
Catalysts of Healing: A Symphony of Synthesis and Clinical Artistry in Small-Molecule Agents for Breast Cancer Alleviation
Abstract
:1. Introduction
2. Small-Molecule Drugs to Treat Breast Cancer
2.1. DNA Inhibitors
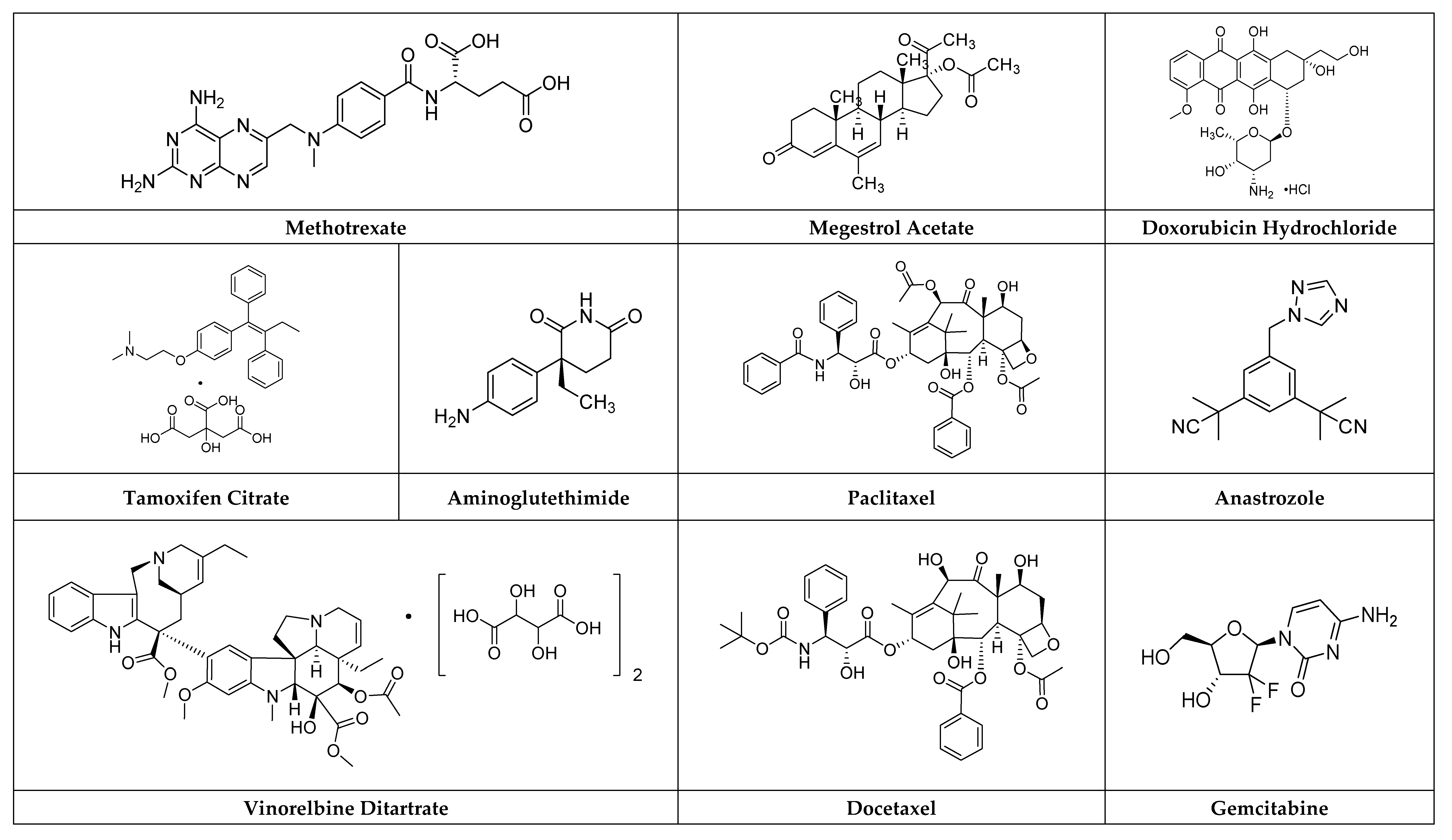
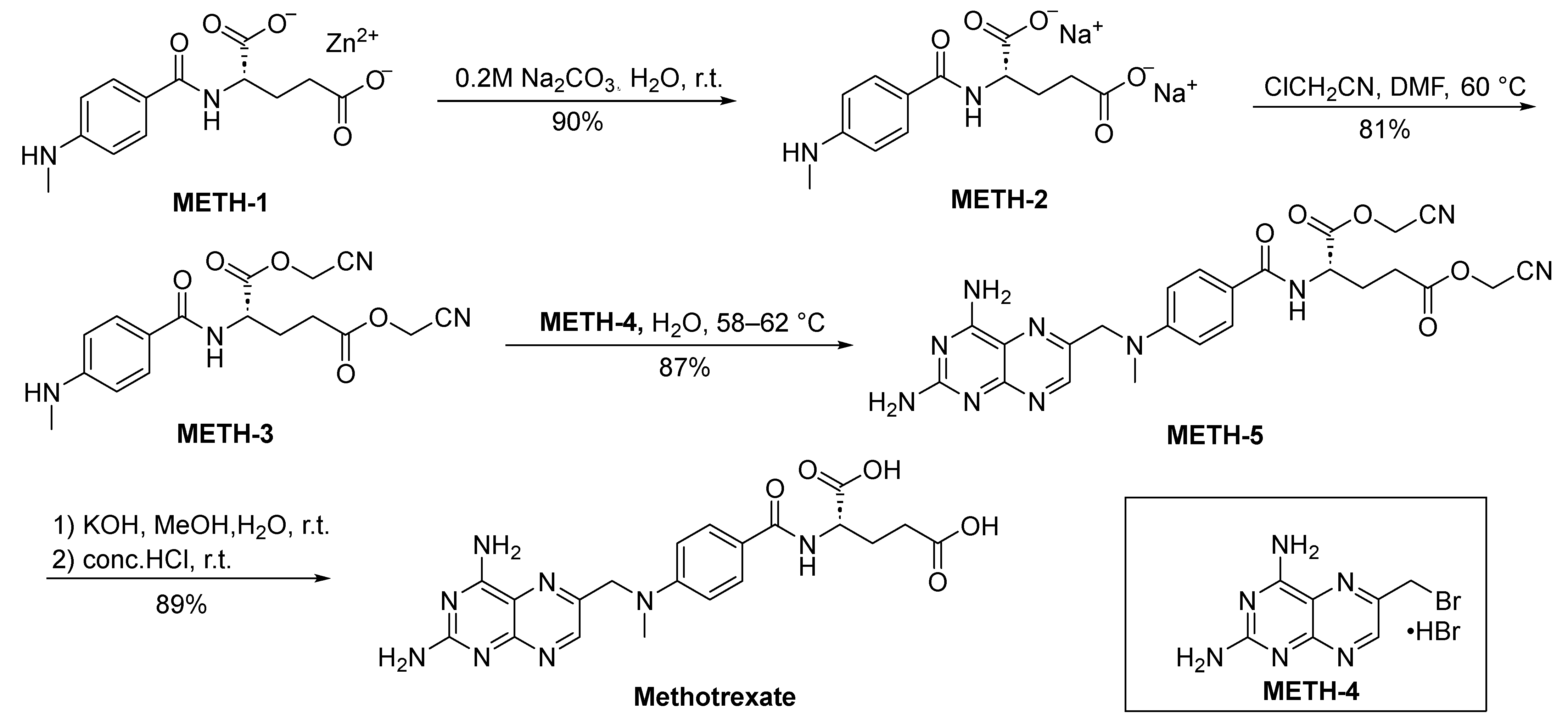
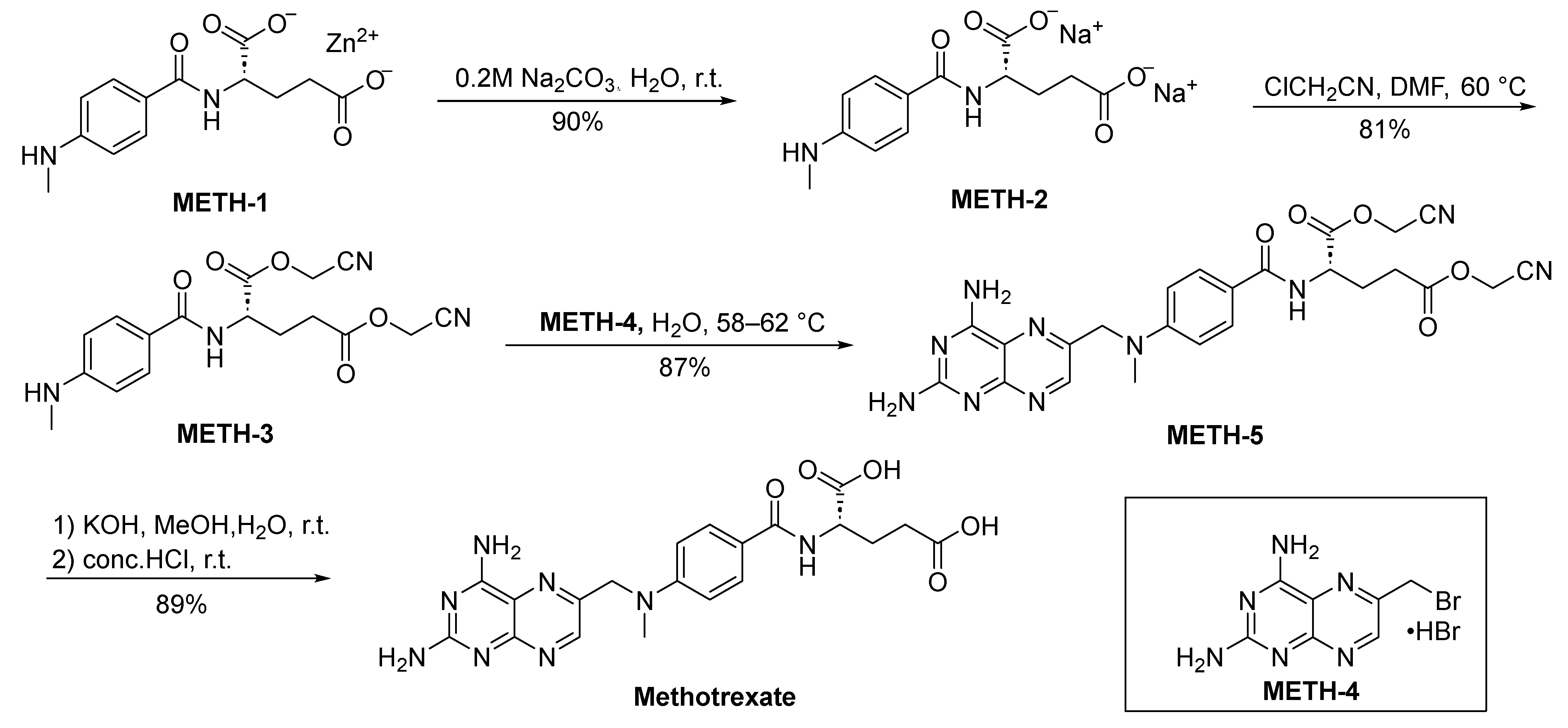
2.1.1. Methotrexate
2.1.2. Gemcitabine
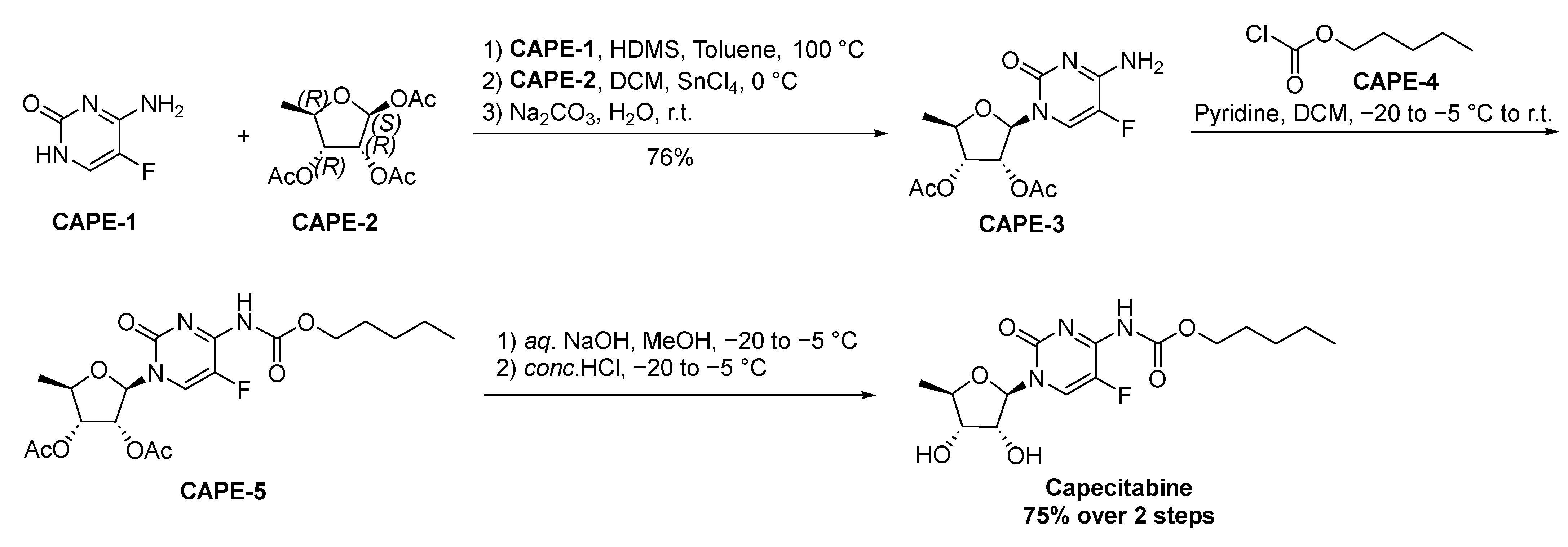
2.1.3. Capecitabine
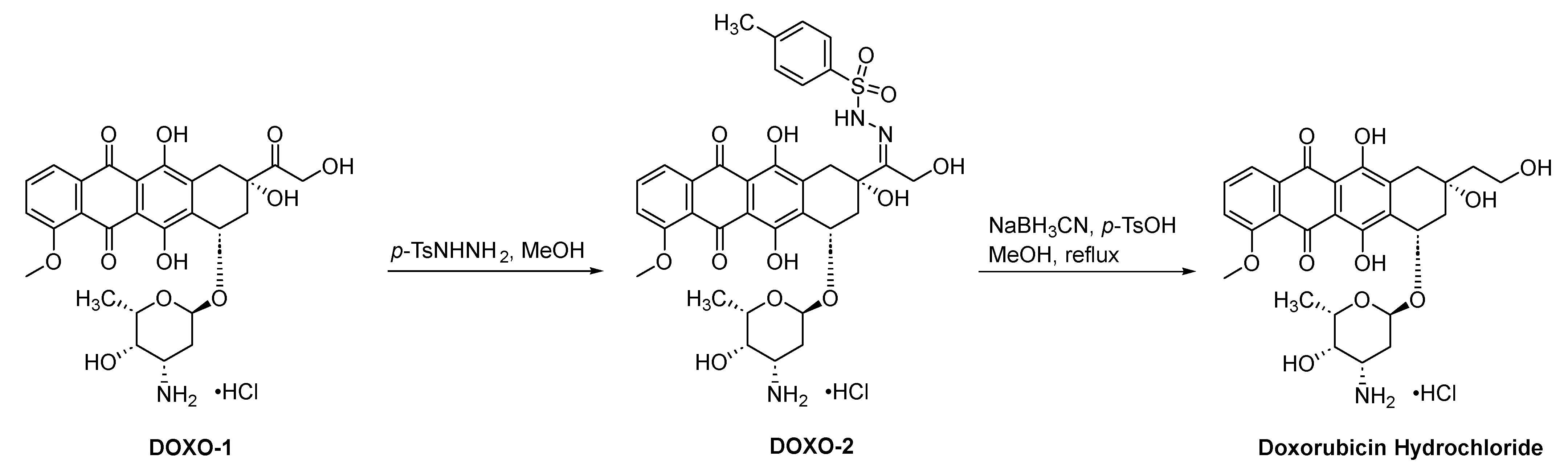
2.1.4. Doxorubicin Hydrochloride
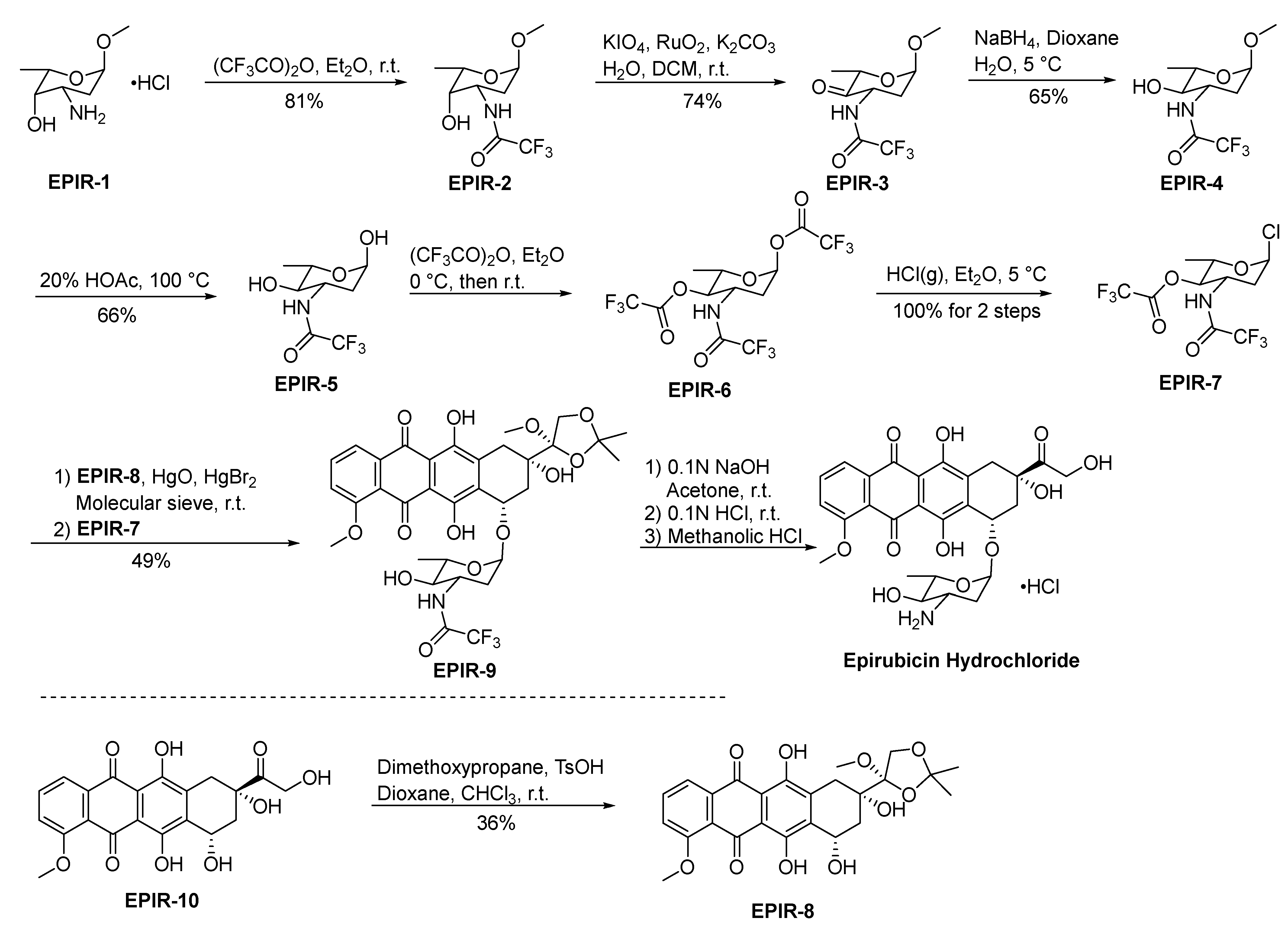
2.1.5. Epirubicin Hydrochloride
2.2. Progesterone Receptor Agonists
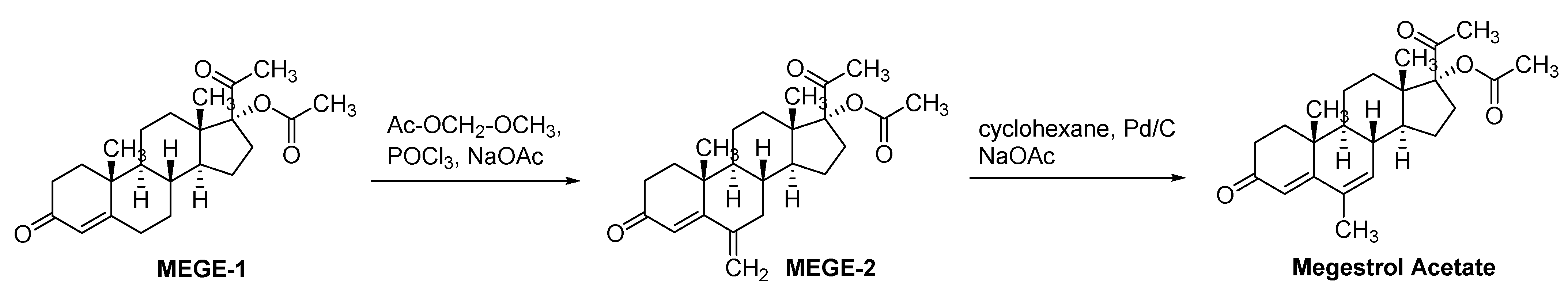
Megestrol Acetate
2.3. Aromatase Inhibitors
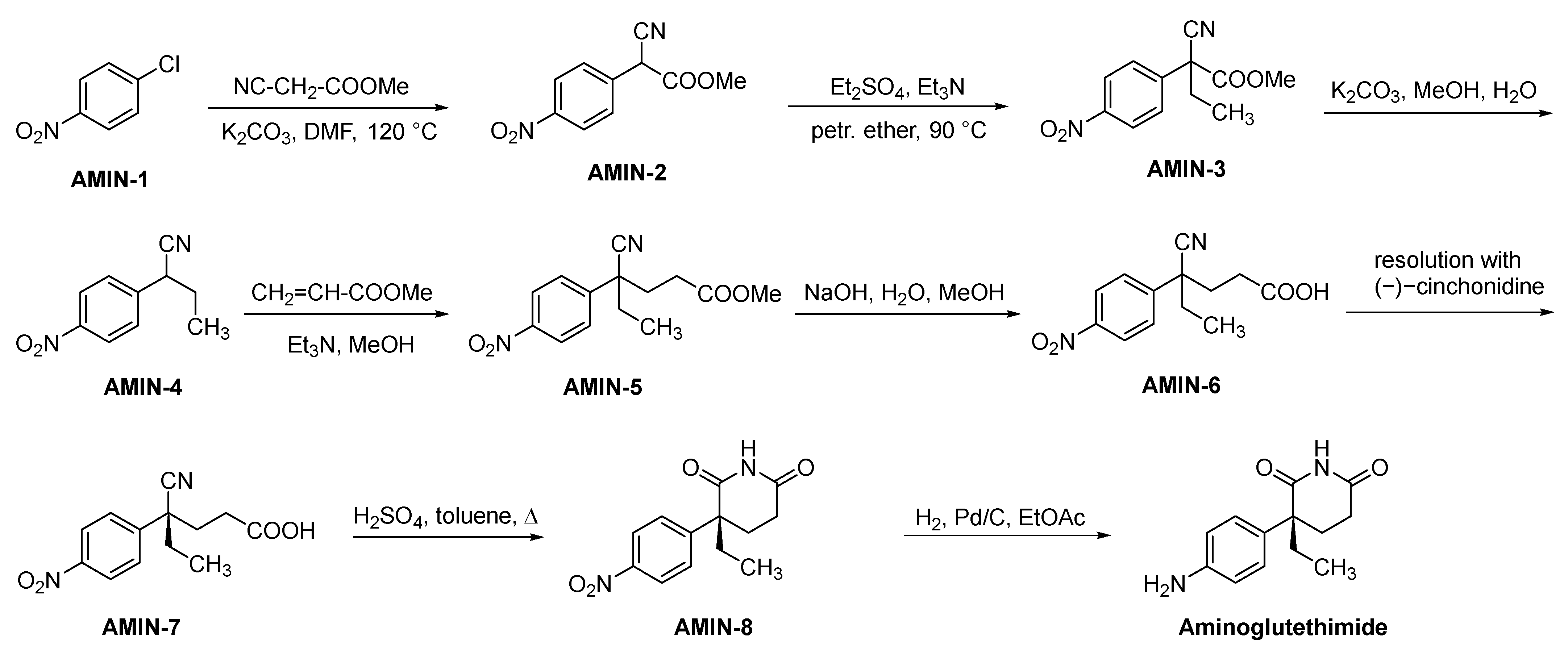
2.3.1. Aminoglutethimide
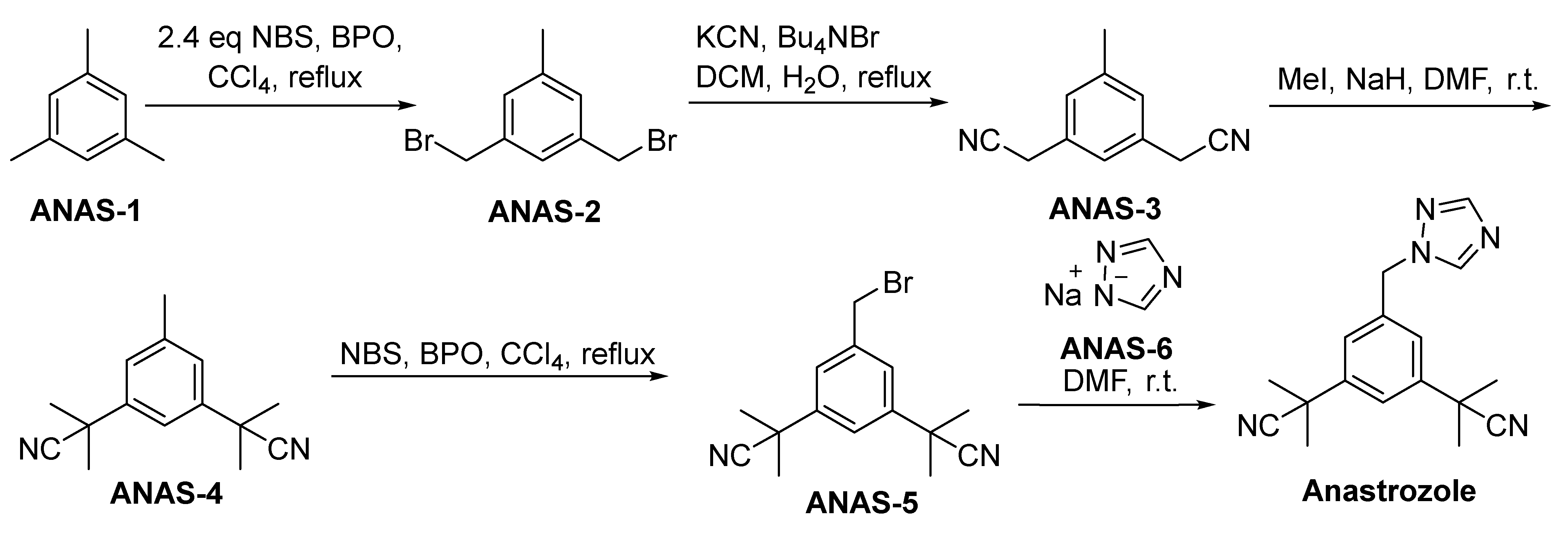
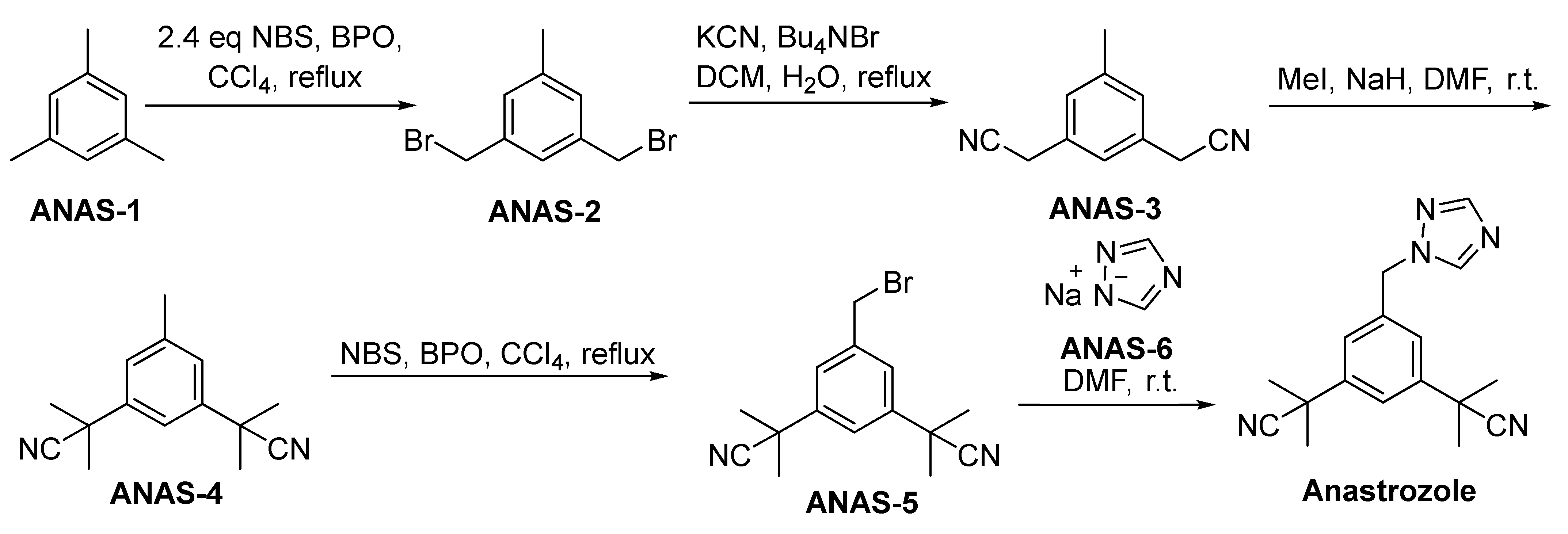
2.3.2. Anastrozole
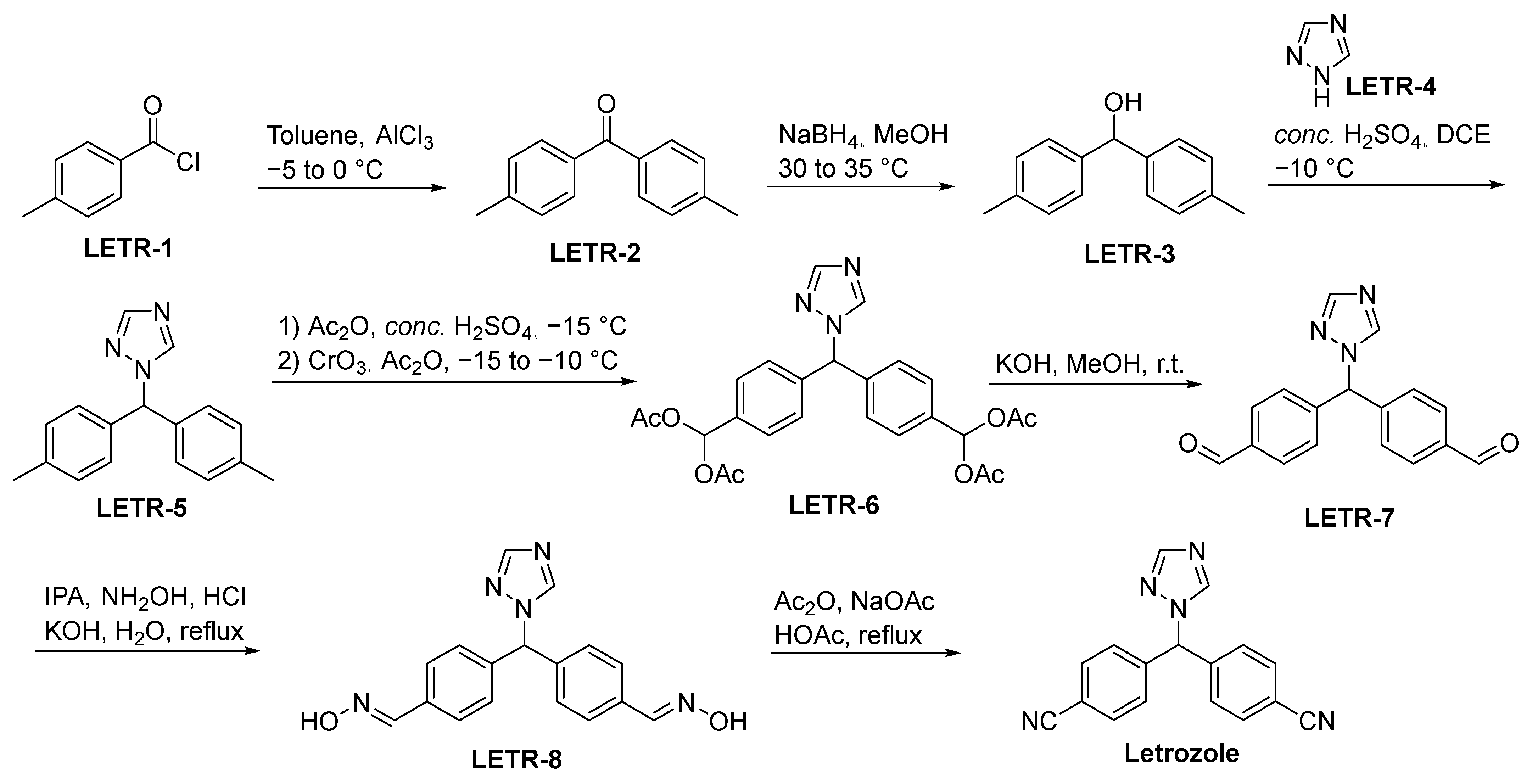
2.3.3. Letrozole
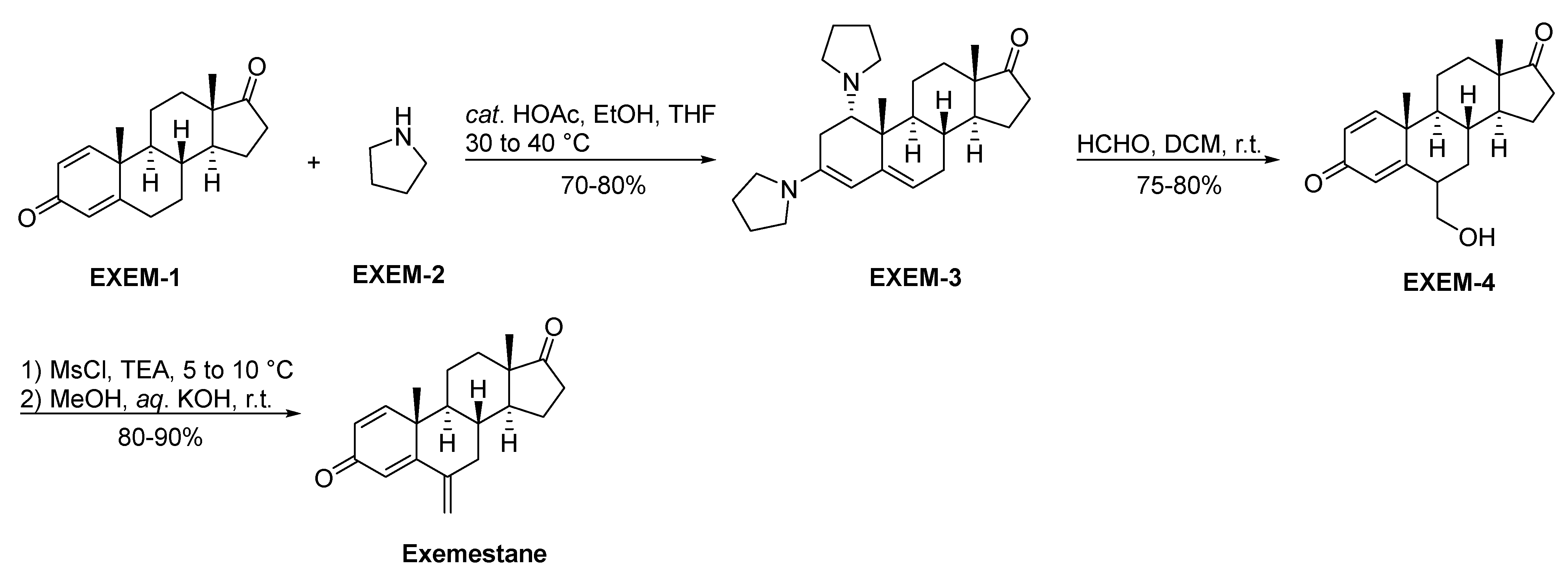
2.3.4. Exemestane
2.4. Selective Estrogen Receptor Modulators (SERMs)
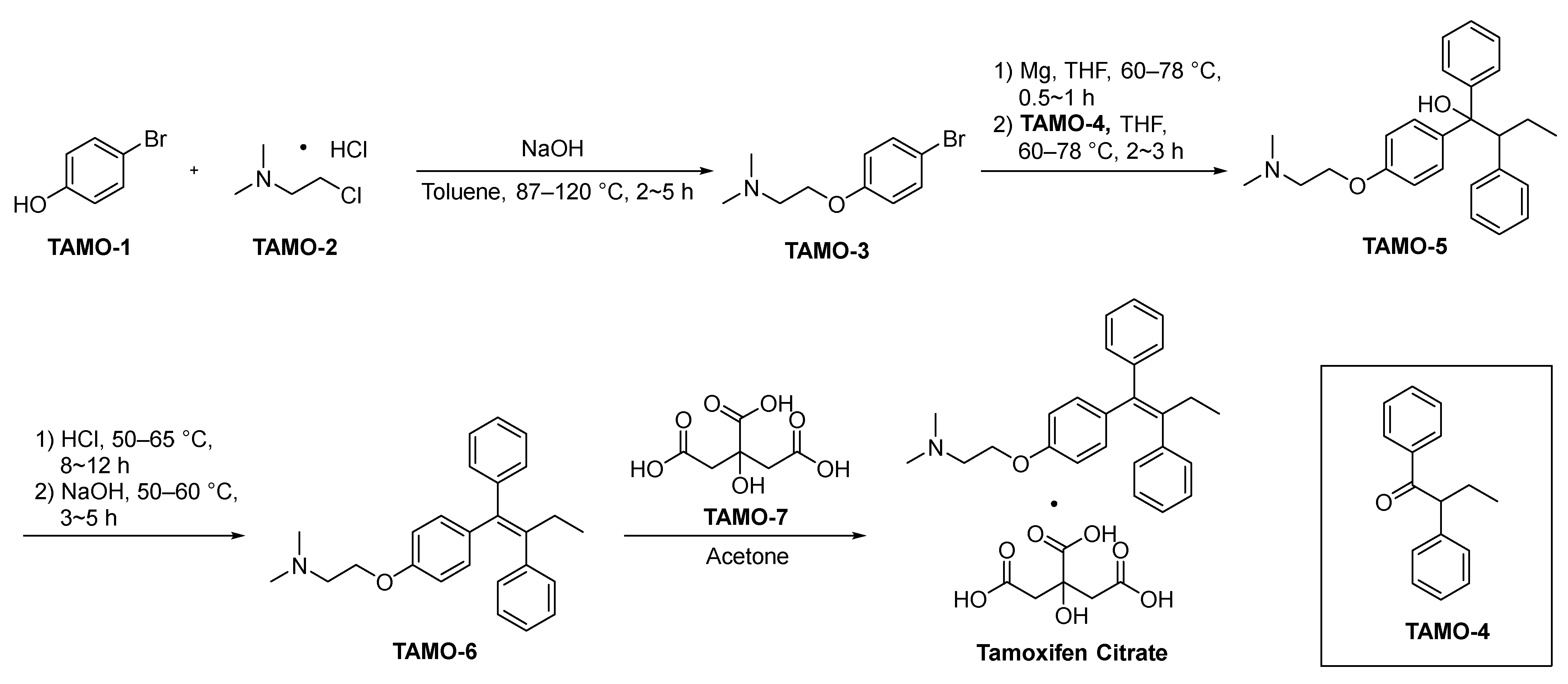
2.4.1. Tamoxifen Citrate
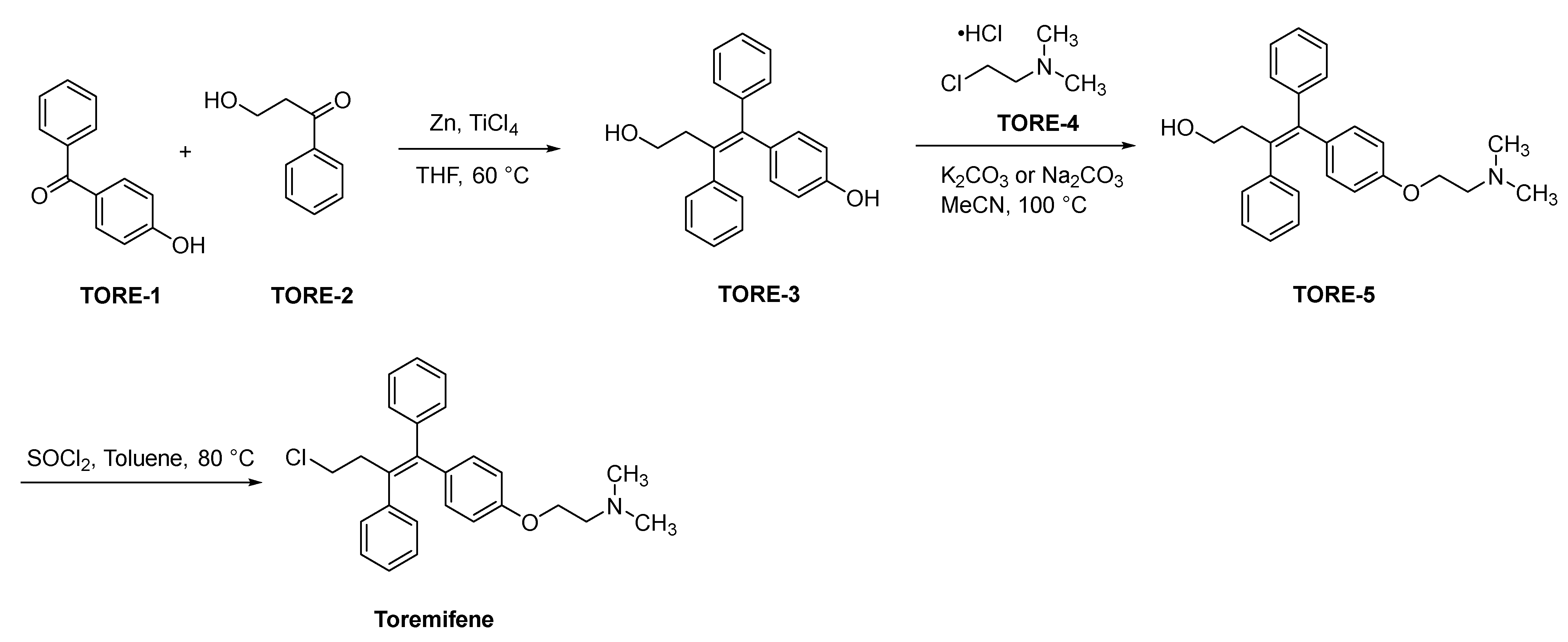
2.4.2. Toremifene
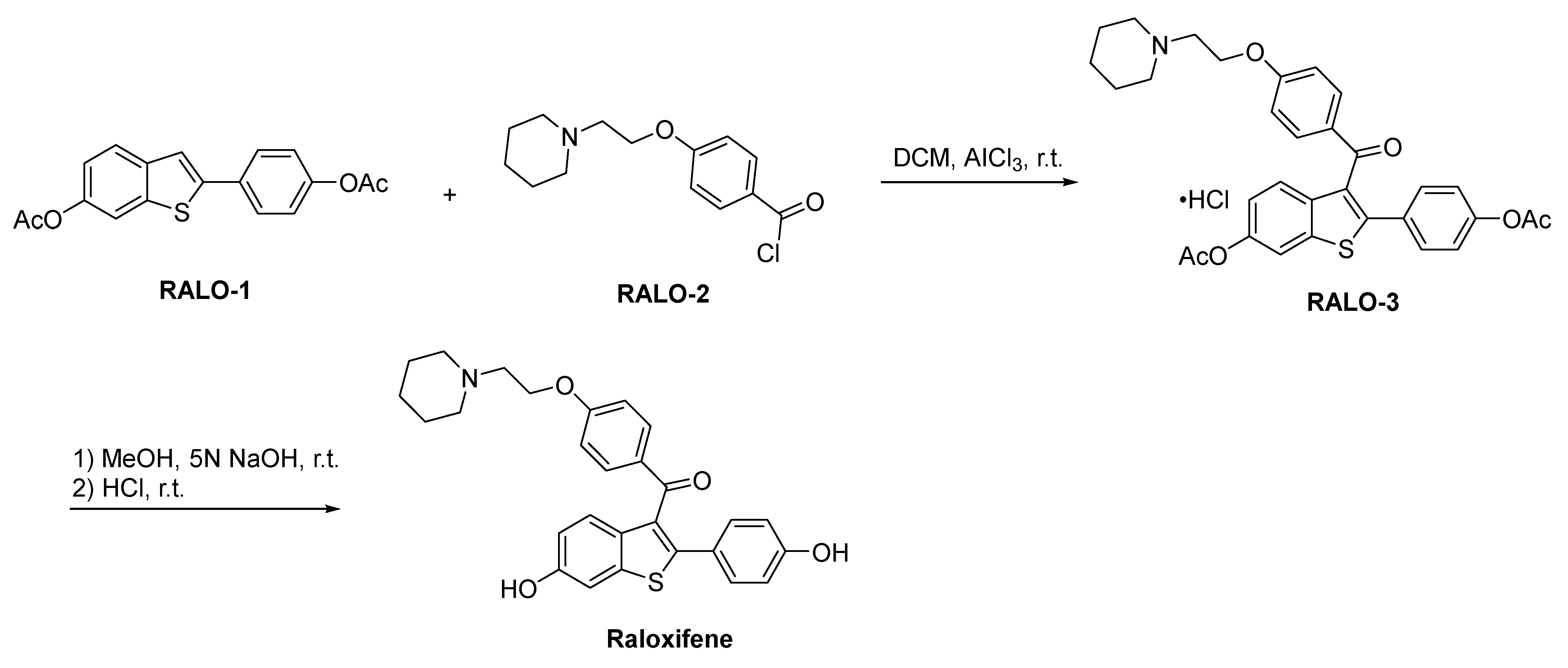
2.4.3. Raloxifene
2.5. Selective Estrogen Receptor Degraders (SERDs)
2.5.1. Fulvestrant
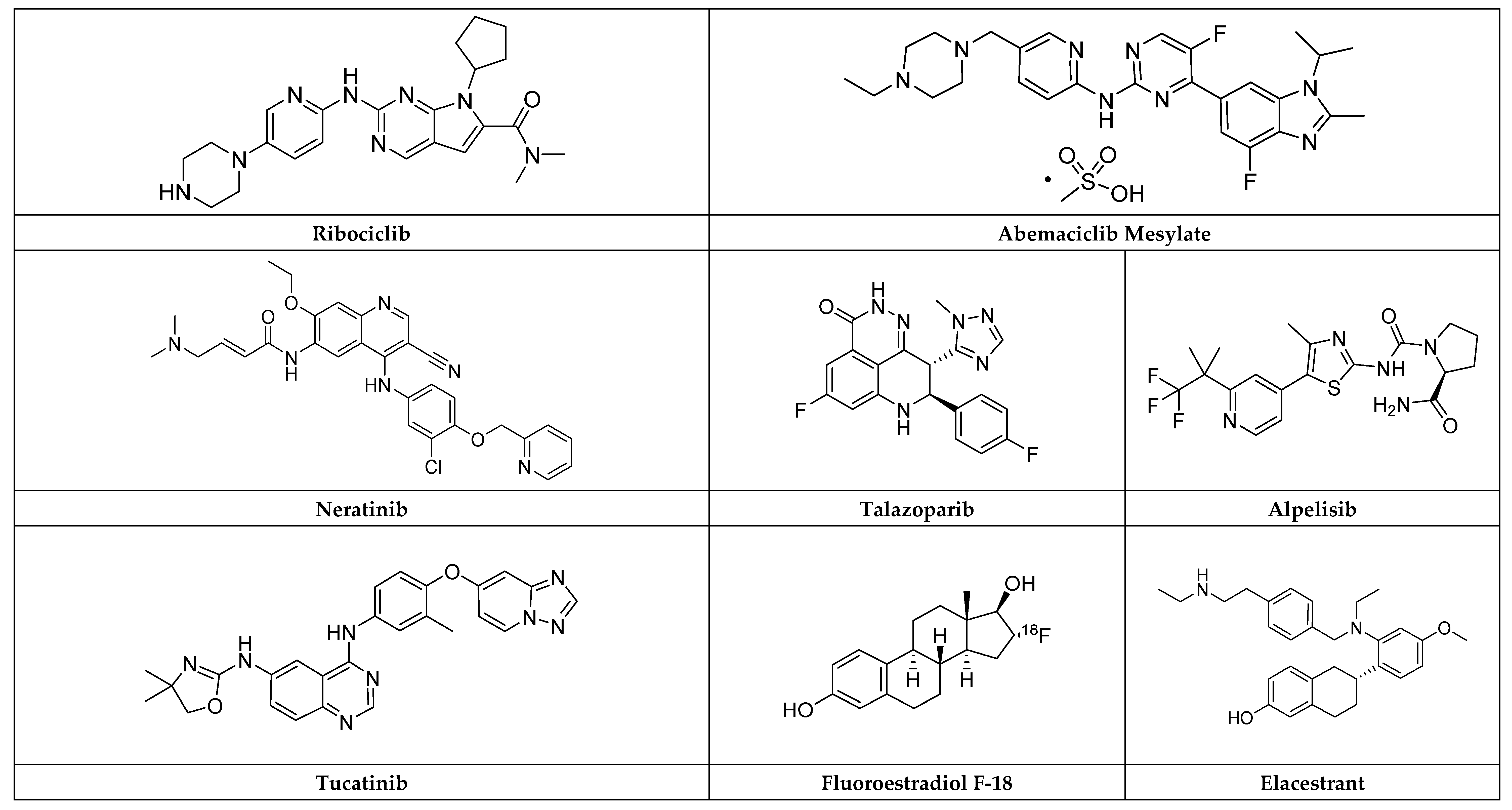
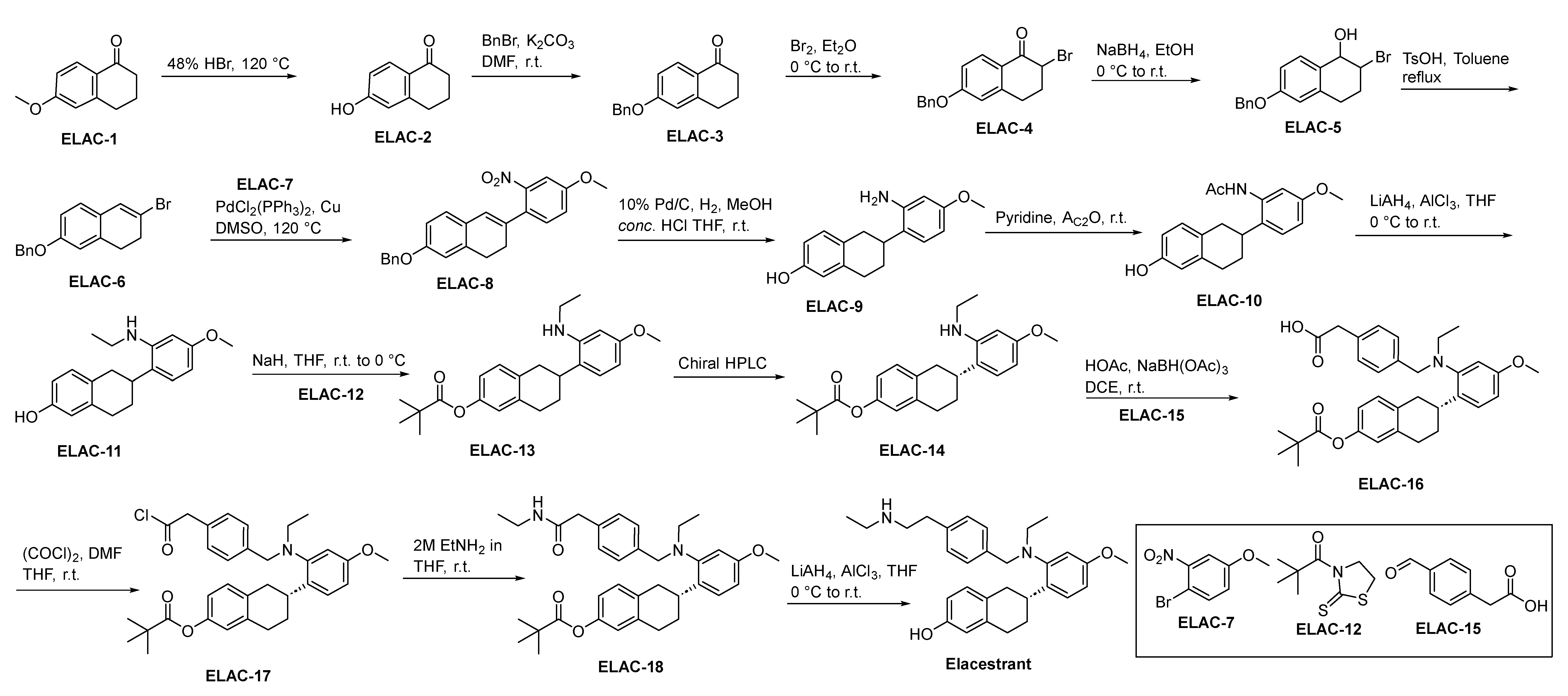
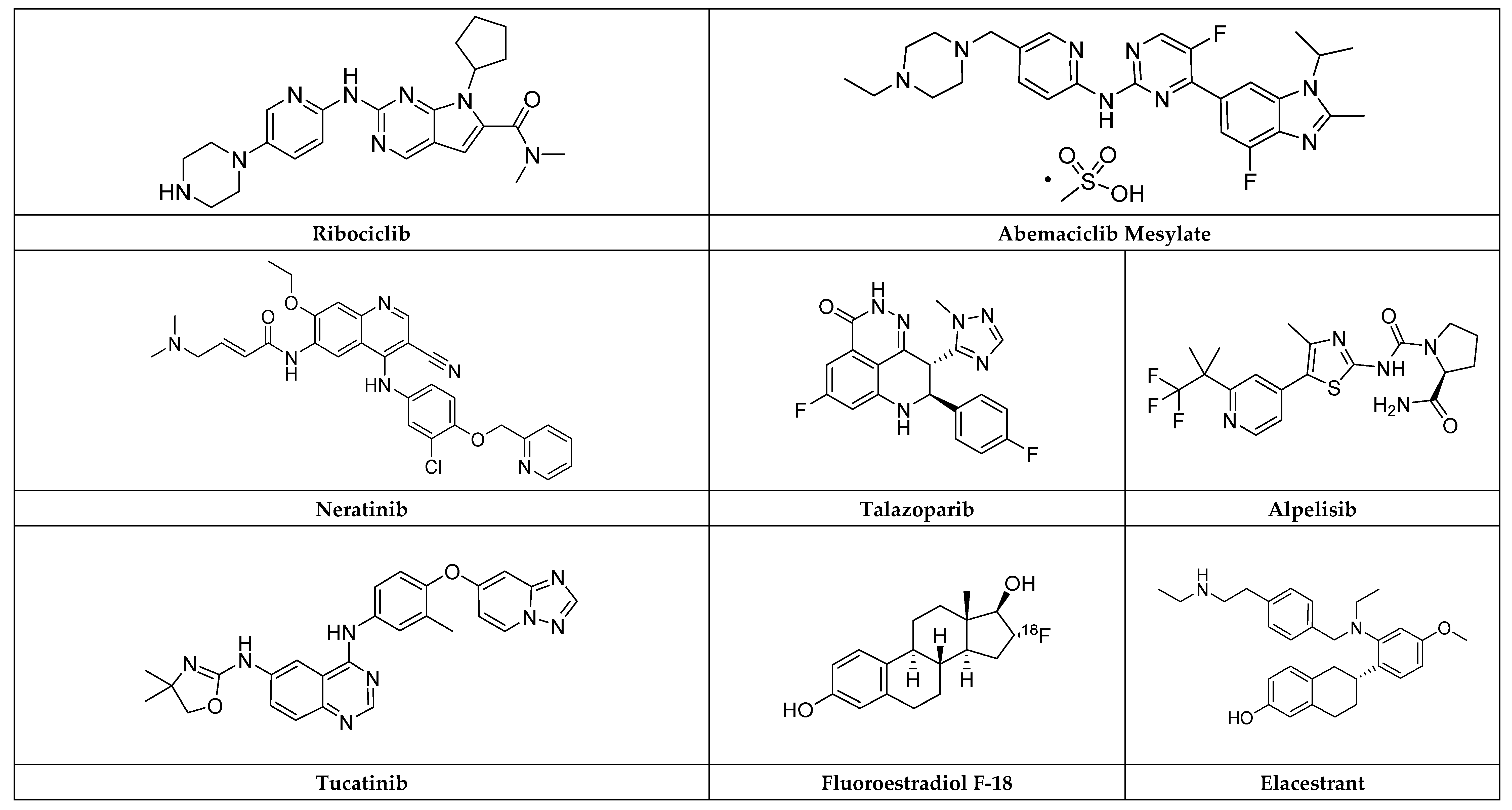
2.5.2. Elacestrant
2.6. Radioactive Diagnostic Agent
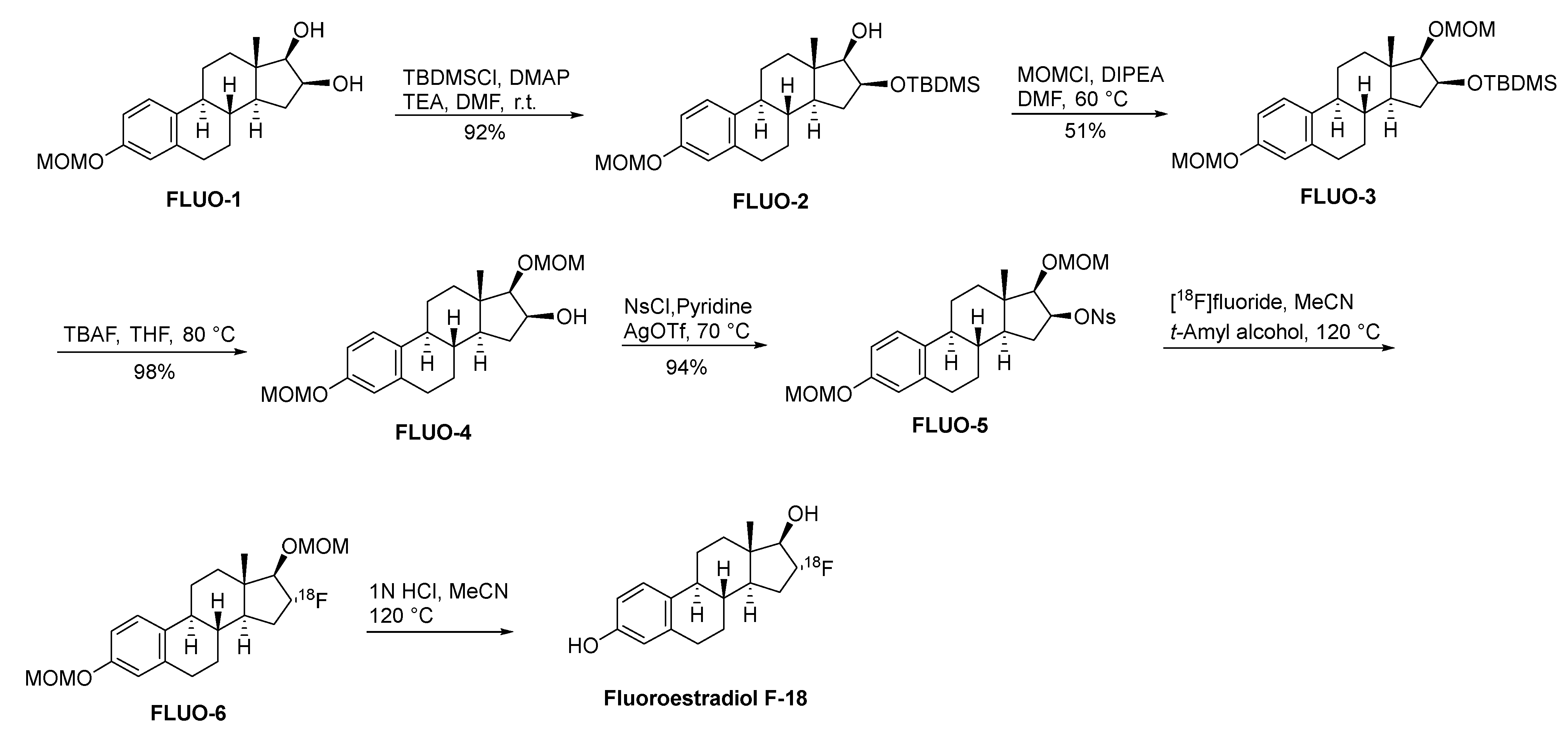
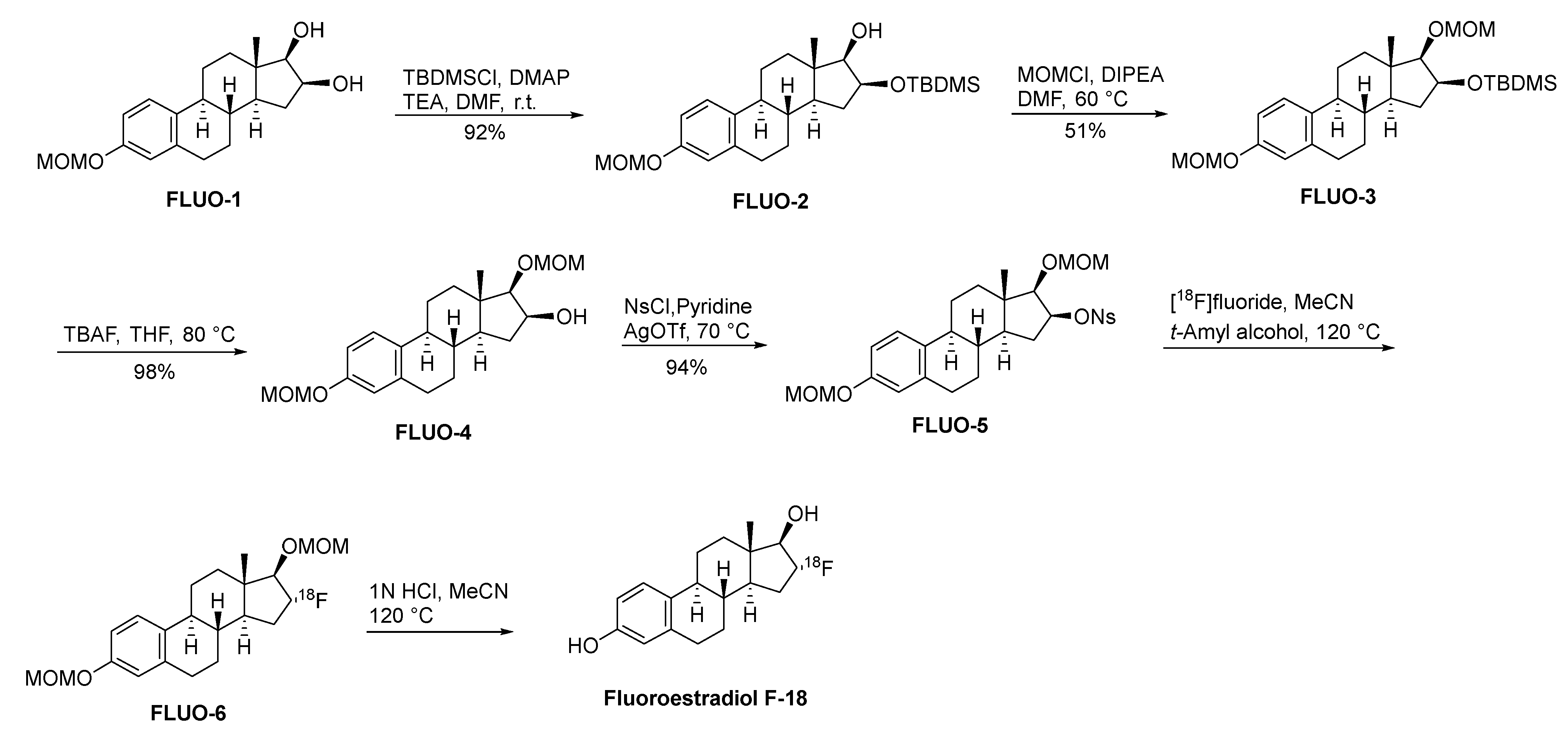
Fluoroestradiol F-18
2.7. HER2 Receptor Tyrosine Kinase Inhibitors
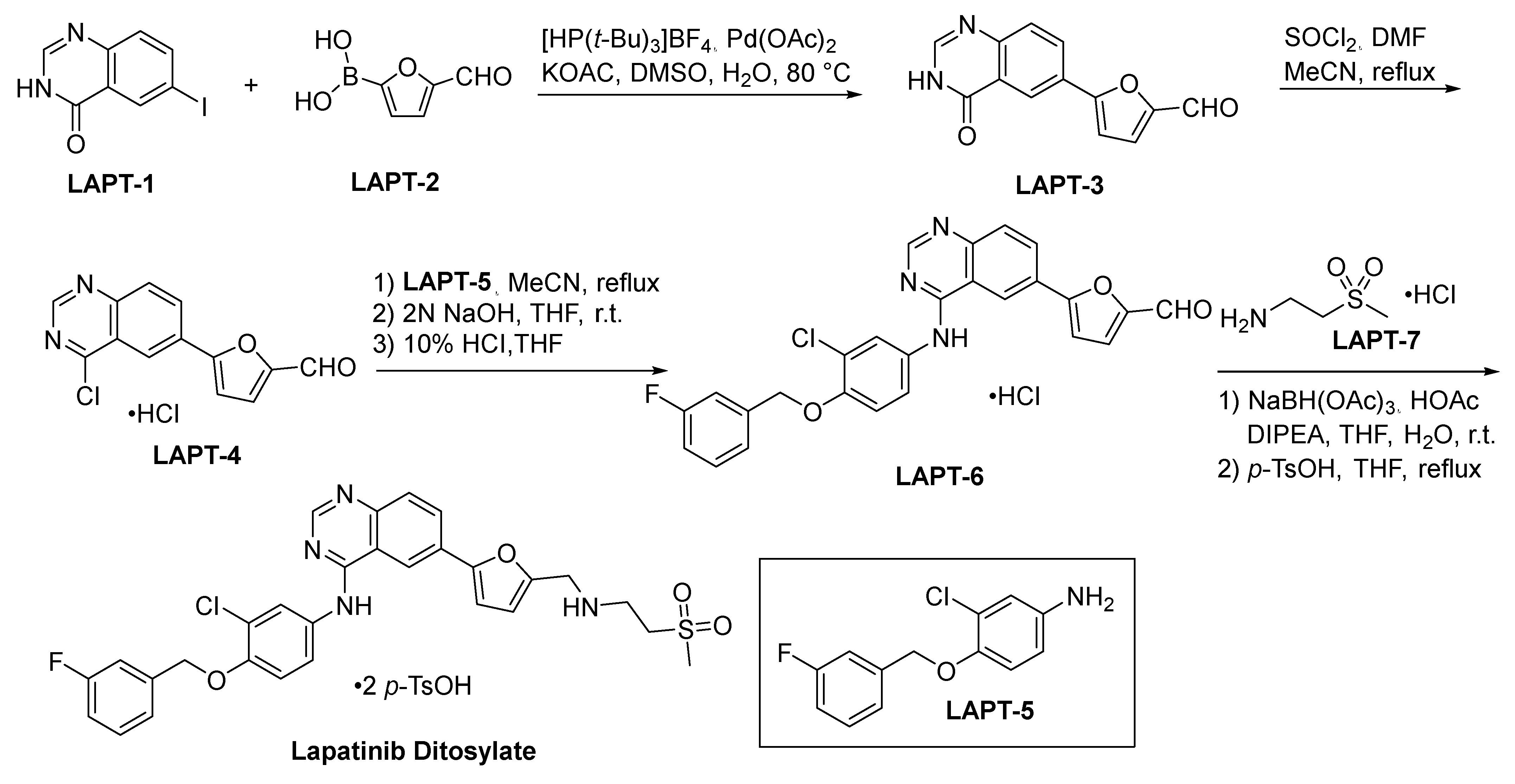
2.7.1. Lapatinib Ditosylate
2.7.2. Neratinib
2.7.3. Tucatinib
2.8. Cyclin-Dependent Kinases 4 and 6 (CDK4/6) Inhibitors
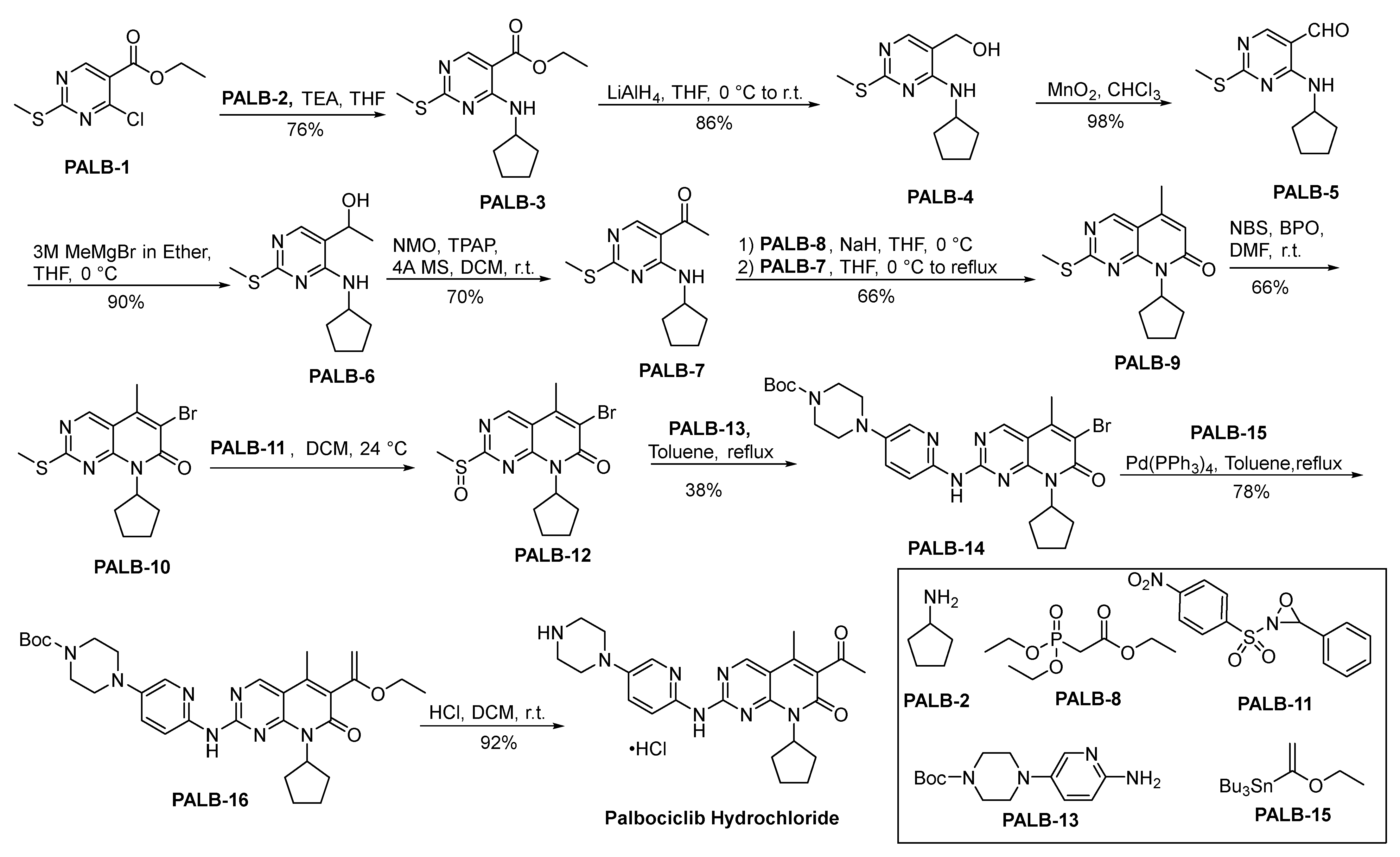
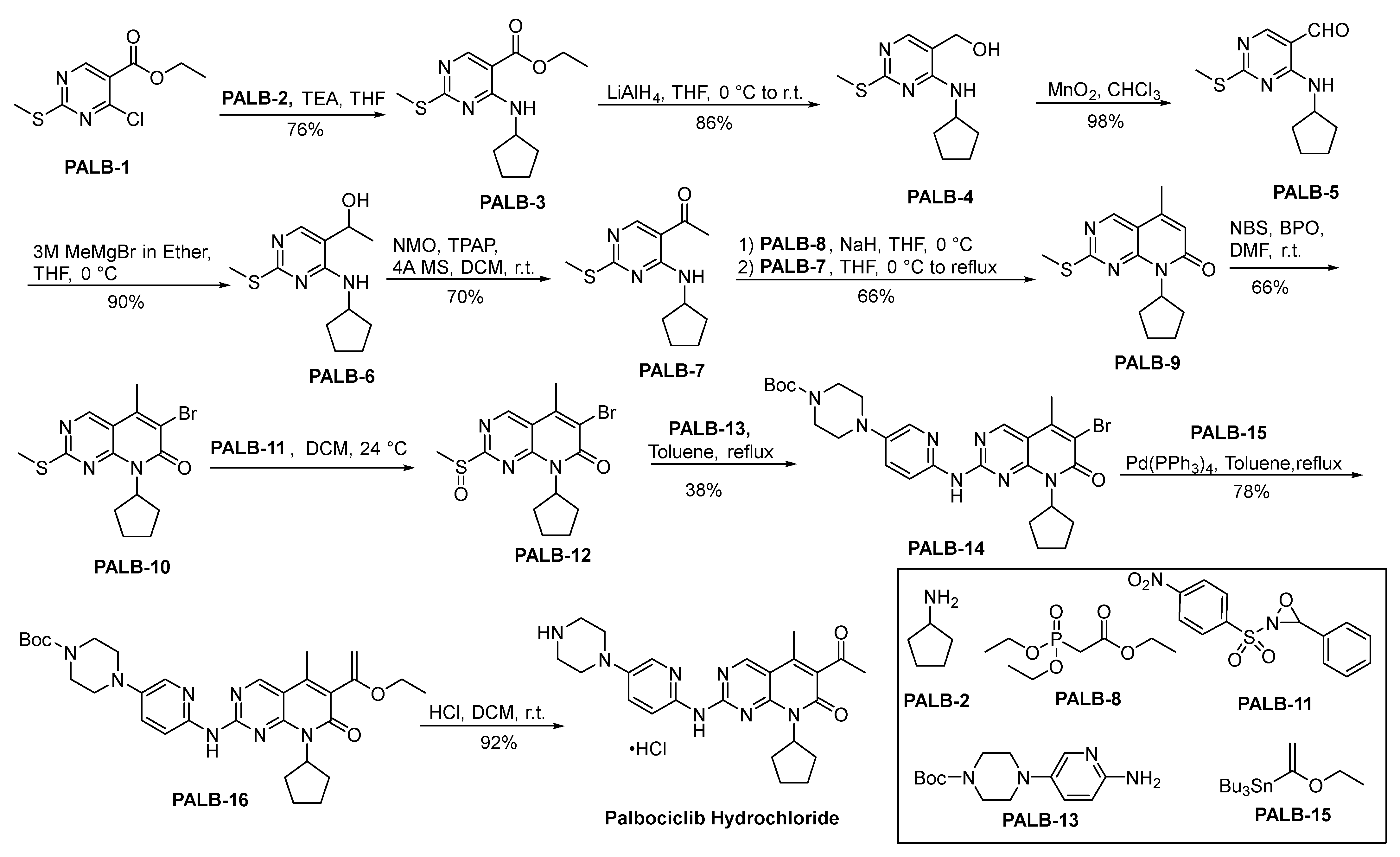
2.8.1. Palbociclib Hydrochloride
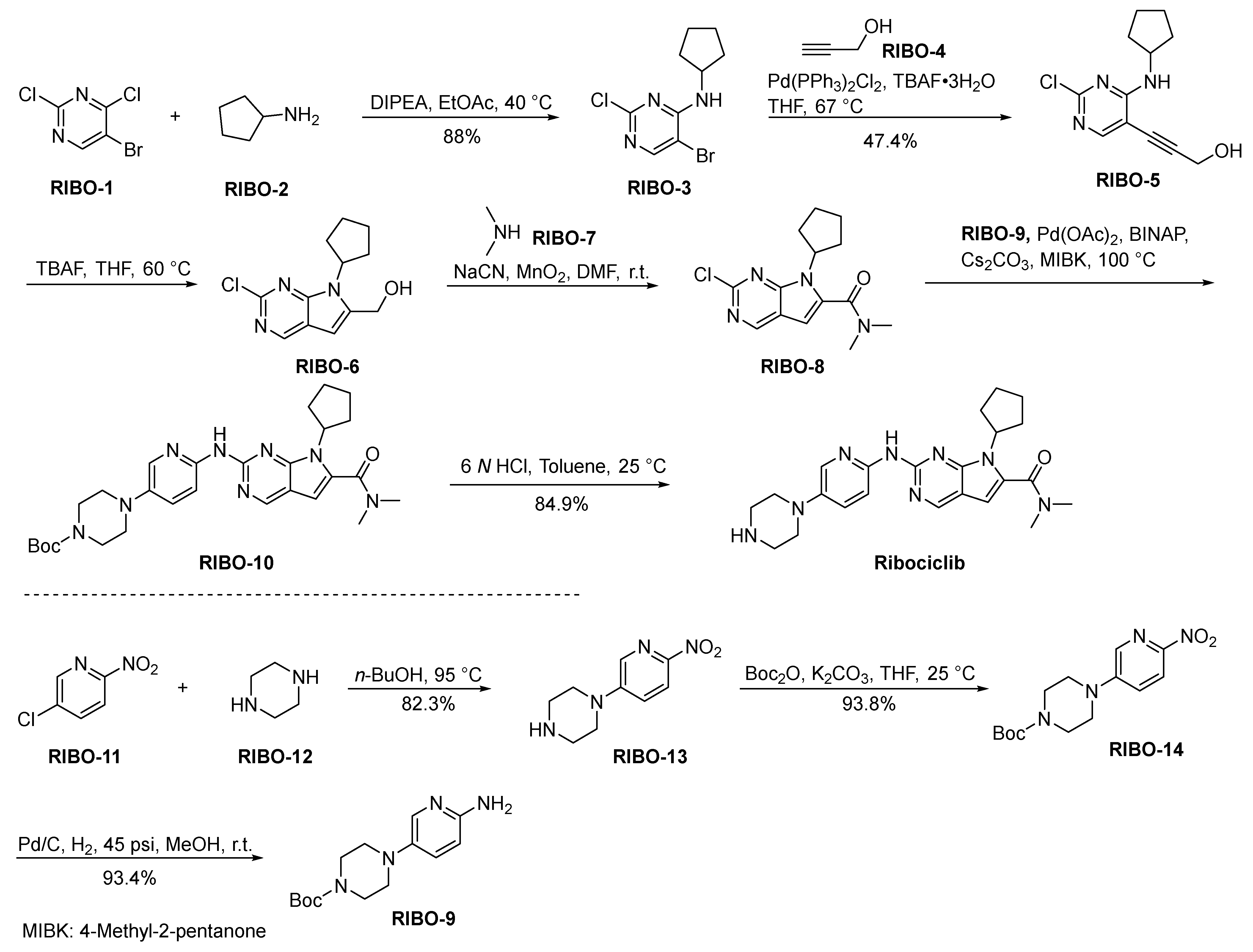
2.8.2. Ribociclib
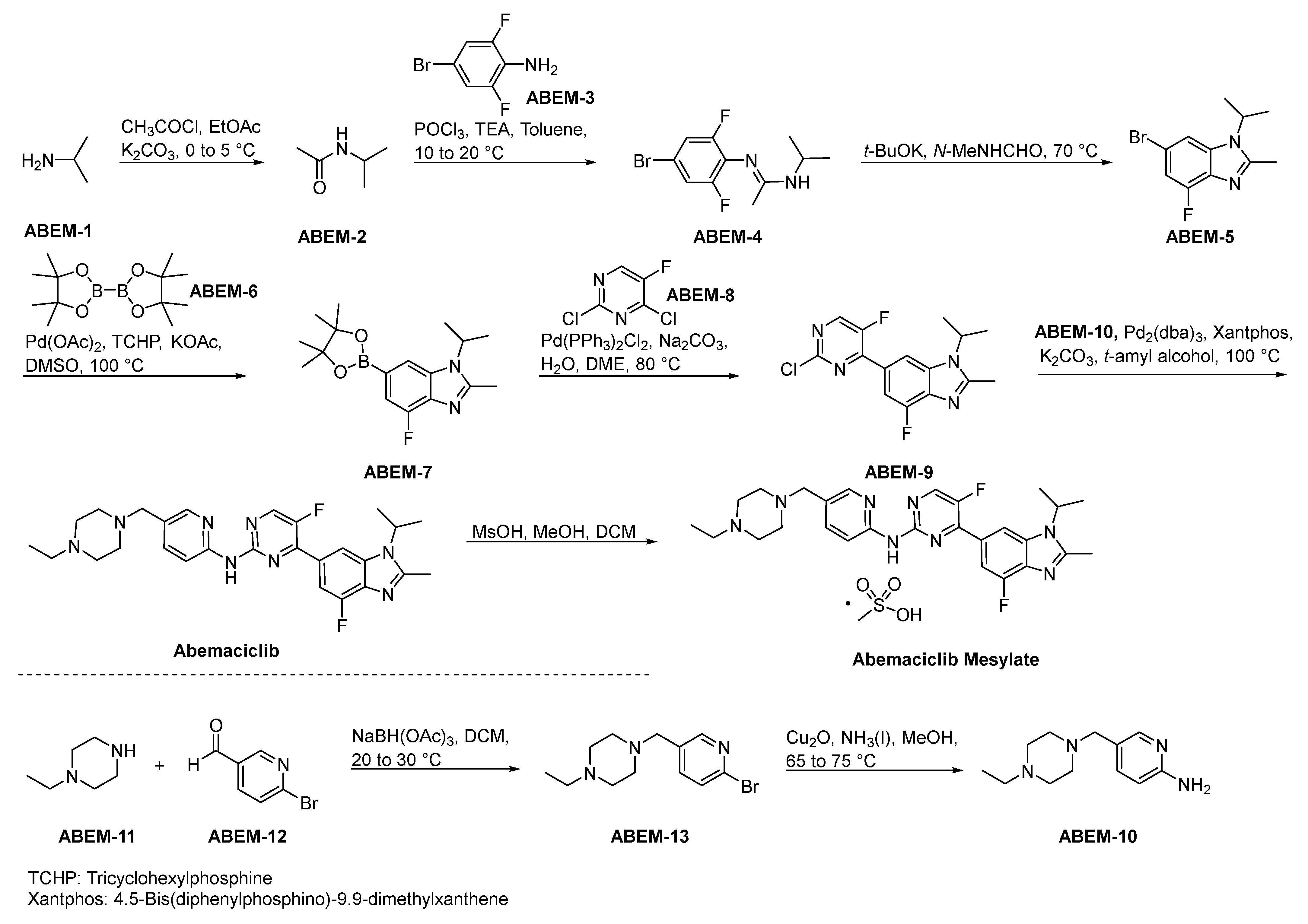
2.8.3. Abemaciclib Mesylate
2.9. Phosphatidylinositol 3-Kinase (PI3K) Inhibitors
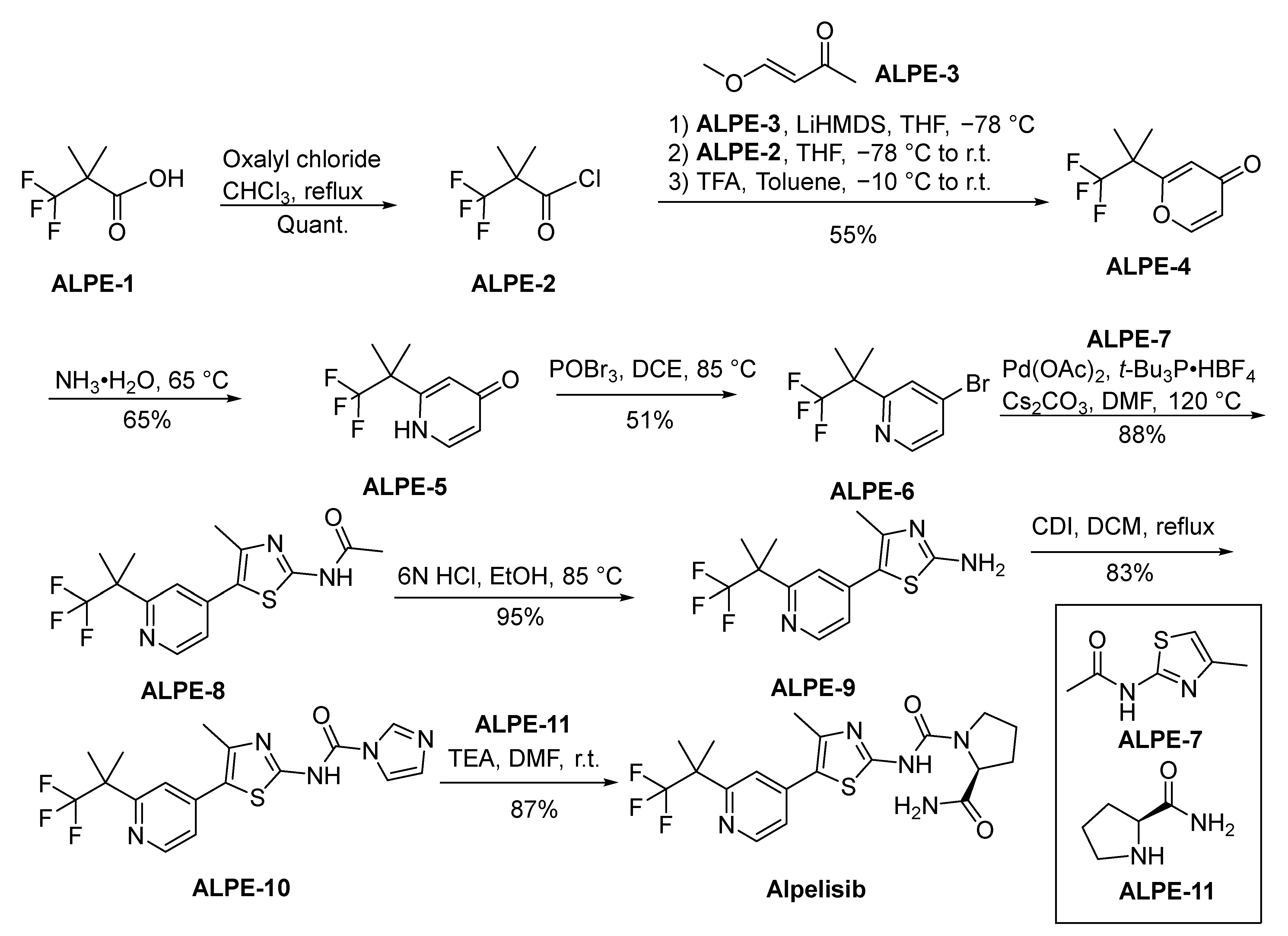
Alpelisib
2.10. Microtubule Inhibitors
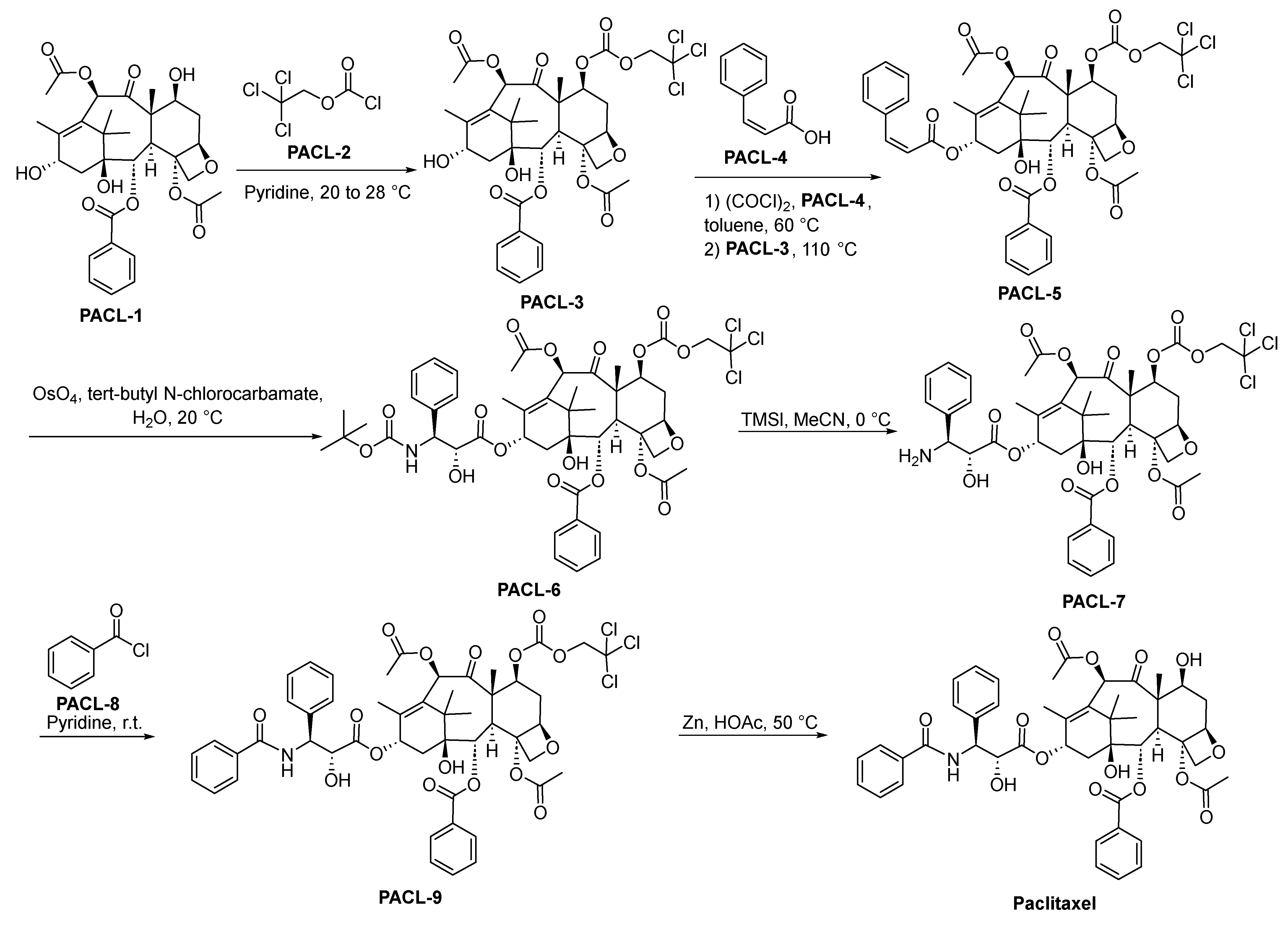
2.10.1. Paclitaxel
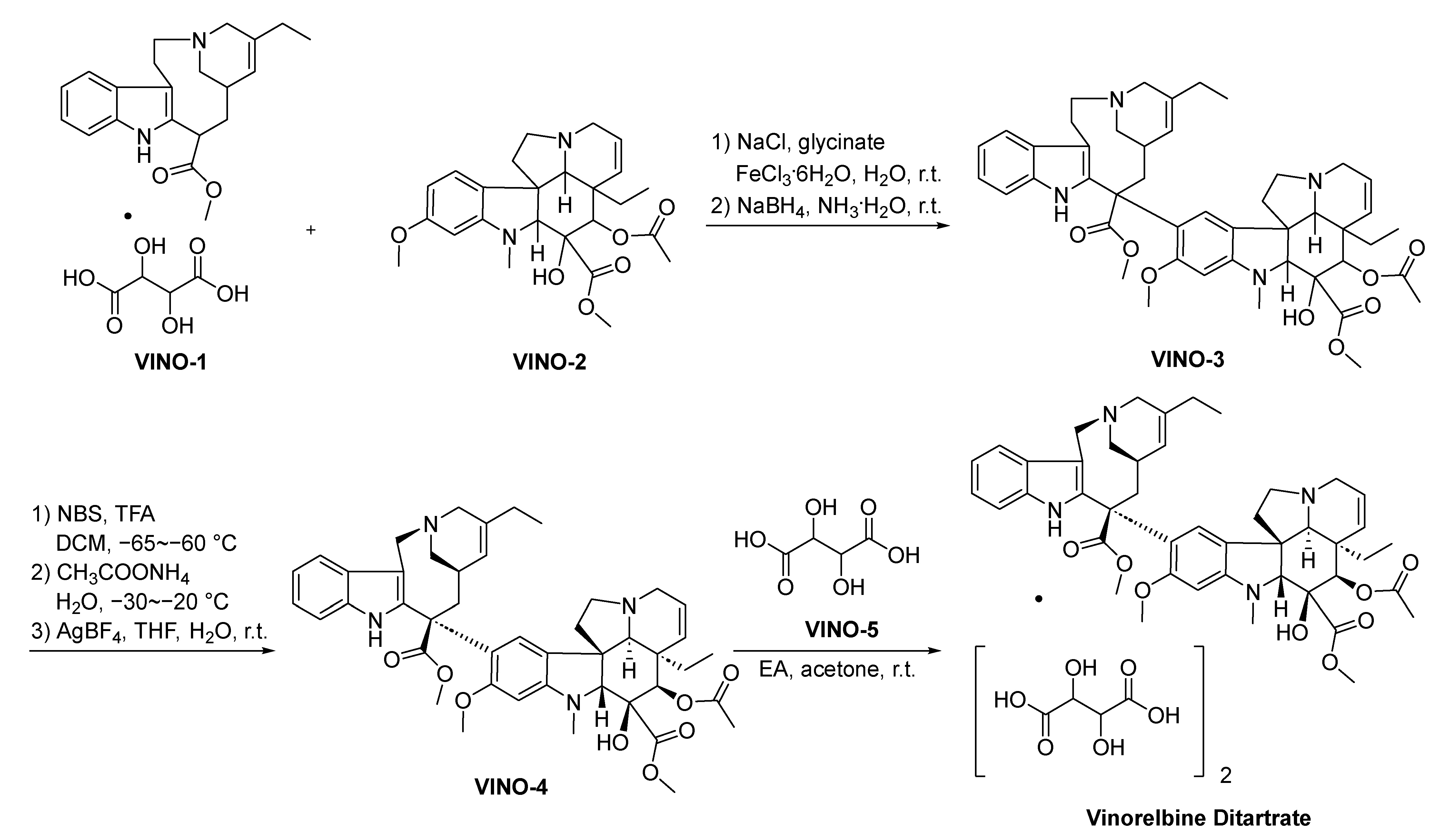
2.10.2. Vinorelbine Ditartrate
2.10.3. Docetaxel
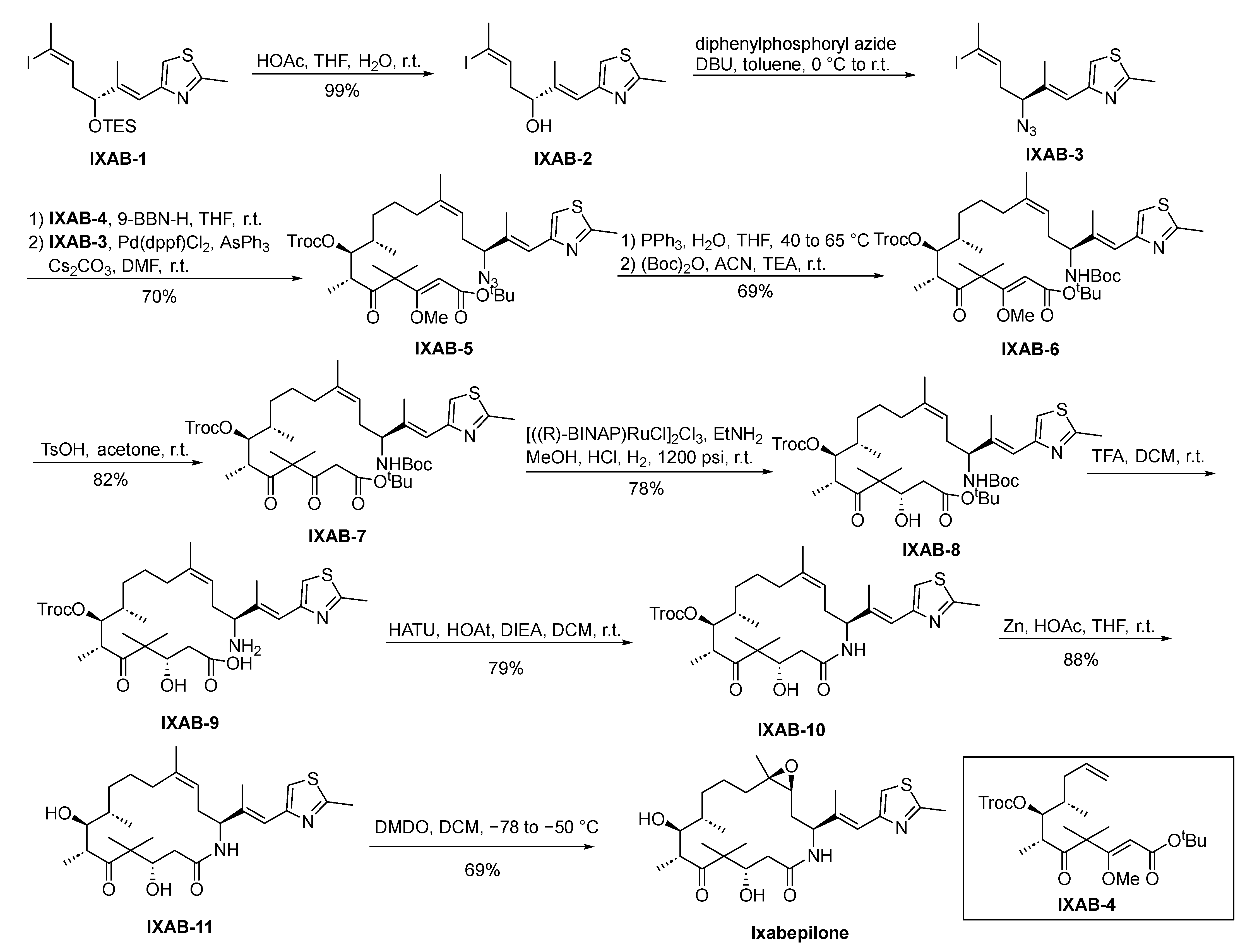
2.10.4. Ixabepilone
2.10.5. Eribulin
2.11. Poly (ADP-Ribose) Polymerase (PARP) Inhibitor
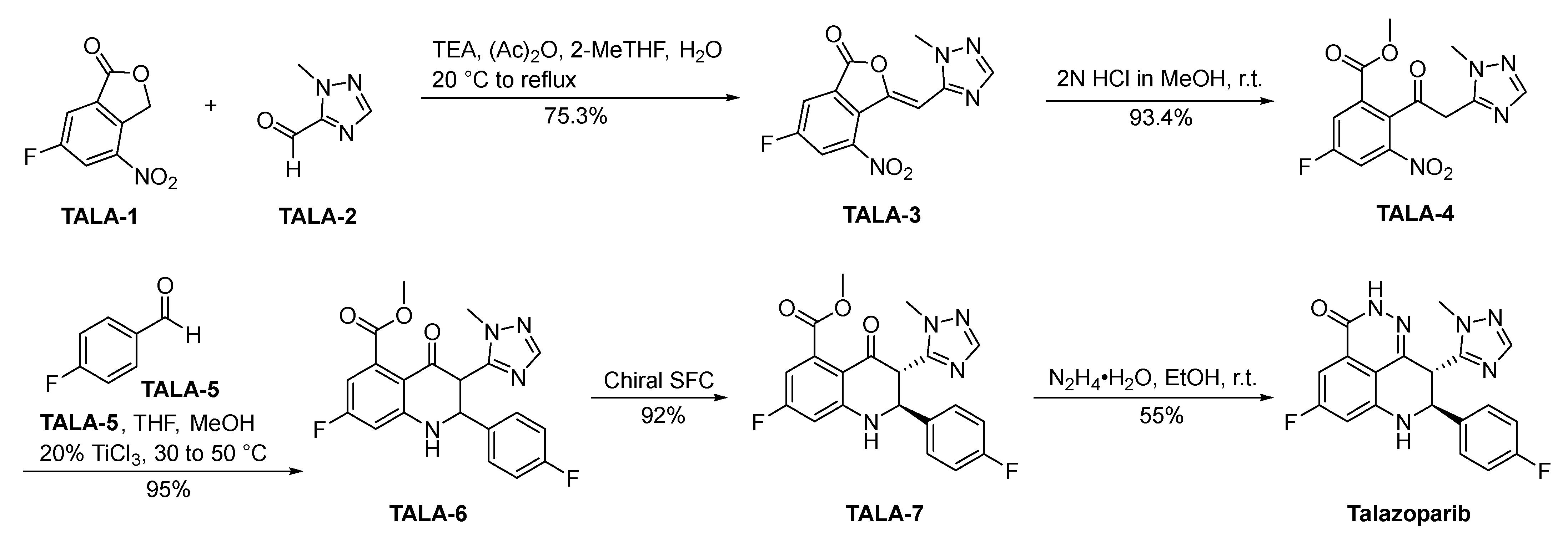
Talazoparib
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Polyak, K. Breast cancer: Origins and evolution. J. Clin. Investig. 2007, 117, 3155–3163. [Google Scholar] [CrossRef]
- Fisher, B.; Anderson, S.; Bryant, J.; Margolese, R.G.; Deutsch, M.; Fisher, E.R.; Jeong, J.H.; Wolmark, N. Twenty-year follow-up of a randomized trial comparing total mastectomy, lumpectomy, and lumpectomy plus irradiation for the treatment of invasive breast cancer. N. Engl. J. Med. 2002, 347, 1233–1241. [Google Scholar] [CrossRef]
- Peto, R.; Davies, C.; Godwin, J.; Gray, R.; Pan, H.C.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Taylor, C.; et al. Comparisons between different polychemotherapy regimens for early breast cancer: Meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 2012, 379, 432–444. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef]
- Zardavas, D.; Baselga, J.; Piccart, M. Emerging targeted agents in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef]
- Drewry, D.H.; Wells, C.I.; Andrews, D.M.; Angell, R.; Al-Ali, H.; Axtman, A.D.; Capuzzi, S.J.; Elkins, J.M.; Ettmayer, P.; Frederiksen, M.; et al. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS ONE 2017, 12, e0181585. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Kang, C.; Syed, Y.Y. Atezolizumab (in combination with nab-paclitaxel): A review in advanced triple-negative breast cancer. Drugs 2020, 80, 601–607. [Google Scholar] [CrossRef]
- Cameron, D.; Casey, M.; Oliva, C.; Newstat, B.; Imwalle, B.; Geyer, C.E. Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: Final survival analysis of a phase III randomized trial. Oncol. 2010, 15, 924–934. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S269–S276. [Google Scholar] [CrossRef]
- Mangini, N.S.; Wesolowski, R.; Ramaswamy, B.; Lustberg, M.B.; Berger, M.J. Palbociclib: A novel cyclin-dependent kinase inhibitor for hormone receptor-positive advanced breast cancer. Ann. Pharmacother. 2015, 49, 1252–1260. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Inoue, K.; Yuasa, H. Molecular basis for pharmacokinetics and pharmacodynamics of methotrexate in rheumatoid arthritis therapy. Drug Metab. Pharmacokinet. 2014, 29, 12–19. [Google Scholar] [CrossRef]
- Tian, H.; Cronstein, B.N. Understanding the mechanisms of action of methotrexate: Implications for the treatment of rheumatoid arthritis. Bull. NYU Hosp. Jt. Dis. 2007, 65, 168–173. [Google Scholar]
- FDA Approved Drug Products: Methotrexate Oral Solution. Available online: Https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208004s002lbl.pdf (accessed on 23 February 2024).
- FDA Approved Drug Products: Methotrexate Subcutaneous Injection. Available online: Https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/205776s002lbl.pdf (accessed on 23 February 2024).
- FDA Approved Drug Products: Methotrexate Prefilled Syringe for Subcutaneous Injection. Available online: Https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/204824s009lbl.pdf (accessed on 23 February 2024).
- FDA Approved Drug Products: Methotrexate Injection. Available online: Https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/011719s125lbl.pdf (accessed on 23 February 2024).
- Ender, K.; Tuncer, A. A New Method for Producing Antifolate Agents Having Glutamic Acid Part in Their Structure. WO2012074496A1, 7 June 2012. [Google Scholar]
- Pandit, B.; Royzen, M. Recent development of prodrugs of gemcitabine. Genes 2022, 13, 466. [Google Scholar] [CrossRef]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of gemcitabine in cancer therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef]
- Ciccolini, J.; Serdjebi, C.; Peters, G.J.; Giovannetti, E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: An EORTC-PAMM perspective. Cancer Chemother. Pharmacol. 2016, 78, 1–12. [Google Scholar] [CrossRef]
- FDA Approved Drug Products: Gemzar (gemcitabine) for Injection, for Intravenous use. Available online: Https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020509s082lbl.pdf (accessed on 23 February 2024).
- Hertel, L.W. Difluoro Antivirals and Intermediate Therefor. US4808614A, 4 June 1987. [Google Scholar]
- Walko, C.M.; Lindley, C. Capecitabine: A review. Clin. Ther. 2005, 27, 23–44. [Google Scholar] [CrossRef]
- Natori, A.; Ethier, J.L.; Amir, E.; Cescon, D.W. Capecitabine in early breast cancer: A meta-analysis of randomised controlled trials. Eur. J. Cancer 2017, 77, 40–47. [Google Scholar] [CrossRef]
- Schellens, J.H. Capecitabine. Oncology 2007, 12, 152–155. [Google Scholar] [CrossRef]
- Andreetta, C.; Puppin, C.; Minisini, A.; Valent, F.; Pegolo, E.; Damante, G.; Di Loreto, C.; Pizzolitto, S.; Pandolfi, M.; Fasola, G.; et al. Thymidine phosphorylase expression and benefit from capecitabine in patients with advanced breast cancer. Ann. Oncol. 2009, 20, 265–271. [Google Scholar] [CrossRef]
- Lou, Y.; Wang, Q.; Zheng, J.; Hu, H.; Liu, L.; Hong, D.; Zeng, S. Possible pathways of capecitabine-induced hand-foot syndrome. Chem. Res. Toxicol. 2016, 29, 1591–1601. [Google Scholar] [CrossRef]
- Arasaki, M.; Ishitsuka, H.; Kuruma, I.; Miwa, M.; Murasaki, C.; Shimma, N. N4-(Substituted-Oxycarbonyl)-5’-Deoxy-5-Fluorocytidine Compounds, Compositions and Methods of Using Same. US5472949A, 14 December 1993. [Google Scholar]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Jawad, B.; Poudel, L.; Podgornik, R.; Steinmetz, N.F.; Ching, W.Y. Molecular mechanism and binding free energy of doxorubicin intercalation in DNA. Phys. Chem. Chem. Phys. PCCP 2019, 21, 3877–3893. [Google Scholar] [CrossRef]
- Xini, Z.; Olson, R.D.; Walsh, G.M. 13-Deoxyanthracycline Derivatives and Processes for Preparing Them. WO9908687A1, 13 August 1998. [Google Scholar]
- FDA Approval of Ellence (Epirubicin Hydrochloride Injection). Available online: Https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/050778s021lbl.pdf (accessed on 23 February 2024).
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Torti, F.M.; Bristow, M.M.; Lum, B.L.; Carter, S.K.; Howes, A.E.; Aston, D.A.; Brown, B.W., Jr.; Hannigan, J.F., Jr.; Meyers, F.J.; Mitchell, E.P.; et al. Cardiotoxicity of epirubicin and doxorubicin: Assessment by endomyocardial biopsy. Cancer Res. 1986, 46, 3722–3727. [Google Scholar]
- Sledge, G.W.; Mamounas, E.P.; Hortobagyi, G.N.; Burstein, H.J.; Goodwin, P.J.; Wolff, A.C. Past, present, and future challenges in breast cancer treatment. J. Clin. Oncol. 2014, 32, 1979–1986. [Google Scholar] [CrossRef]
- Arcamone, F.; Penco, S.; Vigevani, A.; Redaelli, S.; Franchi, G.; DiMarco, A.; Casazza, A.M.; Dasdia, T.; Formelli, F.; Necco, A.; et al. Synthesis and antitumor properties of new glycosides of daunomycinone and adriamycinone. J. Med. Chem. 1975, 18, 703–707. [Google Scholar] [CrossRef]
- Loprinzi, C.L.; Michalak, J.C.; Schaid, D.J.; Mailliard, J.A.; Athmann, L.M.; Goldberg, R.M.; Tschetter, L.K.; Hatfield, A.K.; Morton, R.F. Phase III evaluation of four doses of megestrol acetate as therapy for patients with cancer anorexia and/or cachexia. J. Clin. Oncol. 1993, 11, 762–767. [Google Scholar] [CrossRef]
- Mahayni, H.; Minor, J.R. Megestrol acetate in AIDS-related cachexia. Am. J. Hosp. Pharm. 1991, 48, 2479–2480. [Google Scholar]
- Sedlacek, S.M. An overview of megestrol acetate for the treatment of advanced breast cancer. Semin. Oncol. 1988, 15, 3–13. [Google Scholar]
- Muss, H.B.; Wells, H.B.; Paschold, E.H.; Black, W.R.; Cooper, M.R.; Capizzi, R.L.; Christian, R.; Cruz, J.M.; Jackson, D.V.; Powell, B.L.; et al. Megestrol acetate versus tamoxifen in advanced breast cancer: 5-year analysis—A phase III trial of the Piedmont Oncology Association. J. Clin. Oncol. 1988, 6, 1098–1106. [Google Scholar] [CrossRef]
- Annen, K.; Laurent, H.; Hofmeister, H.; Wiechert, R. Process for the Preparation of 6-Methyl-3-Keto Steroids. US4440684A, 4 February 1983. [Google Scholar]
- Lundgren, S.; Gundersen, S.; Klepp, R.; Lønning, P.E.; Lund, E.; Kvinnsland, S. Megestrol acetate versus aminoglutethimide for metastatic breast cancer. Breast Cancer Res. Treat. 1989, 14, 201–206. [Google Scholar] [CrossRef]
- Santen, R.J.; Misbin, R.I. Aminoglutethimide: Review of pharmacology and clinical use. Pharmacotherapy 1981, 1, 95–120. [Google Scholar] [CrossRef]
- Misbin, R.I.; Canary, J.; Willard, D. Aminoglutethimide in the treatment of Cushing’s syndrome. J. Clin. Pharmacol. 1976, 16, 645–651. [Google Scholar] [CrossRef]
- Bunegar, M.J.; Dyer, U.C.; Evans, G.R.; Hewitt, R.P.; Jones, S.W.; Henderson, N.; Richards, C.J.; Sivaprasad, S.; Skead, B.M.; Stark, M.A.; et al. Production of (R)-aminoglutethimide: a new route from 1-chloro-4-nitrobenzene. Org. Process Res. Dev. 1999, 3, 442–450. [Google Scholar] [CrossRef]
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J.F.; Hoctin-Boes, G.; Houghton, J.; Locker, G.Y.; Tobias, J.S. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005, 365, 60–62. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Jonat, W.; Howell, A.; Jones, S.E.; Blomqvist, C.P.; Vogel, C.L.; Eiermann, W.; Wolter, J.M.; Steinberg, M.; Webster, A.; et al. Anastrozole versus megestrol acetate in the treatment of postmenopausal women with advanced breast carcinoma: Results of a survival update based on a combined analysis of data from two mature phase III trials. Arimidex Study Group. Cancer 1998, 83, 1142–1152. [Google Scholar]
- Plourde, P.V.; Dyroff, M.; Dukes, M. Arimidex: A potent and selective fourth-generation aromatase inhibitor. Breast Cancer Res. Treat. 1994, 30, 103–111. [Google Scholar] [CrossRef]
- Edwards, P.N.; Large, M.S. (Substituted Aralkyl) Heterocyclic Compounds. US4935437A, 10 June 1988. [Google Scholar]
- Mouridsen, H.T. Letrozole in advanced breast cancer: The PO25 trial. Breast Cancer Res. Treat. 2007, 105 (Suppl. S1), 19–29. [Google Scholar] [CrossRef]
- Simpson, E.R.; Clyne, C.; Rubin, G.; Boon, W.C.; Robertson, K.; Britt, K.; Speed, C.; Jones, M. Aromatase—A brief overview. Annu. Rev. Physiol. 2002, 64, 93–127. [Google Scholar] [CrossRef]
- Goss, P.E.; Ingle, J.N.; Martino, S.; Robert, N.J.; Muss, H.B.; Piccart, M.J.; Castiglione, M.; Tu, D.; Shepherd, L.E.; Pritchard, K.I.; et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N. Engl. J. Med. 2003, 349, 1793–1802. [Google Scholar] [CrossRef]
- Kumar, A.S.; Chandra, M.G.; Pradipta, K. Novel Intermediates for Preparation of Letrozole. WO2007074474A1, 27 December 2005. [Google Scholar]
- Kaufmann, M.; Bajetta, E.; Dirix, L.Y.; Fein, L.E.; Jones, S.E.; Zilembo, N.; Dugardyn, J.L.; Nasurdi, C.; Mennel, R.G.; Cervek, J.; et al. Exemestane is superior to megestrol acetate after tamoxifen failure in postmenopausal women with advanced breast cancer: Results of a phase III randomized double-blind trial. The Exemestane Study Group. J. Clin. Oncol. 2000, 18, 1399–1411. [Google Scholar] [CrossRef]
- Geisler, J. Differences between the non-steroidal aromatase inhibitors anastrozole and letrozole—Of clinical importance? Br. J. Cancer 2011, 104, 1059–1066. [Google Scholar] [CrossRef]
- Goss, P.E.; Ingle, J.N.; Pritchard, K.I.; Ellis, M.J.; Sledge, G.W.; Budd, G.T.; Rabaglio, M.; Ansari, R.H.; Johnson, D.B.; Tozer, R.; et al. Exemestane versus anastrozole in postmenopausal women with early breast cancer: NCIC CTG MA.27—A randomized controlled phase III trial. J. Clin. Oncol. 2013, 31, 1398–1404. [Google Scholar] [CrossRef]
- Kunnen, K.; Stehle, N.W.; Weis, S.W.; Pascone, J.M.; Pariza, R.J.; Van Ornum, S.G.; Zizelman, P. Cedarburg Pharmaceuticals Inc. Exemestane and Its Intermediates and Methods of Making the Same. WO2005070951A1, 16 January 2004. [Google Scholar]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213. [Google Scholar] [CrossRef]
- Sinha, D.K.; Pazik, J.E.; Dao, T.L. Prevention of mammary carcinogenesis in rats by pregnancy: Effect of full-term and interrupted pregnancy. Br. J. Cancer 1988, 57, 390–394. [Google Scholar] [CrossRef]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Zhang, J. Industrial Preparation of Tamoxifen Citrate. CN114133334A, 9 November 2021. [Google Scholar]
- Johnston, S.R. New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2010, 16, 1979–1987. [Google Scholar] [CrossRef]
- Mustonen, M.V.; Pyrhönen, S.; Kellokumpu-Lehtinen, P.L. Toremifene in the treatment of breast cancer. World J. Clin. Oncol. 2014, 5, 393–405. [Google Scholar] [CrossRef]
- di Salle, E.; Zaccheo, T.; Ornati, G. Antiestrogenic and antitumor properties of the new triphenylethylene derivative toremifene in the rat. J. Steroid. Biochem. 1990, 36, 203–206. [Google Scholar] [CrossRef]
- Vogel, C.L.; Johnston, M.A.; Capers, C.; Braccia, D. Toremifene for breast cancer: A review of 20 years of data. Clin. Breast Cancer 2014, 14, 1–9. [Google Scholar] [CrossRef]
- Hao, H.; Gage, J.; Li, J.; Zhang, E. Synthesis Method of Toremifene. CN104230723A, 21 August 2014. [Google Scholar]
- Vogel, V.G.; Costantino, J.P.; Wickerham, D.L.; Cronin, W.M.; Cecchini, R.S.; Atkins, J.N.; Bevers, T.B.; Fehrenbacher, L.; Pajon, E.R., Jr.; Wade, J.L., 3rd; et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: The NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006, 295, 2727–2741. [Google Scholar] [CrossRef]
- Jordan, V.C. Fourteenth Gaddum Memorial Lecture. A current view of tamoxifen for the treatment and prevention of breast cancer. Br. J. Pharmacol. 1993, 110, 507–517. [Google Scholar] [CrossRef]
- Turner, C.H.; Sato, M.; Bryant, H.U. Raloxifene preserves bone strength and bone mass in ovariectomized rats. Endocrinology 1994, 135, 2001–2005. [Google Scholar] [CrossRef]
- Ettinger, B.; Black, D.M.; Mitlak, B.H.; Knickerbocker, R.K.; Nickelsen, T.; Genant, H.K.; Christiansen, C.; Delmas, P.D.; Zanchetta, J.R.; Stakkestad, J.; et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: Results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 1999, 282, 637–645. [Google Scholar] [CrossRef]
- Jones, C.D. Antiestrogenic and Antiandrugenic Benzothiophenes. US4418068A, 16 December 1981. [Google Scholar]
- Howell, A.; Pippen, J.; Elledge, R.M.; Mauriac, L.; Vergote, I.; Jones, S.E.; Come, S.E.; Osborne, C.K.; Robertson, J.F. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma: A prospectively planned combined survival analysis of two multicenter trials. Cancer 2005, 104, 236–239. [Google Scholar] [CrossRef]
- Curran, M.; Wiseman, L. Fulvestrant. Drugs 2001, 61, 807–813; discussion 814. [Google Scholar] [CrossRef]
- Osborne, C.K.; Pippen, J.; Jones, S.E.; Parker, L.M.; Ellis, M.; Come, S.; Gertler, S.Z.; May, J.T.; Burton, G.; Dimery, I.; et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J. Clin. Oncol. 2002, 20, 3386–3395. [Google Scholar] [CrossRef]
- Bowler, J.; Tait, B.S. Steroid Derivatives. US4659516A, 1 October 1984. [Google Scholar]
- Bihani, T.; Patel, H.K.; Arlt, H.; Tao, N.; Jiang, H.; Brown, J.L.; Purandare, D.M.; Hattersley, G.; Garner, F. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER(+) breast cancer patient-derived xenograft models. Clin. Cancer Res. 2017, 23, 4793–4804. [Google Scholar] [CrossRef]
- Garner, F.; Shomali, M.; Paquin, D.; Lyttle, C.R.; Hattersley, G. RAD1901: A novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-Cancer Drugs 2015, 26, 948–956. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Wander, S.A.; Hamilton, E.; Razavi, P.; Bardia, A. Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: Current and emerging role. Ther. Adv. Med. Oncol. 2022, 14, 17588359221113694. [Google Scholar] [CrossRef]
- Patel, H.K.; Tao, N.; Lee, K.M.; Huerta, M.; Arlt, H.; Mullarkey, T.; Troy, S.; Arteaga, C.L.; Bihani, T. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. BCR 2019, 21, 146. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: Results from the randomized phase III EMERALD trial. J. Clin. Oncol. 2022, 40, 3246–3256. [Google Scholar] [CrossRef]
- Hamaoka, S.; Kitazawa, N.; Nara, K.; Sasaki, A.; Kamada, A.; Okabe, T. Selective Estrogen Receptor Modulator. US20120004315A1, 15 March 2011. [Google Scholar]
- Linden, H.M.; Kurland, B.F.; Peterson, L.M.; Schubert, E.K.; Gralow, J.R.; Specht, J.M.; Ellis, G.K.; Lawton, T.J.; Livingston, R.B.; Petra, P.H.; et al. Fluoroestradiol positron emission tomography reveals differences in pharmacodynamics of aromatase inhibitors, tamoxifen, and fulvestrant in patients with metastatic breast cancer. Clin. Cancer Res. 2011, 17, 4799–4805. [Google Scholar] [CrossRef]
- Ulaner, G.A. 16α-18F-fluoro-17β-Fluoroestradiol (FES): Clinical applications for patients with breast cancer. Semin. Nucl. Med. 2022, 52, 574–583. [Google Scholar] [CrossRef]
- Sundararajan, L.; Linden, H.M.; Link, J.M.; Krohn, K.A.; Mankoff, D.A. 18F-Fluoroestradiol. Semin. Nucl. Med. 2007, 37, 470–476. [Google Scholar] [CrossRef]
- Kil, H.S.; Cho, H.Y.; Lee, S.J.; Oh, S.J.; Chi, D.Y. Alternative synthesis for the preparation of 16α-[(18) F]fluoroestradiol. J. Label. Compd. Radiopharm. 2013, 56, 619–626. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef]
- Scaltriti, M.; Verma, C.; Guzman, M.; Jimenez, J.; Parra, J.L.; Pedersen, K.; Smith, D.J.; Landolfi, S.; Ramon y Cajal, S.; Arribas, J.; et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2009, 28, 803–814. [Google Scholar] [CrossRef]
- Spector, N.L.; Xia, W.; Burris, H., 3rd; Hurwitz, H.; Dees, E.C.; Dowlati, A.; O’Neil, B.; Overmoyer, B.; Marcom, P.K.; Blackwell, K.L.; et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 2502–2512. [Google Scholar] [CrossRef]
- Chen, Y.; Henschke, J.P.; Liu, Y.; Chu, G.; Zhang, X. Process and Intermediates for Preparing Lapatinib. WO2011116634A1, 23 March 2010. [Google Scholar]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef]
- Canonici, A.; Gijsen, M.; Mullooly, M.; Bennett, R.; Bouguern, N.; Pedersen, K.; O’Brien, N.A.; Roxanis, I.; Li, J.L.; Bridge, E.; et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 2013, 4, 1592–1605. [Google Scholar] [CrossRef]
- Martin, M.; Holmes, F.A.; Ejlertsen, B.; Delaloge, S.; Moy, B.; Iwata, H.; von Minckwitz, G.; Chia, S.K.L.; Mansi, J.; Barrios, C.H.; et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2017, 18, 1688–1700. [Google Scholar] [CrossRef]
- Tsou, H.R.; Overbeek-Klumpers, E.G.; Hallett, W.A.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Michalak, R.S.; Nilakantan, R.; Discafani, C.; Golas, J.; et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005, 48, 1107–1131. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Borges, V.F.; Ferrario, C.; Aucoin, N.; Falkson, C.; Khan, Q.; Krop, I.; Welch, S.; Conlin, A.; Chaves, J.; Bedard, P.L.; et al. Tucatinib combined with ado-trastuzumab emtansine in advanced ERBB2/HER2-positive metastatic breast cancer: A phase 1b clinical trial. JAMA Oncol. 2018, 4, 1214–1220. [Google Scholar] [CrossRef]
- Lin, N.U.; Borges, V.; Anders, C.; Murthy, R.K.; Paplomata, E.; Hamilton, E.; Hurvitz, S.; Loi, S.; Okines, A.; Abramson, V.; et al. Intracranial efficacy and survival with tucatinib plus trastuzumab and capecitabine for previously treated HER2-positive breast cancer with brain metastases in the HER2CLIMB trial. J. Clin. Oncol. 2020, 38, 2610–2619. [Google Scholar] [CrossRef]
- Lyssikatos, J.P.; Hicks, J.M.; Marmsater, F.P.; Qian, Z. N4-Phenyl-Quinazoline-4-Amine Derivatives and Related Compounds as ErbB Type I Receptor Tyrosine Kinase Inhibitors for the Treatment of Hyperproliferative Diseases. US9693989B2, 23 September 2013. [Google Scholar]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. BCR 2009, 11, R77. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Barvian, M.; Booth, R.J.; Iii, J.Q.; Repine, J.T.; Sheehan, D.J.; Toogood, P.L. 2-(Pyridin-2-Ylamino)-Pyrido [2,3-d]Pyrimidin-7-Ones. US6936612B2, 16 January 2003. [Google Scholar]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet. Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Vincent, C.J.; Guangpei, C.; Baoqing, G.; Koteswara, K.P.; Vishal, S. Salt(s) of 7-Cyclopentyl-2-(5-Piperazin-1-yl-Pyridin-2-ylamino)-7h-Pyrrolo [2,3-d]Pyrimidine-6-Carboxylic Acid Dimethylamide and Processes of Making Thereof. WO2012064805A1, 10 November 2010. [Google Scholar]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- David, A.C.; Alfonso, D.D.M.; Ana, D.P.G.; Miriam, F.D.P.C.; Cristina, G.P.M.; Mark, G.L. Protein Kinase Inhibitors. US20100160340A1, 15 December 2009. [Google Scholar]
- Markham, A. Alpelisib: First global approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Royer, B.; Kaderbhaï, C.G.; Schmitt, A. Pharmacokinetics and pharmacodynamic of alpelisib. Clin. Pharmacokinet. 2023, 62, 45–53. [Google Scholar] [CrossRef]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Fairhurst, R.A.; Guagnano, V.; Imbach, P.; Caravatti, G.; Furet, P. Organic Compounds. US20100105711A1, 10 September 2009. [Google Scholar]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Horwitz, S.B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134–136. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Eisenhauer, E.A.; Chaudhry, V.; Arbuck, S.G.; Donehower, R.C. Clinical toxicities encountered with paclitaxel (Taxol). Semin. Oncol. 1993, 20, 1–15. [Google Scholar]
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef]
- Colin, M.; Guenard, D.; Voegelein, F.G.; Potier, P. Process for the Preparation of Taxol and 10-Deacetyltaxol. US4857653A, 14 July 1987. [Google Scholar]
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556. [Google Scholar] [CrossRef]
- Kruczynski, A.; Hill, B.T. Vinflunine, the latest Vinca alkaloid in clinical development. A review of its preclinical anticancer properties. Crit. Rev. Oncol./Hematol. 2001, 40, 159–173. [Google Scholar] [CrossRef]
- Gralla, R.; Harper, P.; Johnson, S.; Delgado, F.M. Vinorelbine (Navelbine) in the treatment of non-small-cell lung cancer: Studies with single-agent therapy and in combination with cisplatin. Ann. Oncol. 1999, 10 (Suppl. S5), S41–S45. [Google Scholar] [CrossRef]
- Marty, M.; Fumoleau, P.; Adenis, A.; Rousseau, Y.; Merrouche, Y.; Robinet, G.; Senac, I.; Puozzo, C. Oral vinorelbine pharmacokinetics and absolute bioavailability study in patients with solid tumors. Ann. Oncol. 2001, 12, 1643–1649. [Google Scholar] [CrossRef]
- Xin, P.; Zhang, S.; Zong, L.; Sun, T. Preparation Method of Vinorelbine. CN104725405A, 20 December 2013. [Google Scholar]
- Ganansia-Leymarie, V.; Bischoff, P.; Bergerat, J.P.; Holl, V. Signal transduction pathways of taxanes-induced apoptosis. Curr. Med. Chem. Anti-Cancer Agents 2003, 3, 291–306. [Google Scholar] [CrossRef]
- Bissery, M.C.; Guénard, D.; Guéritte-Voegelein, F.; Lavelle, F. Experimental antitumor activity of taxotere (RP 56976, NSC 628503), a taxol analogue. Cancer Res. 1991, 51, 4845–4852. [Google Scholar]
- Shepherd, F.A.; Dancey, J.; Ramlau, R.; Mattson, K.; Gralla, R.; O’Rourke, M.; Levitan, N.; Gressot, L.; Vincent, M.; Burkes, R.; et al. Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J. Clin. Oncol. 2000, 18, 2095–2103. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Harada, N.; Ozaki, K.; Hashiyama, T. Synthesis of taxoids 3. A novel and efficient method for preparation of taxoids by employing cis-glycidic acid. Tetrahedron Lett. 1998, 39, 5575–5578. [Google Scholar] [CrossRef]
- Frye, D.K. Advances in breast cancer treatment: The emerging role of ixabepilone. Expert Rev. Anticancer. Ther. 2010, 10, 23–32. [Google Scholar] [CrossRef]
- Lee, F.Y.; Borzilleri, R.; Fairchild, C.R.; Kamath, A.; Smykla, R.; Kramer, R.; Vite, G. Preclinical discovery of ixabepilone, a highly active antineoplastic agent. Cancer Chemother. Pharmacol. 2008, 63, 157–166. [Google Scholar] [CrossRef]
- Higa, G.M.; Abraham, J. Ixabepilone: A new microtubule-targeting agent for breast cancer. Expert Rev. Anticancer. Ther. 2008, 8, 671–681. [Google Scholar] [CrossRef]
- Goodin, S. Ixabepilone: A novel microtubule-stabilizing agent for the treatment of metastatic breast cancer. Am. J. Health Syst. Pharm. AJHP 2008, 65, 2017–2026. [Google Scholar] [CrossRef]
- Thomas, E.S.; Gomez, H.L.; Li, R.K.; Chung, H.C.; Fein, L.E.; Chan, V.F.; Jassem, J.; Pivot, X.B.; Klimovsky, J.V.; de Mendoza, F.H.; et al. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J. Clin. Oncol. 2007, 25, 5210–5217. [Google Scholar] [CrossRef]
- Stachel, S.J.; Lee, C.B.; Spassova, M.; Chappell, M.D.; Bornmann, W.G.; Danishefsky, S.J.; Chou, T.C.; Guan, Y. On the interactivity of complex synthesis and tumor pharmacology in the drug discovery process: Total synthesis and comparative in vivo evaluations of the 15-aza epothilones. J. Org. Chem. 2001, 66, 4369–4378. [Google Scholar] [CrossRef]
- Swami, U.; Shah, U.; Goel, S. Eribulin in cancer treatment. Mar. Drugs 2015, 13, 5016–5058. [Google Scholar] [CrossRef]
- Smith, J.A.; Wilson, L.; Azarenko, O.; Zhu, X.; Lewis, B.M.; Littlefield, B.A.; Jordan, M.A. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry 2010, 49, 1331–1337. [Google Scholar] [CrossRef]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anti-Cancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Diéras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Fabio, S.; Alena, R.; Ming, P.; Boris, G.; Ko, N.T.; Jason, A.B.; Ricardo, O.; Huzaifa, R. Synthetic Process for Preparation of Macrocyclic c1-Keto Analogs of Halichondrin b and Intermediates Useful Therein. WO2013142999A1, 15 May 2012. [Google Scholar]
- Hoy, S.M. Talazoparib: First global approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Hobbs, E.A.; Litton, J.K.; Yap, T.A. Development of the PARP inhibitor talazoparib for the treatment of advanced BRCA1 and BRCA2 mutated breast cancer. Expert Opin. Pharmacother. 2021, 22, 1825–1837. [Google Scholar] [CrossRef]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Wang, B.; Chu, D.; Liu, Y.; Jiang, Q.; Lu, L. Processes of Synthesizing Dihydropyridophthalazinone Derivatives. US2011196153A1, 8 February 2011. [Google Scholar]
- Sahni, J.M.; Gayle, S.S.; Bonk, K.L.; Vite, L.C.; Yori, J.L.; Webb, B.; Ramos, E.K.; Seachrist, D.D.; Landis, M.D.; Chang, J.C.; et al. Bromodomain and extraterminal protein inhibition blocks growth of triple-negative breast cancers through the suppression of aurora kinases. J. Biol. Chem. 2016, 291, 23756–23768. [Google Scholar] [CrossRef]
- Lashen, A.G.; Toss, M.S.; Wootton, L.; Green, A.R.; Mongan, N.P.; Madhusudan, S.; Rakha, E. Characteristics and prognostic significance of polo-like kinase-1 (PLK1) expression in breast cancer. Histopathology 2023, 83, 414–425. [Google Scholar] [CrossRef]
- Núñez Abad, M.; Calabuig-Fariñas, S.; Lobo de Mena, M.; Torres-Martínez, S.; García González, C.; García García, J.; Iranzo González-Cruz, V.; Camps Herrero, C. Programmed death-ligand 1 (PD-L1) as immunotherapy biomarker in breast cancer. Cancers 2022, 14, 307. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Kalampokas, E.; Kalampokas, T.; Spartalis, E.; Daskalopoulou, A.; Valsami, S.; Kontos, M.; Nonni, A.; et al. Histone deacetylases as new therapeutic targets in triple-negative breast cancer: Progress and promises. Cancer Genom. Proteom. 2017, 14, 299–313. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu, S., II; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR signaling pathway in breast cancer: From molecular landscape to clinical aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO | Drug Name | Original Company | Target | Brand Name | Approval Year |
|---|---|---|---|---|---|
| 1 | Methotrexate | Lederle Laboratories | DNA | Emtexate | 1953 |
| 2 | Megestrol Acetate | Bristol-Myers Squibb | Progesterone receptor | Megace | 1971 |
| 3 | Doxorubicin Hydrochloride | Pfizer | DNA | Adriamycin | 1974 |
| 4 | Tamoxifen Citrate | AstraZeneca | Estrogen receptor | Nolvadex | 1977 |
| 5 | Aminoglutethimide | Novartis | Aromatase enzyme | Cytadren | 1980 |
| 6 | Paclitaxel | Bristol-Myers Squibb | Microtubule | Taxol | 1992 |
| 7 | Vinorelbine Ditartrate | GlaxoSmithKline | Microtubule | Navelbine | 1994 |
| 8 | Anastrozole | AstraZeneca | Aromatase enzyme | Arimidex | 1995 |
| 9 | Docetaxel | Sanofi-Aventis | Microtubule | Taxotere | 1996 |
| 10 | Gemcitabine | Eli Lilly | DNA | Gemzar | 1996 |
| 11 | Letrozole | Novartis | Aromatase enzyme | Femara | 1997 |
| 12 | Toremifene | Orion Corporation | Estrogen receptor | Fareston | 1997 |
| 13 | Raloxifene | Eli Lilly | Estrogen receptor | Evista | 1997 |
| 14 | Capecitabine | Roche | DNA | Xeloda | 1998 |
| 15 | Epirubicin Hydrochloride | Pfizer | DNA | Ellence | 1999 |
| 16 | Exemestane | Pfizer | Aromatase enzyme | Aromasin | 1999 |
| 17 | Fulvestrant | AstraZeneca | Estrogen receptor | Faslodex | 2002 |
| 18 | Lapatinib Ditosylate | GlaxoSmithKline | HER2 receptor tyrosine kinase | Tykerb | 2007 |
| 19 | Ixabepilone | Bristol-Myers Squibb | Microtubule | Ixempra | 2007 |
| 20 | Eribulin | Eisai | Microtubule | Halaven | 2010 |
| 21 | Palbociclib Hydrochloride | Pfizer | Cyclin-dependent kinases 4 and 6 (CDK4/6) | Ibrance | 2015 |
| 22 | Ribociclib | Novartis | Cyclin-dependent kinases 4 and 6 (CDK4/6) | Kisqali | 2017 |
| 23 | Abemaciclib Mesylate | Eli Lilly | Cyclin-dependent kinases 4 and 6 (CDK4/6) | Verzenio | 2017 |
| 24 | Neratinib | Puma Biotechnology | HER2 receptor tyrosine kinase | Nerlynx | 2017 |
| 25 | Talazoparib | Pfizer | Poly (ADP-ribose) polymerase (PARP) | Talzenna | 2018 |
| 26 | Alpelisib | Novartis | Phosphatidylinositol 3-kinase (PI3K) | Piqray | 2019 |
| 27 | Tucatinib | Seattle Genetics | HER2 receptor tyrosine kinase | Tukysa | 2020 |
| 28 | Fluoroestradiol F-18 | PETNET Solutions Inc. | Estrogen receptor | Detectnet | 2020 |
| 29 | Elacestrant | Radius Health | Estrogen receptor | Orserdu | 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Zhu, B.-Y.; Niu, Z.-X. Catalysts of Healing: A Symphony of Synthesis and Clinical Artistry in Small-Molecule Agents for Breast Cancer Alleviation. Molecules 2024, 29, 1166. https://doi.org/10.3390/molecules29051166
Hu J, Zhu B-Y, Niu Z-X. Catalysts of Healing: A Symphony of Synthesis and Clinical Artistry in Small-Molecule Agents for Breast Cancer Alleviation. Molecules. 2024; 29(5):1166. https://doi.org/10.3390/molecules29051166
Chicago/Turabian StyleHu, Jing, Bi-Yue Zhu, and Zhen-Xi Niu. 2024. "Catalysts of Healing: A Symphony of Synthesis and Clinical Artistry in Small-Molecule Agents for Breast Cancer Alleviation" Molecules 29, no. 5: 1166. https://doi.org/10.3390/molecules29051166
APA StyleHu, J., Zhu, B.-Y., & Niu, Z.-X. (2024). Catalysts of Healing: A Symphony of Synthesis and Clinical Artistry in Small-Molecule Agents for Breast Cancer Alleviation. Molecules, 29(5), 1166. https://doi.org/10.3390/molecules29051166