
A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates


Abstract
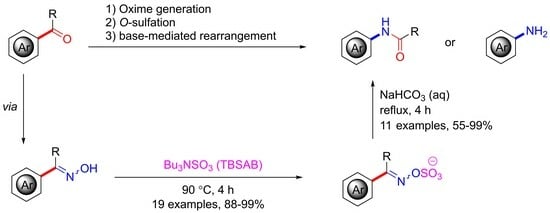
:
1. Introduction
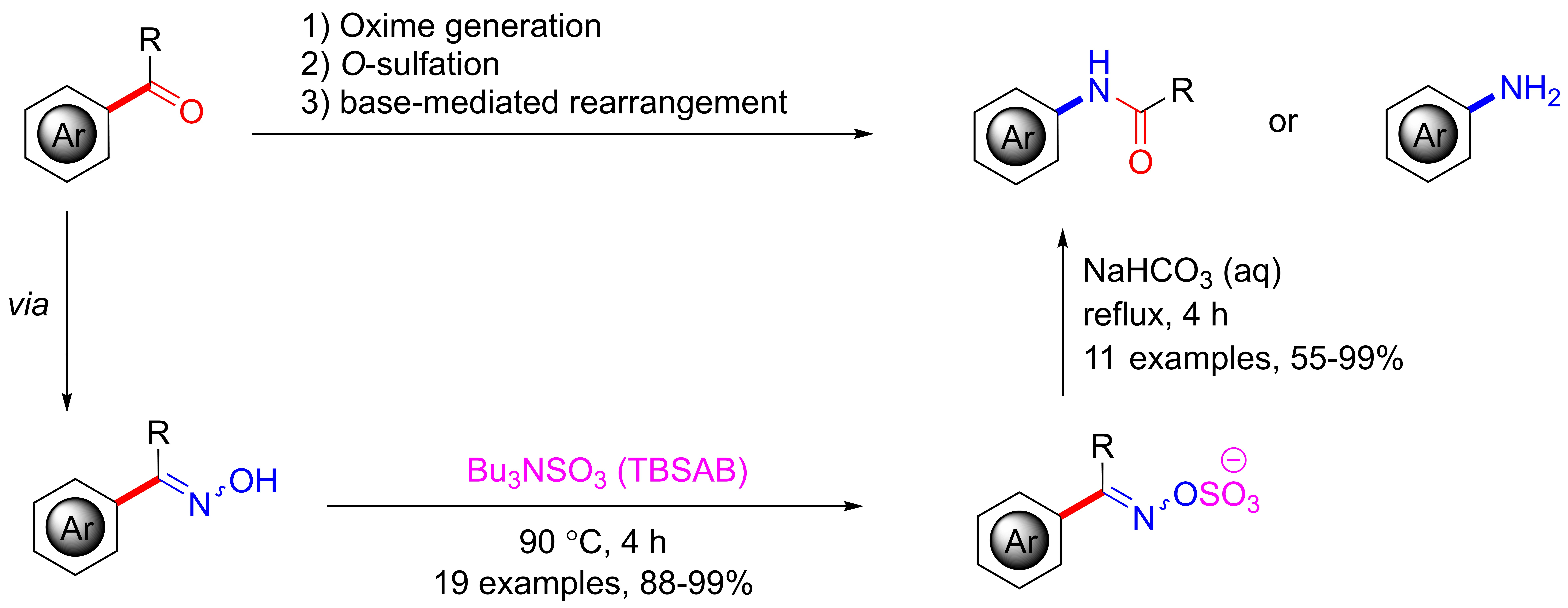
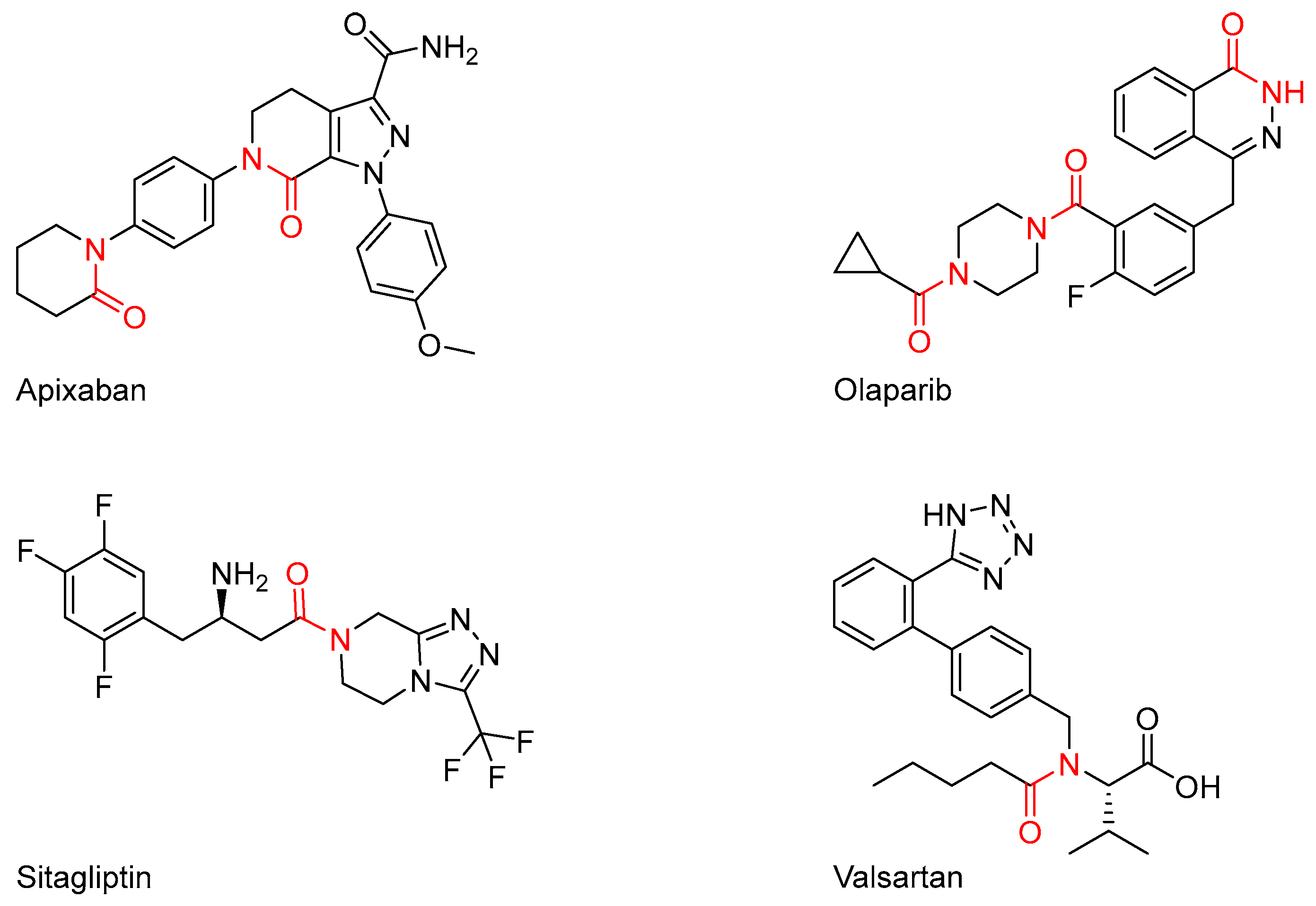
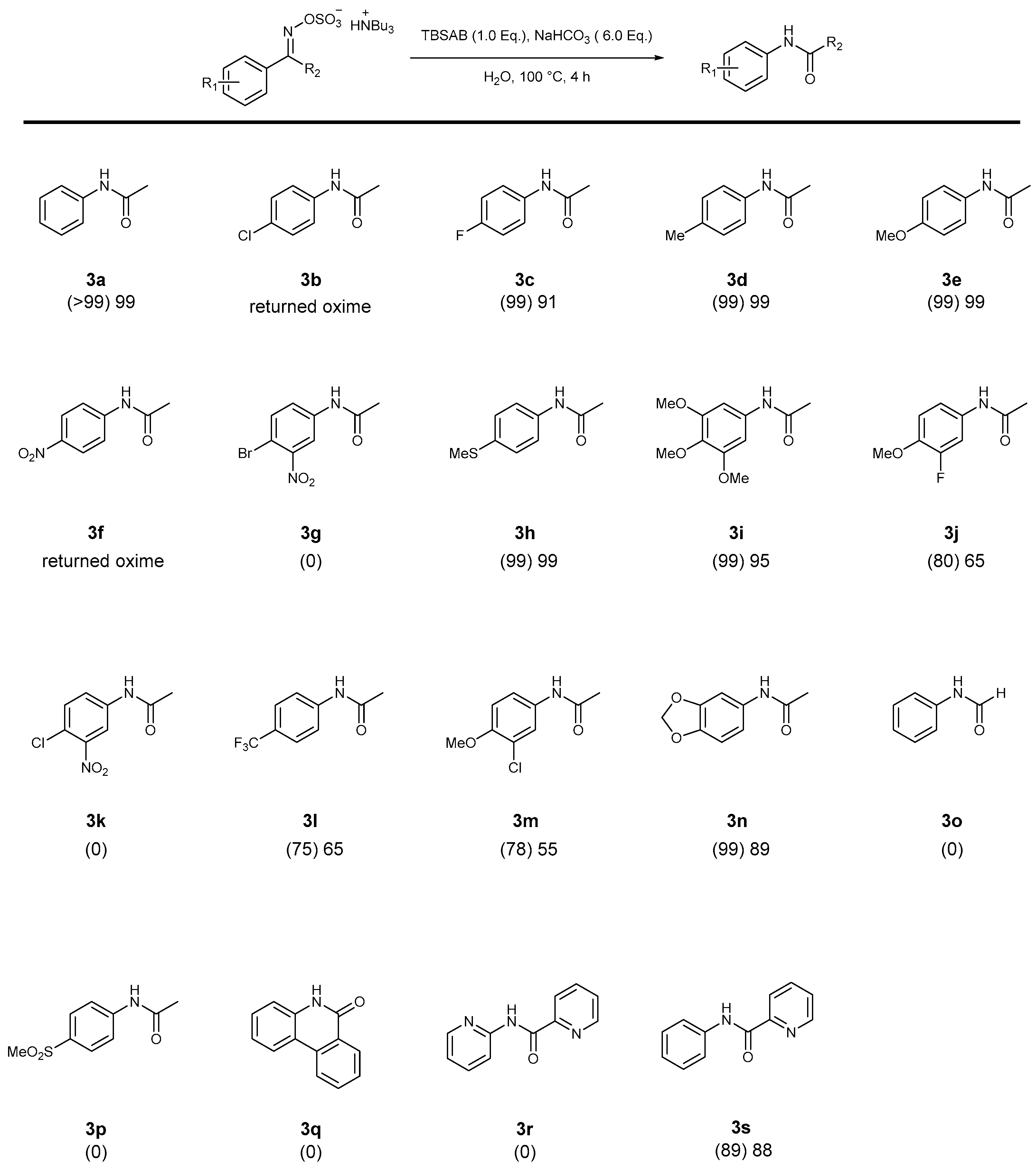
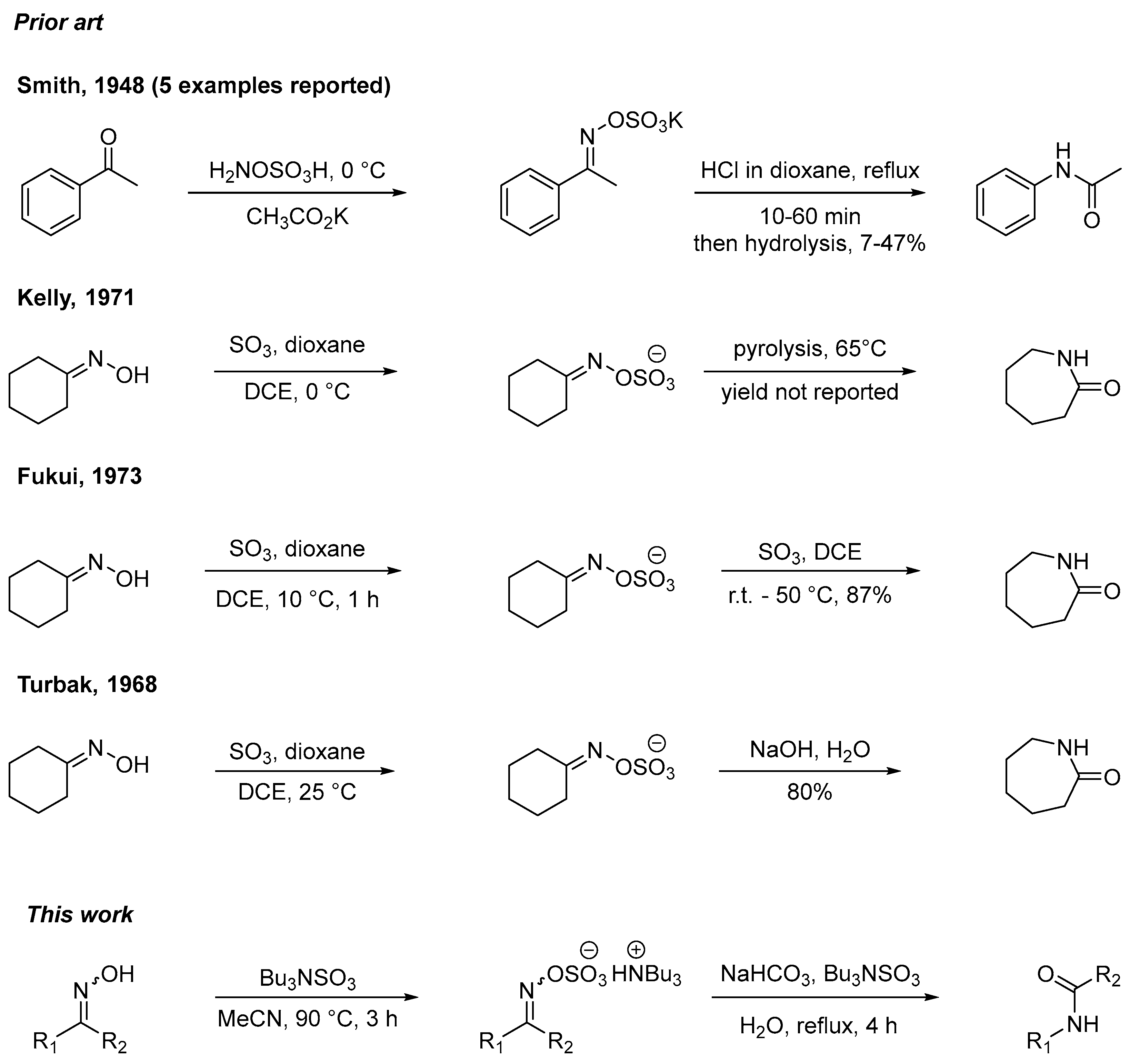
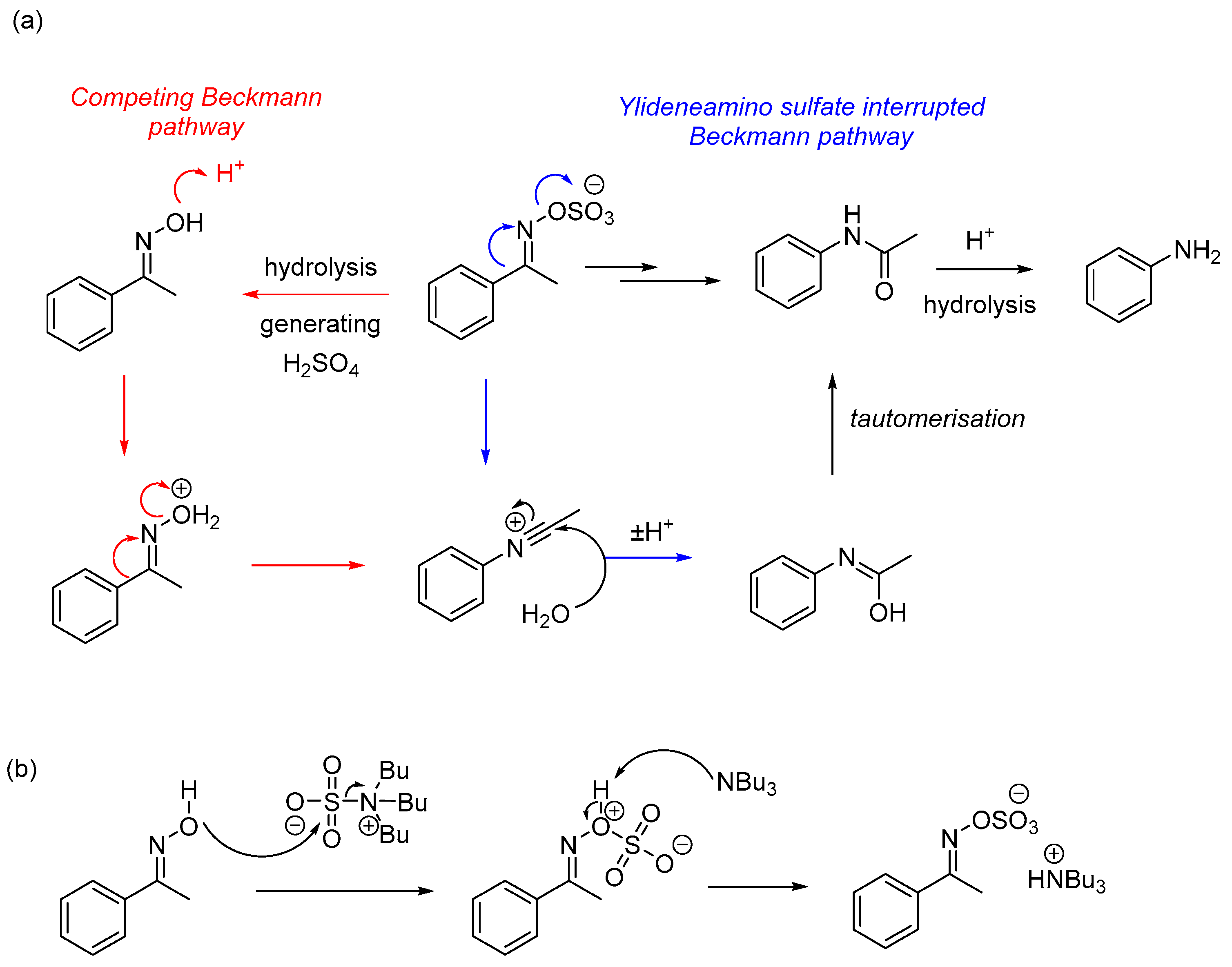
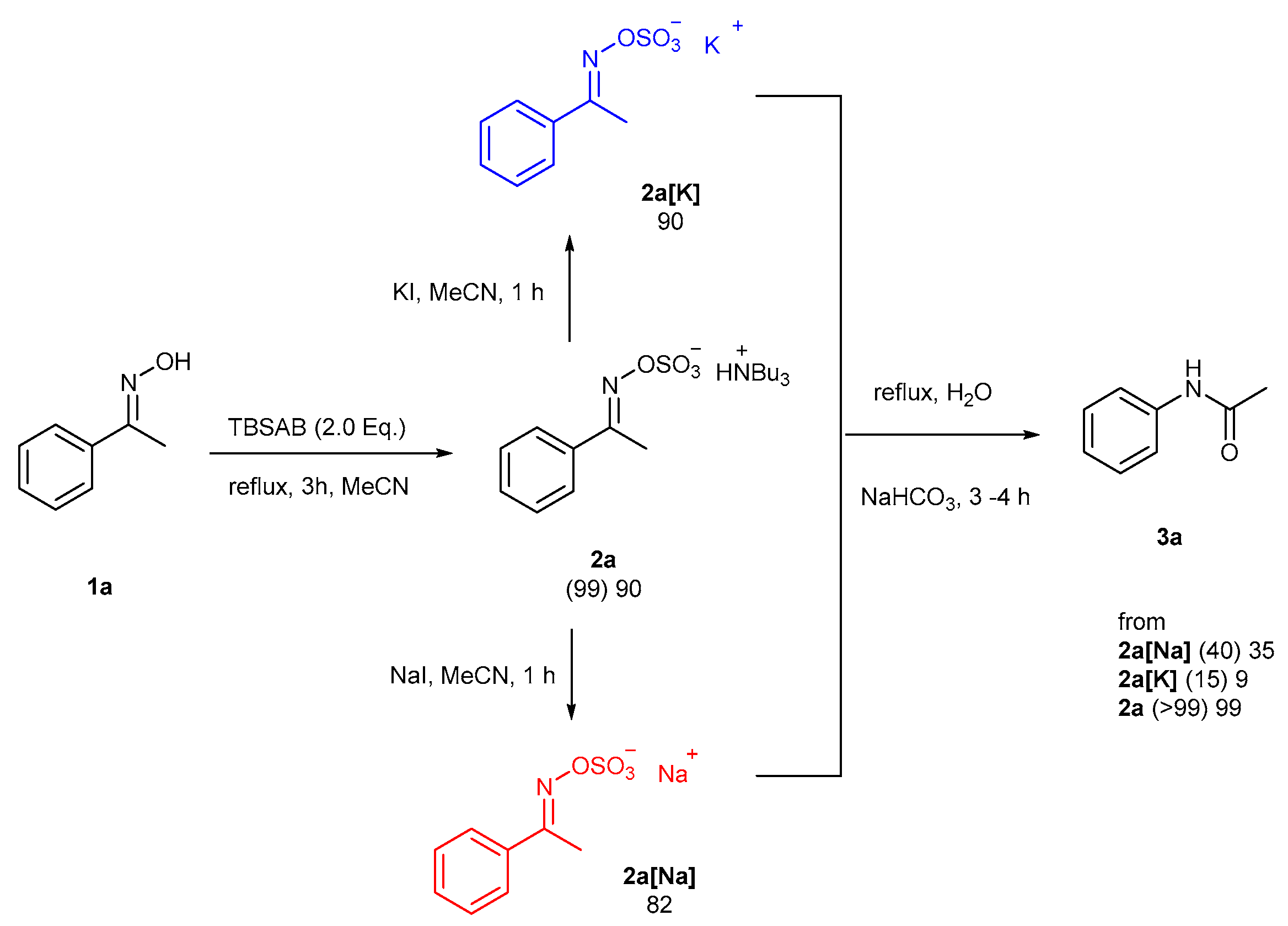
2. Results and Discussion
3. Conclusions
4. Experimental
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salim, H.; Jones, A.M. Angiotensin II receptor blockers (ARBs) and manufacturing contamination: A retrospective National Register Study into suspected associated adverse drug reactions. Br. J. Clin. Pharmacol. 2022, 88, 4812–4827. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Jones, A.M. Suspected adverse drug reactions of the type 2 antidiabetic drug class dipeptidyl-peptidase IV inhibitors (DPP4i): Can polypharmacology help explain? Pharmacol. Res. Perspect. 2022, 10, e01029. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, D.; Antolin, A.A.; Cox, A.R.; Jones, A.M. Identification of different side effects between PARP inhibitors and their polypharmacological multi-target rationale. Br. J. Clin. Pharmacol. 2022, 88, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ferro, C.J.; Solkhon, F.; Jalal, Z.; Al-Hamid, A.M.; Jones, A.M. Relevance of physicochemical properties and functional pharmacology data to predict the clinical safety profile of direct oral anticoagulants. Pharmacol. Res. Perspect. 2020, 8, e00603. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, E. Zur Kenntniss der Isonitrosoverbindungen. Ber. Dtsch. Chem. Ges. 1886, 19, 988–993. [Google Scholar] [CrossRef]
- Kaur, K.; Srivastava, S. Beckmann rearrangement catalysis: A review of recent advances. New J. Chem. 2020, 44, 18530–18572. [Google Scholar] [CrossRef]
- Hashimoto, M.; Obora, Y.; Sakaguchi, S.; Ishii, Y. Beckmann Rearrangement of Ketoximes to Lactams by Triphosphazene Catalyst. J. Org. Chem. 2008, 73, 2894–2897. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Morgan, T.D.R.; Ang, H.T.; Hall, D.G. Scope and Mechanism of a True Organocatalytic Beckmann Rearrangement with a Boronic Acid/Perfluoropinacol System under Ambient Conditions. J. Am. Chem. Soc. 2018, 140, 5264–5271. [Google Scholar] [CrossRef] [PubMed]
- Brzeczek-Szafran, A.; Erfurt, K.; Swadzba-Kwasny, M.; Piotrowski, T.; Chrobok, A. Beckmann Rearrangement with Improved Atom Economy, Catalyzed by Inexpensive, Reusable, Bronsted Acidic Ionic Liquid. ACS Sustain. Chem. Eng. 2022, 10, 13568–13575. [Google Scholar] [CrossRef]
- Hu, H.; Cai, X.; Xu, Z.; Yan, Y.; Zhao, S. Beckmann Rearrangement of Ketoxime Catalyzed by N-methyl-imidazolium Hydrosulfate. Molecules 2018, 23, 1764. [Google Scholar] [CrossRef]
- Munnuri, S.; Verma, S.; Chandra, D.; Anugu, R.R.; Falck, J.R.; Jat, J.L. Cu(OTf)2-Catalyzed Beckmann Rearrangement of Ketones Using Hydroxylamine-O-sulfonic Acid (HOSA). Synthesis 2019, 51, 3709–3714. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.S.; Humne, V.T.; Tanpure, S.D.; Mhaske, S.B. Radical Beckmann Rearrangement and Its Application in the Formal Total Synthesis of Antimalarial Natural Product Isocryptolepine via C–H Activation. Org. Lett. 2016, 18, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cantillo, D.; Kappe, C.O. Visible Light-Promoted Beckmann Rearrangements: Separating Sequential Photochemical and Thermal Phenomena in a Continuous Flow Reactor. Eur. J. Org. Chem. 2019, 2019, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Giacomelli, G.; Porcheddu, A. Beckmann Rearrangement of Oximes under Very Mild Conditions. J. Org. Chem. 2002, 67, 6272–6274. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Ishihara, K.; Yamamoto, H. Cyanuric Chloride as a Mild and Active Beckmann Rearrangement Catalyst. J. Am. Chem. Soc. 2005, 127, 11240–11241. [Google Scholar] [CrossRef]
- Tochette, S.J.; Dunkley, E.M.; Lowder, L.L.; Wu, J. Nucleophile-intercepted Beckmann fragmentation reactions. Chem. Sci. 2019, 10, 7812–7815. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Súarez, P.; Kolliopoulos, A.V.; Smith, J.P.; Banks, C.E.; Jones, A.M. An experimentalist’s guide to electrosynthesis: The Shono oxidation. Tetrahedron Lett. 2015, 56, 6863–6867. [Google Scholar] [CrossRef]
- Bal, M.K.; Banks, C.E.; Jones, A.M. Metabolism Mimicry: An Electrosynthetic Method for the Selective Deethylation of Tertiary Benzamides. ChemElectroChem 2019, 6, 4284–4291. [Google Scholar] [CrossRef]
- Jones, A.M. Dialling-In New Reactivity into the Shono-Type Anodic Oxidation Reaction. Chem. Rec. 2021, 21, 2120–2129. [Google Scholar] [CrossRef]
- Jones, A.M.; Banks, C.E. The Shono-type electroorganic oxidation of unfunctionalised amides. Carbon–carbon bond formation via electrogenerated N-acyliminium ions. Beilstein J. Org. Chem. 2014, 10, 3056–3072. [Google Scholar] [CrossRef]
- Smith, P.A.S. Ketoxime-O-sulfonic Acids. J. Am. Chem. Soc. 1948, 70, 323–326. [Google Scholar] [CrossRef]
- Fukui, K.; Uchida, M.; Masaki, M. Formation and Reaction of Cyclohexanone Oxime Hydrogen Sulfate M. Bull. Jpn Chem. Soc. 1973, 46, 3168–3173. [Google Scholar] [CrossRef]
- Kelly, K.K.; Matthews, J.S. Use of Lewis base-sulfur trioxide complexes as reagents for the Beckmann rearrangement of ketoximes. J. Org. Chem. 1971, 36, 2159–2161. [Google Scholar] [CrossRef]
- Turbak, A.F. A Low Temperature Beckmann Rearrangement with SO3 Including Separation of Caprimidyl Sulfate as a New Composition. Ind. Eng. Chem. Prod. Res. Dev. 1968, 7, 189–191. [Google Scholar] [CrossRef]
- Zhou, Y.; Jones, A.M. Rearrangement of Arylsulfamates and Sulfates to Para-Sulfonyl Anilines and Phenols. Molecules 2024, 29, 1445. [Google Scholar] [CrossRef]
- Alshehri, J.A.; Gill, D.M.; Jones, A.M. A Sulfuryl Group Transfer Strategy to Selectively Prepare Sulfated Steroids and Isotopically Labelled Derivatives. Front. Mol. Biosci. 2021, 8, 776900. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Povinelli, A.P.R.; Zazeri, G.; Mahmoud, A.M.; Shamir, S.A.; Wilkinson, F.L.; Alexander, M.Y.; Cornelio, M.L.; Jones, A.M. The modulatory role of sulfated and non-sulfated small molecule heparan sulfate-glycomimetics in endothelial dysfunction: Absolute structural clarification, molecular docking and simulated dynamics, SAR analyses and ADMET studies. RSC Med. Chem. 2021, 12, 779–790. [Google Scholar] [CrossRef]
- Jones, A.M. Tributylsulfoammonium betaine. In The Encyclopaedia of Reagents for Organic Synthesis (e-EROS); John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2021; Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/047084289X.RN02393 (accessed on 27 March 2024).
- Benedetti, A.M.; Gill, D.M.; Tsang, C.W.; Jones, A.M. Chemical Methods for N- and O-Sulfation of Small Molecules, Amino Acids and Peptides. ChemBioChem 2020, 21, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Male, L.; Jones, A.M. Sulfation made simple: A strategy for synthesising sulfated molecules. Chem. Commun. 2019, 55, 4319–4322. [Google Scholar] [CrossRef]
- Nishijima, K.; Nishida, H.; Yamashita, Y.; Ito, M.; Onuki, Y.; Mizota, M.; Miyano, S. Synthesis and diuretic activity of bicyclic fused heterocycles containing oxime-O-sulfonic acid moiety. Eur. J. Med. Chem. 2000, 35, 227–240. [Google Scholar] [CrossRef]
- Chen, Y.; Gardiner, M.G.; Lan, P.; Banwell, M.G. α-Iodo-α,β-Unsaturated Ketones as Vicinal Dielectrophiles: Their Reactions with Dinucleophiles Provide New Annulation Protocols for the Formation of Carbo- and Heterocyclic Ring Systems. J. Org. Chem. 2022, 87, 6146–6160. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.B.; Boronat, M.; Blasco, T.; Corma, A. Establishing a Molecular Mechanism for the Beckmann Rearrangement of Oximes over Microporous Molecular Sieves. Angew. Chem. Int. Ed. 2005, 44, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Sharghi, H.; Sarvari, M.H. One-step Beckmann rearrangement from carbonyl compounds and hydroxylamine hydrochloride in Al2O3/CH3SO3H (AMA) as a new reagent. J. Chem. Res. 2001, 2001, 446–449. [Google Scholar] [CrossRef]
- Han, Z.; Lv, J.; Zhang, J. One-pot synthesis of 2-amino-3,4-dicyanopyridines from ketoximes and tetracyanoethylene via Cu(I)-catalyzed cyclization. Tetrahedron 2019, 75, 2162–2168. [Google Scholar] [CrossRef]
- Sonn, A. Über einige synthetische Versuche mit O-Trimethyl-gallusaldehyd. (Mitbearbeitet von Ernst Müller, Wolfgang Bülow und Walter Meyer). Ber. Chem. Ges. A/B 1925, 58, 1103–1110. [Google Scholar] [CrossRef]
- Mahajan, P.S.; Mahajan, J.P.; Mhaske, S.B. Malonic ester amide synthesis. An efficient methodology for synthesis of amides. Synth. Commun. 2013, 43, 2508–2516. [Google Scholar] [CrossRef]
- Tang, L.; Wang, Z.-L.; Wan, H.-L.; He, Y.-H.; Guan, Z. Visible-Light-Induced Beckmann Rearrangement by Organic Photoredox Catalysis. Org. Lett. 2020, 22, 6182–6186. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Ghosh, P.; Basu, B. Graphene oxide (GO) catalyzed transamidation of aliphatic amides: An efficient metal-free procedure. Tetrahedron Lett. 2018, 59, 899–903. [Google Scholar] [CrossRef]
- Jat, J.L.; Kumar, P.; Verma, S.; Chandra, D.; Singh, V.; Tiwari, B. Metal-free synthesis of secondary amides using N-Boc-O-tosylhydroxylamine as nitrogen source via Beckmann rearrangement. New J. Chem. 2022, 46, 14782–14785. [Google Scholar] [CrossRef]
- Ito, A.; Asami, Y.; Asato, M.; Fukuda, K.; Yamasaki, R.; Okamoto, I. Synthesis and conformational analysis of N-aryl-N-(3-thienyl)acetamides. Tetrahedron Lett. 2018, 59, 2454–2458. [Google Scholar] [CrossRef]
- Ran, M.; He, J.; Yan, B.; Liu, W.; Li, Y.; Fu, Y.; Li, S.-J.; Yao, Q. Catalyst-free generation of acyl radicals induced by visible light in water to construct C–N bonds. Org. Biomol. Chem. 2021, 19, 1970–1975. [Google Scholar] [CrossRef] [PubMed]
- Chip, G.K.; Grossert, J.S. Aromatic Halogenation with Titanium (IV) Chloride in Presence of Peroxytrifluoroacetic Acid. Can. J. Chem. 1972, 50, 1233–1240. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Sokolova, O.O.; Bower, J.F. Branch-Selective Alkene Hydroarylation by Cooperative Destabilization: Iridium-Catalyzed ortho-Alkylation of Acetanilides. Angew. Chem. Int. Ed. 2015, 54, 14866–14870. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, X.; Tao, S.; Bu, Q.; Wei, D.; Liu, N. Manganese Catalyzed Direct Amidation of Esters with Amines. J. Org. Chem. 2021, 86, 2339–2358. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Entry | Solvent | Temperature [°C] | Time [h] | Conv. [%] |
| 1 | MeCN | 90 | 3 | >99 |
| 2 | MeCN | 60 | 3 | 90 |
| 3 | MeCN | 30 | >24 | <10 |
 | |||||||
| Entry | T (°C) | Time (h) | TBSAB (1.0 Equiv.) | Base [6.0 Equiv.] | Conv. [%] a 1a | Conv. [%] a 3a | Conv. [%] a 4a |
| 1 | 80 | 8 | - | - | 9 | 26 | trace |
| 2 | 100 | 8 | - | - | 6 | 54 | 14 |
| 3 | 120 | 8 | - | - | 7 | 37 | 20 |
| 4 | 80 | 8 | Yes | - | 5 | 40 | trace |
| 5 | 100 | 8 | Yes | - | 10 | 57 | 10 |
| 6 | 120 | 8 | Yes | - | 9 | 37 | 25 |
| 7 b | 100 | 4 | - | NaHCO3 | trace | >99 | 0 |
| 8 b | 100 | 4 | Yes | NaHCO3 | 0 | >99 | 0 |
| 9 b | 100 | 4 | Yes | KHCO3 | 0 | 90 | 0 |
| 10 b | 100 | 4 | Yes | pyridine | 0 | 60 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Jones, A.M. A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates. Molecules 2024, 29, 1667. https://doi.org/10.3390/molecules29071667
Zhou Y, Jones AM. A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates. Molecules. 2024; 29(7):1667. https://doi.org/10.3390/molecules29071667
Chicago/Turabian StyleZhou, Yifei, and Alan M. Jones. 2024. "A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates" Molecules 29, no. 7: 1667. https://doi.org/10.3390/molecules29071667
APA StyleZhou, Y., & Jones, A. M. (2024). A General Method to Access Underexplored Ylideneamino Sulfates as Interrupted Beckmann-Type Rearrangement Intermediates. Molecules, 29(7), 1667. https://doi.org/10.3390/molecules29071667