

Neuroinflammation of Microglial Regulation in Alzheimer’s Disease: Therapeutic Approaches

Abstract
1. Introduction
2. Microglia’s Role in AD
2.1. Main Physiological Functions of Microglia
2.2. Microglial Activation-Mediated Neuroinflammation in AD
3. The Critical Role of Energy Metabolic Disorder of Microglia between Microglial Activation-Mediated Neuroinflammation and AD
4. Anti-Inflammatory Drugs for the Treatment of AD
4.1. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
4.2. TLR4 Antagonists
4.3. p38 Mitogen-Activated Protein Kinase (p38 MAPK) Antagonists
4.4. Microglia Regulators
4.5. Chinese Herbal Polysaccharides
4.6. Other Anti-Inflammatory Drugs
5. Discussion and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Lynch, C. World Alzheimer Report 2019: Attitudes to dementia, a global survey. Alzheimer’s Dement. 2020, 16, e038255. [Google Scholar] [CrossRef]
- Venegas, C.; Heneka, M.T. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 2019, 33, 13075–13084. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s disease? BMJ 2021, 374, n1682. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.; Ji, N.; Zhang, X.; Qiao, W.; Tang, Z.; Gou, X. Drug development for Alzheimer’s disease: Review. J. Drug Target. 2019, 27, 164–173. [Google Scholar] [CrossRef]
- Wojcieszak, J.; Kuczyńska, K.; Zawilska, J.B. Role of Chemokines in the Development and Progression of Alzheimer’s Disease. J. Mol. Neurosci. 2022, 72, 1929–1951. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599. [Google Scholar] [CrossRef]
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Voit-Bak, K.; Rosenthal, P.; Tselmin, S.; Julius, U.; Schatz, U.; Boehm, B.O.; Thuret, S.; Kempermann, G.; Reichmann, H.; et al. Extracorporeal apheresis therapy for Alzheimer disease—Targeting lipids, stress, and inflammation. Mol. Psychiatry 2020, 25, 275–282. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef]
- Allinson, T.M.J.; Parkin, E.T.; Turner, A.J.; Hooper, N.M. ADAMs family members as amyloid precursor protein alpha-secretases. J. Neurosci. Res. 2003, 74, 342–352. [Google Scholar] [CrossRef]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Miao, Y. Structure and mechanism of the γ-secretase intramembrane protease complex. Curr. Opin. Struct. Biol. 2022, 74, 102373. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer’s Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease-one peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Piirainen, S.; Youssef, A.; Song, C.; Kalueff, A.V.; Landreth, G.E.; Malm, T.; Tian, L. Psychosocial stress on neuroinflammation and cognitive dysfunctions in Alzheimer’s disease: The emerging role for microglia? Neurosci. Biobehav. Rev. 2017, 77, 148–164. [Google Scholar] [CrossRef]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Landreth, G.E. Microglial interaction with beta-amyloid: Implications for the pathogenesis of Alzheimer’s disease. Microsc. Res. Tech. 2001, 54, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Yamasaki, T.R.; LaFerla, F.M. Microglia as a potential bridge between the amyloid beta-peptide and tau. Ann. N. Y. Acad. Sci. 2004, 1035, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Tiberi, M.; Matteocci, A.; Fazio, F.; Siffeti, H.; Saracini, S.; Mercuri, N.B.; Sancesario, G. Lipidomics of Bioactive Lipids in Alzheimer’s and Parkinson’s Diseases: Where Are We? Int. J. Mol. Sci. 2022, 23, 6235. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Sun, Y.; Peng, G. Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease. Clin. Interv. Aging 2022, 17, 665–674. [Google Scholar] [CrossRef]
- Wong-Guerra, M.; Calfio, C.; Maccioni, R.B.; Rojo, L.E. Revisiting the neuroinflammation hypothesis in Alzheimer’s disease: A focus on the druggability of current targets. Front. Pharmacol. 2023, 14, 1161850. [Google Scholar] [CrossRef] [PubMed]
- Scipioni, L.; Ciaramellano, F.; Carnicelli, V.; Leuti, A.; Lizzi, A.R.; De Dominicis, N.; Oddi, S.; Maccarrone, M. Microglial Endocannabinoid Signalling in AD. Cells 2022, 11, 1237. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Carrier, M.; Tremblay, M.-È. Physiology of Microglia. Adv. Exp. Med. Biol. 2019, 1175, 129–148. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M.-È. Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef] [PubMed]
- Weinhard, L.; d’Errico, P.; Leng Tay, T. Headmasters: Microglial regulation of learning and memory in health and disease. AIMS Mol. Sci. 2018, 5, 63–89. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Arnò, B.; Grassivaro, F.; Rossi, C.; Bergamaschi, A.; Castiglioni, V.; Furlan, R.; Greter, M.; Favaro, R.; Comi, G.; Becher, B.; et al. Neural progenitor cells orchestrate microglia migration and positioning into the developing cortex. Nat. Commun. 2014, 5, 5611. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Xavier, A.L.; Kress, B.T.; Goldman, S.A.; Lacerda de Menezes, J.R.; Nedergaard, M. A Distinct Population of Microglia Supports Adult Neurogenesis in the Subventricular Zone. J. Neurosci. 2015, 35, 11848–11861. [Google Scholar] [CrossRef] [PubMed]
- Ayata, P.; Badimon, A.; Strasburger, H.J.; Duff, M.K.; Montgomery, S.E.; Loh, Y.-H.E.; Ebert, A.; Pimenova, A.A.; Ramirez, B.R.; Chan, A.T.; et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 2018, 21, 1049–1060. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, A.D.; Wu, L.-J. How microglia sense and regulate neuronal activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef]
- Xiong, X.-Y.; Liu, L.; Yang, Q.-W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Zhang, J. A richer and more diverse future for microglia phenotypes. Heliyon 2023, 9, e14713. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhang, Y.-D.; Chen, Q.; Gao, Q.; Zhu, X.-C.; Zhou, J.-S.; Shi, J.-Q.; Lu, H.; Tan, L.; Yu, J.-T. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Heppner, F.L. Microglia as Dynamic and Essential Components of the Amyloid Hypothesis. Neuron 2013, 78, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Cao, W. Mesenchymal stem cell-mediated immunomodulation in cell therapy of neurodegenerative diseases. Cell. Immunol. 2018, 326, 8–14. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Radford, S.E.; Hewitt, E.W. Modulation of β-Amyloid Fibril Formation in Alzheimer’s Disease by Microglia and Infection. Front. Mol. Neurosci. 2020, 13, 609073. [Google Scholar] [CrossRef]
- Weekman, E.M.; Sudduth, T.L.; Abner, E.L.; Popa, G.J.; Mendenhall, M.D.; Brothers, H.M.; Braun, K.; Greenstein, A.; Wilcock, D.M. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J. Neuroinflamm. 2014, 11, 127. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Commins, C.; DeVos, S.L.; Nobuhara, C.K.; Wegmann, S.; Roe, A.D.; Costantino, I.; Fan, Z.; Nicholls, S.B.; Sherman, A.E.; et al. Seed-competent high-molecular-weight tau species accumulates in the cerebrospinal fluid of Alzheimer’s disease mouse model and human patients. Ann. Neurol. 2016, 80, 355–367. [Google Scholar] [CrossRef]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-D.; Zhu, Y.-G.; Lin, N.; Zhang, J.; Ye, Q.-Y.; Huang, H.-P.; Chen, X.-C. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Rizer, J.; Selenica, M.-L.B.; Reid, P.; Kraft, C.; Johnson, A.; Blair, L.; Gordon, M.N.; Dickey, C.A.; Morgan, D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflamm. 2010, 7, 56. [Google Scholar] [CrossRef]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Singh, R.; Paliwal, S.; Sharma, S. Targeting Neuroinflammation as Disease Modifying Approach toAlzheimer’s Disease: Potential and Challenges. MRMC 2023, 23, 2097–2116. [Google Scholar] [CrossRef]
- Perry, V.H. The influence of systemic inflammation on inflammation in the brain: Implications for chronic neurodegenerative disease. Brain Behav. Immun. 2004, 18, 407–413. [Google Scholar] [CrossRef]
- Aktas, O.; Ullrich, O.; Infante-Duarte, C.; Nitsch, R.; Zipp, F. Neuronal damage in brain inflammation. Arch. Neurol. 2007, 64, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Engl, E.; Attwell, D. Non-signalling energy use in the brain. J. Physiol. 2015, 593, 3417–3429. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef]
- Masoumi, M.; Mehrabzadeh, M.; Mahmoudzehi, S.; Mousavi, M.J.; Jamalzehi, S.; Sahebkar, A.; Karami, J. Role of glucose metabolism in aggressive phenotype of fibroblast-like synoviocytes: Latest evidence and therapeutic approaches in rheumatoid arthritis. Int. Immunopharmacol. 2020, 89, 107064. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, S.; Chen, J.; Sun, J.; Sun, X. PFKFB3 knockdown attenuates Amyloid β-Induced microglial activation and retinal pigment epithelium disorders in mice. Int. Immunopharmacol. 2023, 115, 109691. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef]
- Andersson, A.K.; Rönnbäck, L.; Hansson, E. Lactate induces tumour necrosis factor-alpha, interleukin-6 and interleukin-1beta release in microglial- and astroglial-enriched primary cultures. J. Neurochem. 2005, 93, 1327–1333. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Yang, X.; Wang, J.; Wang, Z.; Wang, Q.; Ding, Y.; Yu, A. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J. Neuroinflamm. 2023, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, B.; Sheng, L.; Zhu, Z.; Sun, H.; Zhou, Y.; Yang, Y.; Xue, D.; Chen, W.; Tian, X.; et al. Hexokinase 2-dependent hyperglycolysis driving microglial activation contributes to ischemic brain injury. J. Neurochem. 2018, 144, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; He, X.; Zhang, Q.; Yuan, H.; Wang, D.; Li, L.; Hui, Y.; Wu, Z.; Li, W.; Zhang, N. Alpha-synuclein induces microglial migration via PKM2-dependent glycolysis. Int. J. Biol. Macromol. 2019, 129, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.-Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022, 34, 634–648.e6. [Google Scholar] [CrossRef]
- Holland, R.; McIntosh, A.L.; Finucane, O.M.; Mela, V.; Rubio-Araiz, A.; Timmons, G.; McCarthy, S.A.; Gun’ko, Y.K.; Lynch, M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018, 68, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 13, 856376. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Piers, T.M.; Cosker, K.; Mallach, A.; Johnson, G.T.; Guerreiro, R.; Hardy, J.; Pocock, J.M. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. 2020, 34, 2436–2450. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Chen, L.; Pan, X.; Li, L.; Zhao, B.; Lan, J. Microglial Metabolic Reprogramming: Emerging Insights and Therapeutic Strategies in Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2023, 43, 3191–3210. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Tatulian, S.A. Challenges and hopes for Alzheimer’s disease. Drug Discov. Today 2022, 27, 1027–1043. [Google Scholar] [CrossRef]
- Elmaleh, D.R.; Farlow, M.R.; Conti, P.S.; Tompkins, R.G.; Kundakovic, L.; Tanzi, R.E. Developing Effective Alzheimer’s Disease Therapies: Clinical Experience and Future Directions. J. Alzheimer’s Dis. 2019, 71, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Ferrucci, L.; Kapogiannis, D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: A systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101339. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2023, 9, e12385. [Google Scholar] [CrossRef] [PubMed]
- Ketabforoush, A.H.M.E.; Chegini, R.; Barati, S.; Tahmasebi, F.; Moghisseh, B.; Joghataei, M.T.; Faghihi, F.; Azedi, F. Masitinib: The promising actor in the next season of the Amyotrophic Lateral Sclerosis treatment series. Biomed. Pharmacother. 2023, 160, 114378. [Google Scholar] [CrossRef] [PubMed]
- Balzano, T.; Esteban-García, N.; Blesa, J. Neuroinflammation, immune response and α-synuclein pathology: How animal models are helping us to connect dots. Expert. Opin. Drug Discov. 2023, 18, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qi, F.; Dong, J.; Tan, Y.; Gao, L.; Liu, F. Application of Baricitinib in Dermatology. J. Inflamm. Res. 2022, 15, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Querfurth, H.; Lee, H.-K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 2021, 16, 44. [Google Scholar] [CrossRef]
- Kopp, K.O.; Greer, M.E.; Glotfelty, E.J.; Hsueh, S.-C.; Tweedie, D.; Kim, D.S.; Reale, M.; Vargesson, N.; Greig, N.H. A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders. Biomolecules 2023, 13, 747. [Google Scholar] [CrossRef]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, e12008. [Google Scholar] [CrossRef]
- Staal, R.G.W.; Weinstein, J.R.; Nattini, M.; Cajina, M.; Chandresana, G.; Möller, T. Senicapoc: Repurposing a Drug to Target Microglia KCa3.1 in Stroke. Neurochem. Res. 2017, 42, 2639–2645. [Google Scholar] [CrossRef] [PubMed]
- Devanand, D.P.; Andrews, H.; Kreisl, W.C.; Razlighi, Q.; Gershon, A.; Stern, Y.; Mintz, A.; Wisniewski, T.; Acosta, E.; Pollina, J.; et al. Antiviral therapy: Valacyclovir Treatment of Alzheimer’s Disease (VALAD) Trial: Protocol for a randomised, double-blind, placebo-controlled, treatment trial. BMJ Open 2020, 10, e032112. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Paquot, N.; Scheen, A.J. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert. Opin. Investig. Drugs 2015, 24, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.; Zuniga, G.; Sun, W.; Beckmann, A.; Ochoa, E.; DeVos, S.L.; Hyman, B.; Chiu, G.; Roy, E.R.; Cao, W.; et al. Pathogenic tau accelerates aging-associated activation of transposable elements in the mouse central nervous system. Prog. Neurobiol. 2022, 208, 102181. [Google Scholar] [CrossRef] [PubMed]
- Porrini, V.; Lanzillotta, A.; Branca, C.; Benarese, M.; Parrella, E.; Lorenzini, L.; Calzà, L.; Flaibani, R.; Spano, P.F.; Imbimbo, B.P.; et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience 2015, 302, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Akirav, E.M.; Chen, W.; Henegariu, O.; Moser, B.; Desai, D.; Shen, J.M.; Webster, J.C.; Andrews, R.C.; Mjalli, A.M.; et al. RAGE ligation affects T cell activation and controls T cell differentiation. J. Immunol. 2008, 181, 4272–4278. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef]
- Shi, S.; Liang, D.; Chen, Y.; Xie, Y.; Wang, Y.; Wang, L.; Wang, Z.; Qiao, Z. Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO, and PGE2 expression and inhibits NF-κB signaling in a mouse model of Alzheimer’s disease. Eur. J. Immunol. 2016, 46, 665–676. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Peng, Y.; Hutchinson, M.R.; Rice, K.C.; Yin, H.; Watkins, L.R. Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br. J. Pharmacol. 2016, 173, 856–869. [Google Scholar] [CrossRef]
- Scheltens, P.; Prins, N.; Lammertsma, A.; Yaqub, M.; Gouw, A.; Wink, A.M.; Chu, H.-M.; van Berckel, B.N.M.; Alam, J. An exploratory clinical study of p38α kinase inhibition in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2018, 5, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Son, S.H.; Lee, N.-R.; Gee, M.S.; Song, C.W.; Lee, S.J.; Lee, S.-K.; Lee, Y.; Kim, H.J.; Lee, J.K.; Inn, K.-S.; et al. Chemical Knockdown of Phosphorylated p38 Mitogen-Activated Protein Kinase (MAPK) as a Novel Approach for the Treatment of Alzheimer’s Disease. ACS Cent. Sci. 2023, 9, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yuan, C.; Wang, G.; Luo, J.; Ma, H.; Xu, L.; Mu, Y.; Li, Y.; Seeram, N.P.; Huang, X.; et al. Urolithins Attenuate LPS-Induced Neuroinflammation in BV2Microglia via MAPK, Akt, and NF-κB Signaling Pathways. J. Agric. Food Chem. 2018, 66, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, H.; Wang, L.; Tao, Y.; Du, G.; Guan, W.; Liu, J.; Brennan, C.; Ho, C.-T.; Li, S. Effects of Selected Resveratrol Analogues on Activation and Polarization of Lipopolysaccharide-Stimulated BV-2 Microglial Cells. J. Agric. Food Chem. 2020, 68, 3750–3757. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.N.; Fisher, D.R.; Rimando, A.M.; Gomes, S.M.; Bielinski, D.F.; Shukitt-Hale, B. Stilbenes and anthocyanins reduce stress signaling in BV-2 mouse microglia. J. Agric. Food Chem. 2013, 61, 5979–5986. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.A.; Fisher, D.R.; Cheng, V.; Rimando, A.M.; Shukitt-Hale, B. Cellular and behavioral effects of stilbene resveratrol analogues: Implications for reducing the deleterious effects of aging. J. Agric. Food Chem. 2008, 56, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.M.; Shen, X.; Ran, C.; Elmaleh, D.R.; et al. Cromolyn Reduces Levels of the Alzheimer’s Disease-Associated Amyloid β-Protein by Promoting Microglial Phagocytosis. Sci. Rep. 2018, 8, 1144. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Lin, C.; Wang, Y.; Wang, H.; Wen, X.; Xiao, P.; Li, X.; Peng, Y.; Sun, J.; Lu, Y.; et al. Cannabidiol Analogue CIAC001 for the Treatment of Morphine-Induced Addiction by Targeting PKM2. J. Med. Chem. 2023, 66, 11498–11516. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Pan, R.-Y.; Ma, J.; Kong, X.-X.; Wang, X.-F.; Li, S.-S.; Qi, X.-L.; Yan, Y.-H.; Cheng, J.; Liu, Q.; Jin, W.; et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 2019, 5, eaau6328. [Google Scholar] [CrossRef]
- Wang, H.; Fu, J.; Xu, X.; Yang, Z.; Zhang, T. Rapamycin Activates Mitophagy and Alleviates Cognitive and Synaptic Plasticity Deficits in a Mouse Model of Alzheimer’s Disease. J. Gerontol. Ser. A 2021, 76, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Nao, J.; Dong, X. Neuroprotective Mechanisms of Salidroside in Alzheimer’s Disease: A Systematic Review and Meta-analysis of Preclinical Studies. J. Agric. Food Chem. 2023, 71, 17597–17614. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, Y.; Pei, G. Polysaccharides from Ganoderma lucidum attenuate microglia-mediated neuroinflammation and modulate microglial phagocytosis and behavioural response. J. Neuroinflamm. 2017, 14, 63. [Google Scholar] [CrossRef]
- Zheng, Y.; Ren, W.; Zhang, L.; Zhang, Y.; Liu, D.; Liu, Y. A Review of the Pharmacological Action of Astragalus Polysaccharide. Front. Pharmacol. 2020, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Qi, J.; Du, D.; Liu, Y.; Jiang, X. Current advances of Dendrobium officinale polysaccharides in dermatology: A literature review. Pharm. Biol. 2020, 58, 664–673. [Google Scholar] [CrossRef]
- Xia, G.; Li, X.; Zhang, Z.; Jiang, Y. Effect of food processing on the antioxidant activity of flavones from Polygonatum odoratum (Mill.) Druce. Open Life Sci. 2021, 16, 92–101. [Google Scholar] [CrossRef]
- Javed, M.A.; Ashraf, N.; Saeed Jan, M.; Mahnashi, M.H.; Alqahtani, Y.S.; Alyami, B.A.; Alqarni, A.O.; Asiri, Y.I.; Ikram, M.; Sadiq, A.; et al. Structural Modification, In Vitro, In Vivo, Ex Vivo, and In Silico Exploration of Pyrimidine and Pyrrolidine Cores for Targeting Enzymes Associated with Neuroinflammation and Cholinergic Deficit in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 4123–4143. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhuang, L.; Luo, X.; Liang, J.; Sun, E.; He, Y. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 2020, 238, 2603–2614. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.E.; Li, Y.-M. Turning the tide on Alzheimer’s disease: Modulation of γ-secretase. Cell Biosci. 2022, 12, 2. [Google Scholar] [CrossRef]
- Choi, S.-H.; Aid, S.; Caracciolo, L.; Minami, S.S.; Niikura, T.; Matsuoka, Y.; Turner, R.S.; Mattson, M.P.; Bosetti, F. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer’s disease. J. Neurochem. 2013, 124, 59–68. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-Steroidal Anti-Inflammatory Drugs and Brain Inflammation: Effects on Microglial Functions. Pharmaceuticals 2010, 3, 1949–1965. [Google Scholar] [CrossRef]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Jia, P.; Zhang, D.; Yao, Z. TTP488 ameliorates NLRP3-associated inflammation, viability, apoptosis, and ROS production in an Alzheimer’s disease cell model by mediating the JAK1/STAT3/NFκB/IRF3 pathway. Cell Biochem. Funct. 2021, 39, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.S.; Haneuse, S.J.P.A.; Walker, R.; Dublin, S.; Crane, P.K.; Gray, S.L.; Larson, E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009, 72, 1899–1905. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar] [CrossRef]
- Wang, Z.; Vilekar, P.; Huang, J.; Weaver, D.F. Furosemide as a Probe Molecule for the Treatment of Neuroinflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2020, 11, 4152–4168. [Google Scholar] [CrossRef]
- Neibert, K.; Gosein, V.; Sharma, A.; Khan, M.; Whitehead, M.A.; Maysinger, D.; Kakkar, A. “Click” dendrimers as anti-inflammatory agents: With insights into their binding from molecular modeling studies. Mol. Pharm. 2013, 10, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- García Bueno, B.; Caso, J.R.; Madrigal, J.L.M.; Leza, J.C. Innate immune receptor Toll-like receptor 4 signalling in neuropsychiatric diseases. Neurosci. Biobehav. Rev. 2016, 64, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Go, M.; Kou, J.; Lim, J.-E.; Yang, J.; Fukuchi, K.-I. Microglial response to LPS increases in wild-type mice during aging but diminishes in an Alzheimer’s mouse model: Implication of TLR4 signaling in disease progression. Biochem. Biophys. Res. Commun. 2016, 479, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K.I. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Zou, Y.; Liu, H.; Chen, T.; Zheng, F.; Huang, Y.; Chen, C.; Zhang, Z. Toll-like receptor 4 deficiency ameliorates β2-microglobulin induced age-related cognition decline due to neuroinflammation in mice. Mol. Brain 2020, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Leitner, G.R.; Wenzel, T.J.; Marshall, N.; Gates, E.J.; Klegeris, A. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert. Opin. Ther. Targets 2019, 23, 865–882. [Google Scholar] [CrossRef] [PubMed]
- Irving, E.A.; Bamford, M. Role of mitogen- and stress-activated kinases in ischemic injury. J. Cereb. Blood Flow Metab. 2002, 22, 631–647. [Google Scholar] [CrossRef]
- Dong, N.; Li, X.; Xue, C.; Zhang, L.; Wang, C.; Xu, X.; Shan, A. Astragalus polysaccharides alleviates LPS-induced inflammation via the NF-κB/MAPK signaling pathway. J. Cell. Physiol. 2020, 235, 5525–5540. [Google Scholar] [CrossRef] [PubMed]
- Prins, N.D.; Harrison, J.E.; Chu, H.-M.; Blackburn, K.; Alam, J.J.; Scheltens, P.; REVERSE-SD Study Investigators. A phase 2 double-blind placebo-controlled 24-week treatment clinical study of the p38 alpha kinase inhibitor neflamapimod in mild Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 106. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Carosi, J.M.; Sargeant, T.J. Rapamycin and Alzheimer disease: A hypothesis for the effective use of rapamycin for treatment of neurodegenerative disease. Autophagy 2023, 19, 2386–2390. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Q.; Sun, S.; Chen, S. Neuroprotective Effects of Salidroside in a Mouse Model of Alzheimer’s Disease. Cell. Mol. Neurobiol. 2020, 40, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, M.; Papi, L.; Gori, F.; Turillazzi, E. Natural Products in Neurodegenerative Diseases: A Great Promise but an Ethical Challenge. Int. J. Mol. Sci. 2019, 20, 5170. [Google Scholar] [CrossRef]
- Cvetkovic, R.S.; Keating, G. Anakinra. BioDrugs 2002, 16, 303–311; discussion 313–314. [Google Scholar] [CrossRef] [PubMed]
- Nair, B.; Raval, G.; Mehta, P. TNF-alpha inhibitor etanercept and hematologic malignancies: Report of a case and review of the literature. Am. J. Hematol. 2007, 82, 1022–1024. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain J. Neurol. 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Akıncıoğlu, H.; Gülçin, İ. Potent Acetylcholinesterase Inhibitors: Potential Drugs for Alzheimer’s Disease. Mini Rev. Med. Chem. 2020, 20, 703–715. [Google Scholar] [CrossRef]
- Bai, H.; Zhang, Q. Activation of NLRP3 Inflammasome and Onset of Alzheimer’s Disease. Front. Immunol. 2021, 12, 701282. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Agent | Type | CADRO Target | Mechanism of Action | Clinical Trial NCT# |
|---|---|---|---|---|
| Masitinib (Phase 3) | small molecule | Inflammation | Tyrosine kinase inhibitor exhibits neuroprotection via inhibition of mast cell and microglia/macrophage activity | NCT05564169 |
| NE3107 (Phase 3) | small molecule | Inflammation | Beta-androstenetriol with anti-inflammatory and insulin signaling effects via ERK 1 and 2 | NCT04669028 |
| Baricitinib (Phase 2) | small molecule | Inflammation | Janus kinase (JAK) inhibitor | NCT05189106 |
| Dasatinib + Quercetin (Phase 2) | small molecule | Inflammation | Dasatinib induces apoptosis in senescent cells to allow their removal; quercetin is a flavonoid | NCT04063124 |
| NCT04685590 | ||||
| NCT04785300 | ||||
| NCT05422885 | ||||
| L-Serine (Phase 2) | small molecule | Inflammation | Naturally occurring dietary amino acid; inhibits toxic misfolding | NCT03062449 |
| Lenalidomide (Phase 2) | small molecule | Inflammation | Immunomodulator | NCT04032626 |
| Montelukast (Phase 2) | small molecule | Inflammation | Leukotriene receptor antagonist (LTRA); anti-inflammatory effects | NCT03402503 |
| Senicapoc (Phase 2) | small molecule | Inflammation | Calcium-activated potassium channel inhibitor | NCT04804241 |
| Valacyclovir (Phase 2) | small molecule | Inflammation | Anti-viral against HSV-1 and −2; reduces vira-related “seeding” of ABP deposition | NCT03282916 |
| Salsalate (Phase 1) | small molecule | Inflammation | Non-steroidal anti-inflammatory (NSAID) | NCT03277573 |
| Emtricitabine (Phase 1) | small molecule | Inflammation | Nucleoside reverse transcriptase inhibitor (NRTI) | NCT04500847 |
| Drug Name | Chemical Structures | Category | Compound Effects |
|---|---|---|---|
| Masitinib | 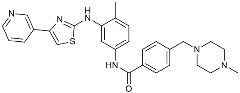 | Tyrosine kinase inhibitor | Downregulated proinflammatory cytokines. Induced neuroprotection [104]. |
| NE3107 |  | NF-κB inhibitor | Decreased activated microglia, Aβ [105]. |
| Baricitinib | 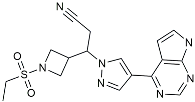 | JAK inhibitor | Blocked intracellular delivery of cytokines via JAK-STAT [106]. |
| Dasatinib + Quercetin | 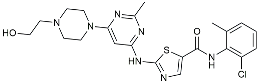 | Tyrosine kinase inhibitor | Alleviated neurodegeneration in AD [107]. |
 | PI3K/Akt inhibitor | ||
| L-Serine |  | mTOR inhibitor | Autophagic clearance of Aβ [108]. |
| Lenalidomide |  | Immunomodulator | Decreased the expression of TNFα, IL-6, IL-8. Increased the expression of anti-inflammatory cytokines [109]. |
| Montelukast |  | Cytochrome P-450 Enzyme Inducers | Increased expression of P450 enzymes [110]. |
| Senicapoc | 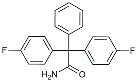 | KCa3.1 inhibitor | Regulated microglia polarization [111]. |
| Valacyclovir | 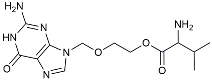 | Anti-virus | Reduced accumulation of Aβ and p-tau [112]. |
| Salsalate | 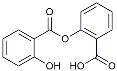 | NASID | Inhibited inflammatory mediators [113]. |
| Emtricitabine | 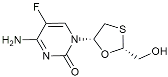 | Anti-virus | Suppressed neuroinflammation [114]. |
| Drug Name | Chemical Structures | Category | Compound Effects |
|---|---|---|---|
| CHF5074 | 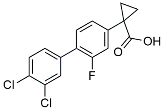 | Improved cognition. Reduced brain inflammation [115]. | |
| Indomethacin |  | Cox-2 inhibitor | Inhibited inflammatory mediators released from microglia [116]. |
| Furosemide | 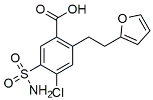 | Inhibited protein expression of COX-2,iNOS. | |
| Azeliragon | 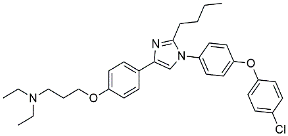 | Mediated the JAK1/STAT3/NF-κB/IRF3 pathway [117]. | |
| TAK242 | 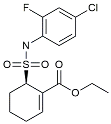 | TLR4 antagonists | Reduced Aβ deposition [118]. |
| Gx-50 | 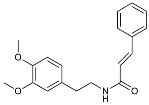 | Suppressed of TLR4-mediated NF-κB/MAPK signaling [119]. | |
| Naloxone | 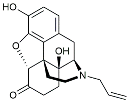 | Inhibited reactive oxygen species production [120]. | |
| VX745 | 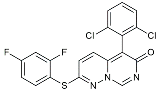 | p38-MAPK antagonists | Reduced IL-1β protein levels in the hippocampus [121]. |
| PRZ-18002 | 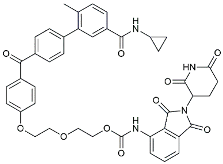 | Degraded phosphorylated p38 MAPK (p-p38) [122]. | |
| Alleviated microglial activation and Aβ deposition [122]. | |||
| Urolithin |  | Reduced NO levels and suppressed pro-inflammatory genes [123]. | |
| TSG | 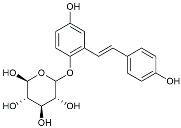 | Microglia regulators | Inhibited PKM2 to adjust microglia polarization [124]. |
| Resveratrol |  | Adjusted microglia polarization [124,125,126]. | |
| Cromolyn | 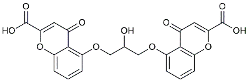 | Induced neuroprotective microglial activation [127]. | |
| CIAC001 | 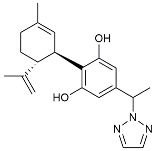 | PKM2 inhibitor | Ameliorated morphine-induced addiction through anti-neuroinflammation [128]. |
| Benserazide | 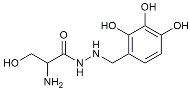 | Inhibited PKM2, thereby blocking aerobic glycolysis and modulating OXPHOS [129]. | |
| Rutin | 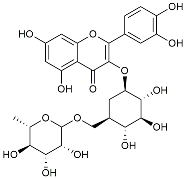 | Promoted a metabolic switch from anaerobic glycolysis to mitochondrial OXPHOS [130]. | |
| Rapamycin |  | mTOR inhibitor | Activated mitophagy and alleviated cognitive impairment [131]. |
| Salidroside | 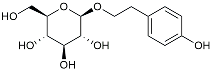 | Inhibited Aβ deposit. Anti-inflammation [132]. | |
| GV-971 |  | Chinese herbal polysaccharides | Remodeled gut microbiota to inhibit AD progression [133]. |
| Ganoderma lucidum polysaccharides | / | Reduced Aβ levels and tau protein hyperphosphorylation [134]. | |
| Astragalus polysaccharides | / | Reduced astrocytic and microglial activation [135]. | |
| Dendrobium orchid polysaccharide | / | Inhibited oxidative stress [136]. | |
| Astragalus polysaccharide | / | Decreased the phosphorylation of p38 MAPK [137]. | |
| Tacrine | 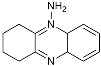 | AChE inhibitor | Inhibited acetylcholinesterase activity [138]. |
| The derivative of tacrine |  | ||
| Donepezil | 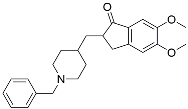 | ||
| Dapansutrile | 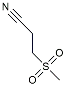 | NLRP3 Inhibitor | Inhibited the associated NLRP3 inflammatory response [120]. |
| MC950 |  | Reduced Aβ deposition and associated neurotoxicity [139]. | |
| JC124 | 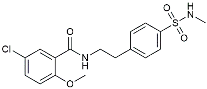 | Reduced CAA, microgliosis and oxidative stress [140]. | |
| CY-09 | 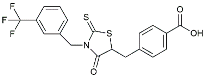 | Inhibited the associated NLRP3 inflammatory response [141]. | |
| Inzomelid | 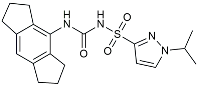 | Inhibited the associated NLRP3 inflammatory response [142]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Zeng, Y.; Wang, D.; Li, Y.; Xing, J.; Zeng, Y.; Liu, Z.; Zhou, X.; Fan, H. Neuroinflammation of Microglial Regulation in Alzheimer’s Disease: Therapeutic Approaches. Molecules 2024, 29, 1478. https://doi.org/10.3390/molecules29071478
Chen H, Zeng Y, Wang D, Li Y, Xing J, Zeng Y, Liu Z, Zhou X, Fan H. Neuroinflammation of Microglial Regulation in Alzheimer’s Disease: Therapeutic Approaches. Molecules. 2024; 29(7):1478. https://doi.org/10.3390/molecules29071478
Chicago/Turabian StyleChen, Haiyun, Yuhan Zeng, Dan Wang, Yichen Li, Jieyu Xing, Yuejia Zeng, Zheng Liu, Xinhua Zhou, and Hui Fan. 2024. "Neuroinflammation of Microglial Regulation in Alzheimer’s Disease: Therapeutic Approaches" Molecules 29, no. 7: 1478. https://doi.org/10.3390/molecules29071478
APA StyleChen, H., Zeng, Y., Wang, D., Li, Y., Xing, J., Zeng, Y., Liu, Z., Zhou, X., & Fan, H. (2024). Neuroinflammation of Microglial Regulation in Alzheimer’s Disease: Therapeutic Approaches. Molecules, 29(7), 1478. https://doi.org/10.3390/molecules29071478