaldehyde Azine Derivatives with Intramolecular Hydrogen Bond. J. Phys. Chem. C 2022, 126, 18429–18438. [Google Scholar] [CrossRef]
- Guan, J.; Shen, C.; Peng, J.; Zheng, J. What leads to aggregation-induced emission? J. Phys. Chem. Lett. 2021, 12, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.B.; Seixas de Melo, J.S. Aggregation-Induced Emission: From Small Molecules to Polymers—Historical Background, Mechanisms and Photophysics. Top. Curr. Chem. 2021, 379, 15. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, L.; Crespo-Otero, R. Aggregation-Induced Emission in the Tetraphenylthiophene Crystal: The Role of Triplet States. J. Phys. Chem. C 2020, 124, 17752–17761. [Google Scholar] [CrossRef]
- Suman, G.; Pandey, M.; Chakravarthy, A.S.J. Review on new horizons of aggregation induced emission: From design to development. Mater. Chem. Front. 2021, 5, 1541–1584. [Google Scholar] [CrossRef]
- Suzuki, S.; Sasaki, S.; Sairi, A.S.; Iwai, R.; Tang, B.Z.; Konishi, G. Principles of Aggregation-Induced Emission: Design of Deactivation Pathways for Advanced AIEgens and Applications. Angew. Chem.—Int. Ed. 2020, 59, 9856–9867. [Google Scholar] [CrossRef]
- Würthner, F. Aggregation-Induced Emission (AIE): A Historical Perspective. Angew. Chem.—Int. Ed. 2020, 59, 14192–14196. [Google Scholar] [CrossRef]
- Chen, B.; Nie, H.; Hu, R.; Qin, A.; Zhao, Z.; Tang, B.Z. Red fluorescent siloles with aggregation-enhanced emission characteristics. Sci. China Chem. 2016, 59, 699–706. [Google Scholar] [CrossRef]
- Chen, B.; Nie, H.; Lu, P.; Zhou, J.; Qin, A.; Qiu, H.; Zhao, Z.; Tang, B.Z. Conjugation versus rotation: Good conjugation weakens the aggregation-induced emission effect of siloles. Chem. Commun. 2014, 50, 4500–4503. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Junquera, M.; Lara, R.; Lalinde, E.; Moreno, M.T. Isomerism, aggregation-induced emission and mechanochromism of isocyanide cycloplatinated(ii) complexes. J. Mater. Chem. C 2020, 8, 7221–7233. [Google Scholar] [CrossRef]
- Zhang, X.; Chi, Z.; Zhang, Y.; Liu, S.; Xu, J. Recent advances in mechanochromic luminescent metal complexes. J. Mater. Chem. C 2013, 1, 3376–3390. [Google Scholar] [CrossRef]
- Alam, P.; Climent, C.; Alemany, P.; Laskar, I.R. “Aggregation-induced emission” of transition metal compounds: Design, mechanistic insights, and applications. J. Photochem. Photobiol. C Photochem. Rev. 2019, 41, 100317. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, G.; Jia, M.; Song, X.; Zhao, J. Exploring and elaborating the excited state mechanism of a novel AIE material 2-(5-(4-carboxyphenyl)-2-hydroxyphenyl)benzothiazole. Struct. Chem. 2018, 29, 1767–1773. [Google Scholar] [CrossRef]
- Quan, C.; Nie, H.; Zhao, Z.; Tang, B.Z. N-type organic luminescent materials based on siloles with aggregation-enhanced emission. In Proceedings of the Organic Light Emitting Materials and Devices XIX, San Diego, CA, USA, 9–13 August 2015; Volume 9566. [Google Scholar] [CrossRef]
- Chen, L.; Nie, H.; Chen, B.; Lin, G.; Luo, W.; Hu, R.; Huang, F.; Qin, A.; Zhao, Z.; Tang, B.Z. 2,5-Dicarbazole-functioned siloles with aggregation-enhanced emission for application in organic light-emitting diodes. J. Photonics Energy 2015, 5, 053598. [Google Scholar] [CrossRef]
- Mei, J.; Wang, J.; Sun, J.Z.; Zhao, H.; Yuan, W.; Deng, C.; Chen, S.; Sung, H.H.Y.; Lu, P.; Qin, A.; et al. Siloles symmetrically substituted on their 2,5-positions with electron-accepting and donating moieties: Facile synthesis, aggregation-enhanced emission, solvatochromism, and device application. Chem. Sci. 2012, 3, 549–558. [Google Scholar] [CrossRef]
- Li, X.F.; Zhou, W.; Liu, Y.C.; Hou, M.; Feng, G.L.; Ji, Y.M.; Zhang, Y.; Xing, G.W. Design and assembly of AIE-active fluorescent organic nanoparticles for anti-counterfeiting fluorescent hydrogels and inks. Chem. Commun. 2022, 58, 11547–11550. [Google Scholar] [CrossRef]
- Chi, W.; Wang, C.; Liu, X. State-crossing from a Locally Excited to an Electron Transfer State(SLEET) Model Rationalizing the Aggregation-induced Emission Mechanism of (Bi)piperidylanthracenes. Chem. Res. Chin. Univ. 2021, 37, 157–161. [Google Scholar] [CrossRef]
- Dai, W.; Bianconi, T.; Ferraguzzi, E.; Wu, X.; Lei, Y.; Shi, J.; Tong, B.; Carlotti, B.; Cai, Z.; Dong, Y. Excited-State Modulation of Aggregation-Induced Emission Molecules for High-Efficiency Triplet Exciton Generation. ACS Mater. Lett. 2021, 3, 1767–1777. [Google Scholar] [CrossRef]
- Liu, S.; Sun, H.; Ma, Y.; Ye, S.; Liu, X.; Zhou, X.; Mou, X.; Wang, L.; Zhao, Q.; Huang, W. Rational design of metallophosphors with tunable aggregation-induced phosphorescent emission and their promising applications in time-resolved luminescence assay and targeted luminescence imaging of cancer cells. J. Mater. Chem. 2012, 22, 22167–22173. [Google Scholar] [CrossRef]
- Huang, S.; Ding, J.; Bi, A.; Yu, K.; Zeng, W. Insights into Optical Probes Based on Aggregation-Induced Emission: From Restriction of Intramolecular Motions to Dark State. Adv. Opt. Mater. 2021, 9, 2100832. [Google Scholar] [CrossRef]
- Tu, Y.; Zhao, Z.; Lam, J.W.Y.; Tang, B.Z. Mechanistic connotations of restriction of intramolecular motions (RIM). Natl. Sci. Rev. 2021, 8, nwaa260. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Lam, J.W.Y.; Tang, B.Z. Restriction of Intramolecular Motion(RIM): Investigating AIE Mechanism from Experimental and Theoretical Studies. Chem. Res. Chin. Univ. 2021, 37, 1–15. [Google Scholar] [CrossRef]
- Zhou, P.; Li, P.; Zhao, Y.; Han, K. Restriction of Flip-flop Motion as a Mechanism for Aggregation-Induced Emission. J. Phys. Chem. Lett. 2019, 10, 6929–6935. [Google Scholar] [CrossRef]
- Li, Q.; Blancafort, L. A conical intersection model to explain aggregation induced emission in diphenyl dibenzofulvene. Chem. Commun. 2013, 49, 5966–5968. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.L.; Ruiz-Barragan, S.; Li, Z.S.; Li, Q.S.; Blancafort, L. Restricted access to a conical intersection to explain aggregation induced emission in dimethyl tetraphenylsilole. J. Mater. Chem. C 2016, 4, 2802–2810. [Google Scholar] [CrossRef]
- Song, Y.; Li, B.; Liu, S.; Qin, M.; Gao, Y.; Zhang, K.; Lin, L.; Wang, C.K.; Fan, J. Theoretical studies on the excited-state properties of thermally activated delayed fluorescence molecules with aggregation induced emission. J. Mater. Chem. C 2022, 10, 9377–9390. [Google Scholar] [CrossRef]
- Tu, Y.; Liu, J.; Zhang, H.; Peng, Q.; Lam, J.W.Y.; Tang, B.Z. Restriction of Access to the Dark State: A New Mechanistic Model for Heteroatom-Containing AIE Systems. Angew. Chem.—Int. Ed. 2019, 58, 14911–14914. [Google Scholar] [CrossRef]
- Wang, H.; Gong, Q.; Wang, G.; Dang, J.; Liu, F. Deciphering the Mechanism of Aggregation-Induced Emission of a Quinazolinone Derivative Displaying Excited-State Intramolecular Proton-Transfer Properties: A QM, QM/MM, and MD Study. J. Chem. Theory Comput. 2019, 15, 5440–5447. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.; Wang, W.; Liu, F. Exploring the Mechanism of Fluorescence Quenching and Aggregation-Induced Emission of a Phenylethylene Derivative by QM (CASSCF and TDDFT) and ONIOM (QM:MM) Calculations. J. Phys. Chem. C 2016, 120, 21850–21857. [Google Scholar] [CrossRef]
- Yamamoto, N. Free Energy Profile Analysis for the Aggregation-Induced Emission of Diphenyldibenzofulvene. J. Phys. Chem. A 2020, 124, 4939–4945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, J.; Zhang, Y.; Fan, J.; Wang, C.K.; Lin, L. Theoretical Study of the Mechanism of Aggregation-Caused Quenching in Near-Infrared Thermally Activated Delayed Fluorescence Molecules: Hydrogen-Bond Effect. J. Phys. Chem. C 2019, 123. [Google Scholar] [CrossRef]
- Alam, P.; Climent, C.; Kaur, G.; Casanova, D.; Roy Choudhury, A.; Gupta, A.; Alemany, P.; Laskar, I.R. Exploring the Origin of Aggregation Induced Emission Activity and Crystallization Induced Emission in Organometallic Iridium(III) Cationic Complexes: Influence of Counterions. Cryst. Growth Des. 2016, 16, 5738–5752. [Google Scholar] [CrossRef]
- Huang, K.; Wu, H.; Shi, M.; Li, F.; Yi, T.; Huang, C. Reply to comment on ’aggregation-induced phosphorescent emission (AIPE) of iridium(III) complexes’: Origin of the enhanced phosphorescence. Chem. Commun. 2009, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Naota, T.; Ehara, M. Origin of the Aggregation-Induced Phosphorescence of Platinum(II) Complexes: The Role of Metal–Metal Interactions on Emission Decay in the Crystalline State. Chem.—Asian J. 2021, 16, 3129–3140. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, L.; Li, F.; Yu, M.; Liu, Z.; Yi, T.; Huang, C. Aggregation-induced phosphorescent emission (AIPE) of iridium(III) complexes. Chem. Commun. 2008, 685–687. [Google Scholar] [CrossRef]
- Ma, L.; Wang, Y.; Wang, X.; Zhu, Q.; Wang, Y.; Li, L.; Cheng, H.B.; Zhang, J.; Liang, X.J. Transition metal complex-based smart AIEgens explored for cancer diagnosis and theranostics. Coord. Chem. Rev. 2022, 473, 214822. [Google Scholar] [CrossRef]
- Prasad, P.; Gupta, A.; Sasmal, P.K. Aggregation-induced emission active metal complexes: A promising strategy to tackle bacterial infections. Chem. Commun. 2021, 57, 174–186. [Google Scholar] [CrossRef]
- Ramdass, A.; Sathish, V.; Thanasekaran, P. AIE or AIE(P)E-active transition metal complexes for highly sensitive detection of nitroaromatic explosives. Results Chem. 2022, 4, 100337. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, X.; Li, X.; Zhang, X.; Deng, J.; Zou, D.; Yang, J. Two Ru(II) compounds with aggregation induced emission as promising photosensitizers for photodynamic therapy. J. Inorg. Biochem. 2020, 212, 111233. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Huh, H.S.; Kim, K.S.; Lee, S.W.; Kim, D.; Park, S.Y. Comment on ’aggregation-induced phosphorescent emission (AIPE) of iridium(III) complexes’: Origin of the enhanced phosphorescence. Chem. Commun. 2008, 3998–4000. [Google Scholar] [CrossRef] [PubMed]
- Che, W.; Li, G.; Liu, X.; Shao, K.; Zhu, D.; Su, Z.; Bryce, M.R. Selective sensing of 2,4,6-trinitrophenol (TNP) in aqueous media with “aggregation-induced emission enhancement” (AIEE)-active iridium(III) complexes. Chem. Commun. 2018, 54, 1730–1733. [Google Scholar] [CrossRef] [PubMed]
- Chao, K.; Shao, K.; Peng, T.; Zhu, D.; Wang, Y.; Liu, Y.; Su, Z.; Bryce, M.R. New oxazoline- and thiazoline-containing heteroleptic iridium(iii) complexes for highly-efficient phosphorescent organic light-emitting devices (PhOLEDs): Colour tuning by varying the electroluminescence bandwidth. J. Mater. Chem. C 2013, 1, 6800–6806. [Google Scholar] [CrossRef]
- Alam, P.; Kaur, G.; Chakraborty, S.; Roy Choudhury, A.; Laskar, I.R. Aggregation induced phosphorescence active rollover iridium(iii) complex as a multi-stimuli-responsive luminescence material. Dalton Trans. 2015, 44, 6581–6592. [Google Scholar] [CrossRef]
- Sheet, S.K.; Sen, B.; Aguan, K.; Khatua, S. A cationic organoiridium(iii) complex-based AIEgen for selective light-up detection of rRNA and nucleolar staining. Dalton Trans. 2018, 47, 11477–11490. [Google Scholar] [CrossRef] [PubMed]
- Vignesh, A.; Zhang, Q.; Khan, A.A.; Li, Z.; Salohiddinov, S.; Ma, Y.; Wang, J.; Sun, W.H. Exploring an aggregation induced emission behaviour of neutral iridium complexes consisting of salicylaldimine ligand with dibenzosuberane core. J. Organomet. Chem. 2021, 949, 121954. [Google Scholar] [CrossRef]
- Wittig, C. The Landau-Zener formula. J. Phys. Chem. B 2005, 109, 8428–8430. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision C.01, 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Azar, Y.T.; Payami, M. Theoretical description of efficiency enhancement in DSSCs sensitized by newly synthesized heteroleptic Ru complexes. Phys. Chem. Chem. Phys. 2015, 17, 29574–29585. [Google Scholar] [CrossRef]
- Le Bahers, T.; Brémond, E.; Ciofini, I.; Adamo, C. The nature of vertical excited states of dyes containing metals for DSSC applications: Insights from TD-DFT and density based indexes. Phys. Chem. Chem. Phys. 2014, 16, 14435–14444. [Google Scholar] [CrossRef] [PubMed]
- Boukabene, M.; Brahim, H.; Hadji, D.; Guendouzi, A. Theoretical study of geometric, optical, nonlinear optical, UV–Vis spectra and phosphorescence properties of iridium(III) complexes based on 5-nitro-2-(2,4-difluorophenyl)pyridyl. Theor. Chem. Accounts 2020, 139, 47. [Google Scholar] [CrossRef]
- Brahim, H.; Haddad, B.; Brahim, S.; Guendouzi, A. DFT/TDDFT computational study of the structural, electronic and optical properties of rhodium (III) and iridium (III) complexes based on tris-picolinate bidentate ligands. J. Mol. Model. 2017, 23, 344. [Google Scholar] [CrossRef] [PubMed]
- Guillemoles, J.F.; Barone, V.; Joubert, L.; Adamo, C. A theoretical investigation of the ground and excited states of selected Ru and Os polypyridyl molecular dyes. J. Phys. Chem. A 2002, 106, 11354–11360. [Google Scholar] [CrossRef]
- Gu, X.; Fei, T.; Zhang, H.; Xu, H.; Yang, B.; Ma, Y.; Liu, X. Theoretical studies of blue-emitting iridium complexes with different ancillary ligands. J. Phys. Chem. A 2008, 112, 8387–8393. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Maldivi, P.; Adamo, C. Predictions of optical excitations in transition-metal complexes with time dependent-Density Functional Theory: Influence of basis sets. J. Chem. Theory Comput. 2005, 1, 953–962. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Calc. | Expt. 1 |
|---|---|---|
| Ir-Ophen | 2.122 | 2.131 |
| Ir-Nox | 2.112 | 2.122 |
| Ir-N1 | 2.026 | 2.006 |
| Ir-N2 | 2.016 | 1.987 |
| Ir-C4 | 2.012 | 2.011 |
| Ir-C5 | 1.998 | 1.996 |
| C1-C2-C3-Nox | −6.9 | −11.9 |
| E (eV) | (nm) | f | Characteristic | Characteristic | (nm) 1 | ||
|---|---|---|---|---|---|---|---|
| Absorption | |||||||
| 3.55 | 348.3 | 0.059 | 0.980 | 370 | |||
| 3.75 | 330.1 | 0.032 | 0.892 | ||||
| 3.80 | 326.2 | 0.015 | 0.919 | ||||
| 3.95 | 313.2 | 0.017 | 0.590 | 0.262 | |||
| 4.00 | 309.9 | 0.026 | 0.583 | 0.336 | 325 | ||
| 4.06 | 304.9 | 0.010 | 0.828 | ||||
| 4.32 | 286.5 | 0.107 | 0.621 | 0.297 | |||
| 4.37 | 283.2 | 0.052 | 0.366 | 0.328 | 290 | ||
| 0.264 | |||||||
| Phosphorescence | |||||||
| 2.30 | 540 | 500 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodowski, P.; Jaworska, M. Theoretical Investigation of Iridium Complex with Aggregation-Induced Emission Properties. Molecules 2024, 29, 580. https://doi.org/10.3390/molecules29030580
Lodowski P, Jaworska M. Theoretical Investigation of Iridium Complex with Aggregation-Induced Emission Properties. Molecules. 2024; 29(3):580. https://doi.org/10.3390/molecules29030580
Chicago/Turabian StyleLodowski, Piotr, and Maria Jaworska. 2024. "Theoretical Investigation of Iridium Complex with Aggregation-Induced Emission Properties" Molecules 29, no. 3: 580. https://doi.org/10.3390/molecules29030580