
Computational Approaches to Evaluate the Acetylcholinesterase Binding Interaction with Taxifolin for the Management of Alzheimer’s Disease

 , , ,
, , ,  ,
,  and
and
Abstract

1. Introduction
2. Results and Discussion
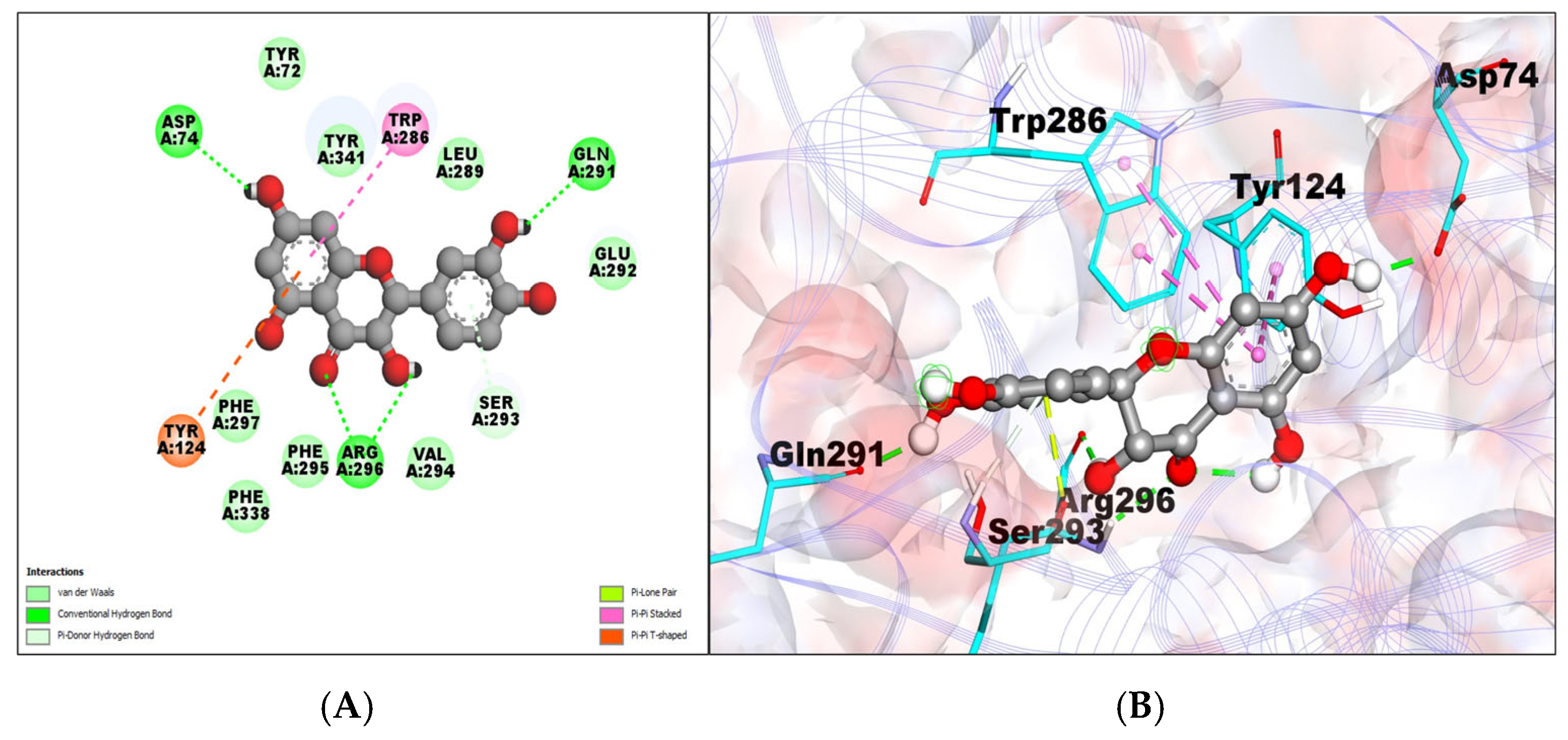
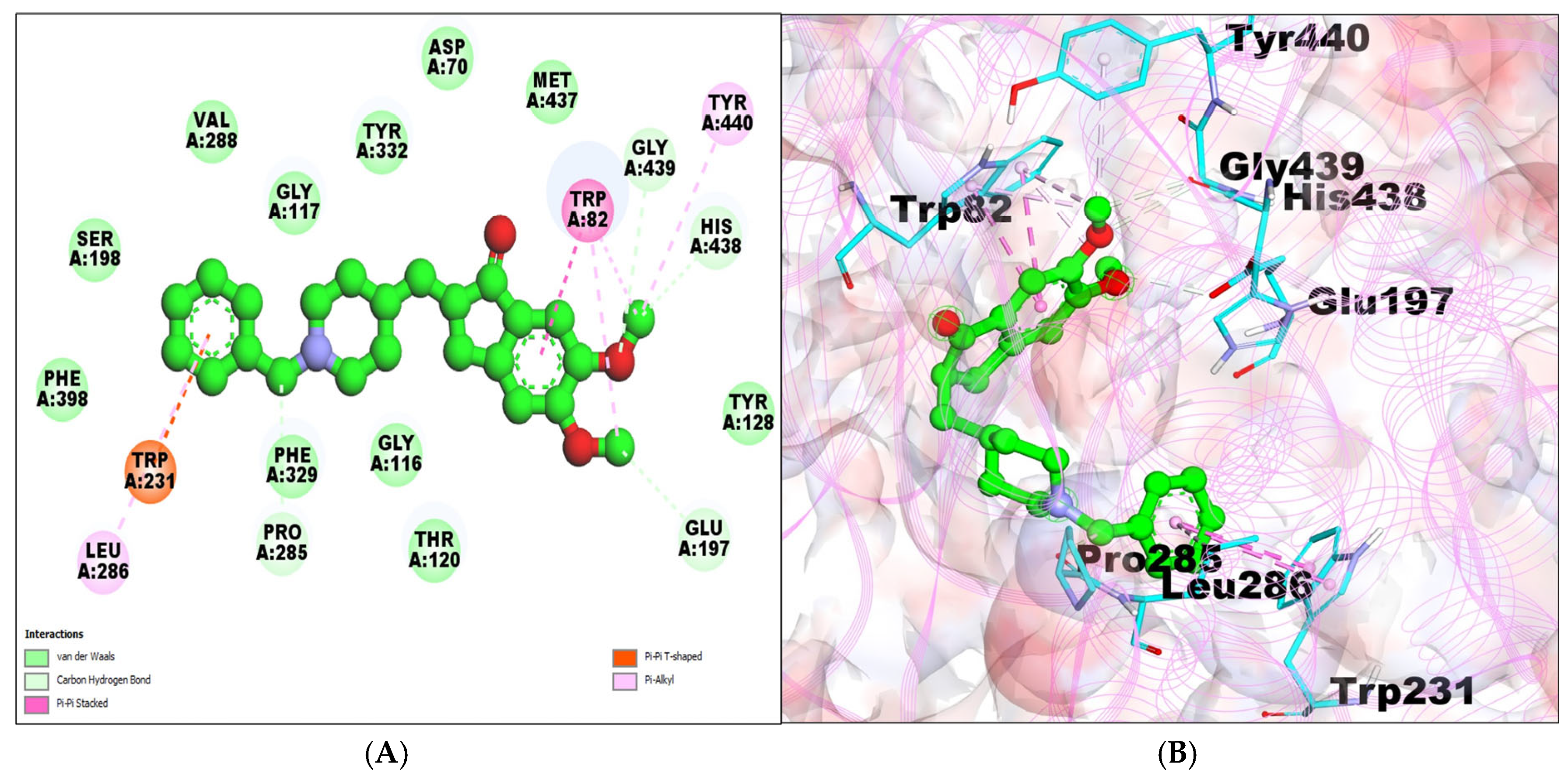
2.1. Molecular Docking Analysis
2.2. Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET) Analysis
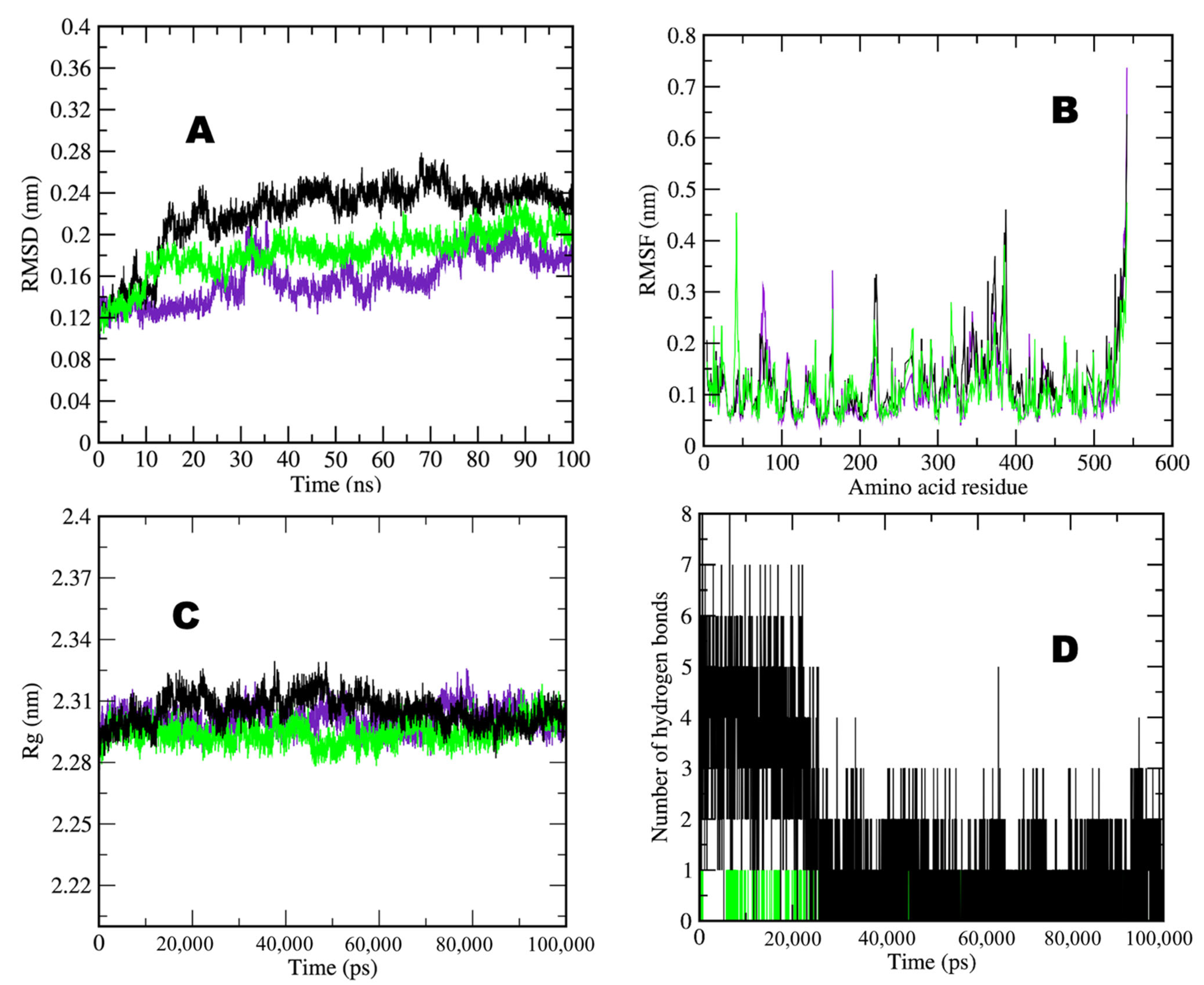
2.3. Molecular Dynamics Simulation(MDS) Analyses
3. Methodology
3.1. Ligand Preparation
3.2. Receptor Preparation
3.3. AutoDock 4.2 Tool Receptor-Ligand Docking
3.4. Drug-Likeness and ADMET
3.5. Molecular Dynamics Simulations (MDS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of Alzheimer’s disease and other neurodegenerative disorders--memantine, a new hope. Pharmacol. Res. 2005, 51, 1–17. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, Y.; Yin, W.; Li, J.; Wang, W.; Bai, F.; Xu, S.; Gong, Q.; Peng, T.; Hong, Y.; et al. Kinetics-Driven Drug Design Strategy for Next-Generation Acetylcholinesterase Inhibitors to Clinical Candidate. J. Med. Chem. 2021, 64, 1844–1855. [Google Scholar] [CrossRef]
- Jamal, Q.M.S.; Khan, M.I.; Alharbi, A.H.; Ahmad, V.; Yadav, B.S. Identification of Natural Compounds of the Apple as Inhibitors against Cholinesterase for the Treatment of Alzheimer’s Disease: An In Silico Molecular Docking Simulation and ADMET Study. Nutrients 2023, 15, 1579. [Google Scholar] [CrossRef]
- Walton, C.C.; Begelman, D.; Nguyen, W.; Andersen, J.K. Senescence as an Amyloid Cascade: The Amyloid Senescence Hypothesis. Front. Cell. Neurosci. 2020, 14, 129. [Google Scholar] [CrossRef]
- Taqui, R.; Debnath, M.; Ahmed, S.; Ghosh, A. Advances on plant extracts and phytocompounds with acetylcholinesterase inhibition activity for possible treatment of Alzheimer’s disease. Phytomed. Plus 2022, 2, 100184. [Google Scholar] [CrossRef]
- Budryn, G.; Grzelczyk, J.; Jaśkiewicz, A.; Żyżelewicz, D.; Pérez-Sánchez, H.; Cerón-Carrasco, J.P. Evaluation of butyrylcholinesterase inhibitory activity by chlorogenic acids and coffee extracts assed in ITC and docking simulation models. Food Res. Int. 2018, 109, 268–277. [Google Scholar] [CrossRef]
- Rani, N.; Bharti, S.; Krishnamurthy, B.; Bhatia, J.; Sharma, C.; Kamal, M.A.; Ojha, S.; Arya, D.S. Pharmacological Properties and Therapeutic Potential of Naringenin: A Citrus Flavonoid of Pharmaceutical Promise. Curr. Pharm. Des. 2016, 22, 4341–4359. [Google Scholar] [CrossRef]
- Jan, S.M.; Fahira, A.; Shi, Y.; Khan, M.I.; Jamal, A.; Mahmood, A.; Shams, S.; Parveen, Z.; Hu, J.; Umair, M.; et al. Integrative Genomic Analysis of m6a-SNPs Identifies Potential Functional Variants Associated with Alzheimer’s Disease. ACS Omega 2023, 8, 13332–13341. [Google Scholar] [CrossRef]
- Cutler, N.R.; Sramek, J.J. Review of the next generation of Alzheimer’s disease therapeutics: Challenges for drug development. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 27–57. [Google Scholar] [CrossRef]
- Jamal, A.; Jahan, S.; Choudhry, H.; Rather, I.A.; Khan, M.I. A Subtraction Genomics-Based Approach to Identify and Characterize New Drug Targets in Bordetella pertussis: Whooping Cough. Vaccines 2022, 10, 1915. [Google Scholar] [CrossRef]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sunil, C.; Xu, B. An insight into the health-promoting effects of taxifolin (dihydroquercetin). Phytochemistry 2019, 166, 112066. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, X.; Tian, Y.; Zhai, S.; Liu, Y.; Xiong, Z.; Chu, S. An insight into novel therapeutic potentials of taxifolin. Front. Pharmacol. 2023, 14, 1173855. [Google Scholar] [CrossRef] [PubMed]
- Topal, F.; Nar, M.; Gocer, H.; Kalin, P.; Kocyigit, U.M.; Gülçin, İ.; Alwasel, S.H. Antioxidant activity of taxifolin: An activity-structure relationship. J. Enzym. Inhib. Med. Chem. 2016, 31, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Saito, S.; Inoue, T.; Satoh-Asahara, N.; Ihara, M. Potential Therapeutic Approaches for Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1992. [Google Scholar] [CrossRef]
- Wadood, A.; Jamal, A.; Riaz, M.; Khan, A.; Uddin, R.; Jelani, M.; Azam, S.S. Subtractive genome analysis for in silico identification and characterization of novel drug targets in Streptococcus pneumonia strain JJA. Microb. Pathog. 2018, 115, 194–198. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Almasoudi, H.H.; Hakami, M.A.; Alhazmi, A.Y.; Makkawi, M.; Alasmari, S.; Alghamdi, Y.S.; Mashraqi, M.M. Unveiling the multitargeted repurposing potential of taxifolin (dihydroquercetin) in cervical cancer: An extensive MM\GBSA-based screening, and MD simulation study. Med. Oncol. 2023, 40, 218. [Google Scholar] [CrossRef]
- Orhan, I.E. Current concepts on selected plant secondary metabolites with promising inhibitory effects against enzymes linked to Alzheimer’s disease. Curr. Med. Chem. 2012, 19, 2252–2261. [Google Scholar] [CrossRef]
- Ishola, A.A.; Oyinloye, B.E.; Ajiboye, B.O.; Kappo, A.P. Molecular docking studies of flavonoids from Andrographis paniculata as potential acetylcholinesterase, butyrylcholinesterase and monoamine oxidase inhibitors towards the treatment of neurodegenerative diseases. Biointerface Res. Appl. Chem. 2021, 11, 9871–9879. [Google Scholar]
- Mittal, A.; Ghai, R.; Srivastava, A.; Ghosh, D.P.; Nagarajan, K. Pharmacokinetics and Pharmacodynamics: Fundamentals and Role (s) in Drug Discovery and Development. In Recent Advances in Pharmaceutical Innovation and Research; Springer: Berlin/Heidelberg, Germany, 2023; pp. 357–393. [Google Scholar]
- Ahmad, V.; Jamal, Q.M.S.; Akhtar, S.; Ahmad, A.; Naseem, Z.; Abuzinadah, M.F.; Karim, S. Marine sources potentiate to inhibit p-glycoprotein for the management of cancers and multidrug resistance. In An Introduction to P-Glycoprotein; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2021; pp. 245–273. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Abuzinadah, M.F.; Ahmad, V.; Al-Thawdi, S.; Zakai, S.A.; Jamal, Q.M.S. Exploring the Binding Interaction of Active Compound of Pineapple against Foodborne Bacteria and Novel Coronavirus (SARS-CoV-2) Based on Molecular Docking and Simulation Studies. Nutrients 2022, 14, 3045. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Duarte, J.M.; Dutta, S.; Fayazi, M.; Feng, Z. RCSB Protein Data Bank: Celebrating 50 years of the PDB with new tools for understanding and visualizing biological macromolecules in 3D. Protein Sci. 2022, 31, 187–208. [Google Scholar] [CrossRef]
- Biovia, D.S.; Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Richmond, T. Dassault systèmes BIOVIA, discovery studio visualizer, v. 17.2, San Diego: Dassault Systèmes, 2016. J. Chem. Phys. 2000, 10, 21–9991. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Dileep, K.V.; Ihara, K.; Mishima-Tsumagari, C.; Kukimoto-Niino, M.; Yonemochi, M.; Hanada, K.; Shirouzu, M.; Zhang, K.Y.J. Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery. Int. J. Biol. Macromol. 2022, 210, 172–181. [Google Scholar] [CrossRef]
- González-García, B.; Olave, M.E.; Ramos-Martínez, E.; González-Horta, C.; Levario-Carrillo, M.; Sánchez-Ramírez, B. Decrease of muscarinic cholinergic receptors expression in placenta from rats exposed to methyl parathion. Hum. Exp. Toxicol. 2008, 27, 241–246. [Google Scholar] [CrossRef]
- Taylor, P.; Camp, S.; Radić, Z. Acetylcholinesterase. In Encyclopedia of Neuroscience; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowski, M.G. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochemistry 2016, 55, 1226–1238. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; Pintro, V.O.; de Azevedo, W.F., Jr. Docking with AutoDock4. Methods Mol. Biol. 2019, 2053, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Binding Energy (Kcal/mol) | Inhibition Constant (Ki) μM: micro molar | Hydrogen Bonds | Hydrogen Bond Length Å(Angstrom) | Van der Waals Interaction | Other Interaction |
|---|---|---|---|---|---|---|
| AChE–donepezil (Control) | −9.33 | 0.144 μM | UNK1:DNP:C28–A:ASP74:OD1 | 3.03318 | TYR72,TYR341, LEU289,GLU292, VAL294,PHE295, PHE338,PHE297 | PI–PI STACKED/ PI–PI T-SHAPED TYR124, TRP286 |
| AChE–taxifolin (PDB:7E3H) | −8.85 | 0.327 μM | A:ARG296:HN–:UNL1:O1 | 2.25805 | TYR72,TYR341, PHE297,PHE338, PHE295,VAL294, GLU292,LEU289, | PI–PI T-SHAPED/PI–PI STACKED TYR124, TRP286 |
| UNL1:H11–A:GLN291:O | 1.87707 | |||||
| UNL1:H10–A:ARG296:O | 2.16795 | |||||
| UNL1:H8–A:ASP74:OD2 | 1.96554 | |||||
| A:SER293:HN–:UNL1 | 2.9256 | |||||
| BChE–donepezil (Control) | −7.67 | 2.390 μM | A:GLY439:CA–UNK1:E20601:O25 | 3.55463 | SER198,PHE398, PHE329,GLY116, THR120,TYR128, MET437,ASP70, TYR332,GLY117, VAL288 | PI–ALKYL LEU286,TRP82, TYR440 PI–PI T-SHAPED/PI–PI STACKED TRP82,TRP231 |
| UNK1:E20601:C17–A:PRO285:O | 3.19904 | |||||
| UNK1:E20601:C26–A:HIS438:O | 2.85071 | |||||
| UNK1:E20601:C28–A:GLU197:OE1 | 3.18095 | |||||
| BChE–taxifolin (PDB:7AIY) | −7.42 | 3.650 μM | A:TYR332:HH–:UNL1:O1 | 2.12817 | ILE69,ASN68, GLY121,SER79, TRP430,MET437, GLY439,TYR440 | PI–ALKYL ALA328 PI–PI STACKED TRP82 |
| UNL1:H12–A:GLN67:OE1 | 1.99252 | |||||
| UNL1:H11–A:ASN83:OD1 | 1.91515 | |||||
| UNL1:H10–A:ASP70:OD1 | 1.83371 | |||||
| UNL1:H8–A:HIS438:O | 2.22385 | |||||
| A:PRO84:CD–:UNL1:O7 | 3.47997 | |||||
| A:HIS438:CD2–:UNL1:O3 | 2.97375 |
| Complex Free Energy Calculation Components (kcal/mol) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Complex | ΔVdwaals | ΔEEL | ΔEPB | ΔENPOLAR | ΔEDISPER | ∆GGas | ∆GSolv | ∆GTotal |
| AChE–donepezil | −4325.87 (±5.40) | −29,859.62 (±21.16) | −4848.75 (±19.85) | 102.59 (±0.23) | 0.00 (±0.0) | −28,931.46 (±22.49) | −4746.16 (±19.71) | −33,677.61 (±11.03) |
| AChE–taxifolin | −4313.25 (±5.17) | −29,618.25 (±16.14) | −5040.32 (±15.79) | 104.17 (±0.24) | 0.00 (±0.0) | −28,477.93 (±18.85) | −4936.15 (±15.64) | −33,414.08 (±9.20) |
| BChE–donepezil | −4372.48 (±5.80) | −32,642.66 (±23.70) | −6031.58 (±22.45) | 112.68 (±0.24) | 0.00 (±0.00) | −29,327.66 (±25.01) | −5918.90 (±22.30) | −35,246.56 (±14.62) |
| BChE–taxifolin | −4398.54 (±4.41) | −32,414.36 (±20.44) | −6199.93 (±20.18) | 110.48 (±0.23) | 0.00 (±0.00) | −29,130.52 (±18.68) | −6089.45 (±20.06) | −35,219.97 (±13.65) |
| Ligand–Receptor Free Energy Calculation Components | ||||||||
|---|---|---|---|---|---|---|---|---|
| Complex | ΔVdwaals | ΔEEL | ΔEPB | ΔENPOLAR | ΔEDISPER | ∆GGas | ∆GSolv | ∆GTotal |
| AChE–donepezil | −56.23 (±0.46) | −256.40 (±1.50) | 282.05 (±2.05) | −5.09 (±0.02) | 0.00 (±0.00) | −312.62 (±1.70) | 276.96 (±2.04) | −35.66 (±0.93) |
| AChE–taxifolin | −35.49 (±0.37) | −10.23 (±0.71) | 24.44 (±0.73) | −3.06 (±0.02) | 0.00 (±0.00) | −45.72 (±0.80) | 21.38 (±0.80) | −24.34 (±0.56) |
| BChE–donepezil | −44.72 (±0.47) | −166.47 (±2.63) | 182.10 (±2.34) | −4.80 (±0.02) | 0.00 (±0.00) | −211.19 (±2.77) | 177.30 (±2.33) | −33.90 (±0.73) |
| BChE–taxifolin | −34.75 (±0.37) | −8.99 (±0.53) | 31.02 (±0.59) | −3.42 (±0.02) | 0.00 (±0.00) | −43.74 (±0.64) | 27.60 (±0.58) | −16.14 (±0.52) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, V.; Alotibi, I.; Alghamdi, A.A.; Ahmad, A.; Jamal, Q.M.S.; Srivastava, S. Computational Approaches to Evaluate the Acetylcholinesterase Binding Interaction with Taxifolin for the Management of Alzheimer’s Disease. Molecules 2024, 29, 674. https://doi.org/10.3390/molecules29030674
Ahmad V, Alotibi I, Alghamdi AA, Ahmad A, Jamal QMS, Srivastava S. Computational Approaches to Evaluate the Acetylcholinesterase Binding Interaction with Taxifolin for the Management of Alzheimer’s Disease. Molecules. 2024; 29(3):674. https://doi.org/10.3390/molecules29030674
Chicago/Turabian StyleAhmad, Varish, Ibrahim Alotibi, Anwar A. Alghamdi, Aftab Ahmad, Qazi Mohammad Sajid Jamal, and Supriya Srivastava. 2024. "Computational Approaches to Evaluate the Acetylcholinesterase Binding Interaction with Taxifolin for the Management of Alzheimer’s Disease" Molecules 29, no. 3: 674. https://doi.org/10.3390/molecules29030674
APA StyleAhmad, V., Alotibi, I., Alghamdi, A. A., Ahmad, A., Jamal, Q. M. S., & Srivastava, S. (2024). Computational Approaches to Evaluate the Acetylcholinesterase Binding Interaction with Taxifolin for the Management of Alzheimer’s Disease. Molecules, 29(3), 674. https://doi.org/10.3390/molecules29030674