
Organic Synthesis and Current Understanding of the Mechanisms of CFTR Modulator Drugs Ivacaftor, Tezacaftor, and Elexacaftor



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ivacaftor (VX-770)
2.1. Synthetic Routes
2.2. Mechanisms of Action
3. Lumacaftor (VX-809), Tezacaftor (VX-661) and Double Combinations with Ivacaftor
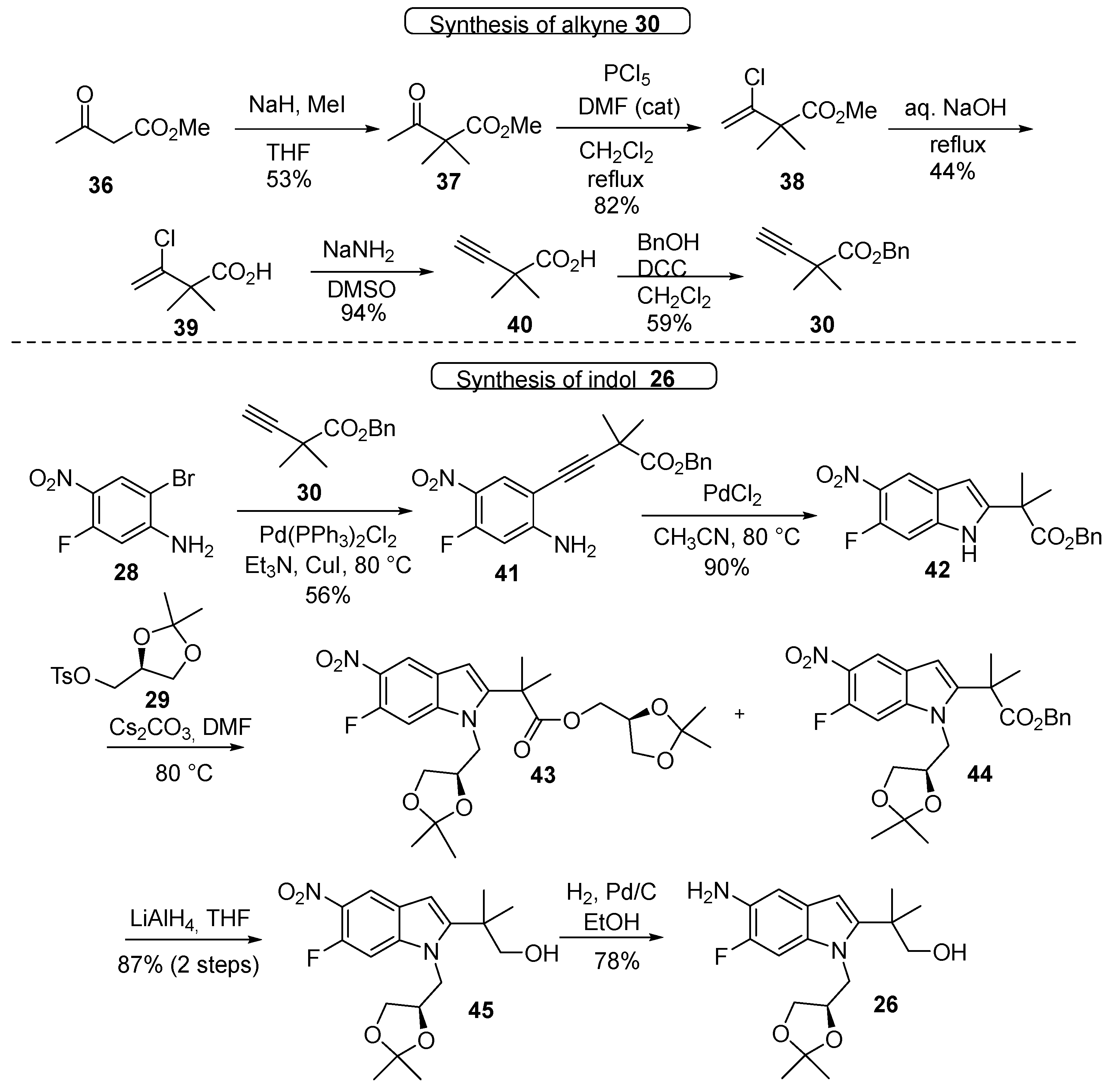
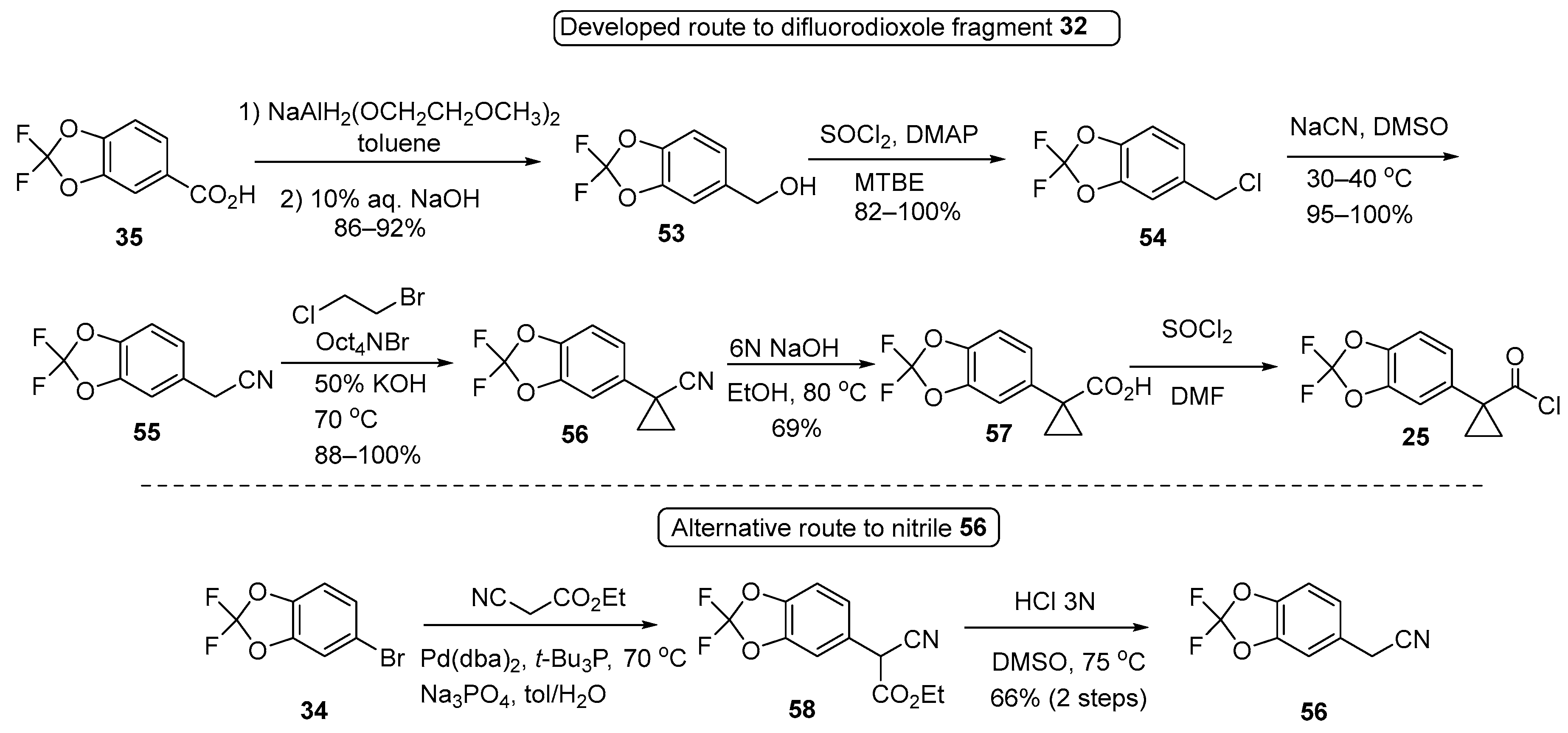
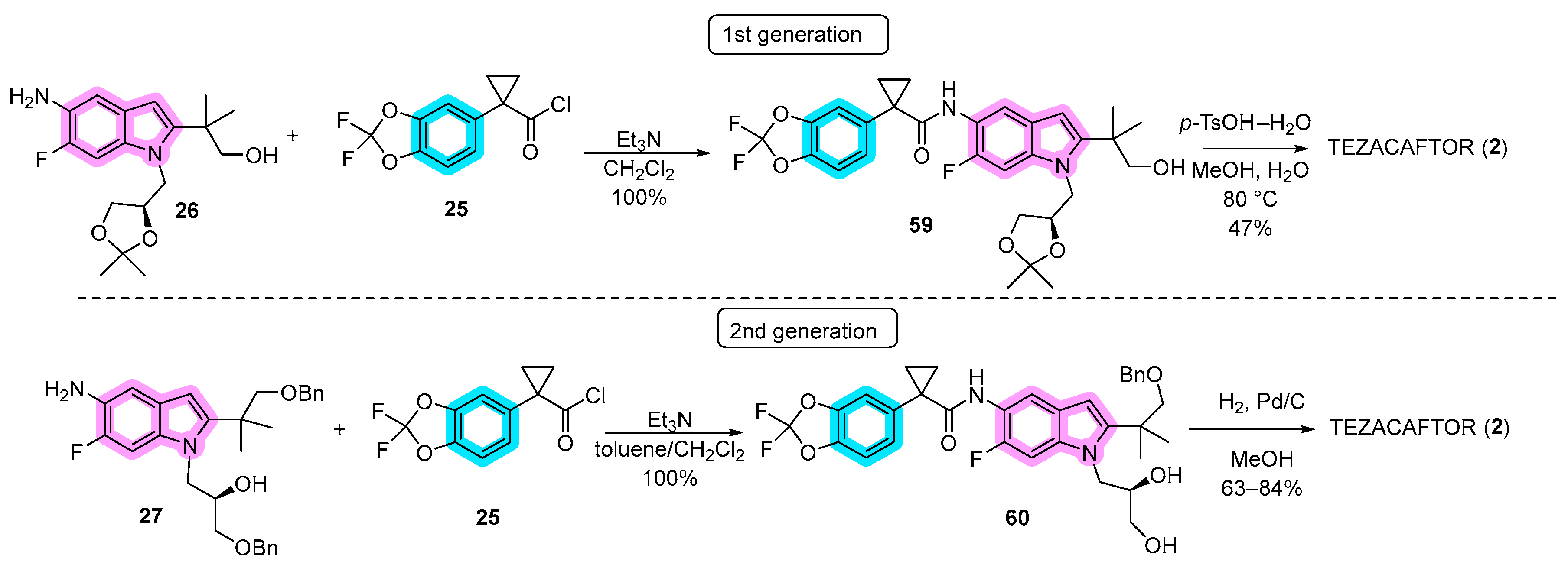
3.1. Synthetic Routes
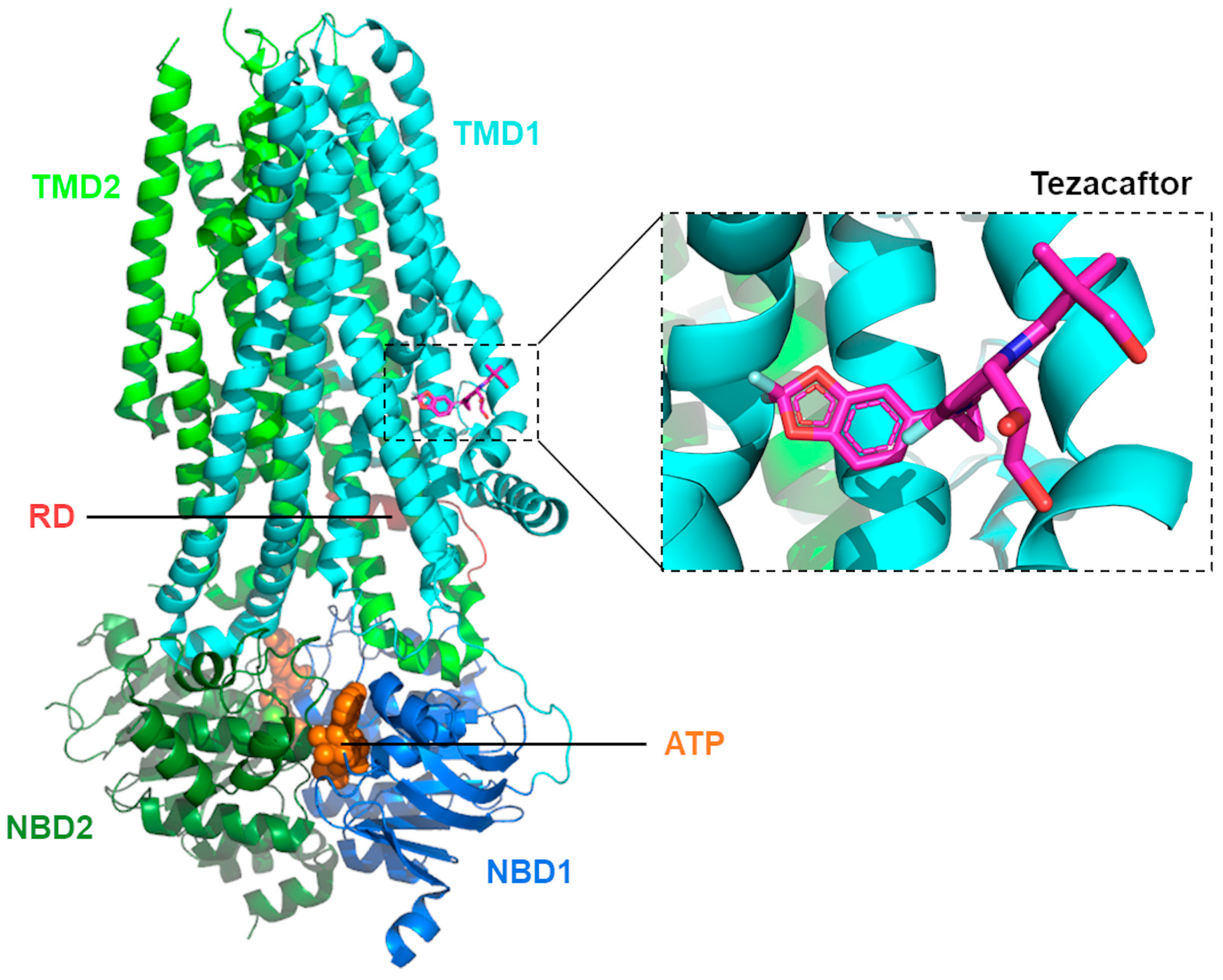
3.2. Mechanisms of Action
4. Elexacaftor (VX-445) and Triple Combination with Tezacaftor and Ivacaftor
4.1. Synthetic Routes
4.2. Mechanisms of Action
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Collins, F.S. Cystic Fibrosis: Molecular Biology and Therapeutic Implications. Science 1992, 256, 774–779. [Google Scholar] [CrossRef]
- Tsui, L.C.; Buchwald, M.; Barker, D.; Braman, J.C.; Knowlton, R.; Schumm, J.W.; Eiberg, H.; Mohr, J.; Kennedy, D.; Plavsic, N. Cystic Fibrosis Locus Defined by a Genetically Linked Polymorphic DNA Marker. Science 1985, 230, 1054–1057. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P. Pharmacological Rescue of Mutant CFTR Function for the Treatment of Cystic Fibrosis. In Top Med Chem; Springer: Berlin/Heidelberg, Jermany, 2008; Volume 3, pp. 91–120. ISBN 978-3-540-79729-6. [Google Scholar]
- Tsui, L.-C.; Dorfman, R. The Cystic Fibrosis Gene: A Molecular Genetic Perspective. Cold Spring Harb. Perspect. Med. 2013, 3, a009472. [Google Scholar] [CrossRef]
- De Boeck, K.; Amaral, M.D. Progress in Therapies for Cystic Fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.N.; Welsh, M.J. Structure and Function of the CFTR Chloride Channel. Physiol. Rev. 1999, 79, S23–S45. [Google Scholar] [CrossRef] [PubMed]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, Gating, and Regulation of the CFTR Anion Channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular Structure of the ATP-Bound, Phosphorylated Human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [PubMed]
- Kidd, J.F.; Ramjeesingh, M.; Stratford, F.; Huan, L.J.; Bear, C.E. A Heteromeric Complex of the Two Nucleotide Binding Domains of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Mediates ATPase Activity. J. Biol. Chem. 2004, 279, 41664–41669. [Google Scholar] [CrossRef]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef]
- Riordan, J.R. Assembly of Functional CFTR Chloride Channels. Annu. Rev. Physiol. 2005, 67, 701–718. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.-S.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.-S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.-C. Identification of the Cystic Fibrosis Gene: Genetic Analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR modulators: Shedding light on precision medicine for cystic fibrosis. Front Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.E.; Carlon, M.S.; Pedemonte, N.; Lopes-Pacheco, M. The Revolution of Personalized Pharmacotherapies for Cystic Fibrosis: What Does the Future Hold? Expert Opin. Pharmacother. 2023, 24, 1545–1565. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR Modulators for the Treatment of Cystic Fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019, 23, 2302–2322. [Google Scholar] [CrossRef]
- Flick, A.C.; Leverett, C.A.; Ding, H.X.; McInturff, E.; Fink, S.J.; Mahapatra, S.; Carney, D.W.; Lindsey, E.A.; DeForest, J.C.; France, S.P.; et al. Synthetic Approaches to the New Drugs Approved during 2019. J. Med. Chem. 2021, 64, 3604–3657. [Google Scholar] [CrossRef] [PubMed]
- Bisacchi, G.S. Origins of the Quinolone Class of Antibacterials: An Expanded “Discovery Story”. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef]
- Dhiman, P.; Arora, N.; Thanikachalam, P.V.; Monga, V. Recent Advances in the Synthetic and Medicinal Perspective of Quinolones: A Review. Bioorg. Chem. 2019, 92, 103291. [Google Scholar] [CrossRef]
- Sun, S.; Jia, Q.; Zhang, Z. Applications of Amide Isosteres in Medicinal Chemistry. Bioorg. Med. Chem. Lett. 2019, 29, 2535–2550. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.G.; Jacobs, W.A. The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc. 1939, 61, 2890–2895. [Google Scholar] [CrossRef]
- Zewge, D.; Chen, C.-Y.; Deer, C.; Dormer, P.G.; Hughes, D.L. A Mild and Efficient Synthesis of 4-Quinolones and Quinolone Heterocycles. J. Org. Chem. 2007, 72, 4276–4279. [Google Scholar] [CrossRef] [PubMed]
- Desi Reddy, S.R.; Rane, D.R.; Velivela, V.S.R. Industrial Process for Making Ivacaftor and Its Intermediates. U.S. Patent 2017/0096397 A1, 6 April 2017. [Google Scholar]
- Zhang, R.; Han, G.; Jiang, L.; Shen, Y.; Yang, R.; Mao, Y.; Wang, H. An Efficient Synthesis of Ivacaftor. J. Heterocycl. Chem. 2017, 54, 3169–3173. [Google Scholar] [CrossRef]
- Thatipally, S.; Reddy, V.K.; Dammalapati, V.L.N.R.; Chava, S. Processes for the Preparation of Ivacaftor. U.S. Patent 2018/0244622 A1, 30 August 2018. [Google Scholar]
- Vasudevan, N.; Jachak, G.R.; Reddy, D.S. Breaking and Making of Rings: A Method for the Preparation of 4-Quinolone-3-carb oxylic Acid Amides and the Expensive Drug Ivacaftor. European J. Org. Chem. 2015, 2015, 7433–7437. [Google Scholar] [CrossRef]
- Reddy, D.S.; Kulkarni, A.A.; Natarajan, V.; Sharma, M.K. An Improved Process for the Synthesis of Ivacaftor. Europe Patent 3464243 A2, 10 April 2019. [Google Scholar]
- Thatipally, S.; Dammalapati, V.L.N.R.; Gorantla, S.R.; Chava, S. A Process for the Preparation of Ivacaftor and Its Intermediates. U.S. Patent WO 2014/125506 A2, 21 August 2014. [Google Scholar]
- Hadida, S.; Van Goor, F.; Zhou, J.; Arumugam, V.; McCartney, J.; Hazlewood, A.; Decker, C.; Negulescu, P.; Grootenhuis, P.D.J. Discovery of N-(2,4-Di-Tert-Butyl-5-Hydroxyphenyl)-4-Oxo-1,4-Dihydroquinoline-3-Carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator. J. Med. Chem. 2014, 57, 9776–9795. [Google Scholar] [CrossRef]
- Hadida-Ruah, S.; Hazelwood, A.; Van Goor, F.; Singh, A.; Zhou, J.; McCartney, J. Modulators of ATP-Binding Cassette Transporters. U.S. Patent 2006/0074075 A1, 6 April 2006. [Google Scholar]
- Hadida-Ruah, S.; Hazelwood, A.; Grootenhuis, P.; Van Goor, F.; Singh, A.; Zhou, J.; McCartney, J. Modulators of ATP-Binding Cassette Transporters. U.S. Patent 7,495,103 B2, 24 February 2009. [Google Scholar]
- Lai, C.K.; Song, T.V.; Li, J.J. Ivacaftor (Kalydeco): A CFTR Potentiator for the Treatment of Cystic Fibrosis. In Innovative Drug Synthesis; Li, J.J., Johnson, D.S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 303–316. ISBN 9781118819951. [Google Scholar]
- Li, J.J. Name Reactions, 3rd ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2006; ISBN 978-3-540-30030-4. [Google Scholar]
- Arekar, S.G.; Johnston, S.C.; Krawiec, M.; Medek, A.; Mudunuri, P.; Sullivan, M.J. Solid Forms of N-[2,4-Bis(1,1-Dimethylethyl)-5-Hydroxyphenyl]-1,4-Dihydro-4-Oxoquinoline-3-Carboxamide. U.S. Patent 2011/0230519 A1, 22 September 2011. [Google Scholar]
- Luisi, B.; Arekar, S.G.; Costache, A.; Dinehart, K.R.; Johnston, S.C.; Neubert-Langille, B.J. Solid Forms of N-[2,4-Bis(1,1-Dimethylethyl)-5-Hydroxyphenyl]-1,4-Dihydro-4-Oxoquinoline-3-Carboxamide. U.S. Patent 9,045,425 B2, 2 June 2015. [Google Scholar]
- European Medicines Agency. European Public Assessment Report: Symkevi (International Non-Proprietary Name: Tezacaftor/Ivacaftor). Procedure No. EMEA/H/C/004682/0000. 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/symkevi-epar-public-assessment-report_en.pdf (accessed on 22 December 2023).
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF Airway Epithelial Cell Function in Vitro by a CFTR Potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and Safety of Ivacaftor in Patients with Cystic Fibrosis and a Non-G551D Gating Mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.; Solomon, G.M.; Baines, A.; Heltshe, S.L.; VanDalfsen, J.; Joseloff, E.; Sagel, S.D.; Rowe, S.M.; GOALe(2) Investigators. Effectiveness of Ivacaftor in Cystic Fibrosis Patients with Non-G551D Gating Mutations. J. Cyst. Fibros. 2019, 18, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Eckford, P.D.W.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-Dependent but ATP-Independent Manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [PubMed]
- Jih, K.-Y.; Hwang, T.-C. Vx-770 Potentiates CFTR Function by Promoting Decoupling between the Gating Cycle and ATP Hydrolysis Cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4404–4409. [Google Scholar] [CrossRef]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator Ivacaftor Abrogates Pharmacological Correction of ΔF508 CFTR in Cystic Fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.-W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.-S.; Finkbeiner, W.E.; et al. Some Gating Potentiators, Including VX-770, Diminish ΔF508-CFTR Functional Expression. Sci. Transl. Med. 2014, 6, 246ra97. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Silva, I.A.L.; Turner, M.J.; Carlile, G.W.; Sondo, E.; Thomas, D.Y.; Pedemonte, N.; Hanrahan, J.W.; Amaral, M.D. Characterization of the Mechanism of Action of RDR01752, a Novel Corrector of F508del-CFTR. Biochem. Pharmacol. 2020, 180, 114133. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric Folding Correction of F508del and Rare CFTR Mutants by Elexacaftor-Tezacaftor-Ivacaftor (Trikafta) Combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Yeh, H.-I.; Yeh, J.-T.; Hwang, T.-C. Modulation of CFTR Gating by Permeant Ions. J. Gen. Physiol. 2015, 145, 47–60. [Google Scholar] [CrossRef]
- Yeh, H.-I.; Sohma, Y.; Conrath, K.; Hwang, T.-C. A Common Mechanism for CFTR Potentiators. J. Gen. Physiol. 2017, 149, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.-I.; Qiu, L.; Sohma, Y.; Conrath, K.; Zou, X.; Hwang, T.-C. Identifying the Molecular Target Sites for CFTR Potentiators GLPG1837 and VX-770. J. Gen. Physiol. 2019, 151, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, L.J.; Xu, Y.; Qiu, X.; Hall, J.D.; West, G.M. Sites Associated with Kalydeco Binding on Human Cystic Fibrosis Transmembrane Conductance Regulator Revealed by Hydrogen/Deuterium Exchange. Sci. Rep. 2018, 8, 4664. [Google Scholar] [CrossRef] [PubMed]
- Csanády, L.; Töröcsik, B. Cystic Fibrosis Drug Ivacaftor Stimulates CFTR Channels at Picomolar Concentrations. Elife 2019, 8, e46450. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of Ivacaftor on CFTR Forms with Missense Mutations Associated with Defects in Protein Processing or Function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Y.; Sohma, Y.; Hwang, T.-C. Synergistic Potentiation of Cystic Fibrosis Transmembrane Conductance Regulator Gating by Two Chemically Distinct Potentiators, Ivacaftor (VX-770) and 5-Nitro-2-(3-Phenylpropylamino) Benzoate. Mol. Pharmacol. 2016, 90, 275–285. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Loizidou, A.; Bugeja, L.A.; Warner, R.; Hawley, B.R.; Cai, Z.; Toye, A.M.; Sheppard, D.N.; Li, H. CFTR Potentiators Partially Restore Channel Function to A561E-CFTR, a Cystic Fibrosis Mutant with a Similar Mechanism of Dysfunction as F508del-CFTR. Br. J. Pharmacol. 2014, 171, 4490–4503. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, Z.; Gosling, M.; Sheppard, D.N. Potentiation of the Cystic Fibrosis Transmembrane Conductance Regulator Cl− Channel by Ivacaftor Is Temperature Independent. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L846–L857. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural Identification of a Hotspot on CFTR for Potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Righetti, G.; Casale, M.; Tonelli, M.; Liessi, N.; Fossa, P.; Pedemonte, N.; Millo, E.; Cichero, E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals 2020, 13, 445. [Google Scholar] [CrossRef]
- Laselva, O.; Qureshi, Z.; Zeng, Z.-W.; Petrotchenko, E.V.; Ramjeesingh, M.; Hamilton, C.M.; Huan, L.-J.; Borchers, C.H.; Pomès, R.; Young, R.; et al. Identification of Binding Sites for Ivacaftor on the Cystic Fibrosis Transmembrane Conductance Regulator. iScience 2021, 24, 102542. [Google Scholar] [CrossRef] [PubMed]
- Levring, J.; Terry, D.S.; Kilic, Z.; Fitzgerald, G.; Blanchard, S.C.; Chen, J. CFTR Function, Pathology and Pharmacology at Single-Molecule Resolution. Nature 2023, 616, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Ersoy, A.; Altintel, B.; Livnat Levanon, N.; Ben-Tal, N.; Haliloglu, T.; Lewinson, O. Computational Analysis of Long-Range Allosteric Communications in CFTR. Elife 2023, 12, RP88659. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Da Fonte, D.F.; Avramescu, R.G.; Premchandar, A.; Bagdany, M.; Xu, H.; Bensinger, D.; Stubba, D.; Schmidt, B.; Matouk, E.; et al. Mutation-Specific Dual Potentiators Maximize Rescue of CFTR Gating Mutants. J. Cyst. Fibros. 2020, 19, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Phuan, P.W.; Son, J.H.; Tan, J.A.; Li, C.; Musante, I.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Kurth, M.J.; Galietta, L.J.; et al. Combination Potentiator (‘co-Potentiator’) Therapy for CF Caused by CFTR Mutants, Including N1303K, That Are Poorly Responsive to Single Potentiators. J. Cyst. Fibros. 2018, 17, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor Co-Potentiates the Activity of F508del and Gating Mutants of CFTR. J. Cyst. Fibros. 2021, 20, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Berg, A.P.; Wang, Y.; Jantarajit, W.; Sutcliffe, K.J.; Stevens, E.B.; Cao, L.; Pregel, M.J.; Sheppard, D.N. A Small Molecule CFTR Potentiator Restores ATP-dependent Channel Gating to the Cystic Fibrosis Mutant G551D-CFTR. Br. J. Pharmacol. 2022, 179, 1319–1337. [Google Scholar] [CrossRef]
- Ensinck, M.M.; De Keersmaecker, L.; Ramalho, A.S.; Cuyx, S.; Van Biervliet, S.; Dupont, L.; Christ, F.; Debyser, Z.; Vermeulen, F.; Carlon, M.S. Novel CFTR Modulator Combinations Maximise Rescue of G85E and N1303K in Rectal Organoids. ERJ Open Res. 2022, 8, 00716–02021. [Google Scholar] [CrossRef]
- Bacalhau, M.; Ferreira, F.C.; Silva, I.A.L.; Buarque, C.D.; Amaral, M.D.; Lopes-Pacheco, M. Additive Potentiation of R334W-CFTR Function by Novel Small Molecules. J. Pers. Med. 2023, 13, 102. [Google Scholar] [CrossRef]
- Talele, T.T. The “Cyclopropyl Fragment” Is a Versatile Player That Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal Chemistry of Indole Derivatives: Current to Future Therapeutic Prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef]
- Ruah, S.H.S.; Grootenhuis, P.D.J.; Van Goor, F.; Miller, M.T.; McCartney, J.; Zhou, J.; Bear, B.; Numa, M.M.D. Modulators of ATP-Binding Cassette Transporters. International Patent 2010/053471 A1, 14 May 2010. [Google Scholar]
- Ruah, S.S.H.; Grootenhuis, P.D.J.; Van Goor, F.F.; Zhou, J.; Bear, B.R.; Miller, M.T.; McCartney, J.; Numa, M.M.D. Modulators of Atp-Binding Cassette Transporters. U.S. Patent 2013/0178471 A1, 11 July 2013. [Google Scholar]
- Ruah, S.S.H.; Grootenhuis, P.D.J.; Van Goor, F.; Zhou, J.; Bear, B.; Miller, M.T.; McCartney, J.; Numa, M.M.D. Indole Derivatives as CFTR Modulators. U.S. Patent 2009/0131492 A1, 21 May 2009. [Google Scholar]
- Alargova, R.G.; Dunbar, C.A.; Kadiyala, I.N. Pharmaceutical Compositions of (R)-1-(2,2-Diflurorbenzo[d][1,3]Dioxol-5-Yl)-N-(1-(2,3-Dihydroxypropyl)-6-Fluoro-2-(1-Hydroxy-2-Methylpropan-2-Yl)-1h-Indol-5-Yl) Cyclopropanecarboxamide and Administration Thereof. International Patent 2014/014841 A1, 23 January 2014. [Google Scholar]
- Tanoury, G.J.; Harrison, C.; Littler, B.J.; Rose, P.J.; Hughes, R.M.; Jung, Y.C.; Siesel, D.A.; Lee, E.C.; Belmont, D.T. Process of Producing Cycloalkylcarboxamido-Indole Compounds. International Patent 2011/133751 A2, 27 October 2011. [Google Scholar]
- Tanoury, G.J.; Harrison, C.; Littler, B.J.; Rose, P.J.; Hughes, R.M.; Jung, Y.C.; Siesel, D.A.; Lee, E.C.; Belmont, D.T.; Nugent, W.A. Process of Producing Cycloalkylcarboxamido-Indole Compounds. U.S. Patent 10,071,979 B2, 11 September 2018. [Google Scholar]
- Jung, J.-C.; Jung, Y.-J.; Park, O.-S. PREPARATION OF 2-CHLOROPYRIDINE. Synth. Commun. 2001, 31, 2507–2511. [Google Scholar] [CrossRef]
- Van Goor, F.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of ΔF508-CFTR Trafficking and Gating in Human Cystic Fibrosis Airway Primary Cultures by Small Molecules. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N. Small-Molecule Correctors of Defective F508-CFTR Cellular Processing Identified by High-Throughput Screening. J. Clin. Investig. 2005, 115, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Robert, R.; Carlile, G.W.; Pavel, C.; Liu, N.; Anjos, S.M.; Liao, J.; Luo, Y.; Zhang, D.; Thomas, D.Y.; Hanrahan, J.W. Structural Analog of Sildenafil Identified as a Novel Corrector of the F508del-CFTR Trafficking Defect. Mol. Pharmacol. 2008, 73, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR Protein Processing Defect in Vitro by the Investigational Drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Sinha, C.; Zhang, W.; Moon, C.S.; Actis, M.; Yarlagadda, S.; Arora, K.; Woodroofe, K.; Clancy, J.P.; Lin, S.; Ziady, A.G.; et al. Capturing the Direct Binding of CFTR Correctors to CFTR by Using Click Chemistry. ChemBioChem 2015, 16, 2017–2022. [Google Scholar] [CrossRef]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of ΔF508 CFTR Restore Global Conformational Maturation without Thermally Stabilizing the Mutant Protein. FASEB J. 2013, 27, 536–545. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-Based Corrector Combination Restores ΔF508-CFTR Folding and Function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, Low Temperature, and Correctors Reveal the Mechanism of F508del-CFTR Rescue by VX-809 and Suggest Multiple Agents for Full Correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Bacalhau, M.; Ramalho, S.S.; Silva, I.A.L.; Ferreira, F.C.; Carlile, G.W.; Thomas, D.Y.; Farinha, C.M.; Hanrahan, J.W.; Amaral, M.D. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells 2022, 11, 136. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 Stabilizes the First Transmembrane Domain of CFTR. Biochem. Pharmacol. 2013, 86, 612–619. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 Corrects Folding Defects in Cystic Fibrosis Transmembrane Conductance Regulator Protein through Action on Membrane-Spanning Domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Eckford, P.D.W.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K.; et al. VX-809 and Related Corrector Compounds Exhibit Secondary Activity Stabilizing Active F508del-CFTR after Its Partial Rescue to the Cell Surface. Chem. Biol. 2014, 21, 666–678. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Boinot, C.; Sabirzhanova, I.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Combination of Correctors Rescue ΔF508-CFTR by Reducing Its Association with Hsp40 and Hsp27. J. Biol. Chem. 2015, 290, 25636–25645. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Sabirzhanova, I.; Rapino, D.; Morales, M.M.; Guggino, W.B.; Cebotaru, L. Correctors Rescue CFTR Mutations in Nucleotide-Binding Domain 1 (NBD1) by Modulating Proteostasis. ChemBioChem 2016, 17, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Corrector VX-809 Promotes Interactions between Cytoplasmic Loop One and the First Nucleotide-Binding Domain of CFTR. Biochem. Pharmacol. 2017, 136, 24–31. [Google Scholar] [CrossRef]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Molinski, S.V.; Shahani, V.M.; Subramanian, A.S.; MacKinnon, S.S.; Woollard, G.; Laforet, M.; Laselva, O.; Morayniss, L.D.; Bear, C.E.; Windemuth, A. Comprehensive Mapping of Cystic Fibrosis Mutations to CFTR Protein Identifies Mutation Clusters and Molecular Docking Predicts Corrector Binding Site. Proteins Struct. Funct. Bioinforma. 2018, 86, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Baatallah, N.; Elbahnsi, A.; Mornon, J.-P.; Chevalier, B.; Pranke, I.; Servel, N.; Zelli, R.; Décout, J.-L.; Edelman, A.; Sermet-Gaudelus, I.; et al. Pharmacological Chaperones Improve Intra-Domain Stability and Inter-Domain Assembly via Distinct Binding Sites to Rescue Misfolded CFTR. Cell. Mol. Life Sci. 2021, 78, 7813–7829. [Google Scholar] [CrossRef] [PubMed]
- Krainer, G.; Treff, A.; Hartmann, A.; Stone, T.A.; Schenkel, M.; Keller, S.; Deber, C.M.; Schlierf, M. A Minimal Helical-Hairpin Motif Provides Molecular-Level Insights into Misfolding and Pharmacological Rescue of CFTR. Commun. Biol. 2018, 1, 154. [Google Scholar] [CrossRef] [PubMed]
- Krainer, G.; Schenkel, M.; Hartmann, A.; Ravamehr-Lake, D.; Deber, C.M.; Schlierf, M. CFTR Transmembrane Segments Are Impaired in Their Conformational Adaptability by a Pathogenic Loop Mutation and Dynamically Stabilized by Lumacaftor. J. Biol. Chem. 2020, 295, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.; De Keersmaecker, L.; Heylen, L.; Ramalho, A.S.; Gijsbers, R.; Farré, R.; De Boeck, K.; Christ, F.; Debyser, Z.; Carlon, M.S. Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells 2020, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Amico, G.; Brandas, C.; Moran, O.; Baroni, D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. Int. J. Mol. Sci. 2019, 20, 5463. [Google Scholar] [CrossRef] [PubMed]
- Uliyakina, I.; Botelho, H.M.; da Paula, A.C.; Afonso, S.; Lobo, M.J.; Felício, V.; Farinha, C.M.; Amaral, M.D. Full Rescue of F508del-CFTR Processing and Function by CFTR Modulators Can Be Achieved by Removal of Two Regulatory Regions. Int. J. Mol. Sci. 2020, 21, 4524. [Google Scholar] [CrossRef] [PubMed]
- Kleizen, B.; van Willigen, M.; Mijnders, M.; Peters, F.; Grudniewska, M.; Hillenaar, T.; Thomas, A.; Kooijman, L.; Peters, K.W.; Frizzell, R.; et al. Co-Translational Folding of the First Transmembrane Domain of ABC-Transporter CFTR Is Supported by Assembly with the First Cytosolic Domain. J. Mol. Biol. 2021, 433, 166955. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR Correction by Type I Folding Correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Apaydın, S.; Török, M. Sulfonamide Derivatives as Multi-Target Agents for Complex Diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [Google Scholar] [CrossRef]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole Containing Natural Products: Synthetic Preview and Biological Significance. Eur. J. Med. Chem. 2013, 69, 735–753. [Google Scholar] [CrossRef]
- Angell, P.T.; Harrison, C.; Hughes, R.M.; Lewandowski, B.; Littler, B.J.; Melilo, V.; Nugent, W.A.; Siesel, D.A.; Smith, D.; Studley, J. Processes for Making Modulators of Cystic Fibrosis Transmembrane Conductance Regulator. International Patent 2019/113476 A2, 13 June 2019. [Google Scholar]
- Angell, P.T.; Lewandowski, B.; Littler, B.J.; Nugent, W.A.; Smith, D.; Studley, J. Processes for Preparing Pyrrolidine Compounds. International Patent 2019/028228 A1, 7 February 2019. [Google Scholar]
- Lai, J.T.; Westfah, J.C. Rearrangement of 2,2,6,6-Tetramethyl-4-Piperidone in Phase-Transfer Catalyzed Reactions. J. Org. Chem. 1980, 45, 1513–1514. [Google Scholar] [CrossRef]
- Abela, A.; Alcacio, T.; Anderson, C.; Angell, P.; Baek, M.; Clemens, J.; Cleveland, T.; Ferris, L.; Grootenhuis, P.D.; Gross, R.; et al. Modulator of Cystic Fibrosis Transmembrane Conductance Regulator, Pharmaceutical Compositions, Methods of Treatment, and Process for Making the Modulator. International Patent 2018/107100 A1, 14 June 2018. [Google Scholar]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-Guided Combination Therapy to Potently Improve the Function of Mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The Impact of FDA and EMA Regulatory Decision-Making Process on the Access to CFTR Modulators for the Treatment of Cystic Fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Velkov, T.; Xu, H.; Vadeboncoeur, N.; Bilodeau, L.; Matouk, E.; Lukacs, G. A Precision Medicine Approach to Optimize Modulator Therapy for Rare CFTR Folding Mutants. J. Pers. Med. 2021, 11, 643. [Google Scholar] [CrossRef] [PubMed]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Mandrup Bertozzi, S.; et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef] [PubMed]
- Becq, F.; Mirval, S.; Carrez, T.; Lévêque, M.; Billet, A.; Coraux, C.; Sage, E.; Cantereau, A. The Rescue of F508del-CFTR by Elexacaftor/Tezacaftor/Ivacaftor (Trikafta) in Human Airway Epithelial Cells Is Underestimated Due to the Presence of Ivacaftor. Eur. Respir. J. 2022, 59, 2100671. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of Multiple Class II CFTR Mutations by Elexacaftor+tezacaftor+ivacaftor Mediated in Part by the Dual Activities of Elexacaftor as Both Corrector and Potentiator. Eur. Respir. J. 2021, 57, 2002774. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, C.A.; Zeitlin, P.L.; Bratcher, P.E. Elexacaftor Is a CFTR Potentiator and Acts Synergistically with Ivacaftor during Acute and Chronic Treatment. Sci. Rep. 2021, 11, 19810. [Google Scholar] [CrossRef] [PubMed]
- Tomati, V.; Costa, S.; Capurro, V.; Pesce, E.; Pastorino, C.; Lena, M.; Sondo, E.; Di Duca, M.; Cresta, F.; Cristadoro, S.; et al. Rescue by Elexacaftor-Tezacaftor-Ivacaftor of the G1244E Cystic Fibrosis Mutation’s Stability and Gating Defects Are Dependent on Cell Background. J. Cyst. Fibros. 2023, 22, 525–537. [Google Scholar] [CrossRef]
- Shaughnessy, C.A.; Zeitlin, P.L.; Bratcher, P.E. Net Benefit of Ivacaftor during Prolonged Tezacaftor/Elexacaftor Exposure in Vitro. J. Cyst. Fibros. 2022, 21, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Fiedorczuk, K.; Chen, J. Molecular Structures Reveal Synergistic Rescue of Δ508 CFTR by Trikafta Modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Z.; Loughlin, B.J.; Xu, H.; Veit, G.; Vorobiev, S.; Clarke, O.B.; Jiang, F.; Li, Y.; Singh, S.; et al. Mechanism of Dual Pharmacological Correction and Potentiation of Human CFTR. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hillenaar, T.; Beekman, J.; van der Sluijs, P.; Braakman, I. Redefining Hypo- and Hyper-Responding Phenotypes of CFTR Mutants for Understanding and Therapy. Int. J. Mol. Sci. 2022, 23, 15170. [Google Scholar] [CrossRef] [PubMed]
- Bongiorno, R.; Ludovico, A.; Moran, O.; Baroni, D. Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2. Int. J. Mol. Sci. 2023, 24, 12838. [Google Scholar] [CrossRef] [PubMed]
- Bacalhau, M.; Ferreira, F.C.; Kmit, A.; Souza, F.R.; da Silva, V.D.; Pimentel, A.S.; Amaral, M.D.; Buarque, C.D.; Lopes-Pacheco, M. Identification of Novel F508del-CFTR Traffic Correctors among Triazole Derivatives. Eur. J. Pharmacol. 2023, 938, 175396. [Google Scholar] [CrossRef]
- Stanke, F.; Pallenberg, S.T.; Tamm, S.; Hedtfeld, S.; Eichhorn, E.M.; Minso, R.; Hansen, G.; Welte, T.; Sauer-Heilborn, A.; Ringshausen, F.C.; et al. Changes in Cystic Fibrosis Transmembrane Conductance Regulator Protein Expression Prior to and during Elexacaftor-Tezacaftor-Ivacaftor Therapy. Front. Pharmacol. 2023, 14, 1114584. [Google Scholar] [CrossRef]
- McKee, A.G.; McDonald, E.F.; Penn, W.D.; Kuntz, C.P.; Noguera, K.; Chamness, L.M.; Roushar, F.J.; Meiler, J.; Oliver, K.E.; Plate, L.; et al. General Trends in the Effects of VX-661 and VX-445 on the Plasma Membrane Expression of Clinical CFTR Variants. Cell Chem. Biol. 2023, 30, 632–642.e5. [Google Scholar] [CrossRef]
- Kim, M.; McDonald, E.F.; Sabusap, C.M.P.; Timalsina, B.; Joshi, D.; Hong, J.S.; Rab, A.; Sorscher, E.J.; Plate, L. Elexacaftor/VX-445-Mediated CFTR Interactome Remodeling Reveals Differential Correction Driven by Mutation-Specific Translational Dynamics. J. Biol. Chem. 2023, 299, 105242. [Google Scholar] [CrossRef]
- Lefferts, J.W.; Bierlaagh, M.C.; Kroes, S.; Nieuwenhuijze, N.D.A.; Sonneveld van Kooten, H.N.; Niemöller, P.J.; Verburg, T.F.; Janssens, H.M.; Muilwijk, D.; van Beuningen, S.F.B.; et al. CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes. Int. J. Mol. Sci. 2023, 24, 14539. [Google Scholar] [CrossRef]
- Im, J.; Hillenaar, T.; Yeoh, H.Y.; Sahasrabudhe, P.; Mijnders, M.; van Willigen, M.; Hagos, A.; de Mattos, E.; van der Sluijs, P.; Braakman, I. ABC-Transporter CFTR Folds with High Fidelity through a Modular, Stepwise Pathway. Cell. Mol. Life Sci. 2023, 80, 33. [Google Scholar] [CrossRef]
- Soya, N.; Xu, H.; Roldan, A.; Yang, Z.; Ye, H.; Jiang, F.; Premchandar, A.; Veit, G.; Cole, S.P.C.; Kappes, J.; et al. Folding Correctors Can Restore CFTR Posttranslational Folding Landscape by Allosteric Domain-Domain Coupling. Nat. Commun. 2023, 14, 6868. [Google Scholar] [CrossRef]
- Ferreira, F.C.; Amaral, M.D.; Bacalhau, M.; Lopes-Pacheco, M. PTI-801 (posenacaftor) shares a common mechanism with VX-445 (elexacaftor) to rescue p.Phe508del-CFTR. Eur. J. Pharmacol. 2024. [Google Scholar] [CrossRef]
- Bacalhau, M.; Camargo, M.; Magalhães-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.Z.; Espinosa, R.; Starner, C.I.; Gleason, P.P. Real-World Outcomes and Direct Care Cost before and after Elexacaftor/Tezacaftor/Ivacaftor Initiation in Commercially Insured Members with Cystic Fibrosis. J. Manag. Care Spec. Pharm. 2023, 29, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Zampoli, M.; Morrow, B.M.; Paul, G. Real-World Disparities and Ethical Considerations with Access to CFTR Modulator Drugs: Mind the Gap! Front. Pharmacol. 2023, 14, 1163391. [Google Scholar] [CrossRef]



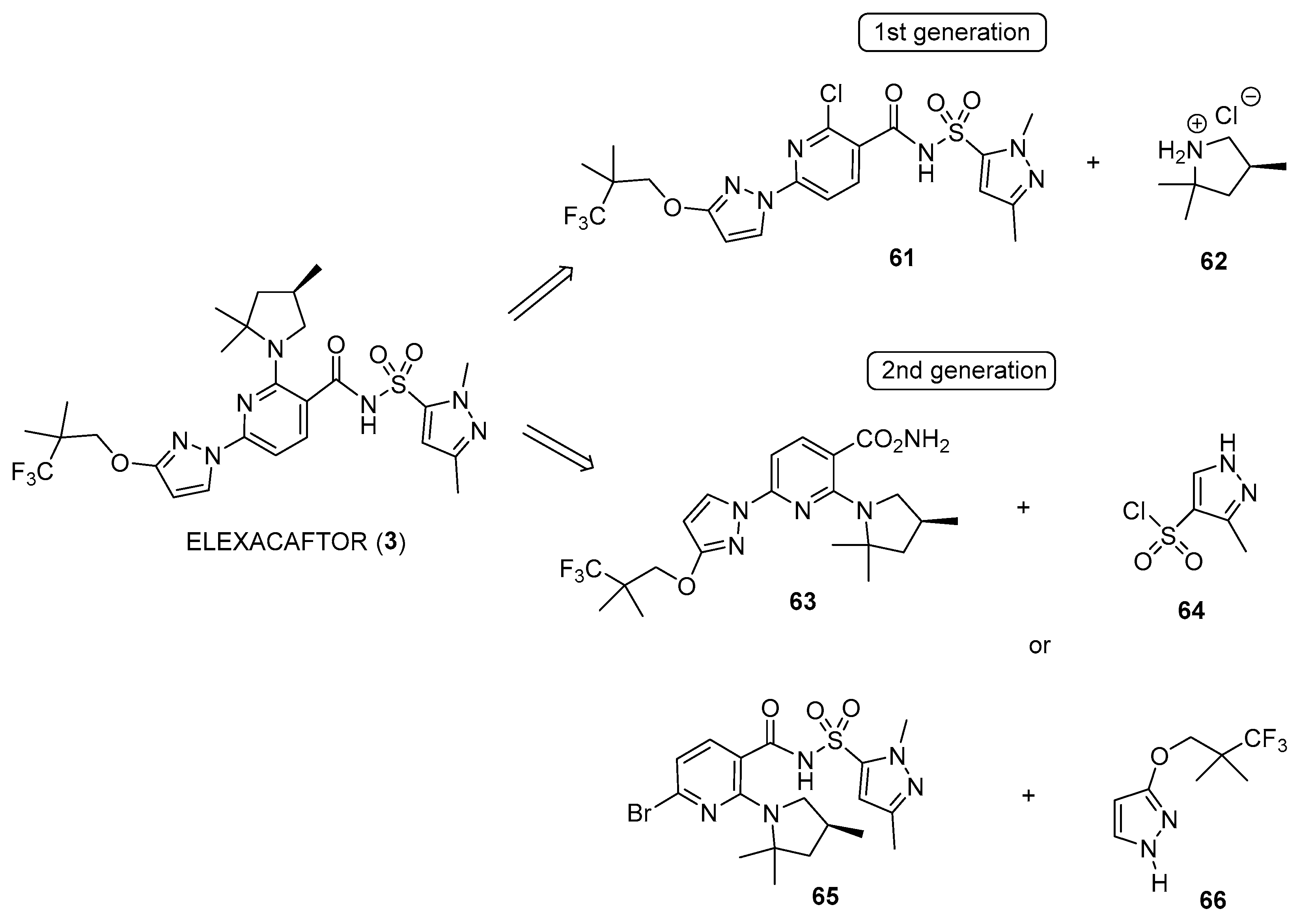

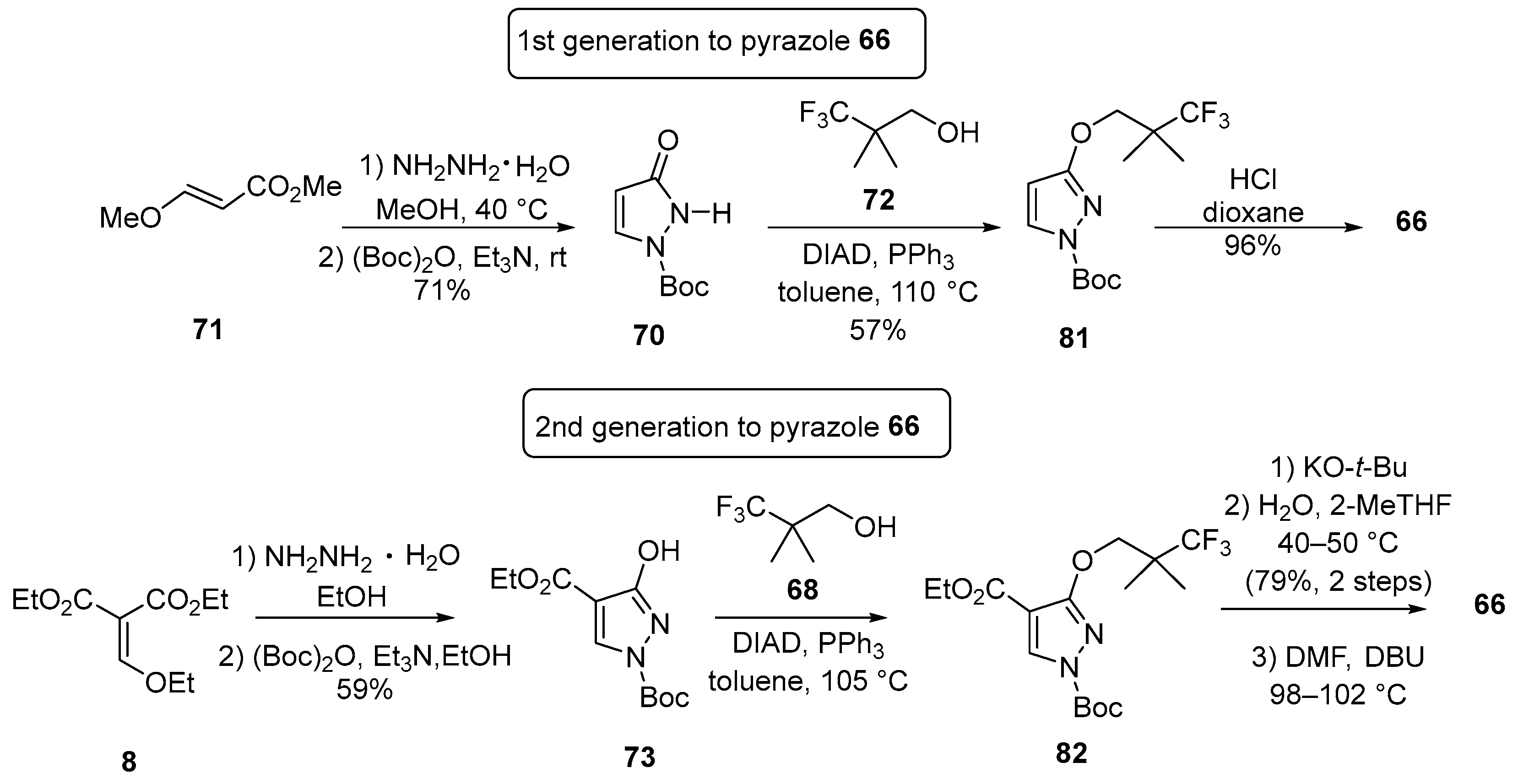

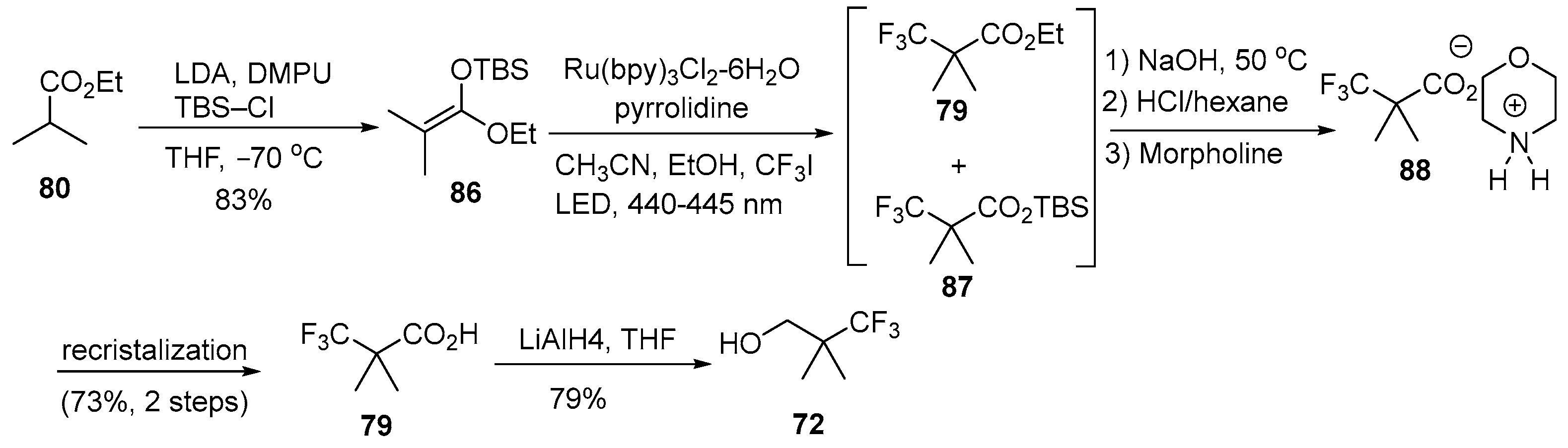
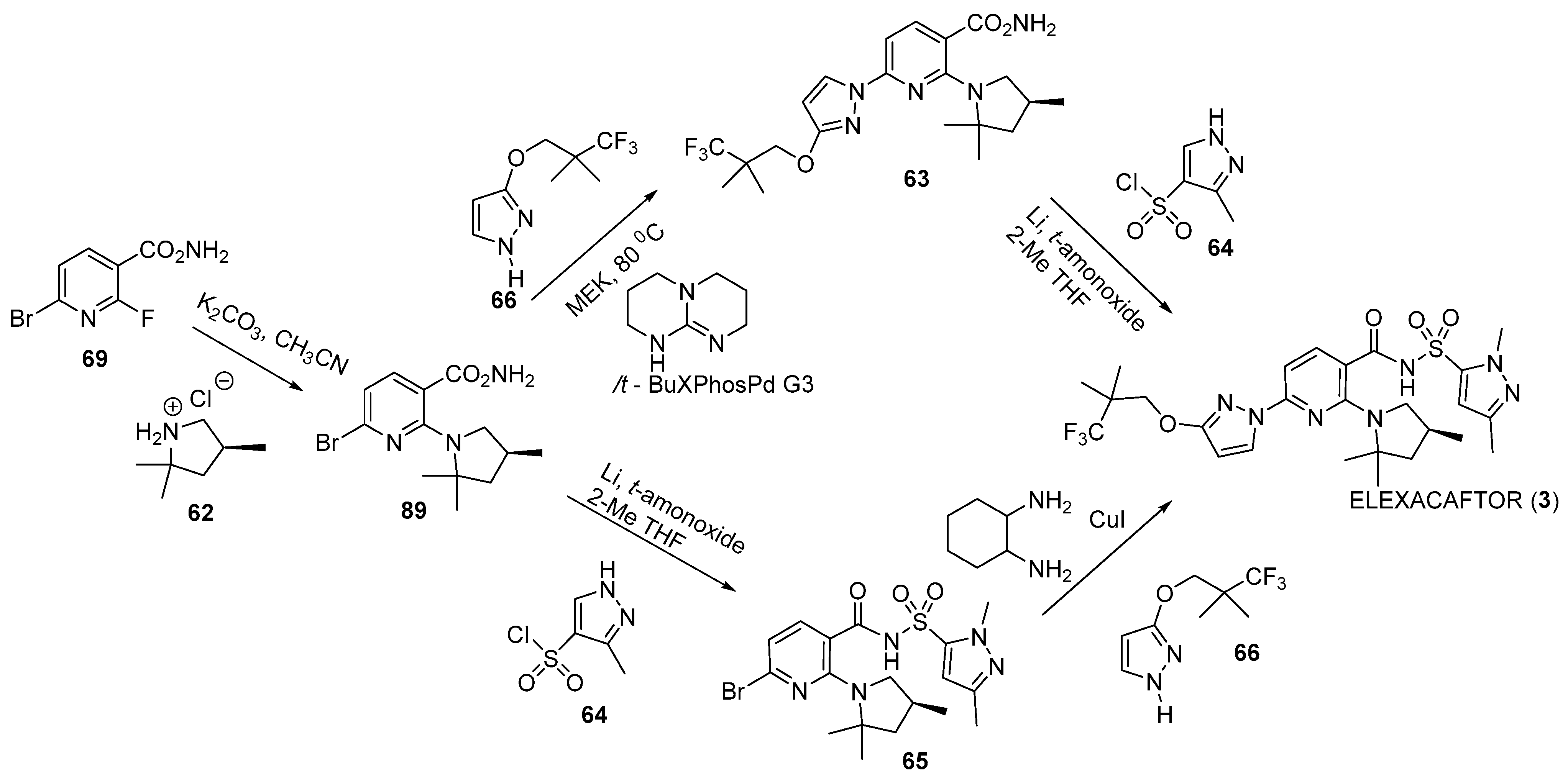
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, F.C.; Buarque, C.D.; Lopes-Pacheco, M. Organic Synthesis and Current Understanding of the Mechanisms of CFTR Modulator Drugs Ivacaftor, Tezacaftor, and Elexacaftor. Molecules 2024, 29, 821. https://doi.org/10.3390/molecules29040821
Ferreira FC, Buarque CD, Lopes-Pacheco M. Organic Synthesis and Current Understanding of the Mechanisms of CFTR Modulator Drugs Ivacaftor, Tezacaftor, and Elexacaftor. Molecules. 2024; 29(4):821. https://doi.org/10.3390/molecules29040821
Chicago/Turabian StyleFerreira, Filipa C., Camilla D. Buarque, and Miquéias Lopes-Pacheco. 2024. "Organic Synthesis and Current Understanding of the Mechanisms of CFTR Modulator Drugs Ivacaftor, Tezacaftor, and Elexacaftor" Molecules 29, no. 4: 821. https://doi.org/10.3390/molecules29040821