Abstract
Substituents at the meso-site of metalloporphyrins profoundly influence the hydrogen evolution reaction (HER) mechanism. This study employs density functional theory (DFT) to computationally analyze NiII-porphyrin and its hydrides derived from tetrakis(pentafluorophenyl)porphyrin molecules, presenting stereoisomers in ortho- or para-positions. The results reveal that the spatial resistance effect of meso-substituted groups at the ortho- and para-positions induces significant changes in Ni-N bond lengths, angles, and reaction dynamics. For ortho-position substituents forming complex I, a favorable 88.88 ų spherical space was created, facilitating proton coordination and the formation of H2 molecules; conversely, para-position substituents forming complex II impeded H2 formation until bimolecular complexes arose. Molecular dynamics (MD) analysis and comparison were conducted on the intermediation products of I-H2 and (II-H)2, focusing on the configuration and energy changes. In the I-H2 products, H2 molecules underwent separation after 150 fs and overcame the 2.2 eV energy barrier. Subsequently, significant alterations in the spatial structure were observed as complex I deformed. In the case of (II-H)2, it was influenced by the distinctive “sandwich” configuration; the spatial structure necessitated overcoming a 6.7 eV energy barrier for H2 detachment and a process observed after 2400 fs.
1. Introduction
Hydrogen has garnered attention as a potential solution for reducing greenhouse gas emissions and promoting sustainability due to its clean, versatile, and renewable properties [1,2,3,4,5]. Among the various methods for hydrogen production, metallic porphyrins have emerged as promising catalysts owing to their unique electrochemical and photophysical properties [6,7,8,9,10,11,12] and their exceptional reactivity in energy-dependent small molecule activation [13]. Notably, the facile modification of porphyrins through substitution reactions at the meso-site [14,15,16,17,18], β-site [19,20,21], and axial coordination site [22,23] provides a means to control proton transfer ability, substrate accessibility to the active center, and product selectivity. Currently, several studies are investigating the hydrogen evolution reaction (HER) mechanism using metallic porphyrins [24,25,26]. The catalytic activity of these metal complexes relies on their ability to participate in proton-coupled electron transfer (PCET) processes, producing intermediates that donate hydrides to free protons and release hydrogen [27]. Furthermore, steric hindrance caused by meso-substituents in metal porphyrin hydrides has been found to affect the HER mechanism [28,29], and computational studies are underway to better understand this hemolysis and heterolysis process.
Metalloporphyrin hydrides are pivotal in catalytic hydrogen production [30,31,32]. Previous studies have shown that the HER mechanism of metal porphyrin hydrides is significantly influenced by meso-substituted groups [33,34]. Recently, Cao et al. [33,35] conducted experimental investigations to determine how the size of meso-substituted groups directly affects the HER mechanism. Two meso-substituted groups were studied: phenyl groups connected by para-positions with amido moieties (Figure 1a) and substituents with the same ortho-positions (Figure 1b). The HER reaction process was characterized for each complex type (Figure 2). The results showed that, in the case of complex I, the bulky pivalamide group led to steric hindrance, forming two intermediate species: NiIII-H, which received one electron, and NiII-H. Finally, heterolysis occurred to produce H2. Conversely, NiIII-H formed a bimetallic complex intermediate for complex II because of the absence of steric hindrance from the substituted group on the active site. The homolysis reaction then took place to produce H2. While it is known that the size of meso-substituted groups has a direct impact on the activity and reaction mechanism of the active center, there is a lack of formation of intermediates, and the protonation reaction, separation of H2 molecules from the system after formation, interaction with meso-position substituents, and charge distribution have not been studied in detail. Therefore, this study comprehensively investigates the effects of substituent steric hindrance on the reactions of complexes I and II and their intermediates. This investigation incorporates multiple aspects, including geometry, electron density distribution, atomic charge, the density of states, and molecular dynamics analysis. The findings of this study provide theoretical underpinnings for the future design and research of novel porphyrin catalysts.
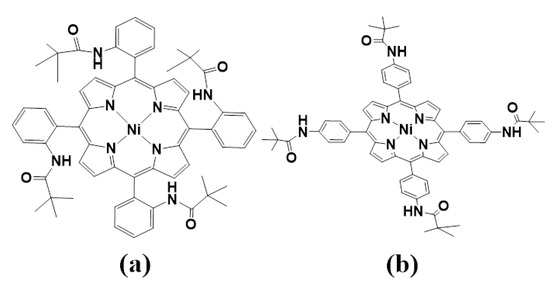
Figure 1.
Structures of the I (a) and II (b) NiII porphyrin complexes.
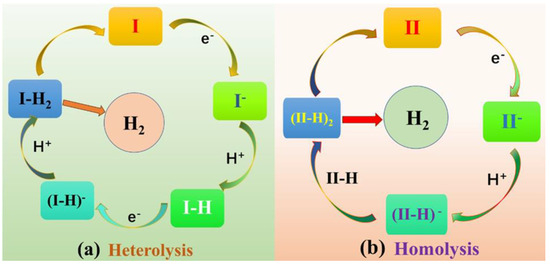
Figure 2.
Catalytic HER mechanisms with I (a) and II (b).
2. Results and Discussion
2.1. Optimized Structure Analysis
The geometrical optimization of the I and II complexes and their related intermediate products was performed using the PBE0/def2-SVP method, as implemented in the Gaussian 09 software package. During the optimization process, the products were carried out at the lowest energy level of the high spin state, and all compounds were optimized to the local minimum of the potential energy surface. The structures of selected I and II complexes and their I-H2 and (II-H)2 compounds were visualized based on the optimized conformations, as shown in Figure S1. The analysis revealed that, in the complex I, where two pivalamido groups replaced the meso-site of the porphyrin ring, the planar structure of the porphyrin ring changed to a “saddle-like” structure due to spatial blockage, whereas in the II complex, the planar structure of the porphyrin ring was less perturbed. The results also show that the bond lengths of NiII ions and pyrrole N (N1) and pyridine N (N2) in I and II complexes underwent slight changes (Table 1). However, the corresponding bond lengths increased gradually when electrons and protons were combined, particularly when comparing the bond lengths of I-H and II-H complexes. This was primarily due to the spatial site resistance, which limited the further expansion of the NiII-porphyrin ring and affected the bond length of NiII-H in complex I. Similarly, this change also caused a corresponding significant change in the atomic charge, which will be discussed in Section 2.2

Table 1.
The selected bond lengths of I, II, and intermediate complexes calculated at the PBE0/def2-SVP level of theory.
2.2. Atomic Charge Analysis
Atomic charges play a significant role in studying electrostatic interactions between molecules and predicting the properties of materials and their interactions with other substances. Accurate atomic charges can aid in designing ligands that interact favorably with proteins and other biological molecules and determine which atoms are likely to participate in catalytic reactions. However, due to the unobservable nature of the atomic charge and the lack of an objective and unique definition, numerous methods exist to calculate atomic charge [36,37]. The atomic dipole corrected Hirshfeld atomic charge (ADCH) method offers a more objective approach to atomic charge analysis, which is essential for predicting chemical reactivity, designing new materials, and understanding electrostatic interactions in biological systems. The ADCH method defines the atomic charge (Equation (1)) as a weighted sum of electron density contributions from neighboring atoms:
where (r), =, where(r) represents the electron density of all atoms in the free state; ∆ρ(r) represents the deformation density, which shows the variation in the electron density during the chirality process after the atoms formed molecules; and is the A-atom weight function, defined as the region in the whole real space belonging to the A-atoms. However, the Hirshfeld charge data are generally small [38], and the dipole moment and electrostatic potential are poorly reproducible [39], mainly because the influence of the atomic dipole moment in the calculation process is neglected. To address this issue, Lu Tian [40] proposed the ADCH method, which defines the atomic dipole moment () as:
In this method, the Hirshfeld charge of each atom and its are calculated first, and then each is expanded into the calibrated positive charge of the surrounding atoms according to Equation (3).
denotes the calibrated positive charge of theof the unfolded A atom on the B atom. Finally, after unfolding theof all atoms into the correctional charge and then accumulating it to the original Hirshfeld charge, the ADCH charge is obtained.
To further analyze the changes in atomic charges or fragment charges of Ni, N1, and N2 atoms, as well as their meso-site substituents in complexes I and II and their intermediate states throughout the entire reaction process, the ADCH method was employed for analysis at each step. The corresponding analysis results are presented in Table 2. The table shows that the up-Sub fragment in complex I carried a charge of −0.009 a.u., while the down-Sub fragment bore a charge of 0.008 a.u. This indicates an uneven charge distribution due to steric hindrance imposed by the substituent at the meso- position. This variation was also reflected in the charge distribution of nitrogen atoms in the porphyrin ring. Pyrrole N1 had a charge of 0.143 a.u., whereas, on pyridine N2, the charge was −0.179 a.u. In complex II, the fragment charges carried by the substituents at the meso-position were equal, and the charges on pyrrole N1 and pyridine N2 were also identical. Moving on to the analysis of the intermediate complexes I− and II−, in Table 2, it can be observed that after receiving an electron, the charges carried by the substituents at the meso-position in complexes I− and II− were both negative. Notably, the negative charge in complex I− was more significant than in II− by approximately 0.015 a.u. Conversely, the charges on pyrrole N1 and pyridine N2 were opposite. Simultaneously, the charges on the Ni ion in complexes I− and II− were identical. From these changes, it can be inferred that in complex I−, due to the greater negative charge carried by the substituent at the meso-position compared to the fragment in II−, a negative electric field was expected to form around complex I−, attracting protons into the coordination center and enhancing the proton-accepting capability.
Table 2.
The selected charges of I, II, and intermediate complexes calculated at the PBE0/def2-SVP level of theory.
Upon further proton coordination with complexes I and II, it was observed that the charges carried by the substituents at the meso-position became positive, while the charges on pyrrole N1 and pyridine N2 essentially remained unchanged and hostile. The primary alteration was detected in the central coordinating metal NiII ion. In the subsequent process, due to the steric hindrance exerted by the substituent at the meso- position in complex I-H, further electron acceptance led to the formation of the intermediate complex (I-H)−. In this intermediate state, the fragment charges carried by the substituent at the meso- position and pyrrole N1 and pyridine N2 all became negative. This change indicated the electron-donating capability of the meso-sited substituent, simultaneously creating a spatial environment with a strong proton-accepting ability around complex (I-H). This environment provided a conducive space for the further binding of protons to form hydrogen molecules. Drawing on earlier research findings and comparing the process of stable H2 formation in complex II-H [33], it was inferred that, since the meso-sited substituents in complex II-H carried positive charges, this might pose challenges for the formation of a stable II-H2 complex. This analysis suggests that II-H is less likely to form a stable diatomic complex under these conditions, which would hinder the formation of H2 molecules.
Alternatively, an ADCH analysis was performed on the diatomic complex (II-H)2, derived from the dimerization of complex II-H. The examination unveiled notable discrepancies in the fragment charges borne by pyrrole N1, pyridine N2, and the meso-sited substituent. Furthermore, variations in the charges of the central ligand NiII were observed. The comprehension of these observed changes is presently in a preliminary exploration stage, and a more comprehensive investigation will be undertaken in subsequent studies.
2.3. Fragment Orbital Interaction Analysis
Based on the previous analysis of the atomic charges and fragment charges of complexes I and II, as well as their corresponding reaction intermediates, it is evident that the bulky pivalamide group positioned at ortho and para sites had a significant impact on the charge distribution of the porphyrin ring and its Ni ions. This impact is primarily attributed to the interaction of molecular orbitals derived from the substituted fragment charges. To further delve into the interaction between substituted fragment molecular orbitals and the molecular orbitals of the porphyrin ring, this study employed the General Charge Decomposition Analysis (GCDA) method proposed by Dapprich and Frenking [41], as refined by Lu Tian [42]. The results of the analysis are presented in Figure 3 and Figure S1.
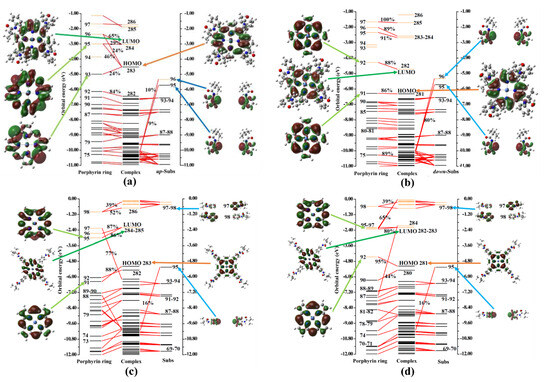
Figure 3.
Fragment orbital interaction diagram of porphyrin rings, up-substituents (up-subs), and down-substituents (down-subs). Black solid and red dashed bars correspond to occupied and unoccupied MOs. (a,c) indicate the major contribution of alpha MOs of the porphyrin ring and up-sub fragments to the I. (b,d) indicate the major contribution of beta MOs of the porphyrin ring and down-sub fragments to the II. The orbital compositions were evaluated using the Mulliken method. Note: up-substituent indicates a para-position substituent close to a proton; down-substituent indicates a distant substituent group.
As depicted in Figure 3, in both complexes I and II, the orbitals constituting their Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), be they alpha or beta molecular orbitals, were primarily composed of the molecular orbital components of the HOMO and LUMO of the porphyrin ring. The molecular orbital components of the substituent groups at the meso-site did not prominently appear in the corresponding orbitals. Additionally, in conjunction with Figure S2, it is evident that when complexes I or II further interacted with electrons or protons to form intermediates, their respective HOMO and LUMO molecular orbitals remained composed of the frontier molecular orbitals of the Ni metal porphyrin ring. The frontier molecular orbital components of the corresponding up-subs or down-subs primarily constituted molecular orbitals lower than HOMO or higher than LUMO.
It was observed that the substituent groups at the meso-position did not significantly participate in the formation of chemical bonds throughout the entire reaction process, but rather acted as steric hindrances.
2.4. Steric Hindrance Analysis
This paper aimed to investigate the impact of steric hindrance from bulky pyramidal group substituents on the active center (NiII) and to understand the mechanism of steric effects on the heterolysis reaction in the formed I-H intermediate. To this end, we employed the Multiwfn software to analyze the geometric structure of the I-H intermediate under a triplet spin state. The resulting structure was analyzed using the visualization software VMD, and the analysis results are presented in Figure 4.
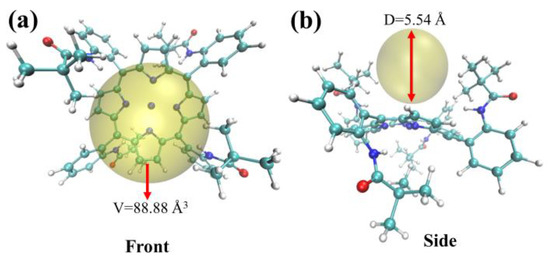
Figure 4.
Steric hindrance analysis of complex I. Note: Yellow sphere shows an unoccupied region inside of complex I. (a) Front (b) Side.
As shown in Figure 4, the yellow sphere represents the unoccupied space in the I-H complex, surrounded by two pivalamido groups. This region had an average diameter of 5.54 Å and a corresponding volume of 88.88 Å3. Protons with diameters of approximately 10−5Å were able to freely enter and exit this region, indicating that they could react with the intermediate (I-H)−. However, (I-H)− was unable to form a bimetallic complex or undergo a homolysis reaction. These findings demonstrate that the size of the substituent directly affects the reaction mechanism.
In conclusion, this study sheds light on the influence of steric hindrance on the heterolysis reaction in the formed I-H intermediate. It provides a reasonable explanation for the observed reaction mechanism.
2.5. Density-of-State Analysis
The density of states (DOS) maps visualize the energy distribution of molecular orbitals (MOs) in a chemical system [43], and the value of the DOS curve reflects the number of MOs at the corresponding energy per unit energy interval. In Figure 5, the total DOS (TDOS) maps for all MOs of I and II and the partial DOS (PDOS) maps contributed by various groups of MOs are plotted. In plotting the DOS and PDOS, the α and β MOs were plotted separately, as both systems of the two complexes used the open-shell layer type. All the α Mos curves are located in the upper half of the graph box (marked by solid lines), and the β MOs curves are in the lower half of the graph box (marked by dotted lines). When the α and β MOs are in perfect symmetry, it means that the electrons in the α and β orbitals are not spin-hybridized in the same energy region, and vice versa, spin polarization occurs.
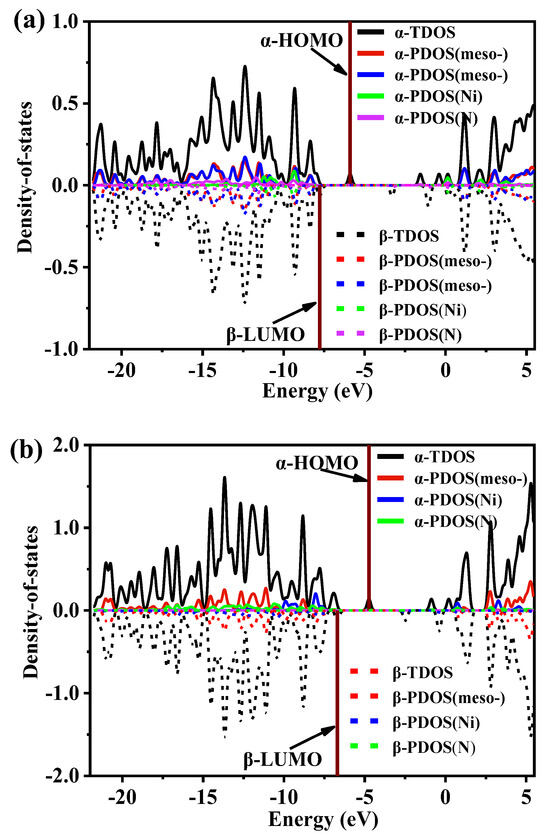
Figure 5.
Density-of-state (DOS) map and MOs degeneracy of the (a) I and (b) II complexes. The location of the HOMO is a wine-colored vertical line.
As can be seen from the curve changes in Figure 5, the TDOS curves for the α and β MOs of each complex were stored in an asymmetric state, mainly due to the different distributions of electrons in the α and β orbitals. This result is significant for the highest energy molecular orbital (highest occupied orbital HOMO). As seen in Figure 5, there was a significant difference in the HOMO energies of the α and β MOs in the I and II complexes, and the HOMO energy in I was lower. To further analyze the effects of various groups in the various complexes on TDOS, the meso-site substituent (abbreviated as meso-), the pyridine N(N) on the porphyrin ring, and the central metal NiII ion were analyzed separately and identified by PDOS. As seen in Figure 5, the meso-site substituents in the individual complexes contributed significantly to the overall orbital energy level. However, differences remained in the I complex due to the presence of two different spatially distributed substituent types, as seen from the PDOS curves in Figure 5a (in red and blue lines). The additional NiII and the four N atoms contributed the most to the molecular front orbitals of HOMO and LUMO. This indicated the location of the catalytically active center.
2.6. Molecules Dynamics Analysis
To investigate the dissociation dynamics of H2 molecules from complexes I-H2 and (II-H)2 and to compare the reaction barrier magnitudes, this study utilized the molecular dynamics (MD) module within CP2K to simulate the dissociation process. The simulation maintained a reaction temperature of 298.15 K, and the MD analysis results are presented in Figure 6. For the I-H2 system, H2 molecules exhibited dissociation from the coordination entity at approximately 150 fs, extending the simulation time to around 500 fs. A notable alteration in the spatial distribution of meso-sited substituents was observed compared to the initial structure. Mainly, amino acid substituents exhibited increased proximity, leading to a gradual contraction of the central region formed by amino acid substituents, albeit without a significant alteration in the overall energy of the complex system.
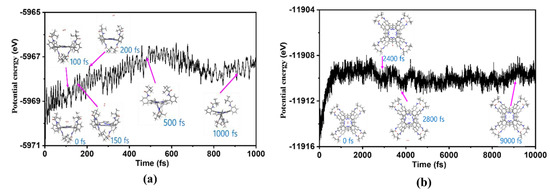
Figure 6.
MD simulations depict the dissociation process of H2 molecules from compounds I-H2 (a) and (II-H)2 (b) at 298.15 K, employing the PBE/GFN1-xTB level of theory. The graph represents the relationship between reaction time and the system’s total energy (electronic energy).
In contrast, under identical simulation conditions for the (II-H)2 system, at 150 fs, H2 molecules remained within the coordination field formed by two NiII ions. Only after extending the simulation time to 2400 fs did H2 molecules dissociate from the complex system. The “sandwich” structure in this system, comprising two complexes II, was found to tightly constrain the spatial configuration, impeding the dissociation of H2 molecules. In Figure 6b, it is evident that, after 240 fs, the total energy of the entire system underwent a relatively modest change. This change implies that, within the initial 2400 femtoseconds, the overall complex system was unstable. However, due to the “sandwich” structure, H2 dissociation was restricted, hindering the anticipated reduction in system energy.
Moreover, upon comparing the energy variations depicted in Figure 6a,b, we observed that the total energy for the I-H2 complex was −5570.6 eV initially and −5568.4 eV at 150 fs; this indicates that the H2 molecule needed to surpass a reaction energy barrier of 2.2 eV to detach from the complex system. Conversely, as shown in Figure 6b, the initial total energy for the (II-H)2 complex was −11,916.5 eV, and after 2400 fs, it increased to −11,909.8 eV. In this scenario, the H2 molecule encountered a higher reaction energy barrier of 6.7 eV for detachment. Notably, the I-H2 system exhibited a more favorable condition for separating H2 molecules, which requires overcoming a lower reaction energy barrier.
3. Computational Methods
This paper details the computational methods employed for optimizing the geometries and conducting single-point calculations for complexes I and II. All calculations were performed using the Gaussian 09 package [44]. Geometry optimization utilized the hybrid functional PBE0 [45] in conjunction with the def2-SVP basis set [46], while single-point calculations employed the def2-TZVP basis set. Chloroform was selected as the solvent for the simulation. To enhance the congruence between calculated results and original data, the simulated NMR spectra were compared with experimental spectra outlined in reference [33] (Figure S3). This comparison aimed to validate the appropriateness of the selected theoretical methods and basis sets during the calculation process, indicating the effective simulation of the transformation process using the current theoretical approach. The optimized structures of I and II obtained at the def2-TZVP level are presented in Figure S2, accompanied by the calculated bond lengths listed in Table 1 and corresponding experimental values. The results from the PBE0/def2-SVP calculations align with the experimental values from reference [33].
To further analyze the geometric structure of steric hindrance in the electron-receiving and protonation processes, as well as the relative electron distribution density changes, the intermediate products were optimized at the def2-SVP level. Their geometric optimization and analysis were carried out using PBE0/def2-SVP. Single-point calculations were conducted at the TZVP level, and the optimized structures of the intermediate products were identified as local minima without imaginary frequencies.
The electronic structure analyses were conducted using the Multiwfn 3.8 (dev) software [47], with isosurface maps of multiple orbitals and real space functions generated using the Visual Molecular Dynamics (VMD) program [48]. The files exported from Multiwfn were employed as an input for VMD to generate the plots.
To further analyze and compare the changes of the meso-substituted groups and binding energies of I and II complexes during the process of forming the intermediate products I-H2 and (II-H)2, where H2 dissociates from the active site of the coordinating metal ion to become an independent H2 compound, density functional theory (DFT) calculations were conducted. The Perdew–Burke–Ernzerh exchange–correlation functional for complexes (PBE) [49,50], along with dispersion correction (DFT-D3), was employed using the Quickstep module of the CP2K program [51,52]. Molecular dynamics studies were conducted for the I-H2 and (II-H)2 complexes. We constructed a three-dimensional periodic framework structure to encapsulate the I-H2 and (II-H)2 complexes. For the framework structure corresponding to the I-H2 complex, a = b = 17.8 Å and c= 15.5 Å, and for the framework structure corresponding to the (II-H)2 complex, a = b = 26.0 Å and c = 15.3 Å. Simulations were performed at 298.15 K using PBE/GFN1-xTB for 1000 fs of I and 10,000 fs of II complexes. The canonical sampling through velocity rescaling (CSVR) ensemble in the NVT (number, volume, temperature) ensemble was employed.
4. Conclusions
In this paper, a density functional theory (DFT) approach was employed on the hybrid functional PBE0 in conjunction with the def2-SVP basis set for the analysis of NiII-porphyrin containing different meso-site substituents. The analysis of NiII-porphyrin and its corresponding hydrides containing different meso-site substituents was carried out on the hybrid functional PBE0 in conjunction with the def2-SVP basis set to understand the role of steric hindrance in the HER reaction mechanism. The structural optimization of the I and II complexes revealed that the four Ni-N bond lengths were equal, but I was slightly shorter than II by 0.01 Å. Upon further binding to the proton, the corresponding Ni-N bond lengths of the two complexes varied more significantly, while the bond lengths of the NiII-H bonds also differed significantly. In addition, the analysis of ADCH showed that the charges of the substituents at opposite positions were also different, which was the main reason for the change in bond length. In addition, the volume of the space surrounded by the two substituents and the porphyrin ring in the I complex was investigated, and it was found that the effective diameter and importance of the area allowed the proton to move freely, but prevented the formation of the bimetallic complex, which led to the heterolysis reaction.
Further analysis by DOS revealed that, in both the I and II complexes, the meso-site substituent contributed to the entire molecular orbital, especially the non-HOMO orbital. Still, NiII and the N atom contributed the most to the HOMO. After GCDA analysis of the I and II complexes and their corresponding intermediates, we found that the HOMO of the porphyrin ring and the alpha or beta molecular orbitals of LUMO were still composed of the frontier molecular orbitals of NiII-porphyrin. The frontier molecular orbital components of the corresponding up-subs or down-subs did not participate in the protonation process and mainly played a steric hindrance role. Furthermore, MD analysis of the I-H2 and (II-H)2 systems formed after protonation was conducted at 298.15 K. The results showed that, for the I-H2 system, H2 molecules separated from complex I at about 150 fs, and the reaction energy barrier of 2.2 eV was overcome. As for the corresponding (II-H)2 system, because of its “sandwich” system, its tight spatial structure limited the separation of H2 molecules to 2400 fs. H2 molecules could be separated from the complex design and overcome the reaction energy barrier of 6.7 electron volts. Complex I was more conducive to the formation of protonation reactions and the separation of H2 molecules. Overall, after accessing the meso-site of the porphyrin ring with different stereoisomerisms and a bulkier substituent, the HER reaction was still able to proceed in the active NiII ion center. However, the reaction mechanism underwent a hemolysis and heterolysis type of distinction.
Supplementary Materials
The following supporting information can be downloaded: https://www.mdpi.com/article/10.3390/molecules29050986/s1. Figure S1: Optimized structure of I, II, I-H2, and (II-H)2; Figure S2: Fragment orbital interaction diagram of porphyrin rings, up-substituents (up-subs), and down-substituents (down-subs). Black solid and red dashed bars correspond to occupied and unoccupied MOs. Figure S3: Simulated Nuclear Magnetic Resonance Hydrogen (HNMR) Spectrum of (a) and (b) complexes I and refer to the intermediates of I−, I-H and (I -H)−; (c) and (d) complexes I and refer to the intermediates of II−, II-H and (II-H)2. Ref. [33] is cited in Supplementary Materials.
Author Contributions
Funding acquisition, A.F.; writing—review and editing, X.L.; conceptualization, Y.Z.; validation, P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 51801001), China Postdoctoral Science Foundation (No. 2016M601878), Provincial Key Research and Development Program of Shaanxi (No. 2019GY-197), Key Project of Baoji University of Arts and Sciences (No. 209040127, No. ZK2018051), Special Scientific Research Project of Shaanxi Education Department (No. 21JK0478, 202310561), and the Natural Science Basic Research Plan in Shanxi Province of China (No. 2015JM5215, 2022JQ-379). Feng AL is supported by The Thousand Talents Plan for Yong Professionals of Shannxi Province and High-level Leading Talents of Scientific and Technological Innovation of Baoji, as well as the Special project of philosophy and Social science of Baoji (BJSKZX-202263).
Data Availability Statement
The raw or processed data necessary to reproduce the findings presented in this study are available upon request. Interested parties may contact the corresponding author for access to the data.
Acknowledgments
We would like to express our gratitude to the Key Laboratory of Materials Physics and Functional Devices in Baoji and the “Sanqin Scholars” Innovation Teams Project of Shaanxi Province (specifically, the Clean Energy Materials and High-Performance Devices Innovation Team of Shaanxi Dongling Smelting Co., Ltd.) for providing a conducive research environment and valuable technical support.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Capurso, T.; Stefanizzi, M.; Torresi, M.; Camporeale, S.M. Perspective of the role of hydrogen in the 21st century energy transition. Energy Convers. Manag. 2022, 251, 114898. [Google Scholar] [CrossRef]
- Faye, O.; Szpunar, J.; Eduok, U. A critical review on the current technologies for the generation, storage, and transportation of hydrogen. Int. J. Hydrogen Energy 2022, 47, 13771–13802. [Google Scholar] [CrossRef]
- Ishaq, H.; Dincer, I.; Crawford, C. A review on hydrogen production and utilization: Challenges and opportunities. Int. J. Hydrogen Energy 2022, 47, 26238–26264. [Google Scholar] [CrossRef]
- Heppe, N.; Gallenkamp, C.; Paul, S.; Segura-Salas, N.; von Rhein, N.; Kaiser, B.; Jaegermann, W.; Jafari, A.; Sergueev, I.; Krewald, V.; et al. Substituent Effects in Iron Porphyrin Catalysts for the Hydrogen Evolution Reaction. Chem. Eur. J. 2023, 29, e202202465. [Google Scholar] [CrossRef]
- Qi, X.W.; Yang, G.; Guo, X.S.; Si, L.P.; Zhang, H.; Liu, H.Y. Electrocatalytic Hydrogen Evolution by Water-Soluble Cobalt (II), Copper (II) and Iron (III) meso-Tetrakis(carboxyl)porphyrin. Eur. J. Inorg. Chem. 2022, 26, e202200613. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Zhang, T.; Zhu, W.; Qin, L.; Kang, S.-Z.; Li, X. Enhanced light absorption and electron transfer in dimensionally matched carbon nitrideporphyrin nanohybrids for photocatalytic hydrogen production. Fuel 2023, 338, e127394. [Google Scholar] [CrossRef]
- Cook, B.J.; Barona, M.; Johnson, S.I.; Raugei, S.; Bullock, R.M. Weakening the N–H Bonds of NH3Ligands: Triple Hydrogen-Atom Abstraction to Form a Chromium(V) Nitride. Inorg. Chem. 2022, 61, 11165–11172. [Google Scholar] [CrossRef] [PubMed]
- Joseph, M.; Haridas, S. Recent progresses in porphyrin assisted hydrogen evolution. Int. J. Hydrogen Energy 2020, 45, 11954–11975. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Li, T.; He, Y.Z.; Han, E.C.; Chen, Y.L.; Jiang, X.Y.; Ni, C.L.; Yang, L.M.; Liu, W. Cobalt-based metalloporphyrins as efficient electro-catalysts for hydrogen evolution from acetic acid and water. Electrocatalysis. 2023, 14, 752–762. [Google Scholar] [CrossRef]
- Zhao, W.; Peng, J.; Wang, W.; Jin, B.; Chen, T.; Liu, S.; Zhao, Q.; Huang, W. Interlayer Hydrogen-Bonded Metal Porphyrin Frameworks/MXene Hybrid Film with High Capacitance for Flexible All-Solid-State Supercapacitors. Small 2019, 15, e1901351. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Han, J.; Li, X.; Liu, G.; Xu, Y.; Peng, Y.; Nie, S.; Li, W.; Li, X.; Chen, Z.; et al. Electrocatalytic hydrogen evolution with a copper porphyrin bearing meso-(o-carborane) substituents. Chem. Commun. 2023, 59, 10777–10780. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, S.; Zhang, Y.; Li, R.; Zhao, B.; Peng, T. Hydrogen-Bond Regulation of the Microenvironment of Ni(II)-Porphyrin Bifunctional Electrocatalysts for Efficient Overall Water Splitting. Adv. Mater. 2023, 35, e2210727. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Li, X.; Lei, H.; Guo, K.; Lv, B.; Guo, H.; Chen, D.; Zhang, W.; Cao, R. Comparing electrocatalytic hydrogen and oxygen evolution activities of first-row transition metal complexes with similar coordination environments. J. Energy Chem. 2021, 63, 659–666. [Google Scholar] [CrossRef]
- Yuasa, M.; Nishihara, R.; Shi, C.; Anson, F.C. Comparison of Several Meso-Tetraalkyl Cobalt Porphyrins as Catalysts for the Electroreduction of Dioxygen. Polym. Adv. Technol. 2001, 12, 266–270. [Google Scholar] [CrossRef]
- Ardakani, M.M.; Rahimi, P.; Dehghani, H.; Karami, P.E.; Zare, H.R.; Karami, S. Electrocatalytic Reduction of Dioxygen on the Surface of Glassy Carbon Electrodes Modified with Cobalt Porphyrin Complexes. Electroanalysis 2007, 19, 2258–2263. [Google Scholar] [CrossRef]
- Qin, H.; Wang, Y.; Wang, B.; Duan, X.; Lei, H.; Zhang, X.; Zheng, H.; Zhang, W.; Cao, R. Cobalt porphyrins supported on carbon nanotubes as model catalysts of metal-N4/C sites for oxygen electrocatalysis. J. Energy Chem. 2021, 53, 77–81. [Google Scholar] [CrossRef]
- Sinha, S.; Aaron, M.S.; Blagojevic, J.; Warren, J.J. Electrocatalytic Dioxygen Reduction by Carbon Electrodes Noncovalently Modified with Iron Porphyrin Complexes: Enhancements from a Single Proton Relay. Chem. —A Eur. J. 2015, 21, 18072–18075. [Google Scholar] [CrossRef]
- Sinha, S.; Ghosh, M.; Warren, J.J. Changing the Selectivity of O2 Reduction Catalysis with One Ligand Heteroatom. ACS Catal. 2019, 9, 2685–2691. [Google Scholar] [CrossRef]
- Su, B.; Hatay, I.; Trojánek, A.; Samec, Z.; Khoury, T.; Gros, C.P.; Barbe, J.-M.; Daina, A.; Carrupt, P.-A.; Girault, H.H. Molecular Electrocatalysis for Oxygen Reduction by Cobalt Porphyrins Adsorbed at Liquid/Liquid Interfaces. J. Am. Chem. Soc. 2010, 132, 2655–2662. [Google Scholar] [CrossRef]
- Shi, F.C.A.C. (5,10,15,20-Tetramethylporphyrinato)cobalt(II): A Remarkably Active Catalyst for the Electroreduction of O2 to H2O. Inorg. Chem. 1998, 37, 1037–1043. [Google Scholar] [CrossRef]
- Lei, H.; Li, X.; Meng, J.; Zheng, H.; Zhang, W.; Cao, R. Structure Effects of Metal Corroles on Energy-Related Small Molecule Activation Reactions. ACS Catal. 2019, 9, 4320–4344. [Google Scholar] [CrossRef]
- Aarabi, M.; Omidyan, R.; Soorkia, S.; Grégoire, G.; Broquier, M.; Crestoni, M.-E.; de la Lande, A.; Soep, B.; Shafizadeh, N. The dramatic effect of N-methylimidazole on trans axial ligand binding to ferric heme: Experiment and theory. Phys. Chem. Chem. Phys. 2019, 21, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Nakatani, N.; Fujii, H.; Hada, M. DFT insight into axial ligand effects on electronic structure and mechanistic reactivity of oxoiron(iv) porphyrin. Phys. Chem. Chem. Phys. 2020, 22, 12173–12179. [Google Scholar] [CrossRef] [PubMed]
- NElgrishi, N.; Kurtz, D.A.; Dempsey, J.L. Reaction Parameters Influencing Cobalt Hydride Formation Kinetics: Implications for Benchmarking H2-Evolution Catalysts. J. Am. Chem. Soc. 2016, 139, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.J.; Gray, H.B.; Winkler, J.R. Hydrogen Generation Catalyzed by Fluorinated Diglyoxime–Iron Complexes at Low Overpotentials. J. Am. Chem. Soc. 2012, 134, 8310–8313. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liang, G.; Reddy, M.R.; Long, M.; Driskill, K.; Lyons, C.; Donnadieu, B.; Bollinger, J.C.; Webster, C.E.; Zhao, X. Electronic and Steric Tuning of Catalytic H2 Evolution by Cobalt Complexes with Pentadentate Polypyridyl-Amine Ligands. J. Am. Chem. Soc. 2018, 140, 9219–9229. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, S.C.; Winkler, J.R.; Gray, H.B. Molecular mechanisms of cobalt-catalyzed hydrogen evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15127–15131. [Google Scholar] [CrossRef]
- Han, Y.; Fang, H.; Jing, H.; Sun, H.; Lei, H.; Lai, W.; Cao, R. Singly versus Doubly Reduced Nickel Porphyrins for Proton Reduction: Experimental and Theoretical Evidence for a Homolytic Hydrogen-Evolution Reaction. Angew. Chem. 2016, 128, 5547–5552. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Chakraborty, S.; Brennessel, W.W.; Chidsey, C.E.D.; Jones, W.D. Rapid oxidative hydrogen evolution from a family of square-planar nickel hydride complexes. Chem. Sci. 2016, 7, 117–127. [Google Scholar] [CrossRef]
- Liberman, I.; Shimoni, R.; Ifraemov, R.; Rozenberg, I.; Singh, C.; Hod, I. Active-Site Modulation in an Fe-Porphyrin-Based Metal–Organic Framework through Ligand Axial Coordination: Accelerating Electrocatalysis and Charge-Transport Kinetics. J. Am. Chem. Soc. 2020, 142, 1933–1940. [Google Scholar] [CrossRef]
- Meng, J.; Lei, H.; Li, X.; Zhang, W.; Cao, R. The Trans Axial Ligand Effect on Oxygen Reduction. Immobilization Method May Weaken Catalyst Design for Electrocatalytic Performance. J. Phys. Chem. C 2020, 124, 16324–16331. [Google Scholar] [CrossRef]
- Samanta, S.; Das, P.K.; Chatterjee, S.; Dey, A. Effect of axial ligands on electronic structure andO2 reduction by iron porphyrin complexes: Towards a quantitative understanding of the “push effect”. J. Porphyrins Phthalocyanines 2015, 19, 92–108. [Google Scholar] [CrossRef]
- Guo, X.; Wang, N.; Li, X.; Zhang, Z.; Zhao, J.; Ren, W.; Ding, S.; Xu, G.; Li, J.; Apfel, U.; et al. Homolytic versus Heterolytic Hydrogen Evolution Reaction Steered by a Steric Effect. Angew. Chem. Int. Ed. 2020, 59, 8941–8946. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lei, H.; Xie, L.; Wang, N.; Zhang, W.; Cao, R. Metalloporphyrins as Catalytic Models for Studying Hydrogen and Oxygen Evolution and Oxygen Reduction Reactions. Accounts Chem. Res. 2022, 55, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lv, X.-L.; Feng, D.; Chen, S.; Sun, J.; Song, L.; Xie, Y.; Li, J.-R.; Zhou, H.-C. Pyrazolate-Based Porphyrinic Metal-Organic Framework with Extraordinary Base-Resistance. J. Am. Chem. Soc. 2016, 138, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Atomic Dipole Moment Corrected Hirshfeld Population Method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Xia-Yu, Z.; Chun-Ying, R.; Tian, L.U.; Shu-Bin, L.I.U. Hirshfeld Charge as a Quantitative Measure of Electrophilicity and Nucleophilicity: Nitrogen-Containing Systems. Acta Phys.-Chim. Sin. 2014, 30, 2055–2062. [Google Scholar] [CrossRef]
- Chakravorty, E.R.D.A.S. A test of the Hirshfeld definition of atomic charges and moments. Theor. Chim. Acta 1992, 83, 319–330. [Google Scholar]
- Wiberg, K.B.; Rablen, P.R. Comparison of Atomic Charges Derived via Different Procedures. J. Comput. Chem. 1993, 14, 1504–1518. [Google Scholar] [CrossRef]
- Tian, L.; Fei-Wu, C. Comparison of Computational Methods for Atomic Charges. Acta Phys. Chim. Sin. 2012, 28, 1–18. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of Donor- Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Xiao, M.; Lu, T. Generalized Charge Decomposition Analysis (GCDA) Method. J. Adv. Phys. Chem. 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Bonding character, electron delocalization, and aromaticity. Carbon 2020, 165, 468–475. [Google Scholar] [CrossRef]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, versions D.01.; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, A.D.W.; Klaus Schulten, V.M.D. Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 13640. [Google Scholar] [CrossRef]
- Fernández, E.M.; Balbás, L.C. GGA versus van der Waals density functional results for mixed gold/mercury molecules and pure Au and Hg cluster properties. Phys. Chem. Chem. Phys. 2011, 13, 20863–20870. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2013, 4, 15–25. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).