
Safety-Catch Linkers for Solid-Phase Peptide Synthesis

Abstract
:
1. Introduction
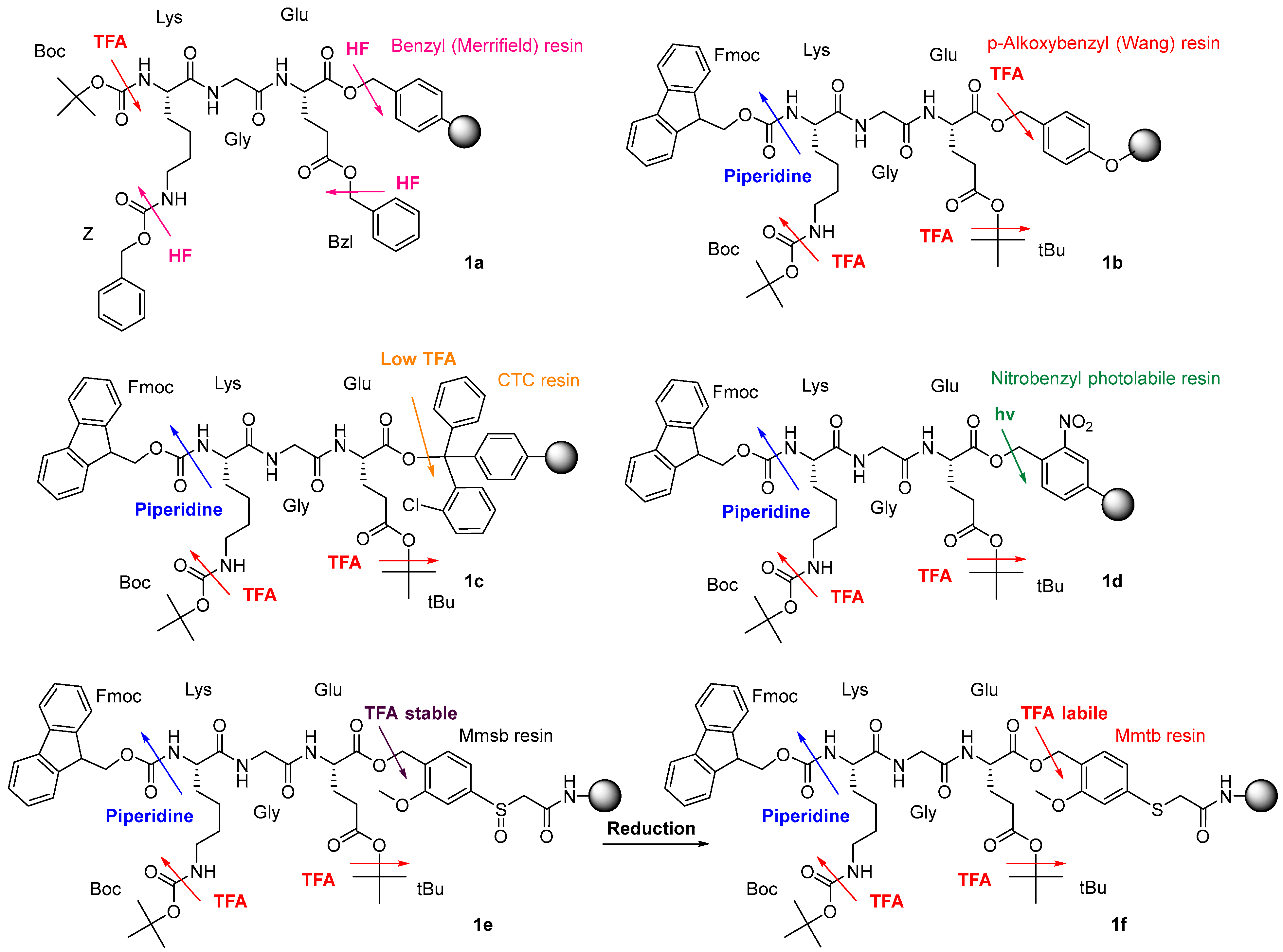
1.1. Safety-Catch Linkers
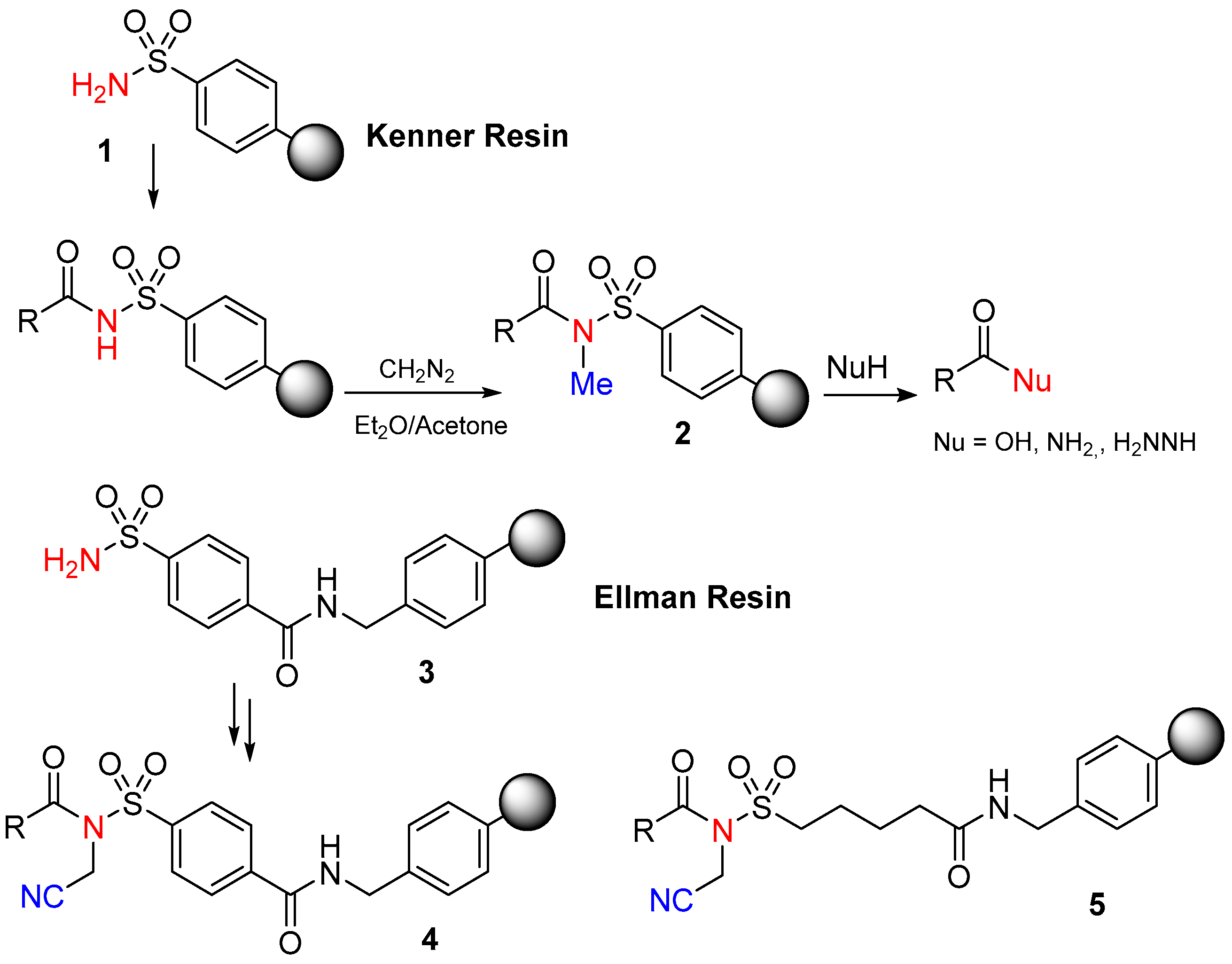
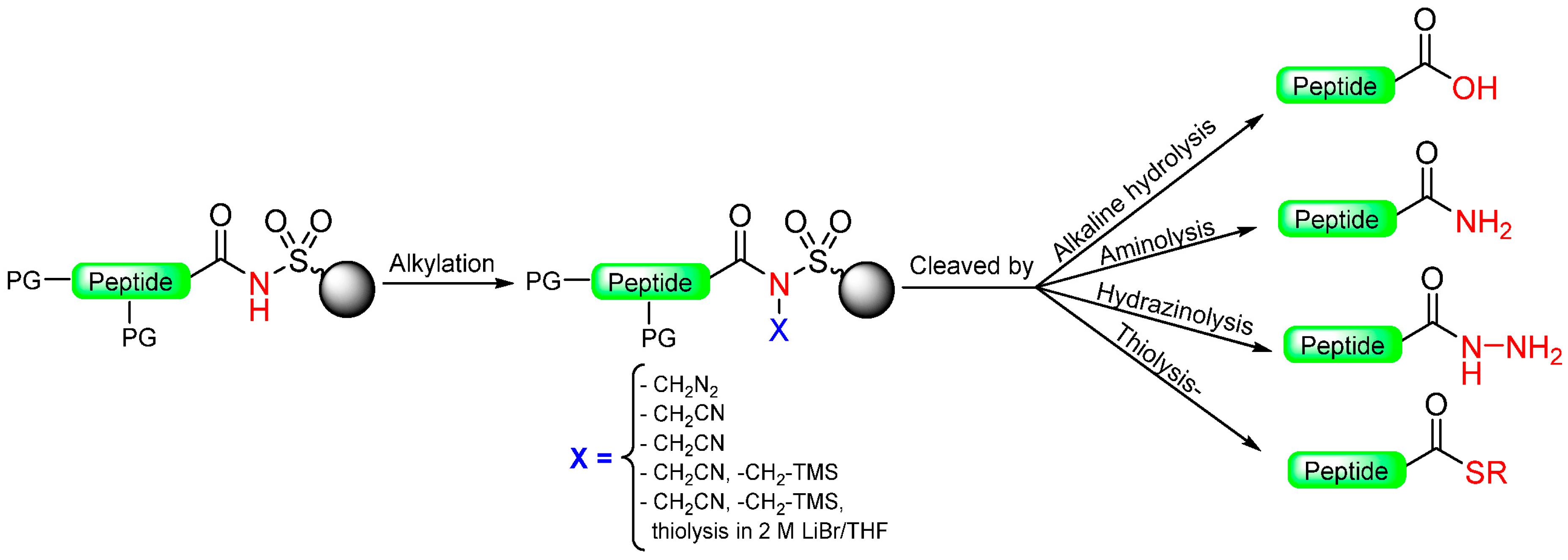
1.2. Kenner and Sulfonamide Safety-Catch Linker
1.3. Isonitrile/Benzamide Safety-Catch Linker
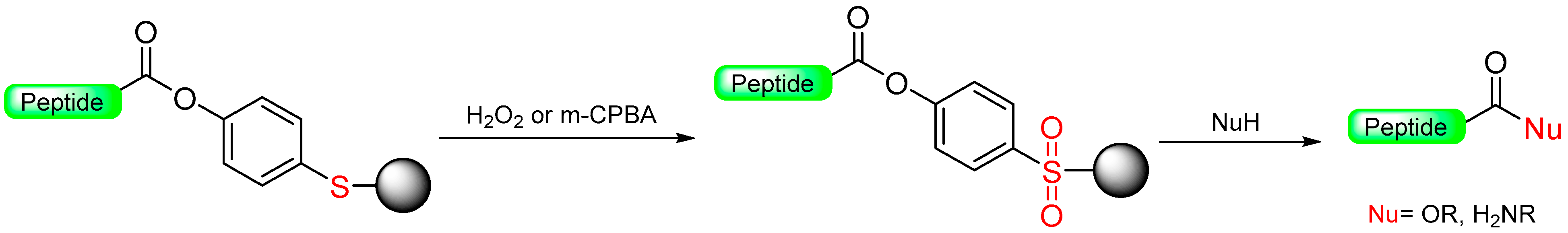
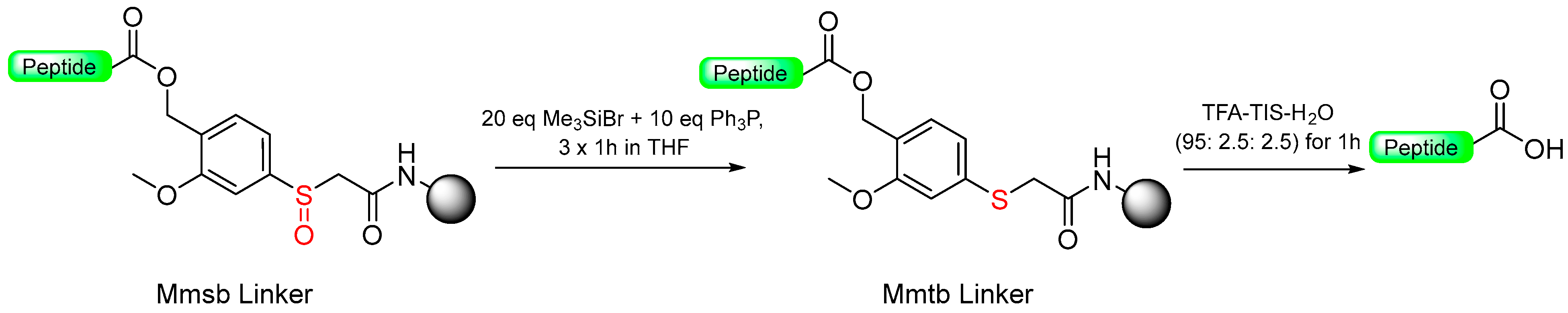
1.4. The Oxidative Safety-Catch Linkers
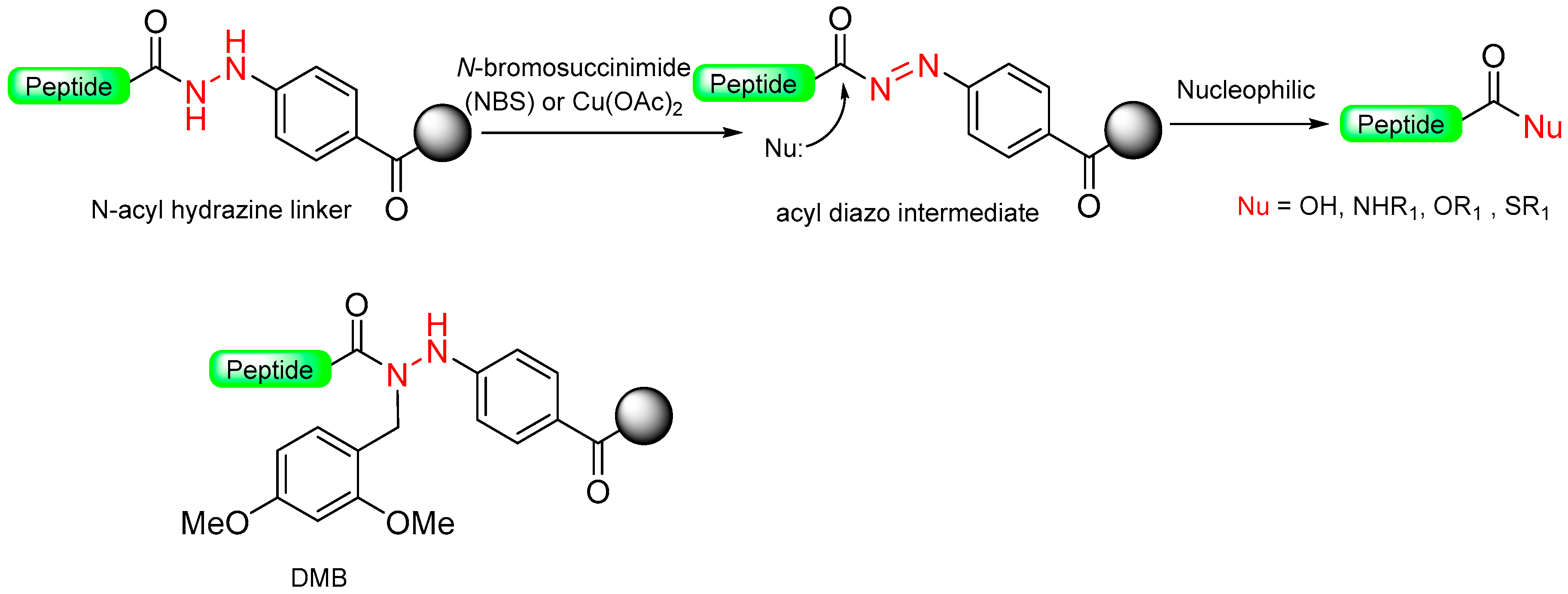
1.5. Aryl Hydrazine
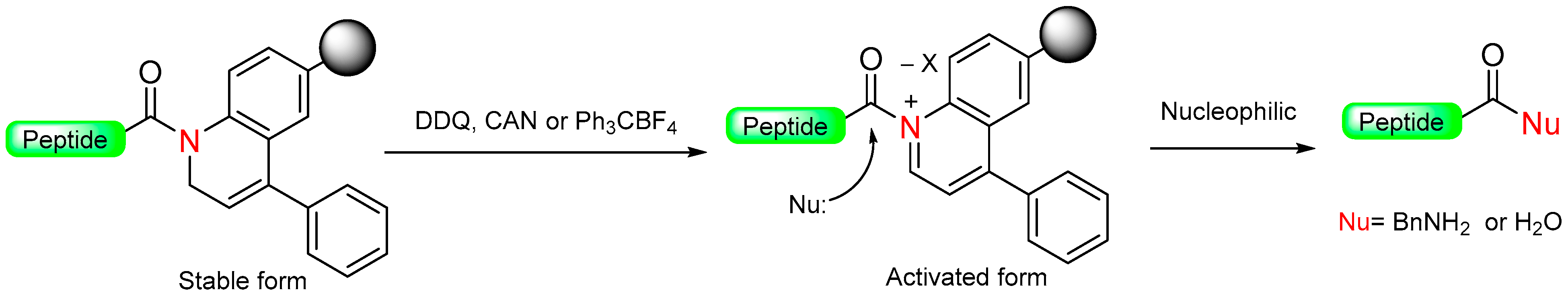
1.6. Dihydroquinoline (DHQ)
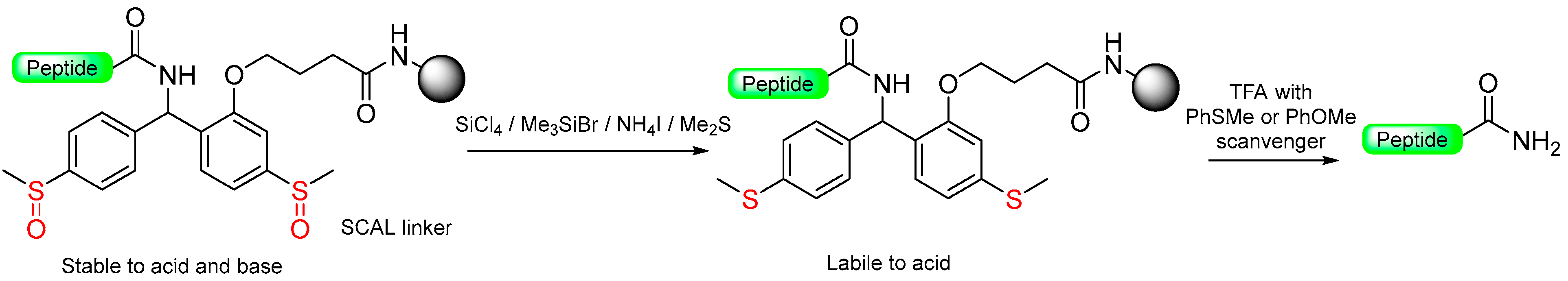
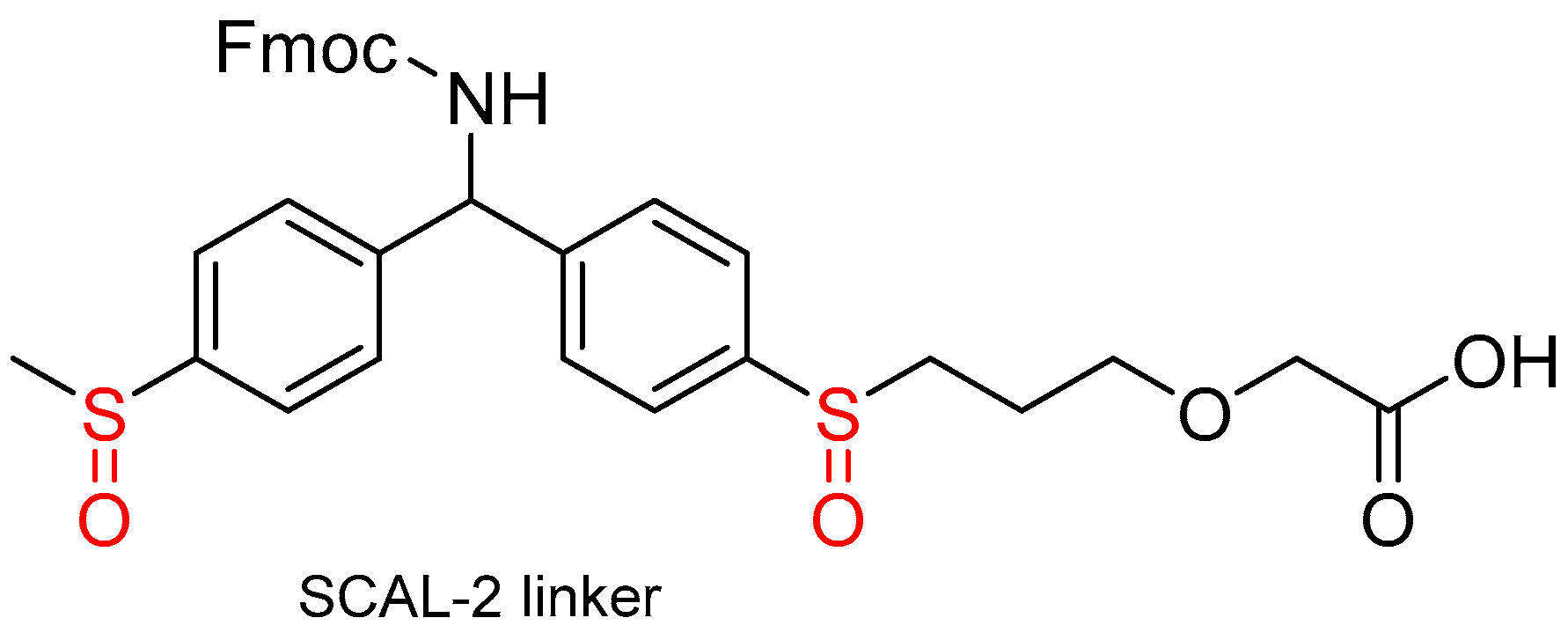
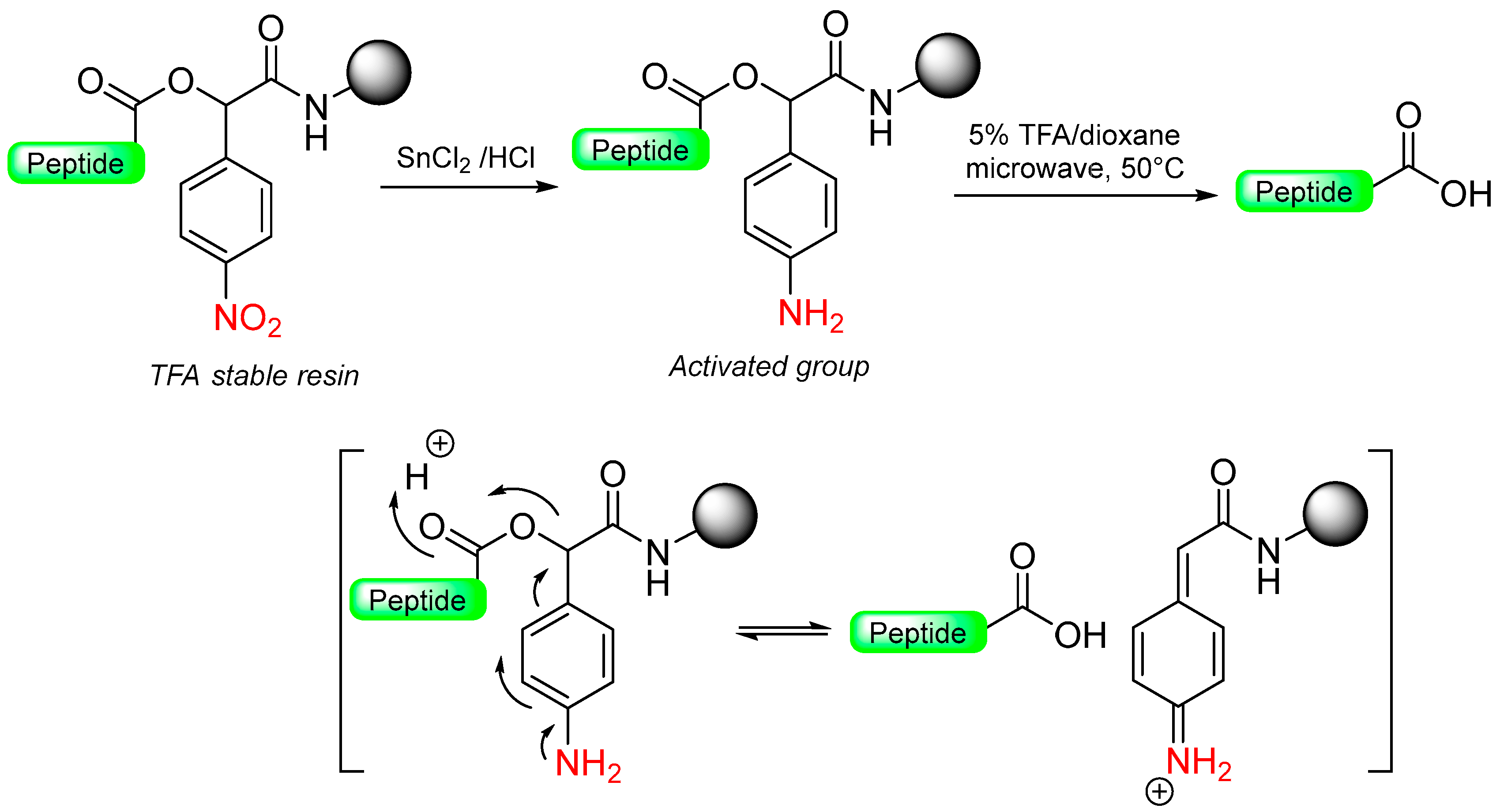
1.7. The Reductive-Acidolytic Safety-Catch Linkers
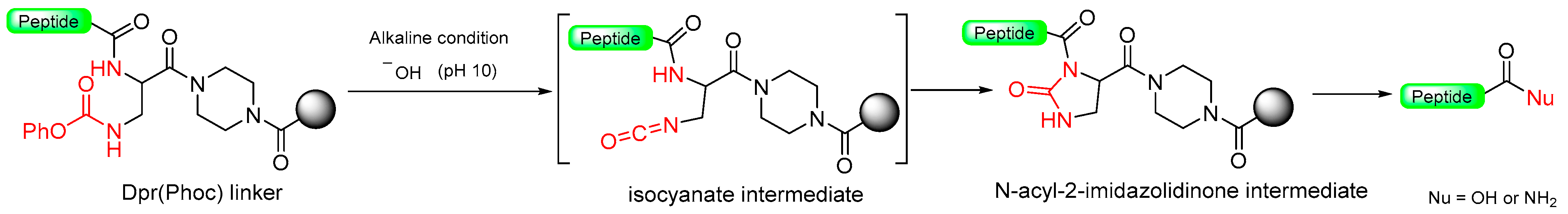
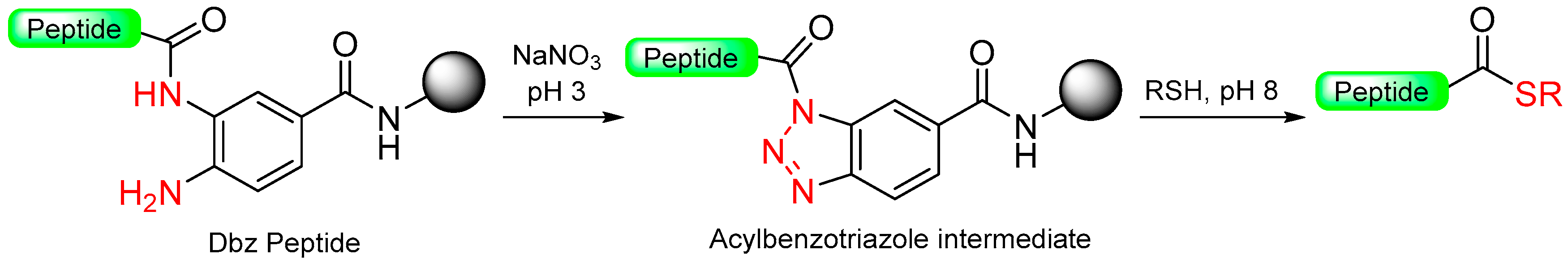
1.8. Heterocycle Formation—Via Intramolecular Cyclization—As Leaving Group
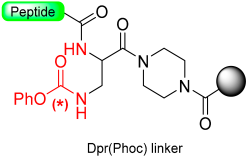
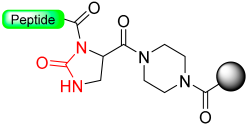
1.9. Dpr(Phoc) Linker–Imidazolidinone (Cyclic Urea)
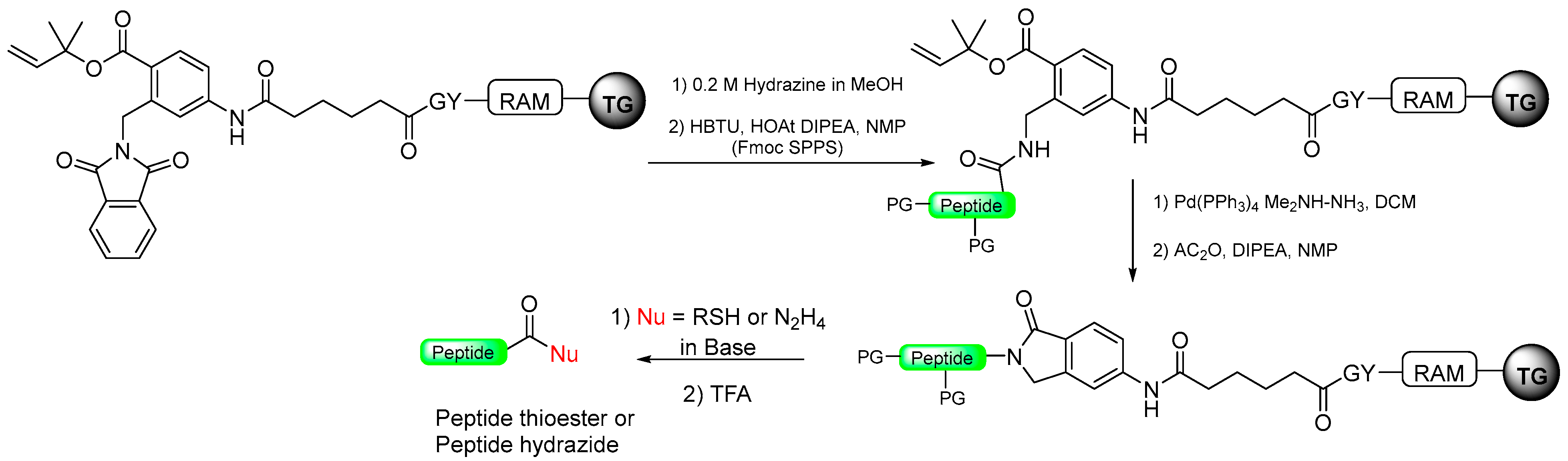
1.10. Diamino Benzoic Acid (Dbz)–Benzimidazolinone (Cyclic Urea)
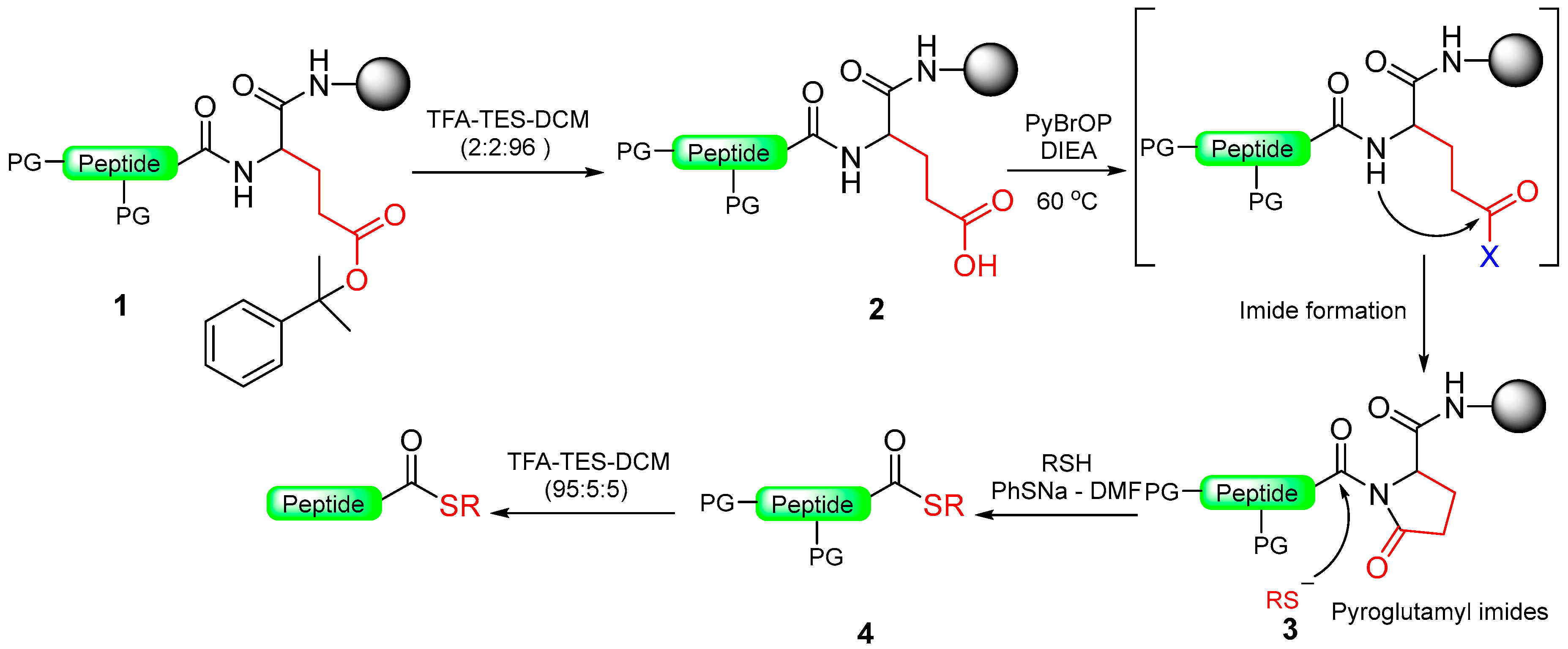
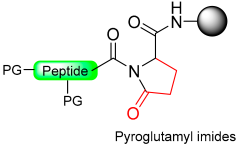
1.11. Pyroglutamyl/Pyrrolidinone Amide
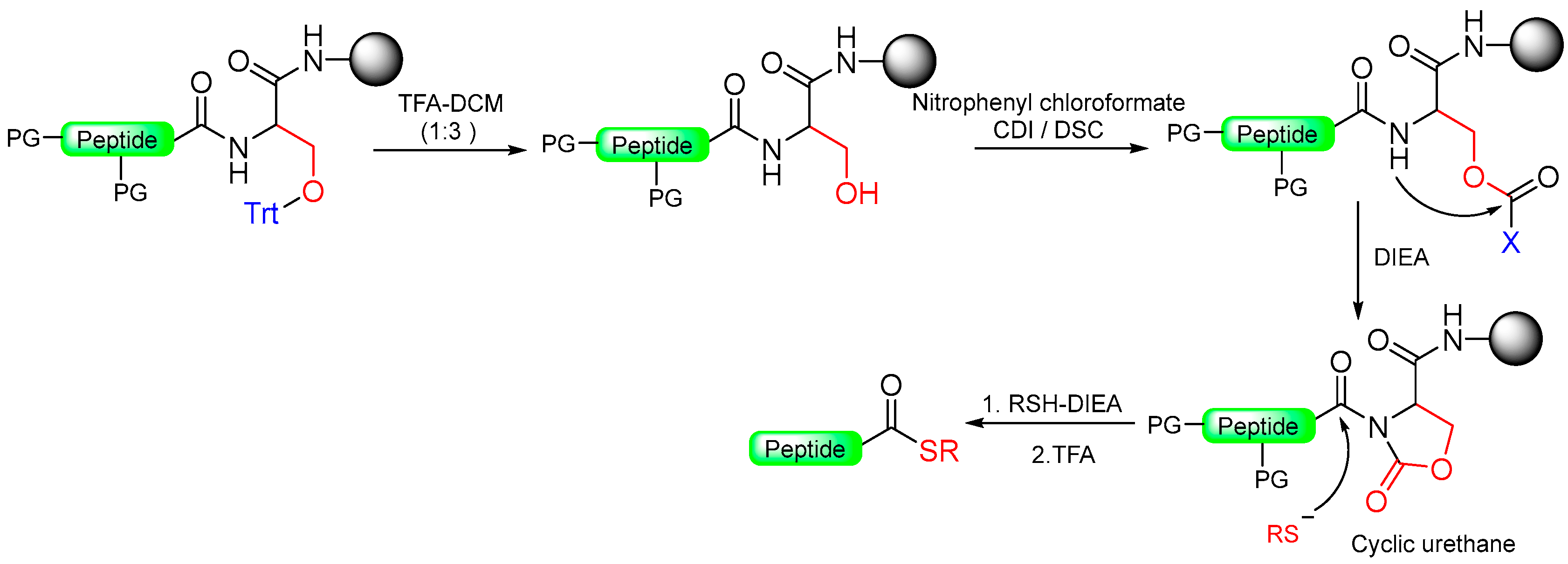
1.12. Cyclic Urethane Moiety
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Lloyd-Williams, P.; Albericio, F.; Giralt, E. Chemical Approaches to the Synthesis of Peptides and Proteins; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Nefkens, G.H.L.; Tesser, G.I. A novel activated ester in peptide syntheses. J. Am. Chem. Soc. 1961, 83, 1263. [Google Scholar] [CrossRef]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.B.H. Total chemical synthesis of proteins. Chem. Soc. Rev. 2009, 38, 338–351. [Google Scholar] [CrossRef]
- Bruckdorfer, T.; Marder, O.; Albericio, F. From Production of Peptides in Milligram Amounts for Research to Multi-Tons Quantities for Drugs of the Future. Curr. Pharm. Biotechnol. 2004, 5, 29–43. [Google Scholar] [CrossRef]
- De la Torre, B.G.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef]
- Albericio, F. Orthogonal protecting groups for Nα-amino and C-terminal carboxyl functions in solid-phase peptide synthesis. Pept. Sci. 2000, 55, 123–139. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Synthesis (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1985, 24, 799–810. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef]
- Barany, G.; Albericio, F. Three-dimensional orthogonal protection scheme for solid-phase peptide synthesis under mild conditions. J. Am. Chem. Soc. 1985, 107, 4936–4942. [Google Scholar] [CrossRef]
- Nandhini, K.P.; Albericio, F.; de la Torre, B.G. 2-Methoxy-4-methylsulfinylbenzyl Alcohol as a Safety-Catch Linker for the Fmoc/tBu Solid-Phase Peptide Synthesis Strategy. J. Org. Chem. 2022, 87, 9433–9442. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, S.; Patek, M. Safety-Catch Linker Units. In Linker Strategies in Solid-Phase Organic Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 195–220. [Google Scholar]
- Sebastian Wiehn, M.; Jung, N.; Bräse, S. Safety-Catch and Traceless Linkers in Solid Phase Organic Synthesis. In The Power of Functional Resins in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 437–465. [Google Scholar]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoët, M.; Monbaliu, J.-C.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef]
- Kenner, G.W.; McDermott, J.R.; Sheppard, R.C. The safety catch principle in solid phase peptide synthesis. J. Chem. Soc. D 1971, 12, 636–637. [Google Scholar] [CrossRef]
- Backes, B.J.; Ellman, J.A. Carbon-Carbon Bond-Forming Methods on Solid Support. Utilization of Kenner’s Safety-Catch” Linker. J. Am. Chem. Soc. 1994, 116, 11171–11172. [Google Scholar] [CrossRef]
- Backes, B.J.; Virgilio, A.A.; Ellman, J.A. Activation Method to Prepare a Highly Reactive Acylsulfonamide “Safety-Catch” Linker for Solid-Phase Synthesis1. J. Am. Chem. Soc. 1996, 118, 3055–3056. [Google Scholar] [CrossRef]
- Suzuki, A. Organoboron compounds in new synthetic reactions. Pure Appl. Chem. 1985, 57, 1749–1758. [Google Scholar] [CrossRef]
- Backes, B.J.; Ellman, J.A. An Alkanesulfonamide “Safety-Catch” Linker for Solid-Phase Synthesis. J. Org. Chem. 1999, 64, 2322–2330. [Google Scholar] [CrossRef]
- He, Y.; Wilkins, J.P.; Kiessling, L.L. N-Acylsulfonamide Linker Activation by Pd-Catalyzed Allylation. Org. Lett. 2006, 8, 2483–2485. [Google Scholar] [CrossRef] [PubMed]
- Sewald, P.D.N. Peptide Synthesis. In Peptides: Chemistry and Biology; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 175–315. [Google Scholar]
- Ugi, I. The α-Addition of Immonium Ions and Anions to Isonitriles Accompanied by Secondary Reactions. Angew. Chem. Int. Ed. Engl. 1962, 1, 8–21. [Google Scholar] [CrossRef]
- Hulme, C.; Peng, J.; Morton, G.; Salvino, J.M.; Herpin, T.; Labaudiniere, R. Novel safety-catch linker and its application with a Ugi/De-BOC/Cyclization (UDC) strategy to access carboxylic acids, 1,4-benzodiazepines, diketopiperazines, ketopiperazines and dihydroquinoxalinones. Tetrahedron Lett. 1998, 39, 7227–7230. [Google Scholar] [CrossRef]
- Marshall, D.L.; Liener, I.E. Modified support for solid-phase peptide synthesis which permits the synthesis of protected peptide fragments. J. Org. Chem. 1970, 35, 867–868. [Google Scholar] [CrossRef]
- Flanigan, E.; Marshall, G.R. Synthesis of cyclic peptides on dual function supports. Tetrahedron Lett. 1970, 11, 2403–2406. [Google Scholar] [CrossRef]
- Fantauzzi, P.P.; Yager, K.M. Synthesis of diverse tetrahydro-β-carboline-3-carboxamides and -2,3-bis-lactams on a versatile 4-hydroxythiophenol-linked solid support. Tetrahedron Lett. 1998, 39, 1291–1294. [Google Scholar] [CrossRef]
- Guy Breitenbucher, J.; Johnson, C.R.; Haight, M.; Christopher Phelan, J. Generation of a piperazine-2-carboxamide library: A practical application of the phenol-sulfide react and release linker. Tetrahedron Lett. 1998, 39, 1295–1298. [Google Scholar] [CrossRef]
- Pátek, M.; Lebl, M. Safety-catch and multiply cleavable linkers in solid-phase synthesis. Pept. Sci. 1998, 47, 353–363. [Google Scholar] [CrossRef]
- Noki, S.; Saneii, H.; de la Torre, B.G.; Albericio, F. Safety-Catch Linkers for Solid Phase Peptide Synthesis. 144281-2, 12 December 2023. [Google Scholar]
- Schwyzer, R.; Felder, E.; Merli-Failli, P. The CAMET and CASET links for the synthesis of protected oligopeptides and oligodeoxynucleotides on solid and soluble supports. Helv. Chim. Acta 1984, 67, 1316–1327. [Google Scholar] [CrossRef]
- García-Echeverría, C. A base labile handle for solid phase organic chemistry. Tetrahedron Lett. 1997, 38, 8933–8934. [Google Scholar] [CrossRef]
- Wade, W.S.; Yang, F.; Sowin, T.J. Application of Base Cleavable Safety Catch Linkers to Solid Phase Library Production. J. Comb. Chem. 2000, 2, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Wieland, T.; Lewalter, J.; Birr, C. Subsequent activation of carboxylic derivatives by oxidation or elimination of water; application for cyclizing peptides. Justus Liebigs Ann. Chem. 1970, 740, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Wolman, Y.; Gallop, P.M.; Patchornik, A. Peptide synthesis via oxidation of hydrazides. J. Am. Chem. Soc. 1961, 83, 1263–1264. [Google Scholar] [CrossRef]
- Berst, F.; Holmes, A.B.; Ladlow, M.; Murray, P.J. A latent aryl hydrazine ‘safety-catch’ linker compatible with N-alkylation. Tetrahedron Lett. 2000, 41, 6649–6653. [Google Scholar] [CrossRef]
- Berst, F.; Holmes, A.B.; Ladlow, M. The development and preparation of the 2,4-dimethoxybenzyl arylhydrazine (DMBAH) “latent” safety-catch linker: Solid phase synthesis of ketopiperazines. Org. Biomol. Chem. 2003, 1, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Canosa, J.B.; Nardone, B.; Albericio, F.; Dawson, P.E. Chemical Protein Synthesis Using a Second-Generation N-Acylurea Linker for the Preparation of Peptide-Thioester Precursors. J. Am. Chem. Soc. 2015, 137, 7197–7209. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef]
- Millington, C.R.; Quarrell, R.; Lowe, G. Aryl hydrazides as linkers for solid phase synthesis which are cleavable under mild oxidative conditions. Tetrahedron Lett. 1998, 39, 7201–7204. [Google Scholar] [CrossRef]
- Camarero, J.A.; Adeva, A.; Muir, T.W. 3-Thiopropionic acid as a highly versatile multidetachable thioester resin linker. Lett. Pept. Sci. 2000, 7, 17–21. [Google Scholar] [CrossRef]
- DeGrado, W.F.; Kaiser, E.T. Polymer-bound oxime esters as supports for solid-phase peptide synthesis. The preparation of protected peptide fragments. J. Org. Chem. 1980, 45, 1295–1300. [Google Scholar] [CrossRef]
- Peters, C.; Waldmann, H. Solid-Phase Synthesis of Peptide Esters Employing the Hydrazide Linker. J. Org. Chem. 2003, 68, 6053–6055. [Google Scholar] [CrossRef]
- Camarero, J.A.; Hackel, B.J.; de Yoreo, J.J.; Mitchell, A.R. Fmoc-Based Synthesis of Peptide α-Thioesters Using an Aryl Hydrazine Support. J. Org. Chem. 2004, 69, 4145–4151. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Welsh, K.; Mitchell, A.R.; Camarero, J.A. Preparation of Peptide p-Nitroanilides Using an Aryl Hydrazine Resin. Org. Lett. 2004, 6, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.-H.; Mitchell, A.R.; Camarero, J.A. The Use of Aryl Hydrazide Linkers for the Solid Phase Synthesis of Chemically Modified Peptides. Int. J. Pept. Res. Ther. 2007, 13, 181–190. [Google Scholar] [CrossRef]
- Arseniyadis, S.; Wagner, A.; Mioskowski, C. Resin-bound 4-phenyl-1,2-dihydroquinoline (DHQ): A new safety-catch linker for solid-phase organic synthesis (SPOS). Tetrahedron Lett. 2004, 45, 2251–2253. [Google Scholar] [CrossRef]
- Tanikaga, R.; Nakayama, K.; Tanaka, K.; Kaji, A.J.C.L. Rapid reaction between sulfonium ion and sulfide. Preparative reduction of sulfoxide to sulfide. Chem. Lett. 1977, 6, 395–396. [Google Scholar] [CrossRef]
- Drabowicz, J.; MikoŁAjczyk, M. A Rapid and Mild Reduction of Sulphoxides with Titanium(II) Chloride. Synthesis 1978, 1978, 138–139. [Google Scholar] [CrossRef]
- Soysa, H.S.D.; Weber, W.P. Facile reduction of sulfoxides by disilthianes. Tetrahedron Lett. 1978, 19, 235–238. [Google Scholar] [CrossRef]
- Numata, T.; Togo, H.; Oae, S. Facile reduction of sulfoxides and sulfimides with thiol/trimethylsilyl chloride system. J. Chem. Lett. 1979, 8, 329–332. [Google Scholar] [CrossRef]
- Samanen, J.M.; Brandeis, E. The p-(methylsulfinyl)benzyl group: A trifluoroacetic acid (TFA)-stable carboxyl-protecting group readily convertible to a TFA-labile group. J. Org. Chem. 1988, 53, 561–569. [Google Scholar] [CrossRef]
- Kiso, Y.; Tanaka, S.; Kimura, T.; Itoh, H.; Akaji, K. New hydroxyl protecting groups of safety-catch removal by reductive acidolysis. Chem. Pharm. Bull. 1991, 39, 3097–3099. [Google Scholar] [CrossRef]
- Beck, W.; Jung, G. Convenient reduction of S-oxides in synthetic peptides, lipopeptides and peptide libraries. Lett. Pept. Sci. 1994, 1, 31–37. [Google Scholar] [CrossRef]
- Erlandsson, M.; Undén, A. 3-(4-Hydroxymethylphenylsulfanyl)propanoic acid (HMPPA) as a new safety catch linker in solid phase peptide synthesis. Tetrahedron Lett. 2006, 47, 5829–5832. [Google Scholar] [CrossRef]
- Kiso, Y.; Fukui, T.; Tanaka, S.; Kimura, T.; Akaji, K. A new reductive acidolysis final deprotection strategy in solid phase peptide synthesis. Use of a new safety-catch linker. Tetrahedron Lett. 1994, 35, 3571–3574. [Google Scholar] [CrossRef]
- Thennarasu, S.; Liu, C.-F. A new safety-catch protecting group and linker for solid-phase synthesis. Tetrahedron Lett. 2010, 51, 3218–3220. [Google Scholar] [CrossRef]
- Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. 2-Methoxy-4-methylsulfinylbenzyl: A Backbone Amide Safety-Catch Protecting Group for the Synthesis and Purification of Difficult Peptide Sequences. Chem. Eur. J. 2014, 20, 15031–15039. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Fukui, T.; Tanaka, S.; Akaji, K.; Kiso, Y. A Reductive Acidolysis Final Deprotection Strategy in Solid Phase Peptide Synthesis Based on Safety-Catch Protection. Chem. Pharm. Bull. 1997, 45, 18–26. [Google Scholar] [CrossRef]
- Fujii, N.; Otaka, A.; Sugiyama, N.; Hatano, M.; Yajima, H. Studies on Peptides. CLV. Evaluation of Trimethylsilyl Bromide as a Hard-Acid Deprotecting Reagent in Peptide Synthesis. Chem. Pharm. Bull. 1987, 35, 3880–3883. [Google Scholar] [CrossRef] [PubMed]
- Pátek, M.; Lebl, M. Safety-catch anchoring linkage for synthesis of peptide amides by Boc/Fmoc strategy. Tetrahedron Lett. 1991, 32, 3891–3894. [Google Scholar] [CrossRef]
- Brust, A.; Tickle, A.E. High-throughput synthesis of conopeptides: A safety-catch linker approach enabling disulfide formation in 96-well format. J. Pept. Sci. 2007, 13, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Noki, S.; Brasil, E.; Zhang, H.; Bruckdorfer, T.; de la Torre, B.G.; Albericio, F. Solid-Phase Peptide Synthesis Using a Four-Dimensional (Safety-Catch) Protecting Group Scheme. J. Org. Chem. 2022, 87, 9443–9453. [Google Scholar] [CrossRef] [PubMed]
- Lebl, M.; Patek, M.; Kocis, P.; Krchnak, V.; Hruby, V.J.; Salmon, S.E.; Lam, K. Multiple release of equimolar amounts of peptides from a polymeric carrier using orthogonal linkage-cleavage chemistry. Int. J. Pept. Protein Res. 1993, 41, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Patek, M. Multistep deprotection for peptide chemistry. Int. J. Pept. Protein Res. 1993, 42, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Patek, M.; Lebl, M. “Safety-Catch” Anchoring Linkages and Protecting Groups in Solid-Phase Peptide Synthesis; Peptides-American Symposium; Escom Science Publishers: Tucson, AZ, USA, 1994; p. 146. [Google Scholar]
- Portal, C.; Hintersteiner, M.; Barbeau, O.; Dodd, P.; Huggett, M.; Pérez-Pi, I.; Evans, D.; Auer, M. Facile Synthesis of a Next Generation Safety-Catch Acid-Labile Linker, SCAL-2, Suitable for Solid-Phase Synthesis, On-Support Display and for Post-Synthesis Tagging. ChemistrySelect 2017, 2, 6658–6662. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Burger, K.; Spengler, J.; Albericio, F. p-Nitromandelic Acid as a Highly Acid-Stable Safety-Catch Linker for Solid-Phase Synthesis of Peptide and Depsipeptide Acids. Org. Lett. 2007, 9, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Pascal, R.; Chauvey, D.; Sola, R. Carboxyl-protecting groups convertible into activating groups. Carbamates of o-aminoanilides are precursors of reactive N-acylureas. Tetrahedron Lett. 1994, 35, 6291–6294. [Google Scholar] [CrossRef]
- Sola, R.; Saguer, P.; David, M.-L.; Pascal, R. A new type of safety-catch linker cleaved by intramolecular activation of an amide anchorage and allowing aqueous processing in solid-phase peptide synthesis. J. Chem. Soc. Chem. Commun. 1993, 23, 1786–1788. [Google Scholar] [CrossRef]
- Sola, R.; Méry, J.; Pascal, R. Fmoc-based solid-phase peptide synthesis using dpr(phoc) linker. Synthesis of a C-terminal proline peptide. Tetrahedron Lett. 1996, 37, 9195–9198. [Google Scholar] [CrossRef]
- Pascal, R.; Sola, R. Calcium-promoted hydrolysis of N-acylureas allows mild release of peptides anchored with the Dpr(Phoc) linker to hydrophilic resins. Tetrahedron Lett. 1997, 38, 4549–4552. [Google Scholar] [CrossRef]
- Mannuthodikayil, J.; Singh, S.; Biswas, A.; Kar, A.; Tabassum, W.; Vydyam, P.; Bhattacharyya, M.K.; Mandal, K. Benzimidazolinone-Free Peptide o-Aminoanilides for Chemical Protein Synthesis. Org. Lett. 2019, 21, 9040–9044. [Google Scholar] [CrossRef]
- Zacharie, B.; Sauvé, G.; Penney, C. Thioacylating agents. Use of thiobenzimidazolone derivatives for the preparation of thiotuftsin analogs. Tetrahedron 1993, 49, 10489–10500. [Google Scholar] [CrossRef]
- Shalaby, M.A.; Grote, C.W.; Rapoport, H. Thiopeptide synthesis. {alpha}-amino thionoacid derivatives of nitrobenzotriazole as thioacylating agents. J. Org. Chem. 1996, 61, 9045–9048. [Google Scholar] [CrossRef]
- Blanco-Canosa, J.B.; Dawson, P.E. An Efficient Fmoc-SPPS Approach for the Generation of Thioester Peptide Precursors for Use in Native Chemical Ligation. Angew. Chem. Int. Ed. 2008, 47, 6851–6855. [Google Scholar] [CrossRef]
- Mahto, S.K.; Howard, C.J.; Shimko, J.C.; Ottesen, J.J. A Reversible Protection Strategy To Improve Fmoc-SPPS of Peptide Thioesters by the N-Acylurea Approach. ChemBioChem 2011, 12, 2386. [Google Scholar] [CrossRef] [PubMed]
- Acosta, G.A.; Royo, M.; de la Torre, B.G.; Albericio, F. Facile solid-phase synthesis of head-side chain cyclothiodepsipeptides through a cyclative cleavage from MeDbz-resin. Tetrahedron Lett. 2017, 58, 2788–2791. [Google Scholar] [CrossRef]
- Abdel Monaim, S.A.H.; Acosta, G.A.; Royo, M.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Solid-phase synthesis of homodetic cyclic peptides from Fmoc-MeDbz-resin. Tetrahedron Lett. 2018, 59, 1779–1782. [Google Scholar] [CrossRef]
- Acosta, G.A.; Murray, L.; Royo, M.; de la Torre, B.G.; Albericio, F. Solid-Phase Synthesis of Head to Side-Chain Tyr-Cyclodepsipeptides Through a Cyclative Cleavage From Fmoc-MeDbz/MeNbz-resins. Front. Chem. 2020, 8, 298. [Google Scholar] [CrossRef]
- Arbour, C.A.; Stamatin, R.E.; Stockdill, J.L. Sequence Diversification by Divergent C-Terminal Elongation of Peptides. J. Org. Chem. 2018, 83, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Arbour, C.A.; Belavek, K.J.; Tariq, R.; Mukherjee, S.; Tom, J.K.; Isidro-Llobet, A.; Kopach, M.E.; Stockdill, J.L. Bringing Macrolactamization Full Circle: Self-Cleaving Head-to-Tail Macrocyclization of Unprotected Peptides via Mild N-Acyl Urea Activation. J. Org. Chem. 2019, 84, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, S.; Uemura, T.; Mochizuki, M.; Nishio, H.; Yoshiya, T. Preparation of Peptide o-Aminoanilides Using a Modified Dawson’s Linker for Microwave-Assisted Peptide Synthesis. Synlett 2017, 28, 1956–1960. [Google Scholar] [CrossRef]
- Bondalapati, S.; Eid, E.; Mali, S.M.; Wolberger, C.; Brik, A. Total chemical synthesis of SUMO-2-Lys63-linked diubiquitin hybrid chains assisted by removable solubilizing tags. Chem. Sci 2017, 8, 4027–4034. [Google Scholar] [CrossRef]
- Weidmann, J.; Dimitrijević, E.; Hoheisel, J.D.; Dawson, P.E. Boc-SPPS: Compatible Linker for the Synthesis of Peptide o-Aminoanilides. Org. Lett. 2016, 18, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-X.; Fang, G.-M.; He, Y.; Qu, D.-L.; Yu, M.; Hong, Z.-Y.; Liu, L. Peptide o-Aminoanilides as Crypto-Thioesters for Protein Chemical Synthesis. Angew. Chem. Int. Ed. 2015, 54, 2194–2198. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, A.; Chen, H.-T.; Ya-Ting Huang, A.; Kao, C.-L. Expedient on-resin modification of a peptide C-terminus through a benzotriazole linker. Chem. Sci 2018, 9, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, J.; Schnölzer, M.; Dawson, P.E.; Hoheisel, J.D. Copying Life: Synthesis of an Enzymatically Active Mirror-Image DNA-Ligase Made of D-Amino Acids. Cell Chem. Biol. 2019, 26, 645–651.e3. [Google Scholar] [CrossRef]
- Tofteng, A.P.; Sørensen, K.K.; Conde-Frieboes, K.W.; Hoeg-Jensen, T.; Jensen, K.J. Fmoc solid-phase synthesis of C-terminal peptide thioesters by formation of a backbone pyroglutamyl imide moiety. Angew. Chem. 2009, 121, 7547–7550. [Google Scholar] [CrossRef]
- Paprocki, M.P.; Rasmussen, J.E.; Sørensen, K.K.; Jensen, K.J. Safety-catch linkers for Fmoc solid-phase synthesis of peptide thioesters and hydrazides by amide-to-imide activation. J. Pept. Sci. 2021, 27, e3364. [Google Scholar] [CrossRef]
- Elashal, H.E.; Raj, M. Site-selective chemical cleavage of peptide bonds. ChemComm 2016, 52, 6304–6307. [Google Scholar] [CrossRef]
- Elashal, H.E.; Sim, Y.E.; Raj, M. Serine promoted synthesis of peptide thioester-precursor on solid support for native chemical ligation. Chem. Sci 2017, 8, 117–123. [Google Scholar] [CrossRef]
- Shin, Y.; Winans, K.A.; Backes, B.J.; Kent, S.B.H.; Ellman, J.A.; Bertozzi, C.R. Fmoc-Based Synthesis of Peptide-αThioesters: Application to the Total Chemical Synthesis of a Glycoprotein by Native Chemical Ligation. J. Am. Chem. Soc. 1999, 121, 11684–11689. [Google Scholar] [CrossRef]
- Heidler, P.; Link, A. N-Acyl-N-alkyl-sulfonamide anchors derived from Kenner’s safety-catch linker: Powerful tools in bioorganic and medicinal chemistry. Bioorg. Med. Chem. 2005, 13, 585–599. [Google Scholar] [CrossRef]
- Zohrabi-Kalantari, V.; Heidler, P.; Larsen, T.; Link, A. O,N,N‘-Trialkylisoureas as Mild Activating Reagents for N-Acylsulfonamide Anchors. Org. Lett. 2005, 7, 5665–5667. [Google Scholar] [CrossRef]
- Ingenito, R.; Bianchi, E.; Fattori, D.; Pessi, A. Solid Phase Synthesis of Peptide C-Terminal Thioesters by Fmoc/t-Bu Chemistry. J. Am. Chem. Soc. 1999, 121, 11369–11374. [Google Scholar] [CrossRef]
- Quaderer, R.; Hilvert, D. Improved Synthesis of C-Terminal Peptide Thioesters on “Safety-Catch” Resins Using LiBr/THF. Org. Lett. 2001, 3, 3181–3184. [Google Scholar] [CrossRef] [PubMed]
- Burlina, F.; Morris, C.; Behrendt, R.; White, P.; Offer, J. Simplifying native chemical ligation with an N-acylsulfonamide linker. ChemComm 2012, 48, 2579–2581. [Google Scholar] [CrossRef] [PubMed]
- Merkx, R.; van Haren, M.J.; Rijkers, D.T.S.; Liskamp, R.M.J. Resin-Bound Sulfonyl Azides: Efficient Loading and Activation Strategy for the Preparation of the N-Acyl Sulfonamide Linker. J. Org. Chem. 2007, 72, 4574–4577. [Google Scholar] [CrossRef] [PubMed]
- Sheppeck Ii, J.E.; Kar, H.; Gosink, L.; Wheatley, J.B.; Gjerstad, E.; Loftus, S.M.; Zubiria, A.R.; Janc, J.W. Synthesis of a statistically exhaustive fluorescent peptide substrate library for profiling protease specificity. Bioorg. Med. Chem. Lett. 2000, 10, 2639–2642. [Google Scholar] [CrossRef] [PubMed]
- Overkleeft, H.S.; Bos, P.R.; Hekking, B.G.; Gordon, E.J.; Ploegh, H.L.; Kessler, B.M. Solid phase synthesis of peptide vinyl sulfone and peptide epoxyketone proteasome inhibitors. Tetrahedron Lett. 2000, 41, 6005–6009. [Google Scholar] [CrossRef]
- Mezzato, S.; Schaffrath, M.; Unverzagt, C. An Orthogonal Double-Linker Resin Facilitates the Efficient Solid-Phase Synthesis of Complex-Type N-Glycopeptide Thioesters Suitable for Native Chemical Ligation. Angew. Chem. Int. Ed. 2005, 44, 1650–1654. [Google Scholar] [CrossRef]
- Dressman, B.A.; Singh, U.; Kaldor, S.W. Solid phase synthesis of urea libraries using a diversifiable thiophenoxy carbonyl linker. Tetrahedron Lett. 1998, 39, 3631–3634. [Google Scholar] [CrossRef]
- Katti, S.B.; Misra, P.K.; Haq, W.; Mathur, K.B. A new base-labile linker for solid-phase peptide synthesis. J. Chem. Soc. Chem. Commun. 1992, 11, 843–844. [Google Scholar] [CrossRef]
- Kang, J.; Macmillan, D. Peptide and protein thioester synthesis via N→S acyl transfer. Org. Biomol. Chem. 2010, 8, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Arbour, C.A.; Saraha, H.Y.; McMillan, T.F.; Stockdill, J.L. Exploiting the MeDbz Linker To Generate Protected or Unprotected C-Terminally Modified Peptides. Chem. Eur. J. 2017, 23, 12484–12488. [Google Scholar] [CrossRef] [PubMed]
- Arbour, C.A.; Kondasinghe, T.D.; Saraha, H.Y.; Vorlicek, T.L.; Stockdill, J.L. Epimerization-free access to C-terminal cysteine peptide acids, carboxamides, secondary amides, and esters via complimentary strategies. Chem. Sci 2018, 9, 350–355. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
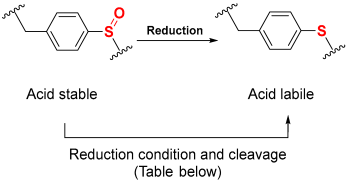 | ||
|---|---|---|
| Entry | Reduction and Cleavage Condition of Sulfoxide to Sulfide | Ref. |
| 1 | 1.1 eq. of TFAA, 2 eq. of Me2S, DCM/THF, 10 min | [50] |
| 2 | 10 eq. TiCl4, 20 eq. of Zn, DCM/THF, 2 h | [51] |
| 3 | 10 eq. of (Me3Si)2S, DCM/THF, 4 h | [52] |
| 4 | 2 eq. Me3SiCl/2 eq. PhSSiMe3, 1/10 eq. Bu4NBr, ether-CHCl3, 1 h | [53] |
| 5 | 10 eq. of Me3SiCl, 20 eq. of Me2S, THF, 2 h | [54] |
| 6 | 10 eq. of ZnCl2, 20 eq. of Me2S, THF, 1 h | [54] |
| 7 | 20 eq. of Me3SiCl, 10 eq. of Ph3P, dioxane | [54] |
| 8 | 10 eq. 1M SiCl4/10 eq. TFA-anisole, 10 eq. thioanisole, 10 eq. EDT, 10 min | [55] |
| 9 | Me3SiBr-EDT in DCM/ACN/DMF/TFA, 15 min-2 h | [56,57] |
| 10 | 10 eq. SiCl4, 10 eq. thioanisole, 10 eq. anisole, TFA-DCM, 3 h | [58] |
| 11 | NH4I/Me2S/TFA-DCM (2 × 30 min) | [59,60] |
| 12 | SiCl4, thioanisole, anisole, EDT, in TFA-DCM, 3 h | [61] |
| 13 | Me3SiBr/thioanisole, TFA-DCM, 2 h | [62,63,64] |
| 14 | 20 eq. Me3SiCl + 10 eq. Ph3P, (3 × 1 h) in THF | [13,65] |
| Structures and Names | Linker Modifications | Cleavage Condition | Compatibility with | C-Terminal Release Form | Ref. |
|---|---|---|---|---|---|
| Kenner and Ellman sulfonamide safety-catch linkers | |||||
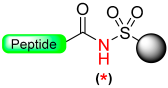 Kenner and Ellman sulphonamide linker | 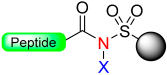 X = CH3, CH2CN Alkylation with CH2N2, TMSCHN2 or ICH2CN Alkylation with Pd (0) Alkylation with O, N, N’- trialkylisoureas |
Nucleophiles (hydroxide, amines, hydrazine, thioles) | Boc/Fmoc chemistry |  Nu = OH, NHR, NHNHR, SR | [18,19,20,22,23,95,96,97,98,99,100,101,102,103,104] |
| The oxidative safety-catch linkers | |||||
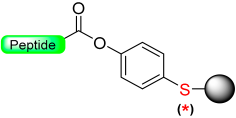 | 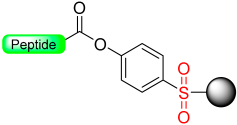 Oxidation with H2O2 or m- CPBA | Nucleophiles (hydroxides, amines) | Boc chemistry | 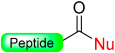 Nu = OR, NHR | [27,28,105] |
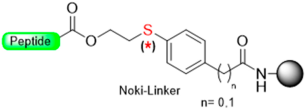 | 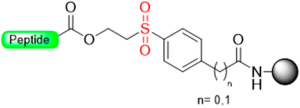 Oxidation with m- CPBA | Nucleophiles (hydroxides, amines) | Boc/Fmoc chemistry |  | [34,35,106] |
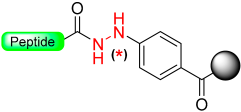 | 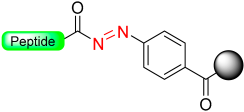 Oxidation with NBS or Cu (OAc)2 | Nucleophiles (amines, alcohols) | Boc/Fmoc chemistry | 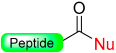 Nu = OR, NHR, SR | [38,39,45,46,47,48] |
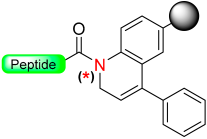 | 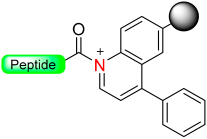 Oxidation with DDQ, CAN or Ph3CBF4 | Nucleophiles (amines, H2O) | Boc/Fmoc chemistry |  Nu = OPhR, NHR, OH | [49] |
| The reductive-acidolytic safety-catch linkers | |||||
 | 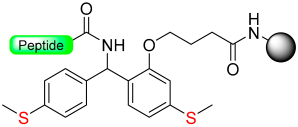 Reduction with SiCl4/Me3SiBr/NH4I/Me2S | TFA, with PhSMe or PhOMe scavenger | Boc/Fmoc chemistry | 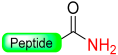 | [63,64] |
 |  Reduction with SiCl4 | TFA with PhSMe/ PhOMe)/EDT scavengers | Boc chemistry |  | [61] |
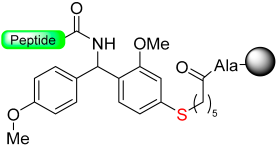
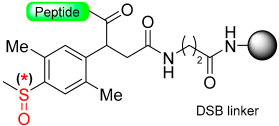 | 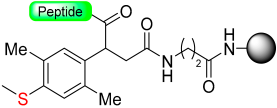 Reduction with SiCl4 | TFA with PhSMe/PhOMe)/EDT scavengers | Boc chemistry | 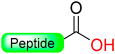 | [58] |
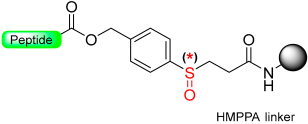 | 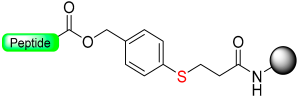 Reduction cleavage with Me3SiBr-EDT-TFA | Reduction cleavage Me3SiBr-EDT-TFA | Boc/Fmoc chemistry |  | [56,57] |
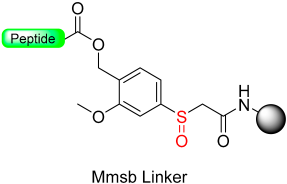 | 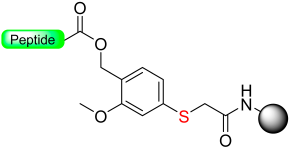 Reduction 20 eq. Me3SiBr–10 eq. Ph3P-THF | Cleavage TFA: TIS: H2O | Boc/Fmoc chemistry |  | [13] |
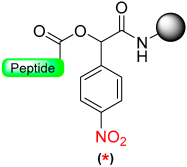 | 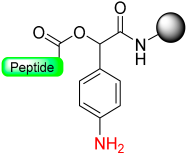 Reduction with SnCl2/HCl | TFA with scavengers | Boc/Fmoc chemistry |  | [70] |
| Isonitrile/benzamide safety-catch linker | |||||
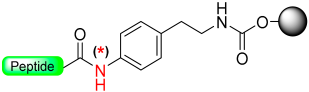 | 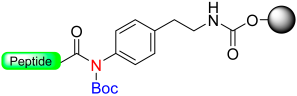 N-Protection with Boc | (i) LiOH, 5% H2O2/H2O/THF (ii) MeONa, MeOH: THF | Fmoc chemistry | 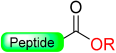 R = H or Me | [26] |
| Heterocycle formation—via intramolecular cyclization—as leaving group | |||||
 |  Acylation NaOH to give isocyanate intermediate | Nucleophiles (NaOH or NH3 in iPrOH) | Boc chemistry |  Nu = OH, NHR | [72,73,74] |
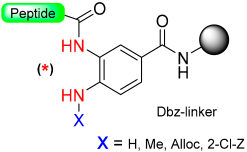 | 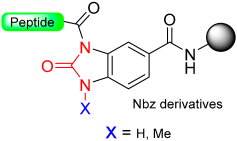 Acylation with p-nitrophenyl chloroformate and DIEA to give Nbz derivatives | (i) TFA with TIS scavengers (ii) Thioestherification | Boc chemistry |  Nu = SR, OR, NHR | [40,78,79,80,81,82,83,84,87,107,108,109] |
 |  Strong activation with PyBrOP/DIEA | (i) Thiolysis (ii) In-solution cleavage TFA with TES scavengers | Boc chemistry |  | [16,91] |
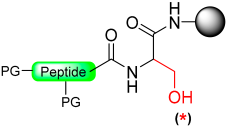 | 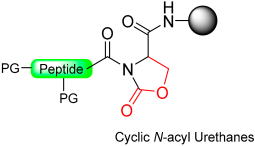 Activation with electrophilic reagent: 4-Nitrophenyl chloroformate/CDI/DSC | (i) Thiolysis (ii) TFA with scavengers | Fmoc chemistry |  | [16,94] |
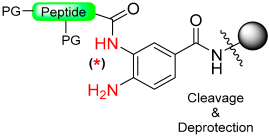 | 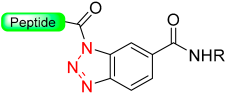 Activation with NaNO2 in aqueous buffer | Thiolysis | Fmoc chemistry |  | [88] |
 | 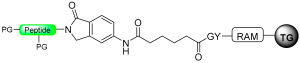 Pd (PPh3)4 Me2NH-NH3, DCM AC2O, DIPEA, NMP | Thiolysis Hydrizide TFA | Fmoc chemistry | 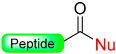 Nu = SR, NHNHR | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noki, S.; de la Torre, B.G.; Albericio, F. Safety-Catch Linkers for Solid-Phase Peptide Synthesis. Molecules 2024, 29, 1429. https://doi.org/10.3390/molecules29071429
Noki S, de la Torre BG, Albericio F. Safety-Catch Linkers for Solid-Phase Peptide Synthesis. Molecules. 2024; 29(7):1429. https://doi.org/10.3390/molecules29071429
Chicago/Turabian StyleNoki, Sikabwe, Beatriz G. de la Torre, and Fernando Albericio. 2024. "Safety-Catch Linkers for Solid-Phase Peptide Synthesis" Molecules 29, no. 7: 1429. https://doi.org/10.3390/molecules29071429