
3-NAntC: A Potent Crotoxin B-Derived Peptide against the Triple-Negative MDA-MB-231 Breast Cancer Cell Line
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
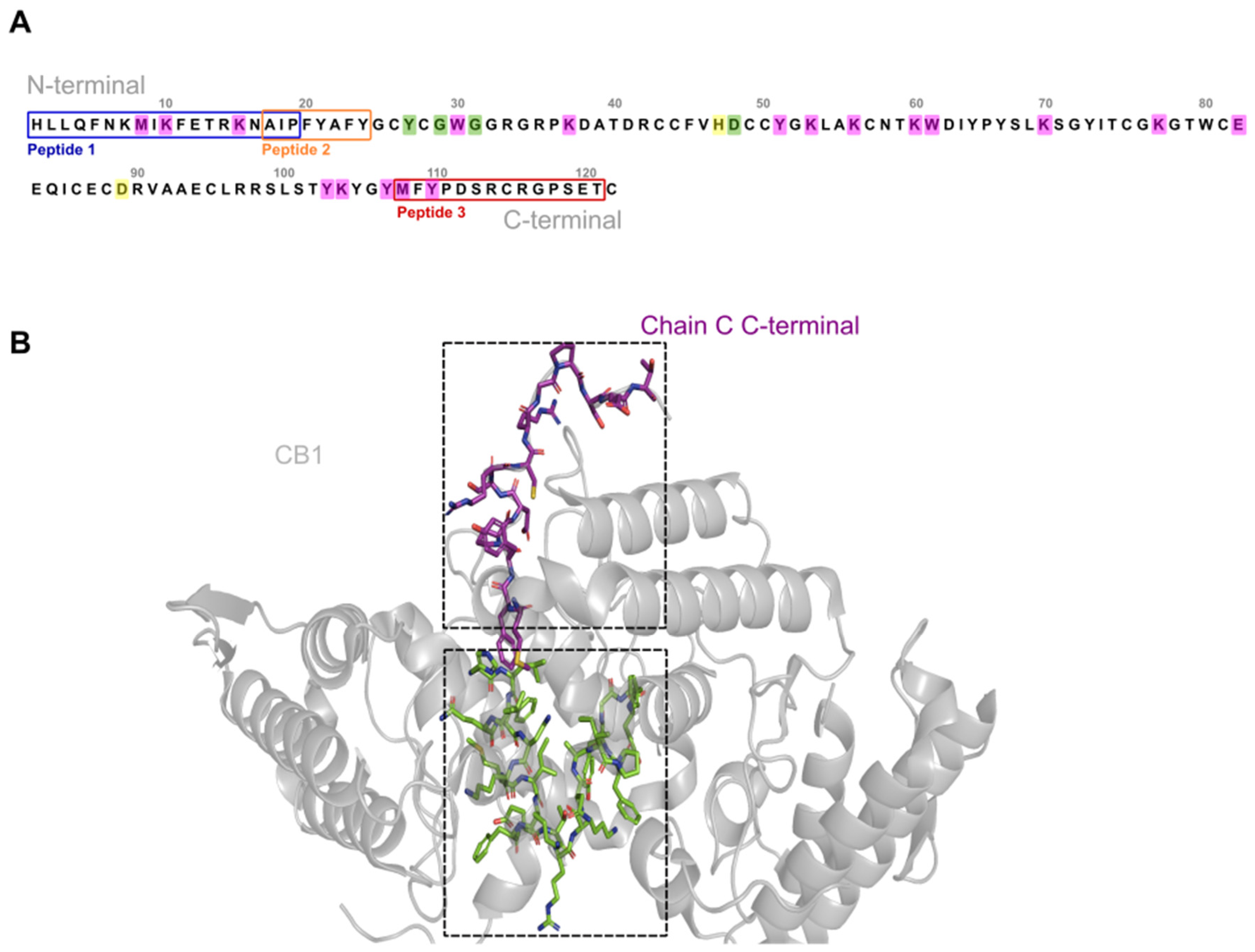
2.1. Synthesis and Characterization of Crotoxin B-Derived Peptides
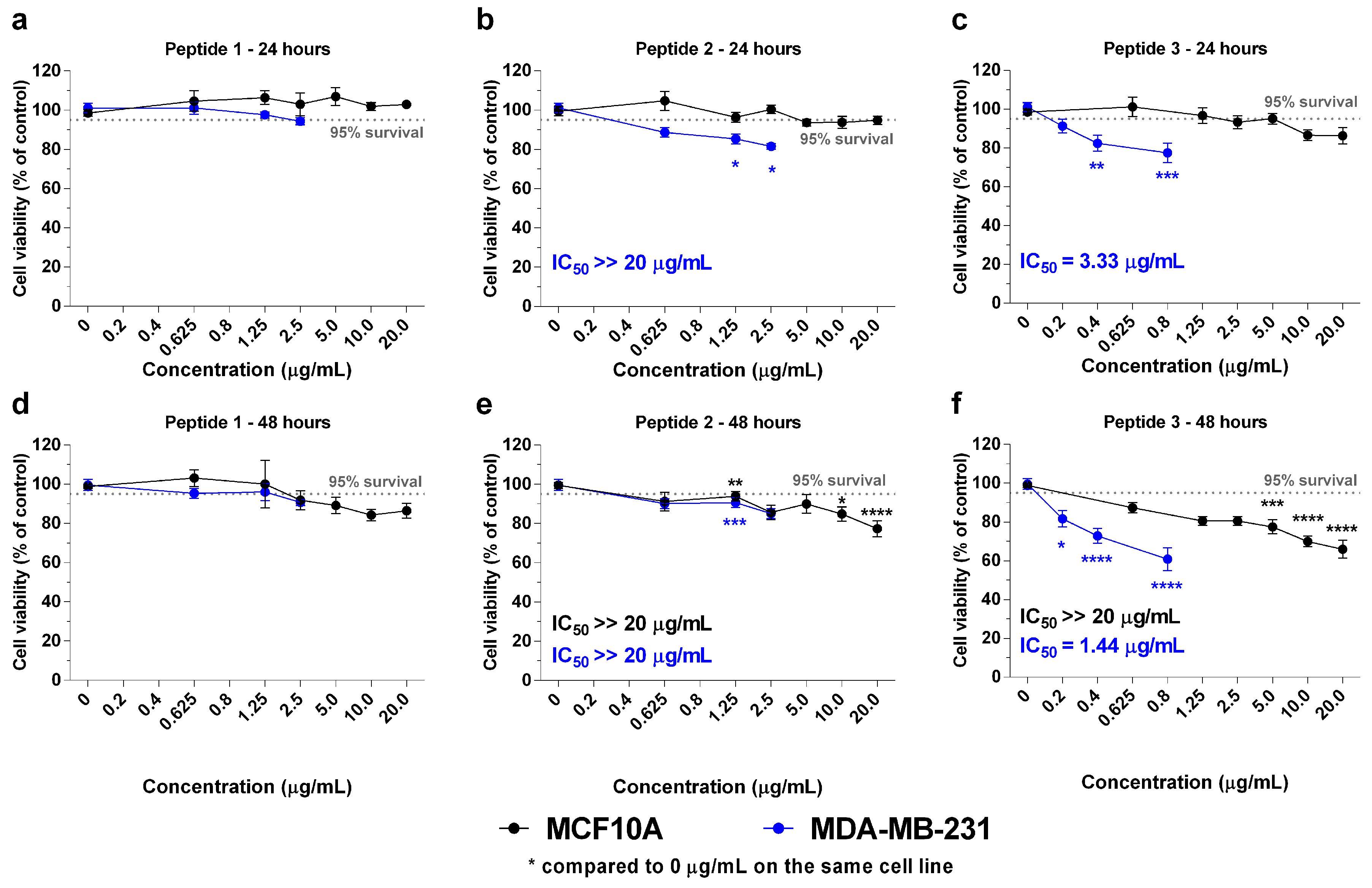
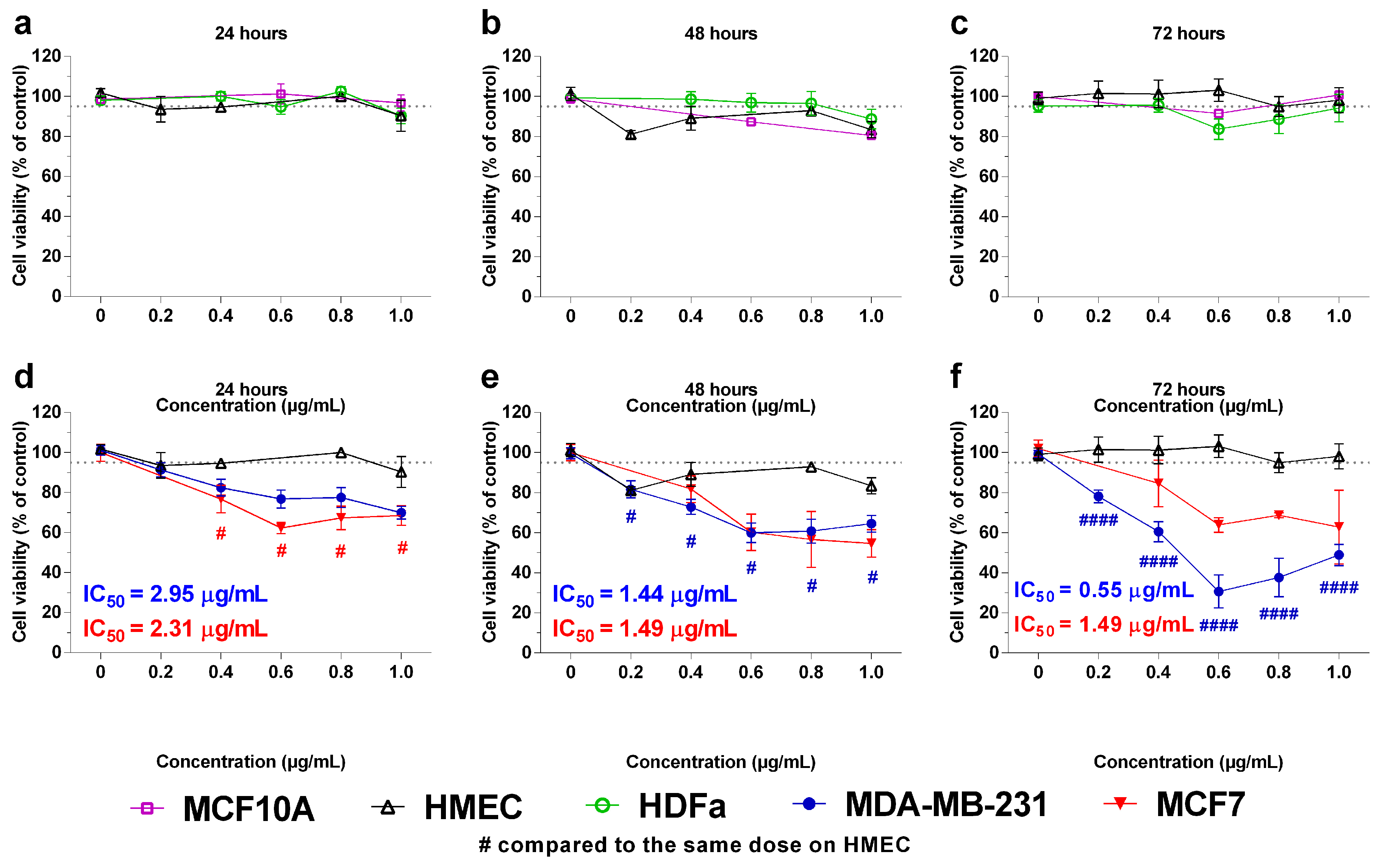
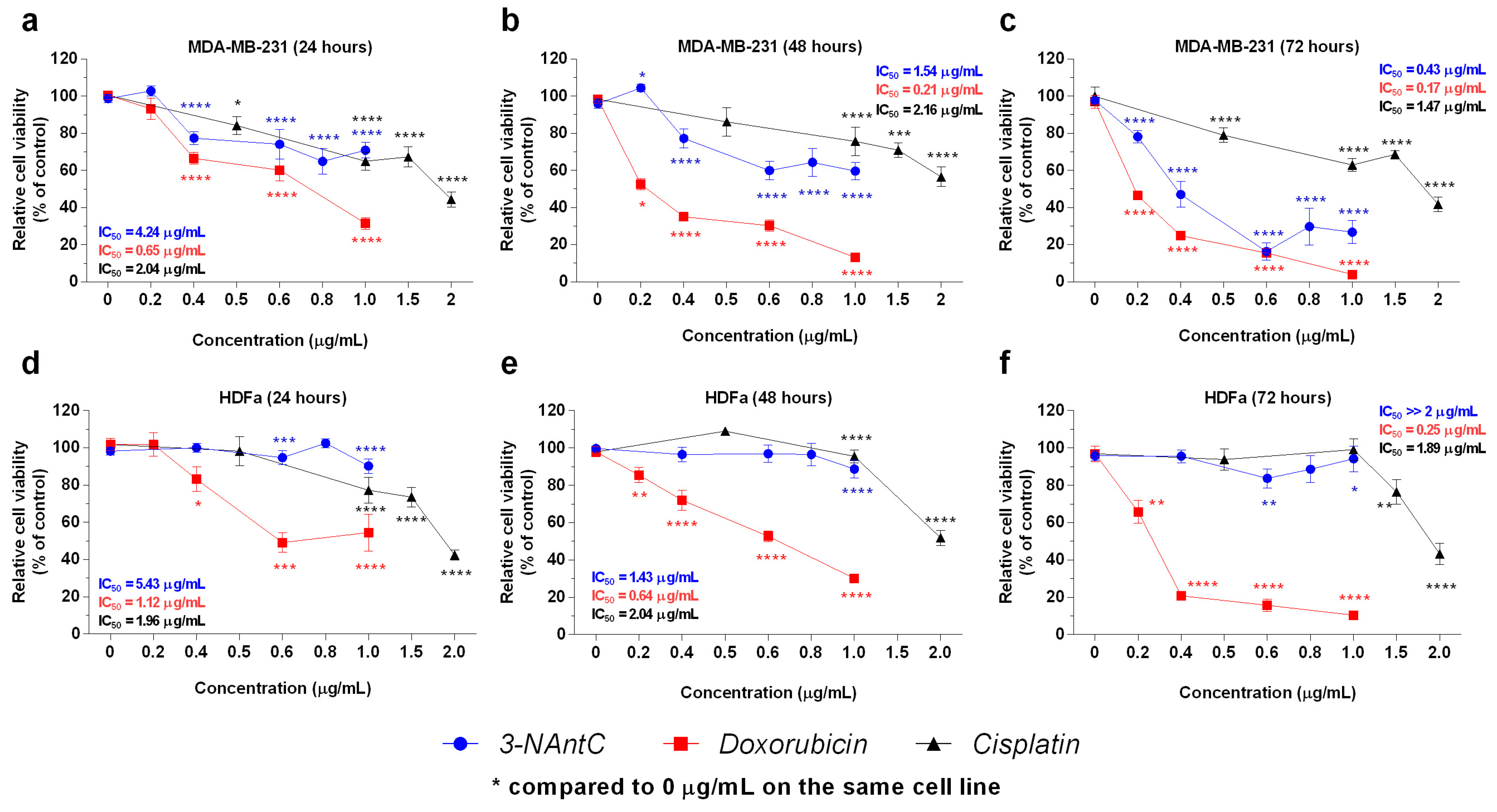
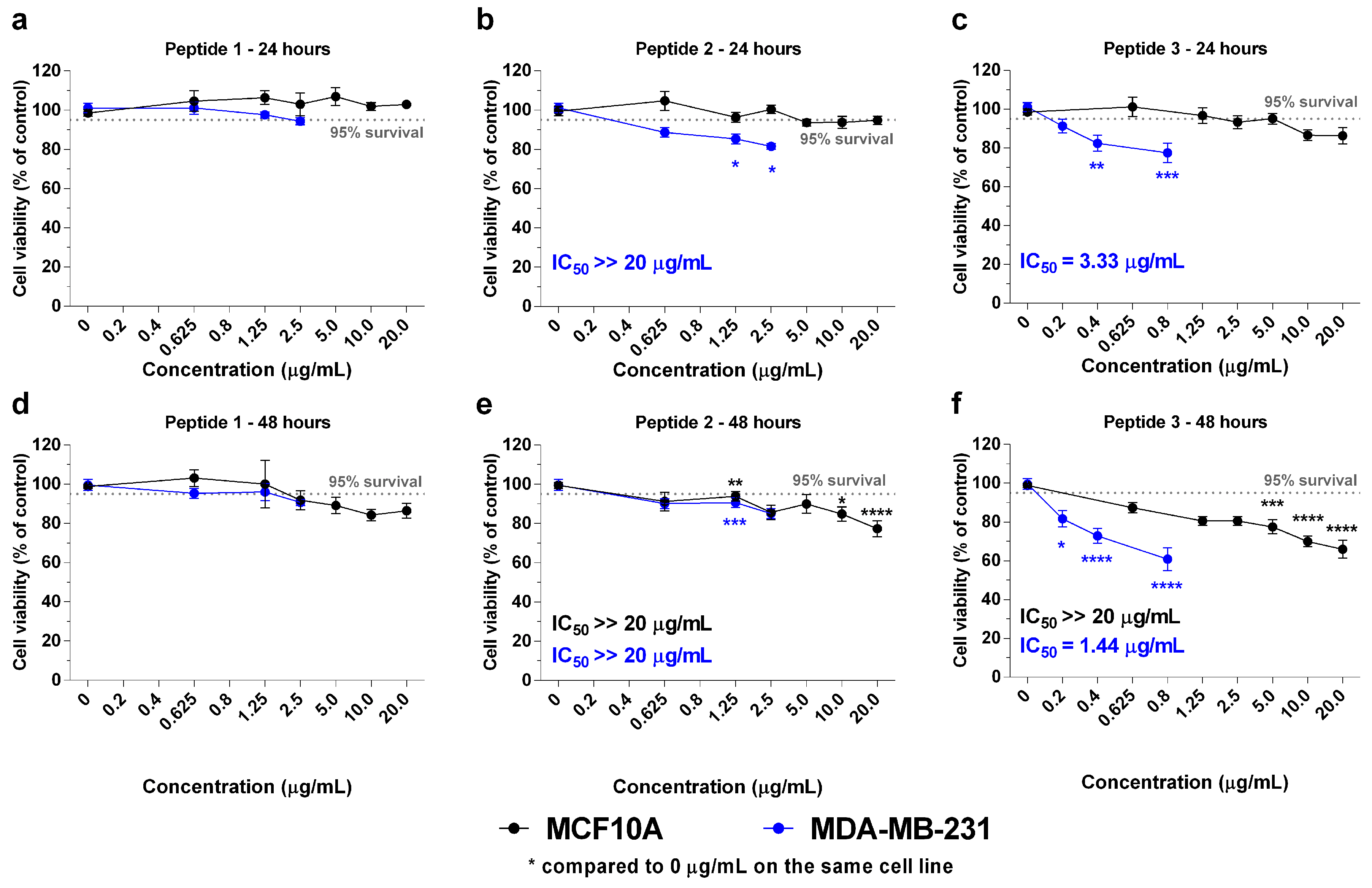
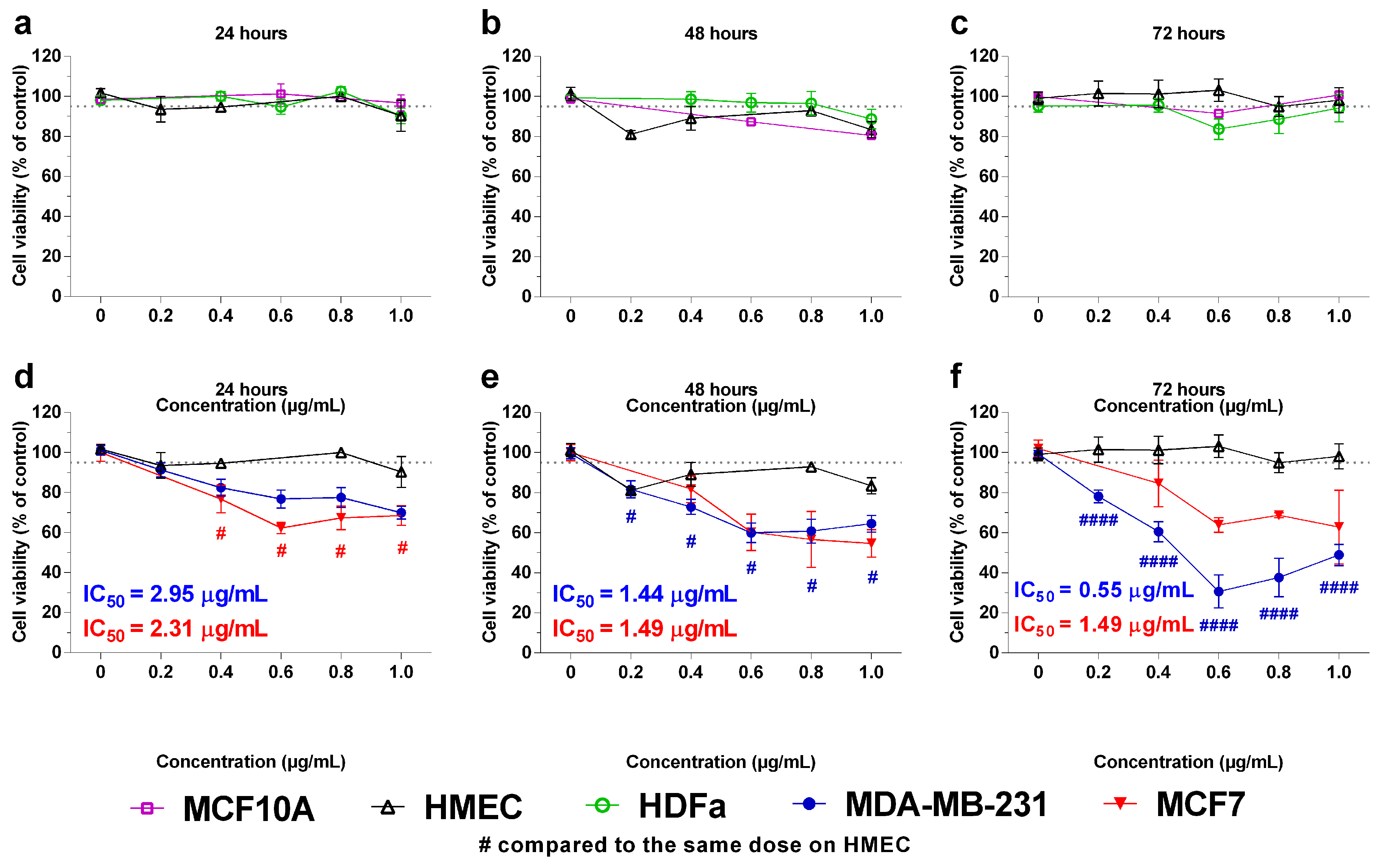
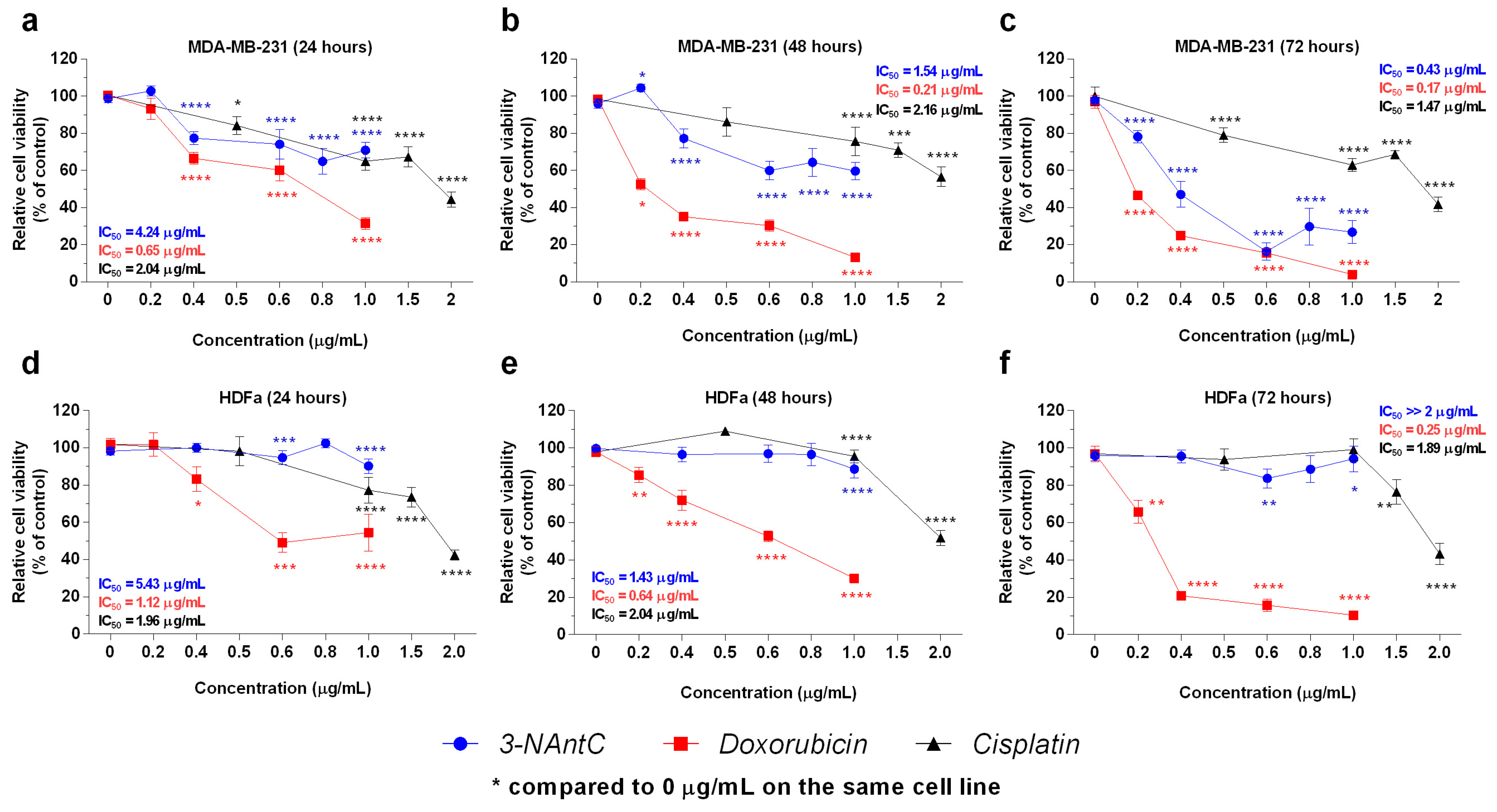
2.2. In Vitro Effects of the Crotoxin B-Derived Peptides in the Viability of Breast Cancer Cell Lines
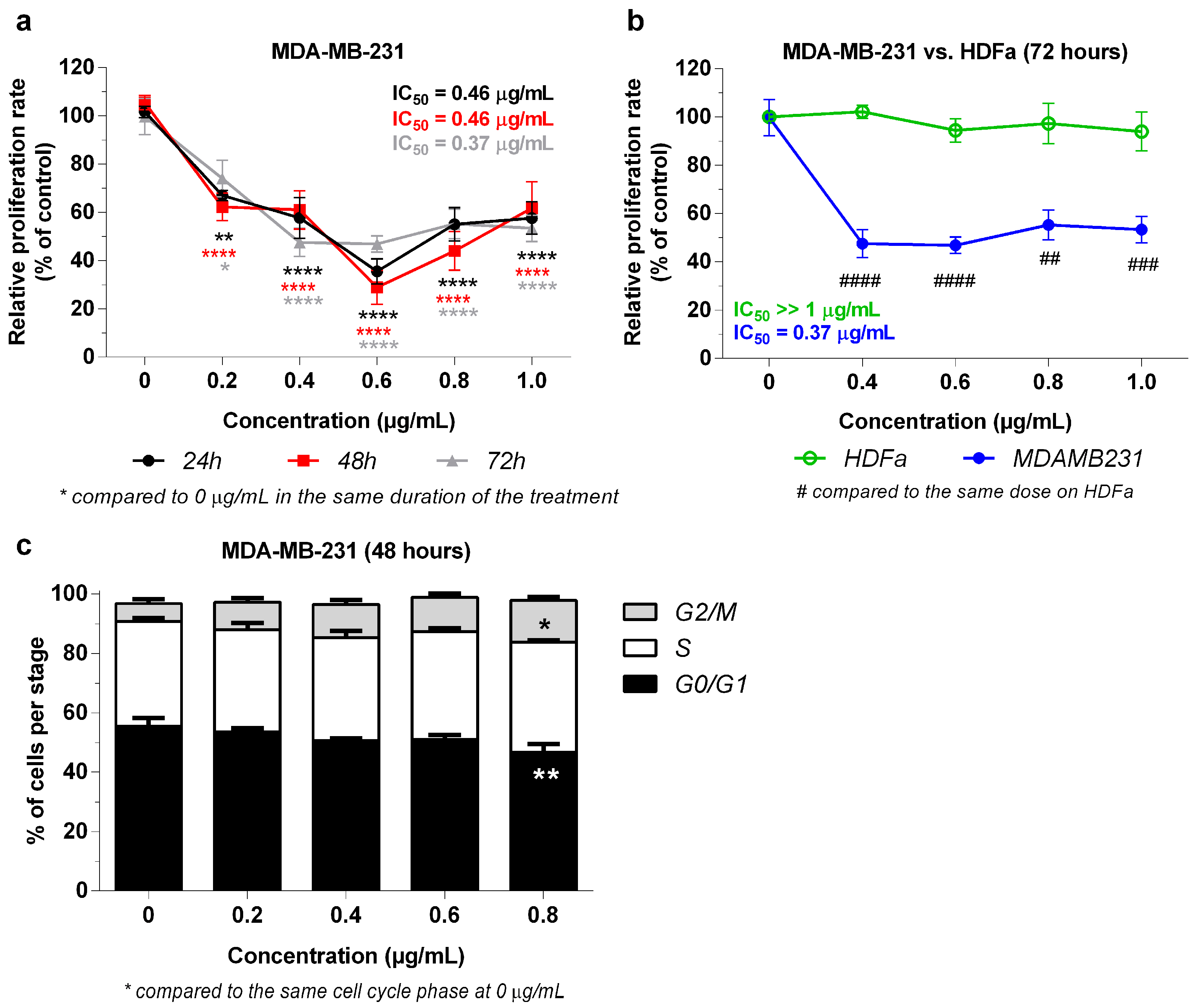
2.3. Effect of 3-NAntC in Cell Proliferation and Cell Cycle Progression
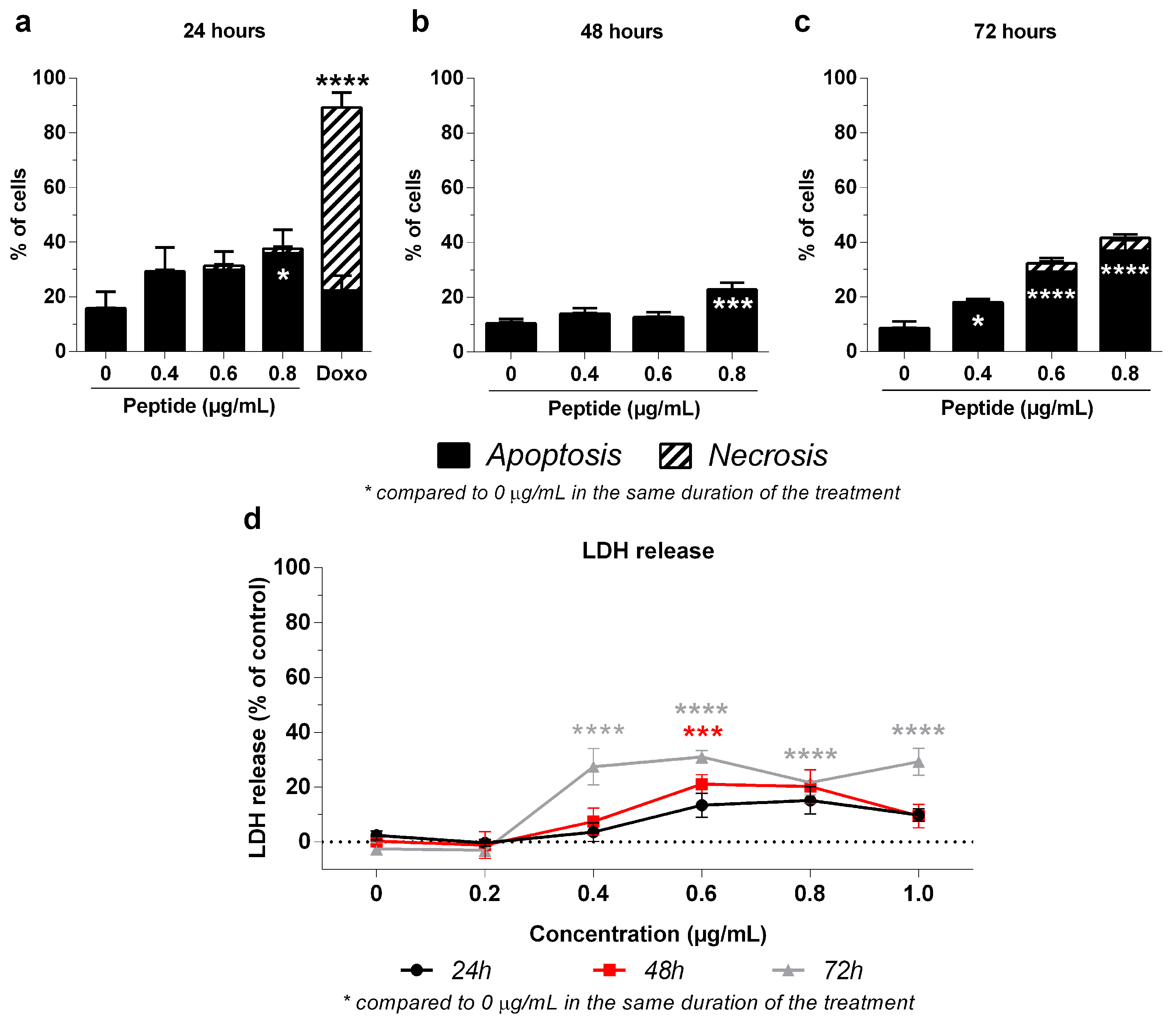
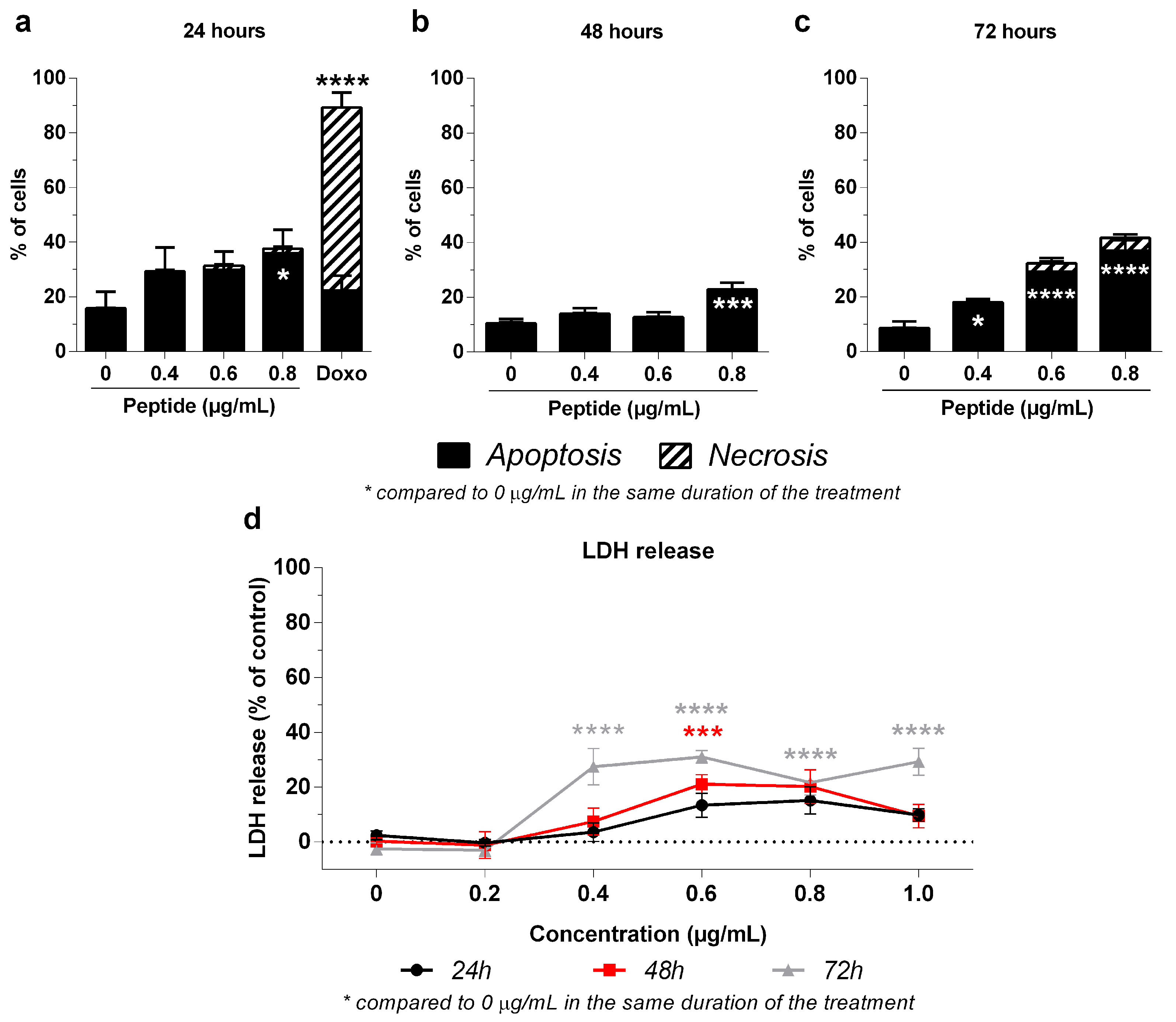
2.4. Mechanism of 3-NAntC-Induced Cellular Death
2.5. Cell Viability in the Presence of the Autophagy Inhibitor 3-MA
2.6. In Vivo Tolerability in Zebrafish
3. Discussion
4. Methods
4.1. Synthesis and Characterization of Crotoxin-Derived Peptides
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Proliferation Assay Using the BrdU Method
4.5. Cell Cycle Progression Assay
4.6. Cellular Death Assessment
4.7. Cellular Death Assay Using the LDH Release Method
4.8. In Vivo Zebrafish Toxicity Study
4.9. Statement of Ethical Approval
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Almansour, N.M. Triple-negative breast cancer: A brief review about epidemiology, risk factors, signaling pathways, treatment and role of artificial intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.; Hernández, J.M.H.; Rotello, V.M.; Ramirez, J.T. Triple-negative breast cancer: A review of conventional and advanced therapeutic strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Van Reesema, L.L.S.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on triple-negative breast cancer: Current treatment strategies, unmet needs, and potential targets for future therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]
- de Castro Figueiredo Bordon, K.; Cologna, C.T.; Fornari-Baldo, E.C.; Pinheiro-Júnior, E.L.; Cerni, F.A.; Amorim, F.G.; Anjolette, F.A.P.; Cordeiro, F.A.; Wiezel, G.A.; Cardoso, I.A.; et al. From animal poisons and venoms to medicines: Achievements, challenges and perspectives in drug discovery. Front. Pharmacol. 2020, 11, 1132. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, S.C.; Hyslop, S.; Fontes, M.R.; Prado-Franceschi, J.; Zambelli, V.O.; Magro, A.J.; Brigatte, P.; Gutierrez, V.P.; Cury, Y. Crotoxin: Novel activities for a classic beta-neurotoxin. Toxicon 2010, 55, 1045–1060. [Google Scholar] [CrossRef]
- He, J.-K.; Wang, J.-H.; Xie, Y.; Wu, J.-C.; Han, R.; Reid, P.F.; Qin, Z.-H. Crotoxin enhances the antitumor activity of gefinitib (Iressa) in SK-MES-1 human lung squamous carcinoma cells. Oncol. Rep. 2012, 27, 1341–1347. [Google Scholar] [CrossRef]
- Wang, J.; Qin, X.; Zhang, Z.; Chen, M.; Wang, Y.; Gao, B. Crotoxin suppresses the tumorigenic properties and enhances the antitumor activity of Iressa® (gefinitib) in human lung adenocarcinoma SPCA-1 cells. Mol. Med. Rep. 2014, 10, 3009–3014. [Google Scholar] [CrossRef]
- Yan, C.-H.; Yang, Y.-P.; Qin, Z.-H.; Gu, Z.-L.; Reid, P.; Liang, Z.-Q. Autophagy is involved in cytotoxic effects of crotoxin in human breast cancer cell line MCF-7 cells. Acta Pharmacol. Sin. 2007, 28, 540–548. [Google Scholar] [CrossRef]
- Ye, B.; Xie, Y.; Qin, Z.-H.; Wu, J.-C.; Han, R.; He, J.-K. Anti-tumor activity of CrTX in human lung adenocarcinoma cell line A549. Acta Pharmacol. Sin. 2011, 32, 1397–1401. [Google Scholar] [CrossRef]
- Cura, J.E.; Blanzaco, D.P.; Brisson, C.; Cura, M.A.; Cabrol, R.; Larrateguy, L.; Mendez, C.; Sechi, J.C.; Silveira, J.S.; Theiller, E.; et al. Phase I and pharmacokinetics study of crotoxin (cytotoxic PLA2, NSC-624244) in patients with advanced cancer. Clin. Cancer Res. 2002, 8, 1033–1041. [Google Scholar] [PubMed]
- Fernandes, C.A.H.; Pazin, W.M.; Dreyer, T.R.; Bicev, R.N.; Cavalcante, W.L.G.; Fortes-Dias, C.L.; Ito, A.S.; Oliveira, C.L.P.; Fernandez, R.M.; Fontes, M.R.M. Biophysical studies suggest a new structural arrangement of crotoxin and provide insights into its toxic mechanism. Sci. Rep. 2017, 7, srep43885. [Google Scholar] [CrossRef] [PubMed]
- Prijatelj, P.; Pražnikar, Z.J.; Petan, T.; Križaj, I.; Pungerčar, J. Mapping the structural determinants of presynaptic neurotoxicity of snake venom phospholipases A2. Toxicon 2008, 51, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Rouault, M.; Rash, L.D.; Escoubas, P.; Boilard, E.; Bollinger, J.; Lomonte, B.; Maurin, T.; Guillaume, C.; Canaan, S.; Deregnaucourt, C.; et al. Neurotoxicity and other pharmacological activities of the snake venom phospholipase A2OS2: The N-terminal region is more important than enzymatic activity. Biochemistry 2006, 45, 5800–5816. [Google Scholar] [CrossRef]
- Soares, A.M.; Mancin, A.C.; Cecchini, A.L.; Arantes, E.C.; França, S.C.; Gutiérrez, J.M.; Giglio, J.R. Effects of chemical modifications of crotoxin B, the phospholipase A2 subunit of crotoxin from Crotalus durissus terrificus snake venom, on its enzymatic and pharmacological activities. Int. J. Biochem. Cell Biol. 2001, 33, 877–888. [Google Scholar] [CrossRef]
- Rayamajhi, M.; Zhang, Y.; Miao, E.A. Detection of pyroptosis by measuring released lactate dehydrogenase activity. Methods Mol. Biol. 2013, 1040, 85–90. [Google Scholar] [PubMed]
- Lekshmi, A.; Varadarajan, S.N.; Lupitha, S.S.; Indira, D.; Mathew, K.A.; Nair, A.C.; Nair, M.; Prasad, T.; Sekar, H.; Gopalakrishnan, A.K.; et al. A quantitative real-time approach for discriminating apoptosis and necrosis. Cell Death Discov. 2017, 3, 16101. [Google Scholar] [CrossRef]
- Almeida, C.F.; Amaral, C.; Augusto, T.V.; Correia-Da-Silva, G.; de Andrade, C.M.; Torqueti, M.R.; Teixeira, N. The anti-cancer potential of crotoxin in estrogen receptor-positive breast cancer: Its effects and mechanism of action. Toxicon 2021, 200, 69–77. [Google Scholar] [CrossRef]
- Medioni, J.; Brizard, M.; Elaidi, R.; Reid, P.F.; Benlhassan, K.; Bray, D. Innovative design for a phase 1 trial with intra-patient dose escalation: The crotoxin study. Contemp. Clin. Trials Commun. 2017, 7, 186–188. [Google Scholar] [CrossRef]
- Kouyoumdjian, J.; Harris, J.; Johnson, M. Muscle necrosis caused by the sub-units of crotoxin. Toxicon 1986, 24, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Salvini, T.; Amaral, A.; Miyabara, E.; Turri, J.; Danella, P.; de Araújo, H.S. Systemic skeletal muscle necrosis induced by crotoxin. Toxicon 2001, 39, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.P.; Silva, V.A.O.; Silvestrini, A.V.P.; de Macedo, L.H.; Caetano, G.F.; Reis, R.M.; Mazzi, M.V. Crotoxin from Crotalus durissus terrificus venom: In vitro cytotoxic activity of a heterodimeric phospholipase A2 on human cancer-derived cell lines. Toxicon 2018, 156, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Mahadevappa, R.; Kwok, H.F. Venom-based peptide therapy: Insights into anti-cancer mechanism. Oncotarget 2017, 8, 100908–100930. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.; Lomonte, B. Antitumor effects of cationic synthetic peptides derived from Lys49 phospholipase A2 homologues of snake venoms. Cell Biol. Int. 2007, 31, 263–268. [Google Scholar] [CrossRef]
- Costa, T.R.; Menaldo, D.L.; Oliveira, C.Z.; Santos-Filho, N.A.; Teixeira, S.S.; Nomizo, A.; Fuly, A.L.; Monteiro, M.C.; de Souza, B.M.; Palma, M.S.; et al. Myotoxic phospholipases A2 isolated from Bothrops brazili snake venom and synthetic peptides derived from their C-terminal region: Cytotoxic effect on microorganism and tumor cells. Peptides 2008, 29, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Gebrim, L.C.; Marcussi, S.; Menaldo, D.L.; de Menezes, C.S.; Nomizo, A.; Hamaguchi, A.; Silveira-Lacerda, E.P.; Homsi-Brandeburgo, M.I.; Sampaio, S.V.; Soares, A.M.; et al. Antitumor effects of snake venom chemically modified lys49 phospholipase A2-like BthTX-I and a synthetic peptide derived from its C-terminal region. Biologicals 2009, 37, 222–229. [Google Scholar] [CrossRef]
- Amjad, M.T.; Chidharla, A.; Kasi, A. Cancer Chemotherapy; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK564367/ (accessed on 8 February 2024).
- DiPaola, R.S. To arrest or not to G2-M cell-cycle arrest: Commentary re: A. K. Tyagi et al., Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G2-M arrest, and apoptosis. Clin. Cancer Res. 2002, 8, 3512–3519, Erratum in Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar]
- He, J.-K.; Wu, X.-S.; Wang, Y.; Han, R.; Qin, Z.-H.; Xie, Y. Growth inhibitory effects and molecular mechanisms of crotoxin treatment in esophageal Eca-109 cells and transplanted tumors in nude mice. Acta Pharmacol. Sin. 2013, 34, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Radwan, E.M.; Abdullah, R.; Al-Qubaisi, M.S.; El Zowalaty, M.E.; Naadja, S.E.; Alitheen, N.B.; Omar, A.R. Effect of recombinant human erythropoietin and doxorubicin in combination on the proliferation of MCF-7 and MDA-MB231 breast cancer cells. Mol. Med. Rep. 2016, 13, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the MTT assay. Cold Spring Harb. Protoc. 2018, 2018, pdb-prot095505. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.M.; Bhattacharya, S.K. The use of bromodeoxyuridine incorporation assays to assess corneal stem cell proliferation. Methods Mol. Biol. 2013, 1014, 65–70. [Google Scholar] [PubMed]
- Darzynkiewicz, Z.; Juan, G.; Bedner, E. Determining cell cycle stages by flow cytometry. Curr. Protoc. Cell Biol. 2001, 1, 8.4.1–8.4.18. [Google Scholar] [CrossRef]
- Sawai, H.; Domae, N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem. Biophys. Res. Commun. 2011, 411, 569–573. [Google Scholar] [CrossRef]
- Ranjitkar, S.; Zhang, D.; Sun, F.; Salman, S.; He, W.; Venkitanarayanan, K.; Tulman, E.R.; Tian, X. Cytotoxic effects on cancerous and non-cancerous cells of trans-cinnamaldehyde, carvacrol, and eugenol. Sci. Rep. 2021, 11, 16281. [Google Scholar] [CrossRef]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezerra, P.; Motti, E.F. 3-NAntC: A Potent Crotoxin B-Derived Peptide against the Triple-Negative MDA-MB-231 Breast Cancer Cell Line. Molecules 2024, 29, 1646. https://doi.org/10.3390/molecules29071646
Bezerra P, Motti EF. 3-NAntC: A Potent Crotoxin B-Derived Peptide against the Triple-Negative MDA-MB-231 Breast Cancer Cell Line. Molecules. 2024; 29(7):1646. https://doi.org/10.3390/molecules29071646
Chicago/Turabian StyleBezerra, Patricia, and Eduardo F. Motti. 2024. "3-NAntC: A Potent Crotoxin B-Derived Peptide against the Triple-Negative MDA-MB-231 Breast Cancer Cell Line" Molecules 29, no. 7: 1646. https://doi.org/10.3390/molecules29071646