3.2. Experimental Synthetic Procedures
(–)-Methyl (S)-2-(benzyloxy)-3-((tert-butyldiphenylsilyl)oxy)propanoate (S2).
![Molecules 29 01647 i007]()
To a solution of alcohol
S1 [
18] (46.2 g, 1.00 equiv, 129 mmol) in anhydrous DMF (0.57 L, 0.23 M) at 0 °C, BnBr (30.6 mL, 2.00 equiv, 258 mmol) and NaH (5.67 g, 1.10 equiv, 142 mmol, 60% oil dispersion) were added. The reaction mixture was stirred at room temperature for 3 h. After cooling to 0 °C, water was added. The aqueous layer was extracted (3× with EtOAc and the combined organic layers were washed with brine, dried over MgSO
4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided methyl ester
S2 (57.1 g, 98% yield).
1H NMR and optical rotation correlate with the previously reported data for this compound [
19]. [α]
D25 −17 (
c 2.9, CHCl
3); literature value [
19] [α]
D23 −19.0 (c = 1.5, CHCl
3);
1H NMR (500 MHz, CDCl
3) δ 7.71–7.64 (m, 4H), 7.44–7.28 (m, 11H), 4.75 (d,
J = 11.9 Hz, 1H), 4.53 (d,
J = 11.9 Hz, 1H), 4.14 (t,
J = 5.1 Hz, 1H), 3.99–3.91 (m, 2H), 3.75 (s, 3H), 1.04 (s, 9H) ppm.
(+)-Methyl (2
S,3
R,4
S)-4-(benzyloxy)-2-bromo-5-((tert-butyldiphenylsilyl)oxy)-3-hydroxy-2-methylpentanoate (
34a) and (+)-Methyl (2
R,3
R,4
S)-4-(benzyloxy)-2-bromo-5-((tert-butyldiphenylsilyl)oxy)-3-hydroxy-2-methylpentanoate (
34b). To a stirred solution of aldehyde
32 [
19] (206 mg, 1.00 equiv, 0.492 mmol) in CH
2Cl
2 (0.74 mL, 0.66 M) at −78 °C, MgBr
2·OEt
2 (635 mg, 5.00 equiv, 2.46 mmol) was added. The reaction mixture was stirred for 15 min at −78 °C followed by slow addition of crude methyl ((2-bromo-1-methoxyprop-1-en-1-yl)oxy)trimethylsilane
33 [
20] (0.19 mL, 2.0 equiv, 0.98 mmol). The reaction mixture was stirred at −78 °C for 1 h. A mixture of 1N HCl/THF (1:1,
v/
v, 2.80 mL) was added followed by gradual warming to room temperature with stirring for 1 h. The aqueous layer was extracted (3×) with Et
2O and the combined organic layers were washed with a saturated solution of NaHCO
3, dried over MgSO
4, filtered, and concentrated under reduced pressure.
1H NMR of the crude reaction indicated a >20:1 ratio of 3,4-
syn:3,4-
anti products with a 1:1 mixture of C2-bromides
34a and
34b. Purification by flash chromatography (Hexanes/EtOAc) allowed for the two C2 diastereomers to be separated providing
34a (126 mg) and
34b (101 mg) for a combined yield of 79%. The 3,4-
syn and C2 stereochemistry were assigned from lactonization of
34a (see below and
Supplementary Information).
34a: R
f = 0.46 (Hexanes/EtOAc, 80:20); [α]
D25 + 11 (c 3.0, CH
2Cl
2); formula: C
30H
37BrO
5Si; MW: 585.61 g/mol; IR (neat) ν
max 3533, 3070, 2999, 2953, 2858, 1742, 1472, 1249, 1112 cm
−1;
1H NMR (500 MHz, CDCl
3) δ 7.72–7.67 (m, 4H), 7.48–7.42 (m, 2H), 7.42–7.38 (m, 4H), 7.33–7.27 (m, 3H), 7.22–7.19 (m, 2H), 4.65 (d,
J = 10.9 Hz, 1H), 4.41 (d,
J = 10.9 Hz, 1H), 4.29 (d,
J = 9.7 Hz, 1H), 3.97 (appt,
J = 6.1 Hz, 1H), 3.86–3.84 (m, 2H), 3.67 (s, 3H), 3.41 (d,
J = 9.7 Hz, 1H), 1.86 (s, 3H), 1.08 (s, 9H) ppm;
13C NMR (126 MHz, CDCl
3) δ 171.3, 137.5, 135.80 (2C), 135.77 (2C), 133.3, 133.1, 130.02, 129.99, 128.5 (2C), 128.14 (2C), 128.10, 128.0 (2C), 127.9 (2C), 76.8, 74.8, 72.8, 63.8, 61.6, 53.2, 27.0 (3C), 23.5, 19.3 ppm; HRMS (ESI)
m/
z: calcd for C
30H
38BrO
5Si [M+H]
+ 585.1666, found 585.1660 (–1.03 ppm).
34b: R
f = 0.95 (Hexanes/EtOAc 50:50); [α]
D25 + 9.2 (c 2.4, CH
2Cl
2); formula: C
30H
37BrO
5Si; MW: 585.6100 g/mol; IR (neat) ν
max 3552, 3070, 2953, 2932, 2858, 1737, 1472, 1428, 1261, 1209, 1112 cm
−1;
1H NMR (500 MHz, CDCl
3) δ 7.75–7.71 (m, 4H), 7.48 (dd,
J = 7.3, 1.3 Hz, 2H), 7.46–7.42 (m, 4H), 7.36–7.29 (m, 3H), 7.27–7.24 (m, 2H), 4.61 (d,
J = 10.9 Hz, 1H), 4.42 (d,
J = 11.0 Hz, 1H), 4.19 (d,
J = 10.4 Hz), 4.05–4.01 (m, 1H), 3.87–3.84 (m, 2H), 3.54 (s, 3H), 3.19 (d,
J = 10.3 Hz, 1H), 2.08 (s, 3H), 1.12 (s, 9H) ppm;
13C NMR (126 MHz, CDCl
3) δ 170.8, 137.7, 135.8 (2C), 135.7 (2C), 133.3, 133.2, 130.0, 129.96, 128.4 (2C), 128.0 (2C), 127.94 (4C), 127.91, 77.5, 76.9, 72.9, 65.7, 63.3, 53.3, 27.2, 27.0 (3C), 19.3 ppm; HRMS (ESI)
m/
z: calcd for C
30H
38BrO
5Si [M+H]
+ 585.1666, found 585.1666 (0 ppm).
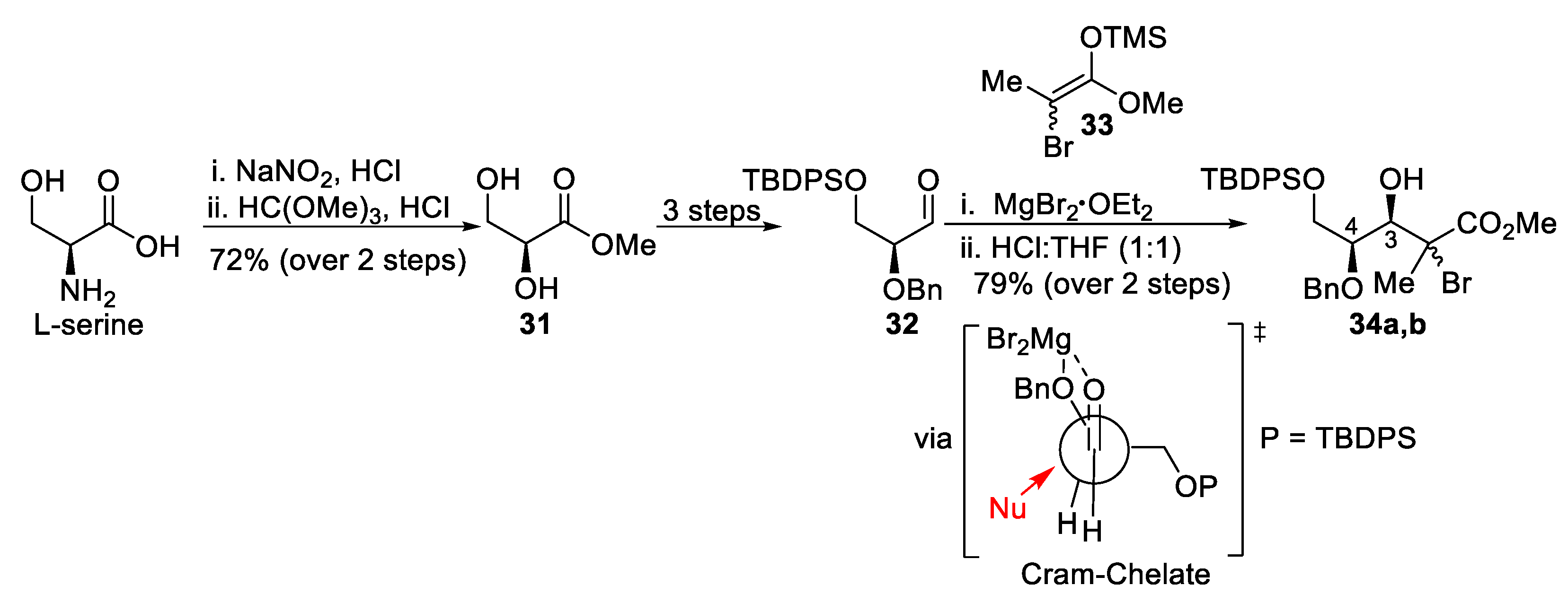
(+)-(3S,4R,5S)-5-(benzyloxy)-3-bromo-4-hydroxy-3-methyltetrahydro-2H-pyran-2-one (S3).
![Molecules 29 01647 i008]()
To a stirred solution of methyl ester 34a (72 mg, 1.0 equiv, 0.12 mmol) in THF (0.49 mL, 0.25 M) at 0 °C, 3HF·NEt3 (0.047 mL, 2.30 equiv, 0.283 mmol) was added. The reaction mixture was stirred at room temperature for 16 h and then quenched by addition of a saturated solution of NaHCO3. The aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided lactone S3 (22 mg, 57% yield). The 3,4 and C2 stereochemistry were determined by the relevant nuclear Overhauser effect (nOe) enhancements, as depicted in the SI. Rf = 0.66 (Hexanes/EtOAc, 50:50); [α]D25 +33 (c 2.2, CH2Cl2); formula: C13H15O4Br; MW: 315.1630 g/mol; IR (neat) νmax 3451, 2925, 2869, 1742, 1455, 1389, 1282, 1228, 1090 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.41–7.32 (m, 5H), 4.71 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 11.7 Hz, 1H), 4.66 (dd, J = 12.2, 5.6 Hz, 1H), 4.19 (dd, J = 12.2, 5.7 Hz, 1H), 3.96 (dt, J = 7.1, 5.6 Hz, 1H), 3.59 (dd, J = 7.0, 5.5 Hz, 1H), 2.67 (d, J = 5.8 Hz, 1H), 2.05 (s, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 168.1, 137.2, 128.9 (2C), 128.5, 128.1 (2C), 76.2, 75.8, 73.0, 68.8, 60.1, 26.3 ppm; HRMS (ESI) m/z: calcd for C13H16O4Br [M+H]+ 315.0226, found 315.0226 (0 ppm).
(–)-Methyl (2S,3R,4S)-4-(benzyloxy)-2-bromo-5-((tert-butyldiphenylsilyl)oxy)-3-((dimethyl (vinyl)silyl)oxy)-2-methylpentanoate (35a). To a stirred solution of alcohol 34a (56 mg, 1.0 equiv, 0.096 mmol) in CH2Cl2 (0.24 mL, 0.40 M) at 0 °C, imidazole (22 mg, 3.4 equiv, 0.33 mmol) was added, followed by chloro(dimethyl)vinylsilane (0.022 mL, 1.5 equiv, 0.14 mmol). The reaction mixture was stirred at room temperature for 19 h and then quenched by the addition of water. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 35a (53 mg, 83% yield). Rf = 0.93 (Hexanes/EtOAc, 30:70); [α]D25 −8.7 (c 4.9, CH2Cl2); formula: C34H45BrO5Si2; MW: 669.8030 g/mol; IR (neat) νmax 3070, 3049, 2999, 2953, 2858, 1742, 1454, 1252, 1127 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.73–7.66 (m, 5H), 7.47–7.43 (m, 2H), 7.43–7.36 (m, 4H), 7.25–7.18 (m, 4H), 6.06 (dd, J = 20.4, 14.9 Hz, 1H), 5.88 (dd, J = 14.9, 3.8 Hz, 1H), 5.66 (dd, J = 20.4, 3.8 Hz, 1H), 4.66 (d, J = 1.6 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 4.41 (d, J = 11.7 Hz, 1H), 4.09 (ddd, J = 7.3, 5.9, 1.6 Hz, 1H), 3.77 (dd, J = 10.7, 5.9 Hz, 1H), 3.74 (s, 3H), 3.72 (dd, J = 10.5, 7.1 Hz, 1H), 1.94 (s, 3H), 1.09 (s, 9H), 0.15 (s, 3H), 0.12 (s, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 171.6, 138.5, 137.9, 136.0 (2C), 135.8 (2C), 133.5, 133.4, 133.0, 130.0, 129.9, 128.3 (2C), 127.89 (4C), 127.85 (2C), 127.5, 78.5, 74.7, 72.6, 63.6, 62.3, 53.2, 27.1 (3C), 22.6, 19.3, −1.1, −1.3 ppm; HRMS (ESI) m/z: calcd for C34H46BrO5Si2 [M+H]+ 669.2062, found 669.2058 (−0.60 ppm).
(+)-Methyl (2R,3R,4S)-4-(benzyloxy)-2-bromo-5-((tert-butyldiphenylsilyl)oxy)-3-((dimethyl (vinyl)silyl)oxy)-2-methylpentanoate (35b). To a stirred solution of alcohol 34b (40 mg, 1.0 equiv, 0.068 mmol) in CH2Cl2 (0.17 mL, 0.40 M) at 0 °C, imidazole (16 mg, 3.4 equiv, 0.23 mmol) was added, followed by chloro(dimethyl)vinylsilane (0.016 mL, 1.5 equiv, 0.10 mmol). The reaction mixture was stirred at room temperature for 19 h and then quenched by the addition of water. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 35b (20 mg, 44% yield). Rf = 0.96 (Hexanes/EtOAc, 30:70); [α]D25 +4.2 (c 2.0, CH2Cl2); formula: C34H45BrO5Si2; MW: 669.8030 g/mol; IR (neat) νmax 3070, 3032, 2953, 2858, 1739, 1472, 1254, 1154, 1110 cm−1; 1H NMR (500 MHz, CDCl3) 7.69–7.63 (m, 5H), 7.47–7.42 (m, 2H), 7.42–7.36 (m, 5H), 7.25–7.23 (m, 1H), 7.16–7.13 (m, 2H), 6.20 (dd, J = 20.5, 14.9 Hz, 1H), 5.94 (dd, J = 14.9, 3.7 Hz, 1H), 5.71 (dd, J = 20.5, 3.7 Hz, 1H), 4.58 (d, J = 2.6 Hz, 1H), 4.41 (d, J = 11.4 Hz, 1H), 4.24 (d, J = 11.4 Hz, 1H), 3.78–3.69 (m, 2H), 3.62 (s, 3H), 3.58 (td, J = 6.0, 2.6 Hz, 1H), 1.92 (s, 3H), 1.07 (s, 9H), 0.29 (s, 3H), 0.25 (s, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 171.0, 137.9, 137.8, 135.8 (2C), 135.7 (2C), 133.39, 133.36, 133.3, 130.01, 129.95, 128.33 (2C), 128.29 (2C), 127.91 (2C), 127.90 (2C), 127.7, 80.2, 77.1, 73.0, 66.7, 63.8, 53.0, 27.0 (3C), 24.3, 19.3, −0.8, −1.3 ppm; HRMS (ESI) m/z: calcd for C34H46BrO5Si2 [M+H]+ 669.2062, found 669.2052 (–1.49 ppm).
(+)-Methyl(2
S,3
S,4
S)-4-(benzyloxy)-5-((tert-butyldiphenylsilyl)oxy)-3-hydroxy-2-methyl-2-vinylpentanoate (
36). To a stirred solution of C2-bromo esters
35a,b (58 mg, 1.0 equiv, 0.087 mmol) in toluene (1.5 mL, 0.060 M) at 0 °C, BEt
3 (0.17 mL, 2.0 equiv, 0.17 mmol, 1.0 M solution in hexanes) was added over 1 h in an open-air system. The reaction mixture was stirred for 1 h at 0 °C followed by the addition of acetic acid (10 μL, 2.0 equiv, 0.17 mmol) and MeOH (1.5 mL), with gradual warming to room temperature and stirring for an additional 30 min. The mixture was concentrated under reduced pressure.
1H NMR analysis of the crude mixture indicated a >20:1 diastereomeric ratio. Purification by flash chromatography (Hexanes/EtOAc) provided methyl ester
36 (26 mg, 57% yield). The 90% yield shown in
Scheme 5 was obtained on a larger 9 g scale. R
f = 0.22 (Hexanes/EtOAc, 90:10); [α]
D25 +7.8 (
c 1.6, MeOH); formula: C
32H
40O
5Si; MW: 532.7520 g/mol; IR (neat) ν
max 3561, 2952, 2858, 1732, 1456, 1192 cm
−1;
1H NMR (500 MHz, CDCl
3) δ 7.68–7.65 (m, 4H), 7.46–7.41 (m, 2H), 7.40–7.36 (m, 4H), 7.31–7.24 (m, 3H), 7.22–7.20 (m, 2H), 6.33 (dd,
J = 17.7, 11.0 Hz, 1H), 5.24 (d,
J = 11.1 Hz, 1H), 5.18 (d,
J = 17.7 Hz, 1H), 4.55 (d,
J = 10.9 Hz, 1H), 4.32 (d,
J = 10.9 Hz, 1H), 3.85–3.76 (m, 3H), 3.59–3.54 (m, 1H), 3.45 (s, 3H), 3.20 (d,
J = 10.2 Hz, 1H), 1.39 (s, 3H), 1.05 (s, 9H) ppm;
13C NMR (126 MHz, CDCl
3) δ 175.6, 138.9, 138.0, 135.8 (4C), 133.5, 133.3, 129.94, 129.91, 128.4 (2C), 128.0 (2C), 127.90 (2C), 127.89 (2C), 127.81, 115.0, 78.0, 76.9, 72.8, 63.6, 52.1, 51.7, 27.0 (3C), 19.4, 19.3 ppm; HRMS (ESI)
m/
z: calcd for C
32H
41O
5Si [M+H]
+ 533.2718, found 533.2720 (+0.38 ppm).
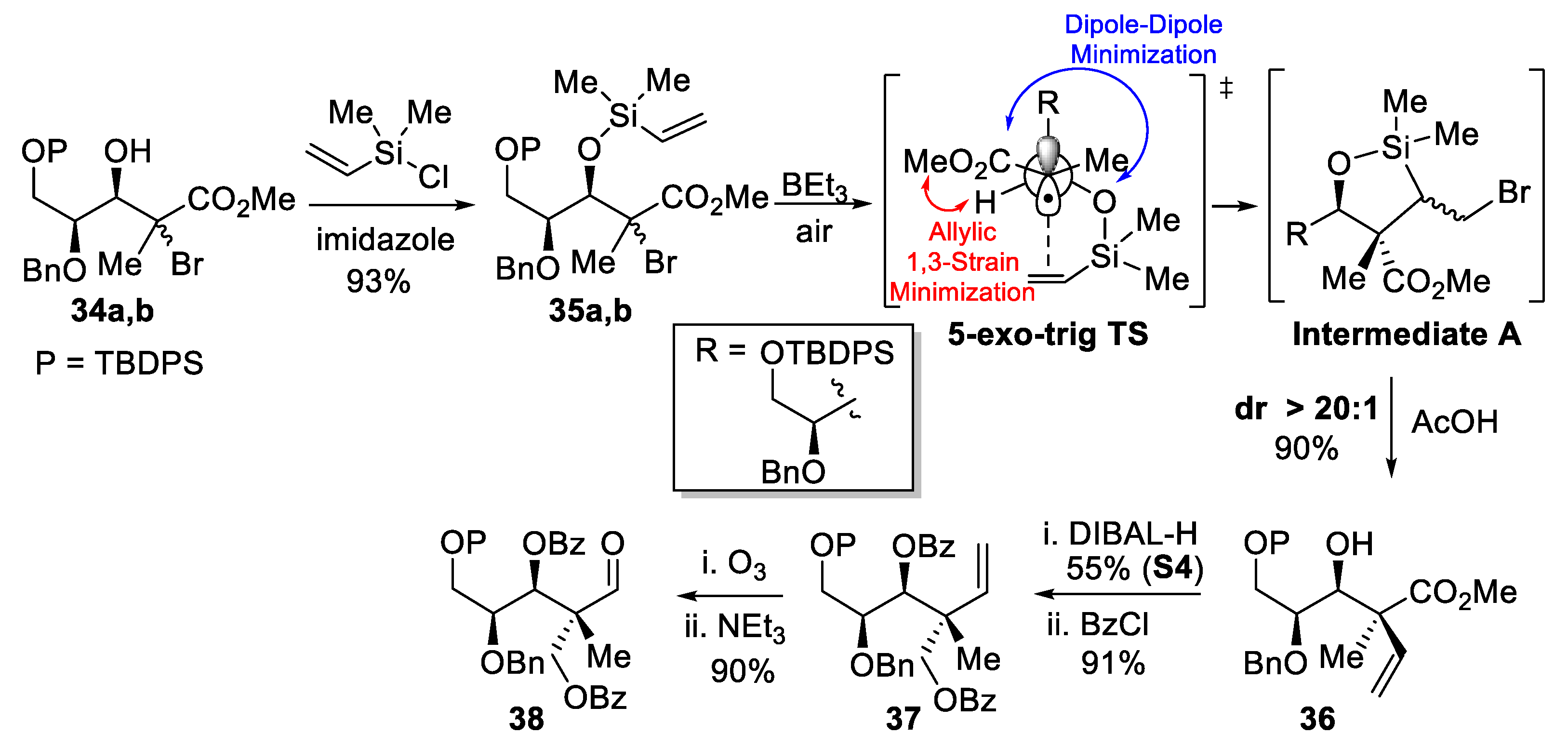
(+)-(2R,3S,4S)-4-(benzyloxy)-5-((tert-butyldiphenylsilyl)oxy)-2-methyl-2-vinylpentane-1,3-diol (S4).
![Molecules 29 01647 i009]()
To a stirred solution of methyl ester 36 (63 mg, 1.0 equiv, 0.12 mmol) in CH2Cl2 (0.91 mL, 0.13 M) at −78 °C, DIBAl-H (1 M in Hexanes, 0.47 mL, 4.0 equiv, 0.47 mmol) was added dropwise. The reaction mixture was stirred at −40 °C for 3 h and then quenched by the addition of a saturated aqueous solution of potassium sodium tartrate at −78 °C, followed by gradual warming to room temperature and stirring for 1 h. The aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided diol S4 (33 mg, 55% yield). Rf = 0.60 (Hexanes/EtOAc, 50:50); [α]D25 +18 (c 2.2, CH2Cl2); formula: C31H40O4Si; MW: 504.7420 g/mol; IR (neat) νmax 3453, 3070, 3032, 2858, 1456, 1112 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.71–7.63 (m, 4H), 7.47–7.42 (m, 2H), 7.41–7.36 (m, 4H), 7.33–7.27 (m, 3H), 7.24–7.22 (m, 2H), 6.06 (dd, J = 17.8, 11.0 Hz, 1H), 5.16 (dd, J = 11.0, 1.5 Hz, 1H), 5.11 (dd, J = 17.7, 1.6 Hz, 1H), 4.53 (d, J = 11.0 Hz, 1H), 4.38 (d, J = 11.0 Hz, 1H), 3.78 (dd, J = 8.6, 4.6 Hz, 1H), 3.75 (dd, J = 8.7, 3.6 Hz, 1H), 3.72 (d, J = 7.8 Hz, 1H), 3.63 (dd, J = 11.0, 7.8 Hz, 1H), 3.59 (dd, J = 10.9, 4.8 Hz, 1H), 3.51 (appt, J = 5.9 Hz, 1H), 3.05 (d, J = 8.1 Hz, 1H), 2.61 (dd, J = 7.8, 4.9 Hz, 1H), 1.07 (s, 9H), 1.02 (s, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 140.4, 137.7, 135.77 (2C), 135.76 (2C), 133.3, 133.2, 130.02, 130.00, 128.5 (2C), 128.1 (2C), 128.0, 127.9 (4C), 114.5, 77.1, 76.7, 72.6, 69.8, 63.9, 45.5, 27.0 (3C), 19.3, 19.0 ppm; HRMS (ESI) m/z: calcd for C31H40NaO4Si [M+Na]+ 527.2588, found 527.2587 (–0.19 ppm).
(–)-(2R,3S,4S)-4-(benzyloxy)-5-((tert-butyldiphenylsilyl)oxy)-2-methyl-2-vinylpentane-1,3-diyl dibenzoate (37). To a stirred solution of diol S4 (115 mg, 1.00 equiv, 0.228 mmol) in CH2Cl2 (1.6 mL, 0.15 M) at room temperature, DMAP (2.8 mg, 0.10 equiv, 0.023 mmol) and pyridine (0.11 mL, 6.0 equiv, 1.4 mmol) were added. The mixture was cooled to 0 °C and BzCl (0.079 mL, 3.0 equiv, 0.68 mmol) was added slowly. The reaction mixture was warmed to room temperature for 16 h. After cooling to 0 °C, ethylenediamine (0.038 mL, 2.5 equiv, 0.57 mmol) was added and stirred for 30 min at 0 °C. Upon warming to room temperature, the aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided alkene 37 (148 mg, 91% yield). Rf = 0.88 (Hexanes/EtOAc, 50:50); [α]D25 −35 (c 7.4, CH2Cl2); formula: C45H48O6Si; MW: 712.9580 g/mol; IR (neat) νmax 3070, 2930, 2857, 1719, 1264, 1106 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.12–8.03 (m, 4H), 7.70–7.62 (m, 4H), 7.59–7.52 (m, 2H), 7.43 (dd, J = 13.0, 7.7 Hz, 5H), 7.40–7.35 (m, 5H), 7.31–7.26 (m, 4H), 7.25–7.23 (m, 1H), 6.22 (dd, J = 17.7, 11.0 Hz, 1H), 5.67 (apps, 1H), 5.24–5.17 (m, 2H), 4.54 (d, J = 11.4 Hz, 1H), 4.49 (d, J = 11.5 Hz, 1H), 4.41 (d, J = 11.1 Hz, 1H), 4.31 (d, J = 11.1 Hz, 1H), 3.87–3.76 (m, 2H), 3.67 (appdd, J = 9.8, 6.3 Hz, 1H), 1.30 (s, 3H), 1.07 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.5, 165.8, 140.1, 138.3, 135.8 (2C), 135.7 (2C), 133.4, 133.2, 133.1, 133.0, 130.4, 130.2, 130.0 (2C), 129.84, 129.77 (2C), 129.69, 128.50 (2C), 128.48 (2C), 128.3 (2C), 127.84 (2C), 127.77 (2C), 127.75 (2C), 127.6, 115.4, 78.3, 75.0, 72.6, 68.9, 63.1, 44.7, 26.9 (3C), 19.3, 19.2 ppm; HRMS (ESI) m/z: calcd for C45H48NaO6Si [M+Na]+ 735.3112, found 735.3124 (+ 1.63 ppm).
(–)-(2R,3S,4S)-4-(benzyloxy)-5-((tert-butyldiphenylsilyl)oxy)-2-formyl-2-methylpentane-1,3-diyl dibenzoate (38). To a stirred solution of alkene 37 (148 mg, 1.00 equiv, 0.208 mmol) in CH2Cl2 (22 mL, 0.010 M) at −78 °C, O3 was added and bubbled under vacuum until the reaction mixture turned blue (about 30 min). The solution was then purged with nitrogen to remove excess ozone. Following the addition of NEt3 (0.029 mL, 1.0 equiv, 0.21 mmol), the reaction was stirred for 1 h while warming to room temperature. After filtering over MgSO4, the mixture was concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided aldehyde 38 (113 mg, 76% yield). Rf = 0.86 (Hexanes/EtOAc, 50:50); [α]D25 −20 (c 5.6, CH2Cl2); formula: C44H46O7Si; MW: 714.9300 g/mol; IR (neat) νmax 2931, 2857, 1721, 1263, 1106 cm−1; 1H NMR (500 MHz, CDCl3) δ 10.02 (s, 1H), 8.11 (d, J = 7.6 Hz, 2H), 8.01 (d, J = 7.6 Hz, 2H), 7.73–7.67 (m, 2H), 7.65–7.61 (m, 1H), 7.61–7.54 (m, 2H), 7.51–7.46 (m, 4H), 7.44–7.40 (m, 4H), 7.39–7.27 (m, 6H), 7.25–7.22 (m, 2H), 5.93 (d, J = 1.6 Hz, 1H), 4.56 (d, J = 11.3 Hz, 1H), 4.46 (appt, J = 11.8 Hz, 2H), 4.37 (d, J = 11.3 Hz, 1H), 3.86–3.83 (m, 1H), 3.81 (dd, J = 10.2, 5.2 Hz, 1H), 3.68 (dd, J = 9.9, 7.9 Hz, 1H), 1.31 (s, 3H), 1.08 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 200.6, 166.1, 165.5, 137.1, 135.68 (2C), 135.67 (2C), 133.5, 133.2, 133.1, 132.8, 130.2 (2C), 130.0, 129.9, 129.8 (2C), 129.7, 129.5, 128.7 (2C), 128.49 (2C), 128.42 (2C), 128.4 (2C), 128.1, 128.0 (2C), 127.8 (2C), 77.4, 75.1, 72.6, 65.7, 61.4, 52.9, 26.9 (3C), 19.2, 15.2 ppm; HRMS (ESI) m/z: calcd for C44H47O7Si [M+H]+ 715.3086, found 715.3076 (–1.40 ppm).
((3R,4S,5S)-4-(benzoyloxy)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(tert-butylthio)-3-methyltetrahydrofuran-3-yl)methyl benzoate (41a,b). To a stirred solution of aldehyde 38 (31 mg, 1.0 equiv, 0.04 mmol) in CH2Cl2 (0.43 mL, 0.10 M) at −60 °C, tert-butylthiol (19 μL, 4.0 equiv, 0.17 mmol) and boron trifluoride diethyletherate (7 μL, 1.3 equiv, 0.06 mmol) were added. The reaction mixture was stirred at −60 °C for 2 h followed by the addition of NEt3 (24 μL, 4.0 equiv, 0.17 mmol) with additional stirring for 15 min at −60 °C. A saturated solution of NaHCO3 was added and the crude mixture was warmed to room temperature. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 41a,b (27 mg, 91% yield) as a 5:1 mixture in favor of the α-L-anomer. Rf = 0.89 (Hexanes/EtOAc, 50:50); formula: C41H48O6SSi; MW: 696.9740 g/mol; IR (neat) νmax 2958, 2930, 2858, 1722, 1266, 1108 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.12–8.08 (m, 2H), 8.02–7.99 (m, 2H), 7.98–7.93 (m, 4H), 7.62–7.60 (m, 3H), 7.58–7.51 (m, 9H), 7.45–7.37 (m, 10H), 7.35 (d, J = 7.6 Hz, 2H), 7.34–7.27 (m, 4H), 7.14 (t, J = 7.7 Hz, 4H), 5.87 (d, J = 4.3 Hz, 1H, major), 5.85 (d, J = 4.9 Hz, 1H, minor), 5.41 (s, 1H, major), 5.21 (s, 1H, minor), 4.65 (ddd, J = 7.8, 5.9, 4.2 Hz, 1H, major), 4.55–4.50 (m, 1H, minor), 4.44 (d, J = 11.2 Hz, 1H, major), 4.41–4.37 (m, 2H, minor), 4.30 (d, J = 11.2 Hz, 1H, major), 4.01–3.96 (m, 1H, minor), 3.82–3.78 (m, 3H, 2× major and 1× minor), 1.47 (s, 3H, minor), 1.38 (s, 12H, major), 1.35 (s, 9H, minor), 0.92 (s, 18H, major and minor) ppm; 13C NMR (126 MHz, CDCl3) δ 166.4 (major), 165.5 (minor), 165.0 (minor), 165.0 (major), 135.73 (2C), 135.69 (2C), 135.56 (2C), 135.54 (2C), 133.32, 133.30, 133.24, 133.15, 133.11, 133.07, 133.04, 130.2, 130.1, 129.9 (2C), 129.81 (4C), 129.79 (2C), 129.76, 129.7, 129.63, 129.57 (2C), 128.61 (2C), 128.59 (2C), 128.5 (2C), 128.4 (2C), 127.82 (2C), 127.77 (2C), 127.65 (2C), 127.60 (2C), 90.3 (minor), 86.6 (major), 81.6 (minor), 80.1 (major), 78.2 (major), 77.0 (minor), 65.7 (major), 65.6 (minor), 62.4 (minor), 61.6 (major), 51.3 (minor), 50.9 (major), 43.7 (minor), 43.6 (major), 31.8 (major, 3C), 31.7 (minor, 3C), 26.9 (minor, 3C), 26.7 (major, 3C), 22.4 (minor), 19.11 (minor), 19.09 (major), 17.6 (major) ppm, due to overlapping carbon signals in the aromatic region, 2 peaks are hidden; HRMS (ESI) m/z: calcd for C41H49O6SSi [M+H]+ 697.3014, found 697.3009 (−0.72 ppm).
(–)-(2R,3S,4S)-4-(benzyloxy)-2-(bis(benzylthio)methyl)-5-((tert-butyldiphenylsilyl)oxy)-2-methylpentane-1,3-diyl dibenzoate (40). To a stirred solution of aldehyde 38 (40 mg, 1.0 equiv, 0.056 mmol) in CH2Cl2 (0.56 mL, 0.10 M) at −60 °C, benzyl mercaptan (27 μL, 4.0 equiv, 0.22 mmol) and boron trifluoride diethyletherate (9.0 μL, 1.3 equiv, 0.073 mmol) were added. The reaction mixture was stirred at −60 °C for 2 h, followed by the addition of NEt3 (31 μL, 4.0 equiv, 0.22 mmol) with an additional 15 min of stirring at −60 °C. A saturated solution of NaHCO3 was added and the crude was warmed to room temperature. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided dithioacetal 40 (46 mg, 87% yield). Rf = 0.57 (Hexanes/EtOAc, 80:20); [α]D25 −11 (c 4.6, CH2Cl2); formula: C58H60O6S2Si; MW: 945.3170 g/mol; IR (neat) νmax 3065, 2930, 2857, 1721, 1494, 1266, 1176, 1108 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.06 (dd, J = 8.3, 1.2 Hz, 2H), 7.93 (dd, J = 8.3, 1.2 Hz, 2H), 7.64 (dd, J = 8.0, 1.3 Hz, 2H), 7.60–7.51 (m, 4H), 7.44 (t, J = 7.8 Hz, 2H), 7.36 (t, J = 7.9 Hz, 3H), 7.30 (t, J = 7.4 Hz, 3H), 7.26–7.17 (m, 14H), 7.16–7.14 (m, 3H), 6.07 (d, J = 2.0 Hz, 1H), 4.70 (d, J = 11.8 Hz, 1H), 4.61 (d, J = 11.8 Hz, 1H), 4.48 (d, J = 11.7 Hz, 1H), 4.35 (d, J = 11.7 Hz, 1H), 3.96 (td, J = 6.4, 1.8 Hz, 1H), 3.84 (s, 1H), 3.80 (d, J = 12.9 Hz, 1H), 3.77 (dd, J = 10.6, 5.8 Hz, 1H), 3.73 (t, J = 12.6 Hz, 2H), 3.68 (d, J = 12.7 Hz, 1H), 3.61 (dd, J = 10.6, 6.6 Hz, 1H), 1.34 (s, 3H), 1.02 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.3, 165.6, 138.4, 137.63, 137.56, 135.8 (2C), 135.7 (2C), 133.4, 133.2, 133.1, 133.0, 130.21, 130.20 (2C), 130.18, 129.9 (2C), 129.73, 129.67, 129.4 (4C), 128.63 (2C), 128.56 (2C), 128.51 (2C), 128.47 (2C), 128.3 (2C), 127.8 (2C), 127.7 (2C), 127.4 (3C), 127.3, 127.2, 78.7, 74.5, 72.2, 67.8, 63.2, 58.6, 48.6, 37.5, 37.4, 26.9 (3C), 19.7, 19.2 ppm; HRMS (ESI) m/z: calcd for C58H60NaO6S2Si [M+Na]+ 967.3493, found 967.3483 (–1.03 ppm).
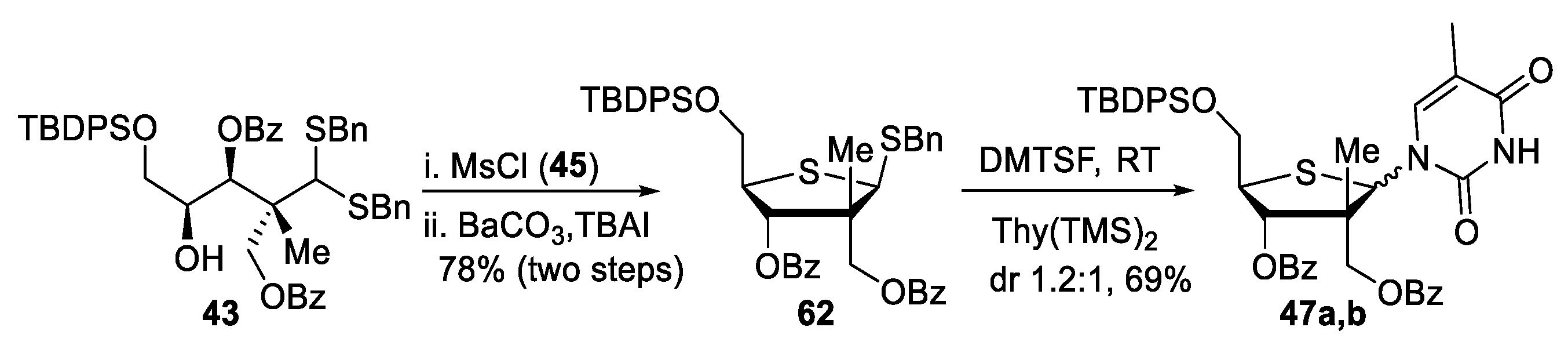
(+)-(2R,3S,4S)-2-(bis(benzylthio)methyl)-5-((tert-butyldiphenylsilyl)oxy)-4-hydroxy-2-methylpentane-1,3-diyl dibenzoate (43). To a stirred solution of C4-protected dithioacetal 40 (38 mg, 1.0 equiv, 0.040 mmol) in CH2Cl2 (0.40 mL, 0.10 M) at −78 °C, boron trichloride (1.0 M in DCM, 52 μL, 1.30 equiv, 0.052 mmol) was added. The reaction mixture was stirred at −78 °C for 1 h followed by the addition of boron trichloride (1.30 equiv). After stirring for another hour at −78 °C, a third addition of boron trichloride (1.30 equiv) was carried out. After one additional hour at −78 °C, the reaction was quenched by the addition of methanol, warmed to room temperature, and was then concentrated under reduced pressure. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided dithioacetal 43 (5 mg, 15% yield). Rf = 0.45 (Hexanes/EtOAc, 80:20); [α]D25 +15 (c 0.3, CH2Cl2); formula: C51H54O6S2Si; MW: 855.1920 g/mol; IR (neat) νmax 3505, 3066, 2929, 2857, 1722, 1268, 1177 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.01 (dd, J = 8.3, 1.2 Hz, 2H), 7.92 (dd, J = 8.3, 1.2 Hz, 2H), 7.58–7.52 (m, 6H), 7.41 (dt, J = 10.1, 7.9 Hz, 5H), 7.32 (t, J = 7.4 Hz, 2H), 7.25–7.21 (m, 8H), 7.19–7.14 (m, 5H), 5.77 (d, J = 0.8 Hz, 1H), 4.71 (d, J = 11.6 Hz, 1H), 4.60 (d, J = 11.6 Hz, 1H), 4.00–3.95 (m, 1H), 3.94 (s, 1H), 3.80 (d, J = 13.0 Hz, 1H), 3.74 (t, J = 12.3 Hz, 2H), 3.71 (d, J = 12.7 Hz, 1H), 3.56 (dd, J = 10.2, 5.3 Hz, 1H), 3.52 (dd, J = 10.2, 7.0 Hz, 1H), 2.52 (d, J = 5.7 Hz, 1H), 1.32 (s, 3H), 0.96 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.3, 165.6, 137.53, 137.51, 135.68 (2C), 135.65 (2C), 133.3, 133.1, 133.00, 132.98, 130.21, 130.17 (2C), 129.9 (2C), 129.83, 129.81, 129.3 (4C), 128.66 (2C), 128.64 (2C), 128.57 (2C), 128.55 (2C), 127.8 (5C), 127.4, 127.3, 74.4, 70.0, 67.4, 65.9, 57.7, 48.4, 37.5, 37.3, 26.9 (3C), 19.31, 19.25 ppm; HRMS (ESI) m/z: calcd for C51H54NaO6S2Si [M+Na]+ 877.3023, found 877.3014 (–1.03 ppm).
((3R,4S,5S)-4-(benzoyloxy)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-2-hydroxy-3-methyltetrahydrofuran-3-yl)methyl benzoate (42a,b). To a stirred solution of aldehyde 38 (50 mg, 1.0 equiv, 0.070 mmol) in THF:iPrOH (3:1, 3.5 mL, 0.020 M) at room temperature, palladium (10 wt.%) on activated carbon (30 mg, 0.40 equiv, 0.028 mmol) was added. The reaction mixture was degassed and flushed using a hydrogen-filled balloon. After stirring for 16 h, the reaction mixture was filtered through Celite®, washed with MeOH, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided a mixture of lactols 42a,b (36 mg, 82% yield) as a 3:1 mixture in favor of the β-L-anomer. Rf = 0.80 (Hexanes/EtOAc, 50:50); formula: C37H40O7Si; MW: 624.8050 g/mol; 1H NMR (500 MHz, CDCl3) δ 7.99 (ddd, J = 7.1, 3.7, 2.1 Hz, 4H), 7.98–7.92 (m, 4H), 7.63–7.57 (m, 3H), 7.56–7.51 (m, 6H), 7.50–7.47 (m, 3H), 7.40 (dtd, J = 16.8, 8.8, 5.7 Hz, 8H), 7.34–7.30 (m, 8H), 7.19 (dt, J = 15.2, 7.6 Hz, 4H), 5.81 (d, J = 4.9 Hz, 1H, minor), 5.78 (d, J = 5.4 Hz, 1H, major), 5.52 (s, 1H, minor), 5.20 (s, 1H, major), 4.67 (q, J = 5.9 Hz, 1H, minor), 4.64–4.61 (m, 1H, major), 4.60 (d, J = 11.2 Hz, 1H, major), 4.49 (d, J = 11.2 Hz, 1H, major), 4.45 (d, J = 11.2 Hz, 1H, minor), 4.36 (d, J = 11.1 Hz, 1H, minor), 3.93 (dd, J = 10.5, 7.1 Hz, 1H, major), 3.84 (dd, J = 10.4, 7.0 Hz, 1H, minor), 3.80 (dt, J = 10.1, 4.8 Hz, 2H, major and minor), 1.39 (s, 3H, minor), 1.32 (s, 3H, major), 0.95 (s, 9H, minor), 0.94 (s, 9H, major) ppm, labile protons were not observed due to exchange; 13C NMR (126 MHz, CDCl3) δ 166.5 (major), 166.4 (minor), 165.34 (minor), 165.31 (major), 135.8 (2C), 135.70 (2C), 135.64 (2C), 135.60 (2C), 133.5, 133.4, 133.2, 133.14, 133.13, 133.11, 133.0, 132.9, 130.03, 129.95 (2C), 129.89, 129.85, 129.82, 129.75 (2C), 129.74, 129.70 (2C), 129.61 (2C), 129.57, 128.7 (2C), 128.6 (2C), 128.51 (2C), 128.48 (2C), 127.8 (2C), 127.79 (2C), 127.72 (2C), 127.68 (2C), 103.2 (major), 100.9 (minor), 81.8 (major), 79.1 (minor), 78.8 (minor), 77.1 (major), 66.2 (minor), 64.5 (major), 62.7 (major), 62.0 (minor), 51.2 (major), 50.5 (minor), 26.79 (major, 3C), 26.75 (minor, 3C), 20.0 (major), 19.14 (minor), 19.09 (major), 16.0 (minor) ppm, due to overlapping carbon signals in the aromatic region 2 peaks are hidden; HRMS (ESI) m/z: calcd for C37H40NaO7Si [M+Na]+ 647.2435, found 647.2427 (–1.24 ppm).
((3R,4S,5S)-4-(benzoyloxy)-2-(benzylthio)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-3-methyltetrahydrofuran-3-yl)methyl benzoate (44a,b). To a stirred solution of lactols 42a,b (36 mg, 1.0 equiv, 0.058 mmol) in CH2Cl2 (0.50 mL, 0.11 M) at −60 °C, benzyl mercaptan (27 μL, 4.0 equiv, 0.23 mmol) and boron trifluoride diethyletherate (18 μL, 2.5 equiv, 0.14 mmol) were added. The reaction mixture was stirred at −40 °C for 2 h. The reaction was quenched by the addition of triethylamine (32 μL, 4.0 equiv, 0.23 mmol) at −60 °C, followed by stirring for 15 min. After the addition of a saturated solution of NaHCO3, the crude was warmed to room temperature. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 44a,b (35 mg, 83% yield) as a 2:1 mixture in favor of the α-L-anomer. Rf = 0.47 (Hexanes/EtOAc, 80:20); formula: C44H46O6SSi; MW: 730.9910 g/mol; IR (neat) νmax 3069, 2931, 2857, 1724, 1270 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 7.1 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 7.1 Hz, 4H), 7.67–7.63 (m, 6H), 7.59–7.48 (m, 7H), 7.46–7.40 (m, 6H), 7.37 (t, J = 5.7 Hz, 6H), 7.32 (d, J = 6.1 Hz, 5H), 7.29–7.21 (m, 6H), 7.18 (t, J = 7.9 Hz, 6H), 5.87 (d, J = 4.9 Hz, 1H, minor), 5.79 (d, J = 4.4 Hz, 1H, major), 5.25 (s, 1H, major), 4.97 (s, 1H, minor), 4.69–4.59 (m, 2H, major and minor), 4.38 (d, J = 3.7 Hz, 2H, minor), 4.35 (d, J = 11.0 Hz, 1H, major), 4.17 (d, J = 11.2 Hz, 1H, major), 4.09–4.01 (m, 1H, minor), 3.89 (s, 2H), 3.84 (appd, J = 8.2 Hz, 5H), 1.38 (s, 3H, major), 1.36 (s, 3H, minor), 0.97 (s, 9H, major), 0.94 (s, 9H, minor) ppm; 13C NMR (126 MHz, CDCl3) δ 166.25 (major), 166.24 (minor), 165.1 (major), 165.0 (minor), 138.0, 137.7, 135.739 (2C), 135.733 (2C), 135.62 (2C), 135.56 (2C), 133.4, 133.34, 133.33, 133.25, 133.22, 133.21, 133.17, 133.069, 133.066, 130.2, 129.9, 129.83 (4C), 129.82 (4C), 129.79 (2C), 129.7, 129.62, 129.61, 129.552, 129.548, 129.225, 129.214 (2C), 128.7 (2C), 128.63 (2C), 128.58 (4C), 128.5 (2C), 128.4 (2C), 127.8 (2C), 127.7 (2C), 127.6 (2C), 127.3 (2C), 127.1, 91.0 (minor), 88.8 (major), 82.1 (minor), 79.7 (major), 79.0 (major), 76.7 (minor), 66.1 (major), 64.8 (minor), 62.6 (minor), 61.6 (major), 51.5 (minor), 50.8 (major), 35.7 (major), 35.4 (minor), 26.8 (major, 3C), 26.7 (minor, 3C), 22.3 (minor), 19.2 (major), 19.1 (minor), 18.0 (major) ppm; HRMS (ESI) m/z: calcd for C44H46NaO6SSi [M+Na]+ 753.2676, found 753.2662 (–1.86 ppm).
(+)-(2R,3S,4S)-2-(bis(benzylthio)methyl)-5-((tert-butyldiphenylsilyl)oxy)-4-hydroxy-2-methylpentane-1,3-diyl dibenzoate (43). To a stirred solution of lactols 42a,b (273 mg, 1.00 equiv, 0.437 mmol) in CH2Cl2 (4.4 mL, 0.10 M) at −40 °C, benzyl mercaptan (0.25 mL, 4.8 equiv, 2.1 mmol) and TiCl4 (1.2 mL, 2.6 equiv, 1.2 mmol, 1M DCM) were added. The reaction mixture was stirred at −20 °C for 4 h. The reaction was quenched by the addition of triethylamine (0.3 mL, 4.8 equiv, 2.1 mmol) at −60 °C followed by stirring for 15 min. After the addition of a saturated solution of NaHCO3, the mixture was warmed to room temperature. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 43 (299 mg, 80% yield), which corresponds to the same product obtained from C4-OBn deprotection of dithioacetal 40, as reported above.
(2R,3S,4S)-2-(bis(benzylthio)methyl)-5-((tert-butyldiphenylsilyl)oxy)-2-methyl-4-((methylsulfonyl)oxy)pentane-1,3-diyl dibenzoate (45). To a stirred solution of dithioacetal 43 (434 mg, 1.00 equiv, 0.507 mmol) in pyridine (8.7 mL, 0.06 M) at 0 °C, methanesulfonyl chloride (80 μL, 2.0 equiv, 1.0 mmol) was added. The reaction mixture was stirred at room temperature for 3 h. The reaction was concentrated and then diluted with CH2Cl2. The aqueous layer was extracted (3×) with CH2Cl2 and the combined organic layers were washed with HCl (0.1N), a saturated solution of NaHCO3, and with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. C4-mesylated dithioacetal 45 was used without further purification. 1H NMR (500 MHz, CDCl3) δ 8.01 (dd, J = 8.3, 1.3 Hz, 2H), 7.94 (dd, J = 8.3, 1.2 Hz, 2H), 7.64–7.51 (m, 7H), 7.45 (t, J = 7.8 Hz, 2H), 7.40–7.28 (m, 7H), 7.20 (dd, J = 10.6, 4.9 Hz, 4H), 7.17–7.13 (m, 3H), 7.12 (d, J = 4.4 Hz, 3H), 6.01 (d, J = 3.2 Hz, 1H), 5.30–5.26 (m, 1H), 4.68 (d, J = 12.0 Hz, 1H), 4.39 (d, J = 11.9 Hz, 1H), 4.00 (dd, J = 11.2, 7.0 Hz, 1H), 3.89 (s, 1H), 3.79 (d, J = 12.8 Hz, 1H), 3.73 (dd, J = 11.3, 5.4 Hz, 1H), 3.71 (d, J = 12.8 Hz, 1H), 3.62 (s, 2H), 2.96 (s, 3H), 1.39 (s, 3H), 1.00 (s, 9H) ppm.
Preparation of silylated thymine. To a suspension of thymine (0.80 g, 1.0 equiv., 6.4 mmol) in HMDS (4.0 mL, 3.0 equiv., 19 mmol) under inert atmosphere, (NH4)2SO4 (18 mg, 0.022 equiv., 0.14 mmol) was added. The reaction mixture was refluxed until a clear solution was obtained (3 h). Upon cooling to room temperature, the solution was placed under high vacuum for approximately 1 h to remove excess HMDS. A 0.78 M solution of the silylated nucleobase was made in CH2Cl2.
(+)-(2R,3S,4S)-2-((R)-(benzylthio)(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-5-((tert-butyldiphenylsilyl)oxy)-2-methyl-4-((methylsulfonyl)oxy)pentane-1,3-diyl dibenzoate (46a) and (–)-(2R,3S,4R)-2-((S)-(benzylthio)(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl) methyl)-5-((tert-butyldiphenylsilyl)oxy)-2-methyl-4-((methylsulfonyl)oxy)pentane-1,3-diyl dibenzoate (46b). To a stirred solution of crude C4-Ms dithioacetal 45 (200 mg, 1.00 equiv, 0.214 mmol) in anhydrous CH2Cl2 (1.1 mL, 0.20 M), silylated thymine (0.82 mL, 3.0 equiv, 0.64 mmol, 0.78 M in CH2Cl2) was added. The resulting solution was cooled to 0 °C and iodide (114 mg, 2.10 equiv, 0.450 mmol) was added, followed by stirring at room temperature for 3 h. After cooling to 0 °C, the reaction was quenched by theaddition of a saturated solution of Na2S2O3 and dissolved in EtOAc. The aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. 1H NMR analysis of the crude mixture indicated a 1.1:1 ratio of thioaminals. Purification by flash chromatography (Hexanes/EtOAc) provided thioaminals 46a,b (154 mg, 77% yield over 2 steps). A pure fraction of each isomer was obtained for characterization. 46a (1′,2′-anti): Rf = 0.17 (Hexanes/EtOAc, 60:40); [α]D25 +53 (c 0.9, CH2Cl2); formula: C50H54N2O10S2Si; MW: 935.1910 g/mol; IR (neat) νmax 3182, 3032, 2931, 2857, 1724, 1683, 1264, 1105 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.09 (dd, J = 8.3, 1.2 Hz, 2H), 7.69 (ddd, J = 7.8, 6.4, 1.4 Hz, 4H), 7.62 (ddd, J = 16.1, 5.5, 4.3 Hz, 2H), 7.54 (dd, J = 8.0, 1.3 Hz, 2H), 7.47 (ddd, J = 14.3, 7.5, 2.2 Hz, 4H), 7.41 (dt, J = 13.7, 6.7 Hz, 3H), 7.30 (dd, J = 9.5, 6.2 Hz, 3H), 7.21–7.14 (m, 7H), 6.22 (s, 1H), 6.14 (d, J = 1.9 Hz, 1H), 5.45–5.40 (m, 1H), 4.69 (s, 2H), 4.05 (dd, J = 10.5, 5.3 Hz, 1H), 3.82–3.76 (m, 2H), 3.71 (dd, J = 10.5, 8.3 Hz, 1H), 3.30 (s, 3H), 1.69 (s, 3H), 1.26 (s, 3H), 1.08 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 165.9, 165.3, 162.4, 151.2, 137.8, 136.5, 135.7 (2C), 135.6 (2C), 133.9, 133.5, 132.6, 132.5, 130.3 (2C), 130.1, 129.9, 129.25, 129.24 (2C), 129.0, 128.9 (2C), 128.8 (2C), 128.64 (2C), 128.58(2C), 128.0 (2C), 127.8 (2C), 127.6, 110.2, 77.5, 73.3, 66.6, 66.0, 62.3, 48.5, 39.4, 37.1, 26.8 (3C), 19.3, 16.2, 12.7 ppm; HRMS (ESI) m/z: calcd for C50H55N2O10S2Si [M+H]+ 935.3062, found 935.3049 (–1.39 ppm). 46b (1′,2′-syn): Rf = 0.77 (Hexanes/EtOAc, 30:70); [α]D25 −8 (c 0.3, CH2Cl2); formula: C50H54N2O10S2Si; MW: 934.1910 g/mol; IR (neat) νmax 2955, 2922, 2852, 1726, 1686, 1261, 1107 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 7.1 Hz, 2H), 7.96 (d, J = 7.1 Hz, 2H), 7.68 (s, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.55 (d, J = 6.6 Hz, 2H), 7.50 (q, J = 7.1 Hz, 4H), 7.39–7.34 (m, 4H), 7.32–7.28 (m, 3H), 7.27 (d, J = 3.4 Hz, 1H), 7.18 (t, J = 7.5 Hz, 2H), 6.99 (dt, J = 15.1, 7.4 Hz, 4H), 6.91 (t, J = 7.2 Hz, 1H), 6.13 (s, 1H), 5.80 (d, J = 2.9 Hz, 1H), 5.21 (ddd, J = 7.8, 5.0, 3.1 Hz, 1H), 4.56 (d, J = 12.8 Hz, 1H), 4.10 (d, J = 12.9 Hz, 1H), 4.01 (dd, J = 11.4, 7.6 Hz, 1H), 3.75 (dd, J = 11.4, 5.0 Hz, 1H), 3.53 (d, J = 14.2 Hz, 1H), 3.49 (d, J = 14.2 Hz, 1H), 2.95 (s, 3H), 1.50 (s, 3H), 1.33 (s, 3H), 0.96 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 165.6, 165.2, 162.6, 151.4, 138.0, 136.5, 135.54 (2C), 135.50 (2C), 133.8, 133.5, 132.5, 132.4, 130.12 (2C), 130.09, 130.05 (3C), 129.2, 129.1, 128.8 (2C), 128.7 (2C), 128.6 (2C), 128.4 (2C), 128.0 (2C), 127.9 (2C), 127.3, 111.0, 79.7, 70.9, 65.5, 65.2, 64.2, 47.8, 39.5, 37.1, 26.9 (3C), 19.3, 15.1, 12.4 ppm; HRMS (ESI) m/z: calcd for C50H54N2NaO10S2Si [M+Na]+ 957.2881, found 957.2884 (+0.31 ppm).
(–)-(2R,3S,4R)-2-((S)-(benzylthio)(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-5-((tert-butyldiphenylsilyl)oxy)-4-hydroxy-2-methylpentane-1,3-diyl dibenzoate (48). To a 1.1:1 mixture of thioaminals 46a,b (266 mg, 1.00 equiv, 0.284 mmol) in a high-pressure flask, anhydrous 2,6-lutidine (2.9 mL, 0.10 M), and sodium iodide (426 mg, 10.0 equiv, 2.84 mmol) were added. The reaction mixture was stirred at 160 °C for 16 h in a sand bath. After cooling to room temperature, the volatiles were removed under reduced pressure. 1H NMR analysis of the crude mixture indicated a 7:1 ratio of β:α thiofuranosides 47a,b, along with unreacted 1′,2′-syn thioaminal 46b and side-product 48. Purification by flash chromatography (Hexanes/EtOAc) provided thiofuranoside 47a (106 mg, 49% yield), 47b (8 mg, 5%), 1′,2′-syn thioaminal 46b (94 mg, 35%) and side-product 48 (14 mg, 6%). Thiofuranosides 47a,b corresponded to those characterized below for thymine addition onto the cyclic thiofuranoside 62. 48: Rf = 0.84 (Hexanes/EtOAc, 30:70); [α]D25 −27 (c 3.3, CH2Cl2); formula: C49H52N2O8SSi; MW: 857.1060 g/mol; IR (neat) νmax 3069, 2930, 2857, 1679, 1260, 1105 cm−1; 1H NMR (500 MHz, CDCl3) 8.17 (s, 1H), 8.11 (dd, J = 8.4, 1.3 Hz, 2H), 7.93 (dd, J = 8.4, 1.3 Hz, 2H), 7.62–7.57 (m, 5H), 7.50 (dd, J = 8.1, 1.4 Hz, 2H), 7.47–7.43 (m, 2H), 7.43–7.35 (m, 5H), 7.30 (t, J = 7.1 Hz, 2H), 7.15–7.11 (m, 4H), 7.09 (d, J = 8.2 Hz, 2H), 6.34 (s, 1H), 5.63 (d, J = 8.5 Hz, 1H), 4.71 (d, J = 12.0 Hz, 1H), 4.50 (d, J = 12.1 Hz, 1H), 4.11–4.00 (m, 1H), 3.88 (d, J = 8.3 Hz, 1H), 3.70–3.66 (m, 1H), 3.66–3.60 (m, 2H), 3.50 (d, J = 14.0 Hz, 1H), 1.71 (s, 3H), 1.20 (s, 3H), 1.00 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.2, 165.4, 162.8, 151.9, 138.4, 136.8, 135.7 (2C), 135.6 (2C), 133.5, 133.4, 132.9, 132.7, 130.0 (2C), 129.93 (2C), 129.85, 129.83, 129.7, 129.5, 128.68 (2C), 128.66 (2C), 128.6 (2C), 128.5 (2C), 127.8 (2C), 127.7 (2C), 127.4, 110.9, 72.5, 71.8, 66.8, 66.0, 65.1, 47.9, 37.6, 26.9 (3C), 19.2, 16.0, 12.7 ppm; HRMS (ESI) m/z: calcd for C49H52N2NaO8SSi [M+Na]+ 879.3106, found 879.3111 (+0.57 ppm).
(–)-(2R,3S,4R)-2-((S)-(benzylthio)(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-5-((tert-butyldiphenylsilyl)oxy)-2-methyl-4-((methylsulfonyl)oxy)pentane-1,3-diyl dibenzoate (49). To a stirred solution of alcohol 48 (20 mg, 1.0 equiv, 0.023 mmol) in pyridine (0.4 mL, 0.06 M) at 0 °C, MsCl (4 μL, 2 equiv, 0.05 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 3 h. After completion of the reaction, the mixture was concentrated under reduced pressure, diluted in CH2Cl2, washed with a solution of HCl (0.1N), a saturated solution of NaHCO3, and with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided the pure product 49 (13 mg, 60% yield). Rf = 0.55 (Hexanes/EtOAc, 30:70); [α]D25 −18 (c 1.0, CH2Cl2); formula: C50H54N2O10S2Si; MW: 935.1910 g/mol; IR (neat) νmax 3177, 3070, 2932, 2857, 1726, 1688, 1261, 1105 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.03 (dd, J = 7.3, 1.1 Hz, 2H), 7.94 (dd, J = 7.3, 1.2 Hz, 2H), 7.76 (s, 1H), 7.59 (dd, J = 8.0, 1.2 Hz, 4H), 7.53 (dd, J = 6.8, 1.2 Hz, 2H), 7.48–7.42 (m, 4H), 7.39 (dd, J = 14.1, 6.9 Hz, 1H), 7.33 (dd, J = 14.8, 7.3 Hz, 3H), 7.28 (d, J = 7.4 Hz, 1H), 7.25 (d, J = 8.2 Hz, 2H), 6.98 (d, J = 4.6 Hz, 4H), 6.94–6.89 (m, 1H), 6.10 (s, 1H), 5.94 (d, J = 2.6 Hz, 1H), 5.20 (dt, J = 7.6, 2.8 Hz, 1H), 4.40 (d, J = 12.6 Hz, 1H), 4.16 (d, J = 12.6 Hz, 1H), 3.95 (dd, J = 12.0, 3.2 Hz, 1H), 3.83 (dd, J = 12.0, 7.6 Hz, 1H), 3.50 (d, J = 14.2 Hz, 1H), 3.41 (d, J = 14.2 Hz, 1H), 2.98 (s, 3H), 1.45 (s, 3H), 1.13 (s, 3H), 0.99 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 165.7, 164.9, 162.5, 151.4, 137.8, 136.5, 135.7 (2C), 135.6 (2C), 133.8, 133.6, 132.4, 132.3, 130.3, 130.2 (2C), 130.1, 129.9 (2C), 129.1, 129.0, 128.9 (2C), 128.7 (2C), 128.51 (2C), 128.45 (2C), 128.1 (2C), 128.0 (2C), 127.4, 111.2, 83.0, 74.5, 65.9, 65.2, 63.4, 47.4, 39.2, 37.1, 26.9 (3C), 19.2, 15.6, 12.3 ppm; HRMS (ESI) m/z: calcd for C50H55N2O10S2Si [M+H]+ 935.3062, found 935.3069 (+0.75 ppm).
(–)-((2S,3R,4S,5S)-4-(benzoyloxy)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-3-methyl-2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrothiophen-3-yl)methyl benzoate (50). To a stirred solution of C4′-OMs thioaminal 49 (13 mg, 1.0 equiv, 0.014 mmol) in a high-pressure flask, 2,6-lutidine (0.13 mL, 0.11 M) and NaI (22 mg, 10 equiv, 0.14 mmol) were added. The reaction mixture was stirred at 160 °C for 16 h in a sand bath. After completion of the reaction, the mixture was concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided the β-L-thiofuranoside 50 (8 mg, 77% yield). Rf = 0.64 (Hexanes/EtOAc, 30:70); [α]D25 −65 (c 0.6, CH2Cl2); formula: C42H44N2O7SSi; MW: 748.9660 g/mol; IR (neat) νmax 3194, 3070, 2931, 2857, 1725, 1691, 1274, 1263, 1107 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.02 (s, 1H), 7.98 (d, J = 1.3 Hz, 1H), 7.93 (dd, J = 8.1, 1.5 Hz, 2H), 7.77 (dd, J = 8.2, 1.5 Hz, 2H), 7.64 (dd, J = 8.1, 1.5 Hz, 2H), 7.60 (t, J = 7.5 Hz, 1H), 7.53 (dd, J = 7.8, 1.1 Hz, 2H), 7.45–7.33 (m, 9H), 7.27 (t, J = 7.4 Hz, 2H), 6.30 (s, 1H), 5.84 (d, J = 4.3 Hz, 1H), 4.28–4.23 (m, 2H), 4.22–4.12 (m, 2H), 3.81 (dd, J = 9.6, 7.4 Hz, 1H), 1.85 (s, 3H), 1.54 (s, 3H), 1.00 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.0, 165.4, 162.9, 151.1, 138.3, 135.8 (2C), 135.6 (2C), 133.9, 133.3, 132.8, 132.7, 130.1, 130.0, 129.8 (2C), 129.6 (2C), 129.4, 129.0, 128.9 (2C), 128.5 (2C), 127.92 (2C), 127.90 (2C), 110.5, 80.0, 68.3, 64.0, 63.1, 55.8, 53.9, 26.8 (3C), 23.3, 19.3, 13.0 ppm; HRMS (ESI) m/z: calcd for C42H45N2O7SSi [M+H]+ 749.2711, found 749.2694 (–2.27 ppm).
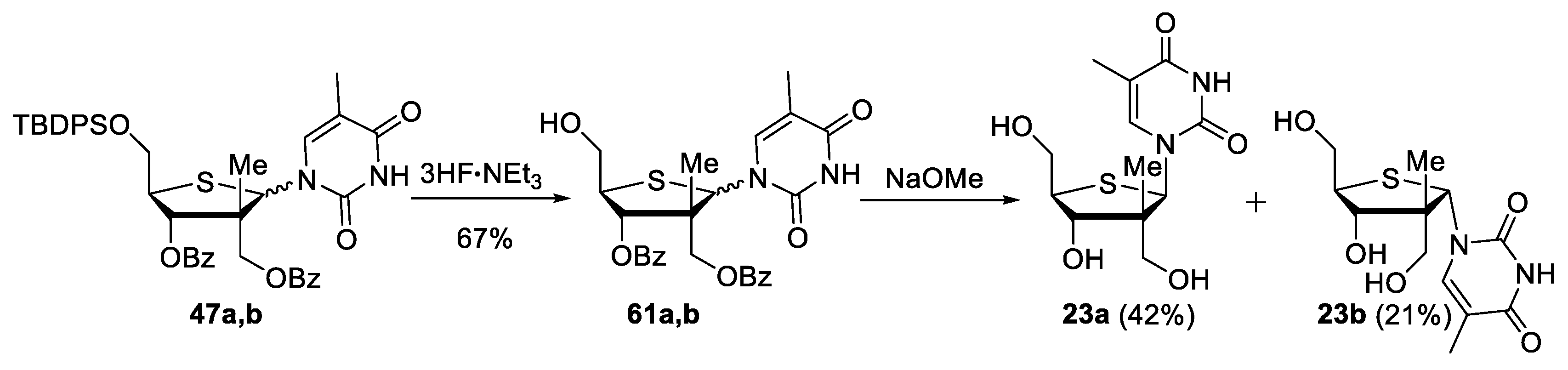
((3R,4S,5R)-4-(benzoyloxy)-5-(hydroxymethyl)-3-methyl-2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrothiophen-3-yl)methyl benzoate (61a,b). To a stirred solution of C5′-protected thiofuranosides 47a,b (241 mg, 1.00 equiv, 0.322 mmol) in anhydrous THF (1.3 mL, 0.25 M) at 0 °C, 3HF·NEt3 (0.13 mL, 2.5 equiv, 0.80 mmol) was added. The reaction mixture was stirred at room temperature for 18 h. After dilution with EtOAc, a saturated solution of NaHCO3 was added and the mixture was concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 61a,b (110 mg, 67% yield) as a 5:1 mixture in favor of the β-D-anomer. Rf = 0.49 and 0.53 (CH2Cl2/MeOH, 90:10); formula: C26H26N2O7S; MW: 510.5610 g/mol; 1H NMR (500 MHz, CDCl3) δ 9.62 (s, 1H, α-anomer), 9.51 (s, 1H, β-anomer), 8.60 (s, 1H, β-anomer), 8.24 (d, J = 8.3 Hz, 2H, β-anomer), 8.04 (d, J = 8.4 Hz, 2H, β-anomer), 7.90 (dd, J = 13.9, 8.3 Hz, 4H, α-anomer), 7.74 (s, 1H, α-anomer), 7.64–7.58 (m, 5H, α-anomer), 7.58–7.52 (m, 2H, β-anomer and α-anomer), 7.46 (t, J = 7.2 Hz, 4H, β-anomer), 7.39 (dt, J = 22.1, 7.4 Hz, 1H, β-anomer), 6.54 (s, 1H, β-anomer), 6.36 (s, 1H, α-anomer), 5.66 (d, J = 9.5 Hz, 1H, β-anomer), 5.52 (d, J = 3.9 Hz, 1H, α-anomer), 4.70 (d, J = 11.2 Hz, 1H, β-anomer), 4.64 (d, J = 11.2 Hz, 1H, β-anomer), 4.53 (d, J = 11.4 Hz, 1H, α-anomer), 4.40 (d, J = 11.5 Hz, 1H, α-anomer), 4.04 (appt, J = 3.3 Hz, 2H, α-anomer), 3.99 (s, 1H, α-anomer), 3.97–3.89 (m, 2H, β-anomer), 3.85 (appt, J = 6.7 Hz, 1H, α-anomer), 3.67 (appd, J = 9.5 Hz, 1H, β-anomer), 3.45 (s, 1H, β-anomer), 2.00 (s, 3H, β-anomer), 1.74 (s, 3H, α-anomer), 1.55 (s, 3H, α-anomer), 1.15 (s, 3H, β-anomer) ppm; 13C NMR (126 MHz, CDCl3) δ 166.71, 166.67, 165.9, 165.8, 164.0, 163.6, 151.5, 151.4, 138.0 (β-anomer), 137.8 (α-anomer), 134.3, 134.1, 133.5, 133.3, 130.0 (4C), 129.7 (2C), 129.6, 129.5, 129.2 (2C), 128.9 (2C), 128.7 (2C), 128.60 (2C), 128.57 (2C), 128.47, 128.36, 111.4 (β-anomer), 111.0 (α-anomer), 81.8 (α-anomer), 77.1 (β-anomer), 68.0 (α-anomer), 66.2 (β-anomer), 64.2 (2C, α-anomer), 62.7 (β-anomer), 59.9 (β-anomer), 56.5 (α-anomer), 54.3 (α-anomer), 53.7 (β-anomer), 50.8 (β-anomer), 23.1 (α-anomer), 17.3 (β-anomer), 12.9 (β-anomer), 12.8 (α-anomer) ppm; HRMS (ESI) m/z: calcd for C26H26N2NaO7S [M+Na]+ 533.1353, found 533.1356 (+0.56 ppm).
(+)-1-((2R,3R,4S,5R)-4-hydroxy-3,5-bis(hydroxymethyl)-3-methyltetrahydrothiophen-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (23a) and (+)-1-((2S,3R,4S,5R)-4-hydroxy-3,5-bis(hydroxymethyl)-3-methyltetrahydrothiophen-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (23b). To a stirred solution of 2′,3′-protected thiofuranosides 61a,b (24 mg, 1.0 equiv, 0.047 mmol) in MeOH (0.24 mL, 0.20 M), a solution of NaOMe (11 μL, 1.0 equiv, 0.047 mmol, 4.4 M in MeOH) was added. The reaction mixture was stirred at room temperature for 3 h. After the addition of formic acid until a neutral pH was reached, the mixture was concentrated under reduced pressure. Purification by C18 reverse-phase flash chromatography (H2O/MeOH) provided thiofuranosides 23a (6 mg, 42% yield) and 23b (3 mg, 21% yield). 23a: Rf= 0.13 (DCM/MeOH, 90:10); [α]25D +23 (c 0.25, MeOH); formula: C12H18N2O5S; MW: 302.3450 g/mol; IR (neat) νmax 3376, 2925, 1683, 1470 cm−1; 1H NMR (500 MHz, CD3OD): δ 8.44 (s, 1H), 6.17 (s, 1H), 4.04 (d, J = 9.6 Hz, 1H), 3.95 (dd, J = 11.9, 3.6 Hz, 1H), 3.90 (dd, J = 11.8, 2.6 Hz, 1H), 3.77 (d, J = 11.3 Hz, 1H), 3.70 (d, J = 11.3 Hz, 1H), 3.34 (ddd, J = 9.5, 3.6, 2.6 Hz, 1H), 1.90 (d, J = 1.2 Hz, 3H), 0.98 (s, 3H) ppm, labile protons were not observed due to exchange; 13C NMR (126 MHz, CD3OD) δ 166.3, 153.0, 140.8, 110.9, 78.7, 65.0, 64.1, 61.0, 55.5, 54.6, 17.3, 12.5 ppm; HRMS (ESI) m/z: calcd for C12H18N2NaO5S [M+Na]+ 325.0829, found 325.0840 (+3.4 ppm). 23b: Rf= 0.13 (DCM/MeOH, 90:10); [α]25D +18 (c 0.18, MeOH); formula: C12H18N2O5S; MW: 302.3450 g/mol; IR (neat) νmax 3368, 2926, 1682, 1468 cm−1; 1H NMR (500 MHz, CD3OD): δ 8.16 (d, J = 1.2 Hz, 1H), 6.01 (s, 1H), 3.96 (d, J = 5.1 Hz, 1H), 3.91 (dd, J = 10.9, 5.7 Hz, 1H), 3.87–3.82 (m, 1H), 3.62 (dd, J = 10.9, 7.5 Hz, 1H), 3.59 (d, J = 2.8 Hz, 2H), 1.89 (d, J = 1.2 Hz, 3H), 1.23 (s, 3H) ppm, labile protons were not observed due to exchange; 13C NMR (126 MHz, CD3OD) δ 166.3, 153.5, 141.9, 110.1, 82.2, 68.8, 65.8, 63.4, 59.1, 55.4, 22.9, 12.6. ppm; HRMS (ESI) m/z: calcd for C12H18N2NaO5S [M+Na]+ 325.0829, found 325.0841 (+3.7 ppm).
(–)-((2
R,3
R,4
S,5
R)-4-(benzoyloxy)-2-(benzylthio)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-3-methyltetrahydrothiophen-3-yl)methyl benzoate (
62). To a solution of crude C4-OMs dithioacetal
45 (432 mg, 1.00 equiv, 0.463 mmol) in pyridine (4.3 mL, 0.10 M), tetrabutylammonium iodide (188 mg, 1.10 equiv, 0.509 mmol) and barium carbonate (112 mg, 1.23 equiv, 0.569 mmol) were added [
28]. The reaction mixture was stirred at 80 °C for 3 h. After cooling to room temperature, the volatiles were removed under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided the pure product
62 (271 mg, 78% yield over two steps). R
f = 0.61 (Hexanes/EtOAc, 80:20); [α]
D25 −56 (
c 1.9, CH
2Cl
2); formula: C
44H
46O
5S
2Si; MW: 747.0520 g/mol; IR (neat) ν
max 3069, 2931, 2857, 1723, 1265, 1108 cm
−1;
1H NMR (500 MHz, CDCl
3) δ 7.92 (dd,
J = 8.4, 1.3 Hz, 2H), 7.83 (dd,
J = 8.4, 1.3 Hz, 2H), 7.66 (ddd,
J = 8.1, 2.6, 1.5 Hz, 4H), 7.59–7.53 (m, 2H), 7.43–7.29 (m, 15H), 5.70 (d,
J = 6.1 Hz, 1H), 4.55 (d,
J = 11.0 Hz, 1H), 4.41 (s, 1H), 4.31 (d,
J = 11.0 Hz, 1H), 4.03 (dd,
J = 10.4, 5.7 Hz, 1H), 3.90 (d,
J = 6.0 Hz, 2H), 3.87–3.81 (m, 1H), 3.65 (dt,
J = 7.8, 5.9 Hz, 1H), 1.29 (s, 3H), 1.02 (s, 9H) ppm;
13C NMR (126 MHz, CDCl
3) δ 166.3, 165.1, 137.3, 135.84 (2C), 135.75 (2C), 133.4, 133.3, 133.2, 129.9 (2C), 129.83, 129.82, 129.7 (2C), 129.5, 129.3 (2C), 128.7 (2C), 128.6 (2C), 128.5 (2C), 127.82 (2C), 127.78 (2C), 127.4, 81.1, 67.3, 66.4, 55.1, 52.9, 52.8, 37.3, 26.9 (3C), 19.3, 18.7 ppm, due to overlapping carbon signals in the aromatic region 2 peaks are hidden; HRMS (ESI)
m/
z: calcd for C
44H
46NaO
5S
2Si [M+Na]
+ 769.2448, found 769.2453 (+0.65 ppm).
((3R,4S,5R)-4-(benzoyloxy)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-3-methyl-2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrothiophen-3-yl)methyl benzoate (47a,b). To a stirred solution of thiofuranoside 62 (32 mg, 1.0 equiv, 0.043 mmol) in anhydrous DCE (0.43 mL, 0.10 M) at room temperature, silylated thymine (0.78 M in MeCN, 0.16 mL, 3.0 equiv, 0.13 mmol) was added. The resulting solution was cooled to 0 °C and dimethyl(methylthio)sulfonium tetrafluoroborate (34 mg, 4.0 equiv, 0.17 mmol) was added. The reaction mixture was stirred at room temperature for 3 h. After cooling to 0 °C, the reaction was quenched by the addition of H2O and dissolved in EtOAc. The aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. 1H NMR analysis of the crude mixture indicated a 1.2:1 ratio of nucleosides. Purification by flash chromatography (Hexanes/EtOAc) provided a mixture of products 47a,b (22 mg, 69% yield) in a 1.3:1 (β:α) ratio. Rf = 0.79 (Hexanes/EtOAc, 30:70); formula: C42H44N2O7SSi; MW: 748.9660 g/mol; IR (neat) νmax 3190, 3069, 2930, 2857, 1720, 1686, 1263, 1104 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.35 (s, 1H), 8.33 (s, 1H), 8.23 (dd, J = 8.3, 1.1 Hz, 2H), 7.98 (dd, J = 8.3, 1.2 Hz, 2H), 7.89 (ddd, J = 14.0, 8.3, 1.2 Hz, 3H), 7.77 (s, 1H, β-anomer), 7.74 (d, J = 1.2 Hz, 1H, α-anomer), 7.71–7.64 (m, 8H), 7.64–7.51 (m, 5H), 7.50–7.35 (m, 15H), 7.35–7.28 (m, 5H), 6.56 (s, 1H, β-anomer), 6.27 (s, 1H, α-anomer), 5.65 (d, J = 9.8 Hz, 1H, β-anomer), 5.42 (d, J = 3.8 Hz, 1H, α-anomer), 4.61 (d, J = 11.3 Hz, 1H, β-anomer), 4.54 (d, J = 11.3 Hz, 1H, β-anomer), 4.47 (d, J = 11.4 Hz, 1H, α-anomer), 4.32 (d, J = 11.4 Hz, 1H, α-anomer), 4.19 (dd, J = 10.2, 5.2 Hz, 1H, α-anomer), 4.07–4.01 (m, 2H, β-anomer and α-anomer), 3.90 (dd, J = 10.9, 6.4 Hz, 1H, β-anomer), 3.84 (ddd, J = 11.3, 7.4, 4.4 Hz, 2H, β-anomer and α-anomer), 1.80 (d, J = 1.1 Hz, 3H, α-anomer), 1.76 (d, J = 0.9 Hz, 3H, β-anomer), 1.41 (s, 3H, α-anomer), 1.09 (s, 3H, β-anomer), 1.07 (s, 9H, α-anomer), 1.05 (s, 9H, β-anomer) ppm; 13C NMR (126 MHz, CDCl3) δ 166.7, 165.9, 165.4, 165.1, 163.3, 163.2, 151.10, 151.09, 138.1, 137.0, 135.9 (2C), 135.7 (4C), 135.6 (2C), 133.99, 133.96, 133.54, 133.50, 133.1, 133.0, 132.8, 132.7, 130.12, 130.10, 130.07 (2C), 130.06, 130.04, 130.02 (2C), 129.7 (2C), 129.62 (2C), 129.61, 129.4, 128.92, 128.87 (2C), 128.82 (2C), 128.76, 128.73 (2C), 128.6 (2C), 127.99 (2C), 127.96 (2C), 127.95 (2C), 127.92 (2C), 111.7, 110.8, 81.3 (α-anomer), 77.6 (β-anomer), 68.3 (α-anomer), 66.4 (α-anomer), 66.1 (β-anomer), 64.3 (α-anomer), 64.2 (β-anomer), 62.2 (β-anomer), 57.7 (β-anomer), 54.5, 53.2, 51.7 (α-anomer), 27.0 (β-anomer, 3C), 26.9 (α-anomer, 3C), 23.1 (α-anomer), 19.5 (β-anomer), 19.4 (α-anomer), 17.3 (β-anomer), 12.9 (α-anomer), 12.8 (β-anomer) ppm; HRMS (ESI) m/z: calcd for C42H45N2NaO7SSi [M+Na]+ 771.2531, found 771.2534 (+0.39 ppm).
(2R,3S,4R)-5-acetoxy-4-((benzoyloxy)methyl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-4-methyltetrahydrothiophen-3-yl benzoate (63a,b). To a stirred solution of thiofuranoside 62 (156 mg, 1.00 equiv, 0.209 mmol) in acetic acid (1.6 mL, 0.13 M), mercury acetate (133 mg, 2.00 equiv, 0.418 mmol) was added. After stirring at room temperature for 2 h, the mixture was concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc) provided 63a,b (129 mg, 91% yield) as a 3:1 mixture in favor of the β-anomer. Rf = 0.43 and 0.40 (Hexanes/EtOAc, 80:20); formula: C39H42O7SSi; MW: 682.2421 g/mol; 1H NMR (500 MHz, CDCl3) δ 8.06–8.05 (m, 4H), 8.04 (d, J = 1.2 Hz, 4H), 8.02 (d, J = 1.1 Hz, 1H), 8.00 (d, J = 1.4 Hz, 1H), 7.97 (d, J = 1.2 Hz, 1H), 7.95 (d, J = 1.4 Hz, 1H), 7.69 (t, J = 1.3 Hz, 1H), 7.68 (dd, J = 3.3, 1.5 Hz, 2H), 7.67 (d, J = 1.6 Hz, 1H), 7.64–7.63 (m, 2H), 7.63–7.61 (m, 2H), 7.60–7.57 (m, 5H), 7.56 (dd, J = 3.0, 1.7 Hz, 1H), 7.47–7.42 (m, 5H), 7.41–7.38 (m, 1H), 7.38–7.34 (m, 4H), 7.33–7.27 (m, 4H), 6.13 (s, 1H, β-anomer), 6.06 (s, 1H, α-anomer), 5.84 (d, J = 9.0 Hz, 1H, β-anomer), 5.46 (d, J = 2.7 Hz, 1H, α-anomer), 4.64 (d, J = 11.3 Hz, 1H, β-anomer), 4.61 (d, J = 11.5 Hz, 1H, β-anomer), 4.58 (d, J = 11.0 Hz, 1H, α-anomer), 4.49 (d, J = 11.0 Hz, 1H, α-anomer), 4.09 (dd, J = 10.1, 5.8 Hz, 1H, α-anomer), 3.99 (ddd, J = 8.6, 5.7, 2.8 Hz, 1H, α-anomer), 3.90 (dd, J = 10.7, 4.7 Hz, 1H, β-anomer), 3.84–3.81 (m, 1H, α-anomer), 3.79 (dd, J = 10.7, 6.6 Hz, 1H, β-anomer), 3.66 (ddd, J = 9.1, 6.5, 4.7 Hz, 1H, β-anomer), 2.15 (s, 3H, α-anomer), 2.11 (s, 3H, β-anomer), 1.31 (s, 3H, α-anomer), 1.28 (s, 3H, β-anomer), 1.05 (s, 9H, α-anomer), 1.00 (s, 9H, β-anomer) ppm; 13C NMR (126 MHz, CDCl3) δ 170.2 (β-anomer), 170.1 (α-anomer), 166.5 (β-anomer), 166.3 (α-anomer), 165.5 (β-anomer), 165.3 (α-anomer), 135.9 (2C), 135.80 (2C), 135.77 (2C), 135.73 (2C), 133.7, 133.6, 133.4, 133.3, 133.2, 133.1, 133.04, 133.01, 130.0 (2C), 129.94, 129.92, 129.87 (2C), 129.84 (2C), 129.81, 129.79 (2C), 129.73 (2C), 129.69, 129.3 (2C), 128.7 (4C), 128.64, 128.62, 127.88 (2C), 127.85 (2C), 127.80 (2C), 127.75 (2C), 86.5 (α-anomer), 81.4 (α-anomer), 81.3 (β-anomer), 79.1 (β-anomer), 77.4 (α-anomer), 66.2 (β-anomer), 65.6 (β-anomer), 64.9 (α-anomer), 57.4 (α-anomer), 53.5 (α-anomer), 52.5 (β-anomer), 50.9 (β-anomer), 26.9 (3C, α-anomer), 26.8 (3C, β-anomer), 21.7 (α-anomer), 21.40 (β-anomer), 21.37 (α-anomer), 19.34 (α-anomer), 19.28 (β-anomer), 17.0 (β-anomer) ppm, due to overlapping carbon signals in the aromatic region 2 peaks are hidden; HRMS (ESI) m/z: calcd for C39H42NaO7SSi [M+Na]+ 705.2313, found 705.2290 (−3.26 ppm).
(–)-((2R,3S,4R,5R)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-((benzoyloxy)methyl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-4-methyltetrahydrothiophen-3-yl benzoate (64a) and (–)-(2R,3S,4R,5S)-5-(6-amino-2-chloro-9H-purin-9-yl)-4-((benzoyloxy)methyl)-2-(((tert-butyldiphenylsilyl)oxy)methyl)-4-methyltetrahydrothiophen-3-yl benzoate (64b). To a suspension of 2-chloroadenine (23 mg, 2.0 equiv, 0.14 mmol) in anhydrous DCE (0.67 mL, 0.20 M), BSA (0.11 mL, 6.5 equiv, 0.44 mmol) was added. The reaction mixture was refluxed at 84 °C until a clear solution was obtained. After cooling to −10 °C, the mixture was added to a solution of thiofuranosides 63a,b (46 mg, 1.0 equiv, 0.067 mmol) in anhydrous DCE (0.67 mL, 0.10 M), followed by dropwise addition of TMSOTf (25 μL, 2.0 equiv, 0.14 mmol). The resulting solution was stirred at 84 °C for 2 h. The crude was dissolved in EtOAc, and a saturated solution of NaHCO3 was added. The aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. 1H NMR analysis of the crude mixture indicated a 1.0:1.1 ratio of β:α N9-thionucleosides. Purification by flash chromatography (Hexanes/EtOAc) provided thionucleosides 64a (13 mg, 24% yield) and 64b (23 mg, 43% yield). 64a: Rf = 0.86 (Hexanes/EtOAc, 30:70); [α]D25 −33 (c 0.8, CH2Cl2); formula: C42H42ClN5O5SSi; MW: 792.4230 g/mol; IR: (neat) νmax 3323, 3171, 2958, 2859, 1727, 1644, 1263, 1108 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.58 (s, 1H), 8.37 (d, J = 7.4 Hz, 2H), 8.00 (d, J = 7.6 Hz, 2H), 7.69 (t, J = 6.9 Hz, 4H), 7.63 (dd, J = 14.0, 7.2 Hz, 2H), 7.57 (t, J = 7.5 Hz, 2H), 7.47 (t, J = 7.7 Hz, 2H), 7.44–7.35 (m, 4H), 7.29 (t, J = 7.4 Hz, 2H), 6.42 (s, 1H), 5.99 (s, 2H), 5.97 (d, J = 9.7 Hz, 1H), 4.68 (d, J = 11.1 Hz, 1H), 4.63 (d, J = 11.1 Hz, 1H), 3.99 (dd, J = 11.1, 3.1 Hz, 1H), 3.90 (dd, J = 11.1, 5.5 Hz, 1H), 3.84 (m, 1H), 1.11 (s, 9H), 0.88 (s, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 166.8, 165.2, 156.3, 154.6, 151.9, 141.0, 135.9 (2C), 135.7 (2C), 134.0, 133.7, 132.7, 132.4, 130.2 (2C), 130.12, 130.10, 130.0 (2C), 129.5, 129.0 (2C), 128.82 (2C), 128.80, 128.1 (2C), 128.0 (2C), 118.2, 77.1, 66.0, 63.5, 60.3, 53.7, 51.2, 27.0 (3C), 19.3, 17.5 ppm; HRMS (ESI) m/z: calcd for C42H43ClN5O5SSi [M+H]+ 792.2437, found 792.2434 (–0.38 ppm). 64b: Rf = 0.74 (Hexanes/EtOAc, 30:70); [α]D25 −4 (c 0.7, CH2Cl2); formula: C42H42ClN5O5SSi; MW: 792.4230 g/mol; IR (neat) νmax 3320, 3168, 3071, 2931, 2858, 1725, 1266, 1110 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.38 (s, 1H), 7.91–7.87 (m, 2H), 7.84 (dd, J = 8.1, 1.0 Hz, 2H), 7.68 (ddd, J = 7.8, 3.7, 1.4 Hz, 4H), 7.60 (t, J = 7.4 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.48–7.33 (m, 10H), 6.19 (s, 1H), 5.82 (s, 2H), 5.53 (d, J = 4.4 Hz, 1H), 4.34 (d, J = 11.7 Hz, 1H), 4.21–4.13 (m, 3H), 3.91–3.86 (m, 1H), 1.48 (s, 3H), 1.06 (s, 9H) ppm; 13C NMR (126 MHz, CDCl3) δ 165.9, 165.2, 156.0, 154.6, 151.7, 141.2, 135.9 (2C), 135.8 (2C), 134.0, 133.5, 133.0, 132.8, 130.1 (2C), 129.9 (2C), 129.7 (2C), 129.2, 128.9 (2C), 128.8, 128.7 (2C), 128.0 (2C), 127.9 (2C), 117.9, 81.3, 65.9, 65.5, 64.0, 57.1, 54.4, 26.9 (3C), 22.5, 19.4 ppm; HRMS (ESI) m/z: calcd for C42H43ClN5O5SSi [M+H]+ 792.2437, found 792.2433 (−0.51 ppm).
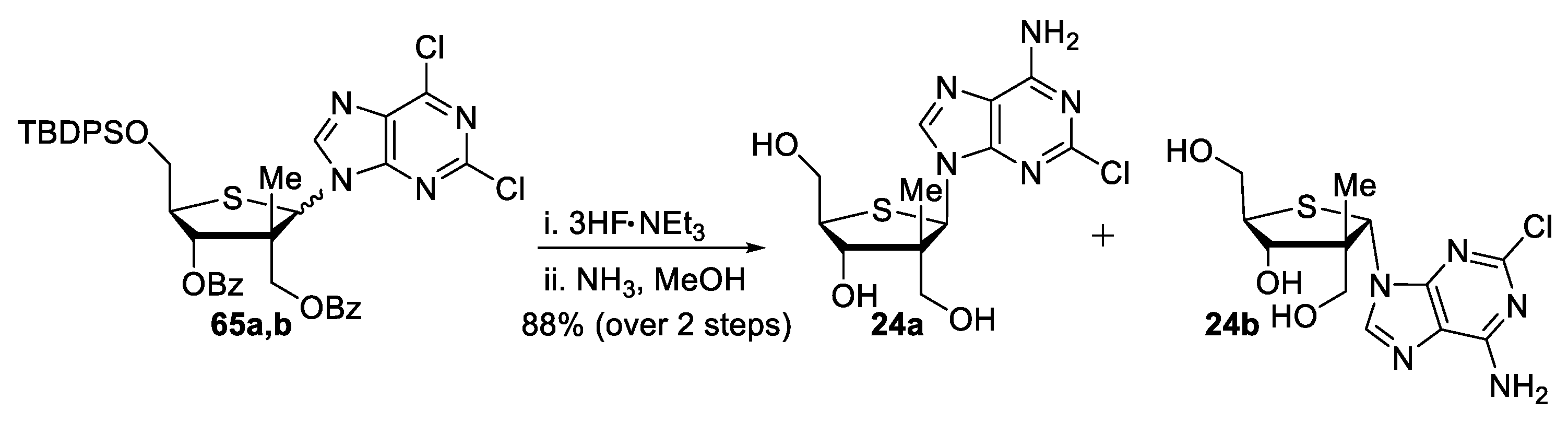
((3R,4S,5R)-4-(benzoyloxy)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-2-(2,6-dichloro-9H-purin-9-yl)-3-methyltetrahydrothiophen-3-yl)methyl benzoate (65a,b). To a stirred solution of thiofuranosides 63a,b (33 mg, 1.0 equiv, 0.048 mmol) in anhydrous MeCN (2.8 mL, 0.25 M), 2,6-dichloropurine (10 mg, 1.1 equiv, 0.053 mmol) was added. The resulting solution was cooled to −10 °C and DBU (22 μL, 3.0 equiv, 0.15 mmol) was added, followed by dropwise addition of TMSOTf (36 μL, 4.0 equiv, 0.19 mmol). The reaction mixture was stirred at room temperature for 16 h. The crude was dissolved in EtOAc, and a saturated solution of NaHCO3 was added. The aqueous layer was extracted (3×) with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. 1H NMR analysis of the crude mixture indicated a 1.3:1 ratio of β:α N9-thionucleosides. Purification by flash chromatography (Hexanes/EtOAc) provided a mixture of thionucleosides 65a,b (34 mg, 87% yield) in a 1.3:1 ratio in favor of the β-anomer. Rf = 0.31 (Hexanes/EtOAc, 80:20); formula: C42H40Cl2N4O5SSi; MW: 811.8500 g/mol; 1H NMR (500 MHz, CDCl3) δ 8.91 (s, 1H, β-anomer), 8.69 (s, 1H, α-anomer), 8.33 (d, J = 7.0 Hz, 2H, β-anomer), 8.01 (d, J = 7.1 Hz, 2H, β-anomer), 7.88 (d, J = 7.1 Hz, 2H, α-anomer), 7.81 (d, J = 7.2 Hz, 2H, α-anomer), 7.71–7.66 (m, 9H), 7.65–7.61 (m, 2H), 7.59–7.54 (m, 4H), 7.48 (td, J = 7.9, 3.3 Hz, 4H), 7.44–7.35 (m, 11H), 7.29 (t, J = 7.3 Hz, 2H), 6.48 (s, 1H, β-anomer), 6.23 (s, 1H, α-anomer), 5.98 (d, J = 9.6 Hz, 1H, β-anomer), 5.56 (d, J = 4.3 Hz, 1H, α-anomer), 4.71 (d, J = 11.2 Hz, 1H, β-anomer), 4.64 (d, J = 11.1 Hz, 1H, β-anomer), 4.30 (d, J = 11.7 Hz, 1H, α-anomer), 4.22–4.19 (m, 2H, α-anomer), 4.19 (d, J = 11.8 Hz, 1H, α-anomer), 4.00 (dd, J = 11.0, 3.1 Hz, 1H, β-anomer), 3.95–3.85 (m, 3H), 1.51 (s, 3H, α-anomer), 1.12 (s, 9H, β-anomer), 1.07 (s, 9H, α-anomer), 0.89 (s, 3H, β-anomer) ppm; 13C NMR (126 MHz, CDCl3) δ 166.6 (β-anomer), 165.7 (α-anomer), 165.2 (α-anomer), 165.1 (β-anomer), 153.6, 153.44, 153.42 (β-anomer), 153.3 (α-anomer), 152.5, 152.2, 146.2 (α-anomer), 145.9 (β-anomer), 135.9 (2C), 135.8 (2C), 135.7 (2C), 134.11, 134.08, 133.8, 133.7, 132.9, 132.7, 132.6, 132.3, 131.1 (β-anomer), 130.8 (α-anomer), 130.18, 130.14, 130.13, 130.11, 130.10 (2C), 130.0 (2C), 129.8 (2C), 129.6 (2C), 129.4, 129.04 (2C), 128.95 (2C), 128.90, 128.85 (2C), 128.76 (2C), 128.64, 128.60, 128.1 (2C), 127.99 (2C), 127.98 (2C), 127.96 (2C), 81.4 (α-anomer), 76.9 (β-anomer), 66.4 (α-anomer), 65.9 (β-anomer), 65.8 (α-anomer), 63.8 (α-anomer), 63.4 (β-anomer), 61.1 (β-anomer), 57.6 (α-anomer), 54.7 (α-anomer), 53.7 (β-anomer), 51.5 (β-anomer), 27.0 (3C, β-anomer), 26.9 (3C, α-anomer), 22.6 (α-anomer), 19.34 (α-anomer), 19.29 (β-anomer), 17.5 (β-anomer) ppm; HRMS (ESI) m/z: calcd for C42H41Cl2N4O5SSi [M+H]+ 811.1939, found 811.1938 (−0.12 ppm).
(+)-((2R,3S,4R,5R)-5-(6-Amino-2-chloro-9H-purin-9-yl)-3-hydroxy-4-methyltetrahydrothiophene-2,4-diyl)dimethanol (24a) and (+)-((2R,3S,4R,5S)-5-(6-Amino-2-chloro-9H-purin-9-yl)-3-hydroxy-4-methyltetrahydrothiophene-2,4-diyl)dimethanol (24b). To a mixture of thionucleosides 65a,b (226 mg, 1.00 equiv, 0.278 mmol) in anhydrous THF (1.1 mL, 0.25 M) at 0 °C, 3HF.NEt3 (0.11 mL, 2.5 equiv, 0.69 mmol) was added. The reaction mixture was stirred at room temperature for 16 h. After dilution with EtOAc, a saturated solution of NaHCO3 was added and the mixture was concentrated under reduced pressure. The reaction mixture was passed through a pad of silica allowing for a 1H NMR of each C5′-OH product to be obtained. Major isomer: Rf = 0.61 (CH2Cl2/MeOH, 90:10); formula: C26H22Cl2N4O5S; MW: 573.4450 g/mol; 1H NMR (500 MHz, CDCl3) δ 9.79 (s, 1H), 8.37 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.65 (t, J = 6.8 Hz, 1H), 7.62–7.55 (m, 3H), 7.43 (t, J = 7.2 Hz, 2H), 6.47 (s, 1H), 5.95 (d, J = 9.5 Hz, 1H), 5.08 (apps, 1H), 4.77 (d, J = 11.1 Hz, 1H), 4.69 (d, J = 11.1 Hz, 1H), 4.20 (dd, J = 12.6, 6.0 Hz, 1H), 4.04 (dd, J = 12.5, 4.9 Hz, 1H), 3.84 (appd, J = 9.5 Hz, 1H), 0.95 (s, 3H) ppm; HRMS (ESI) m/z: calcd for C26H23Cl2N4O5S [M+H]+ 573.0761, found 573.0750 (−1.75 ppm). Minor isomer: Rf = 0.55 (CH2Cl2/MeOH, 90:10); formula: C26H22Cl2N4O5S; MW: 573.4450 g/mol; 1H NMR (500 MHz, CDCl3) δ 8.71 (s, 1H), 7.94 (d, J = 7.9 Hz, 2H), 7.76 (d, J = 8.0 Hz, 2H), 7.62 (t, J = 7.4 Hz, 1H), 7.56 (t, J = 7.4 Hz, 1H), 7.51–7.45 (m, 2H), 7.43–7.39 (m, 2H), 6.30 (s, 1H), 5.62 (d, J = 5.1 Hz, 1H), 4.44 (d, J = 11.8 Hz, 1H), 4.32 (d, J = 11.8 Hz, 1H), 4.19 (appq, J = 5.7 Hz, 1H), 4.08 (dd, J = 11.4, 5.4 Hz, 1H), 3.95–3.89 (m, 1H), 2.78 (s, 1H), 1.62 (s, 3H) ppm; HRMS (ESI) m/z: calcd for C26H22Cl2N4NaO5S [M+Na]+ 595.0580, found 595.0573 (−1.18 ppm). To a mixture of C5′-alcohols (34 mg, 1.0 equiv, 59 umol) in anhydrous MeOH (1.5 mL, 0.040 M) in a high-pressure flask, NH3 was bubbled until saturation. The reaction mixture was warmed to 80 °C for 48 h. The mixture was then concentrated under reduced pressure. Purification by flash chromatography (Hexanes/EtOAc/MeOH) provided the pure products 24a and 24b (18 mg, 88% yield over two steps). 24a: Rf= 0.25 (DCM/MeOH, 9:1); [α]25D +45 (c 0.11, MeOH); IR (neat) νmax 3316, 2881, 2509, 2327, 1630 cm−1; formula: C12H16ClN5O3S; MW: 345.8020 g/mol; 1H NMR (500 MHz, CD3OD): δ 8.65 (s, 1H), 6.00 (s, 1H), 4.32 (apps, 1H), 4.05 (dd, J = 11.3, 4.6 Hz, 1H), 3.99 (dd, J = 11.5, 2.9 Hz, 1H), 3.84 (s, 2H), 3.45 (ddd, J = 9.7, 5.0, 2.8 Hz, 1H), 0.74 (s, 3H) ppm, OH and NH2 signals are missing due to exchange; 13C NMR (126 MHz, CD3OD) δ 158.2, 155.3, 152.3, 143.2, 118.8, 78.9, 64.5, 62.8, 62.3, 55.5, 54.7, 17.5 ppm; HRMS (ESI) m/z: calcd for C12H17ClN5O3S [M+H]+ 346.0735; found 346.0740 (+1.44 ppm). 24b: Rf= 0.15 (DCM/MeOH, 9:1); [α]25D +4 (c 0.8, MeOH) IR (neat) νmax 3346, 2931, 2384, 1615 cm−1; formula: C12H16ClN5O3S; MW: 345.8020 g/mol; 1H NMR (500 MHz, CD3OD): δ 8.56 (s, 1H), 5.90 (s, 1H), 4.03 (d, J = 5.1 Hz, 1H), 3.97 (m, 2H), 3.67 (dd, J = 12.7, 9.3 Hz, 1H), 3.55 (d, J = 11.2 Hz, 1H), 3.47 (d, J = 11.2 Hz, 1H), 1.29 (s, 3H); OH and NH2 signals are missing due to exchange; 13C NMR (126 MHz, CD3OD) δ 158.0, 155.1, 152.4, 144.0, 118.4, 82.0, 66.6, 65.7, 63.2, 59.0, 55.2, 22.2 ppm; HRMS (ESI) m/z: calcd for C12H17ClN5O3S [M+H]+ 346.0735; found 346.0731 (–1.16 ppm).



 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





