
Synthesis, Anti-Inflammatory Activities, and Molecular Docking Study of Novel Pyxinol Derivatives as Inhibitors of NF-κB Activation


Abstract
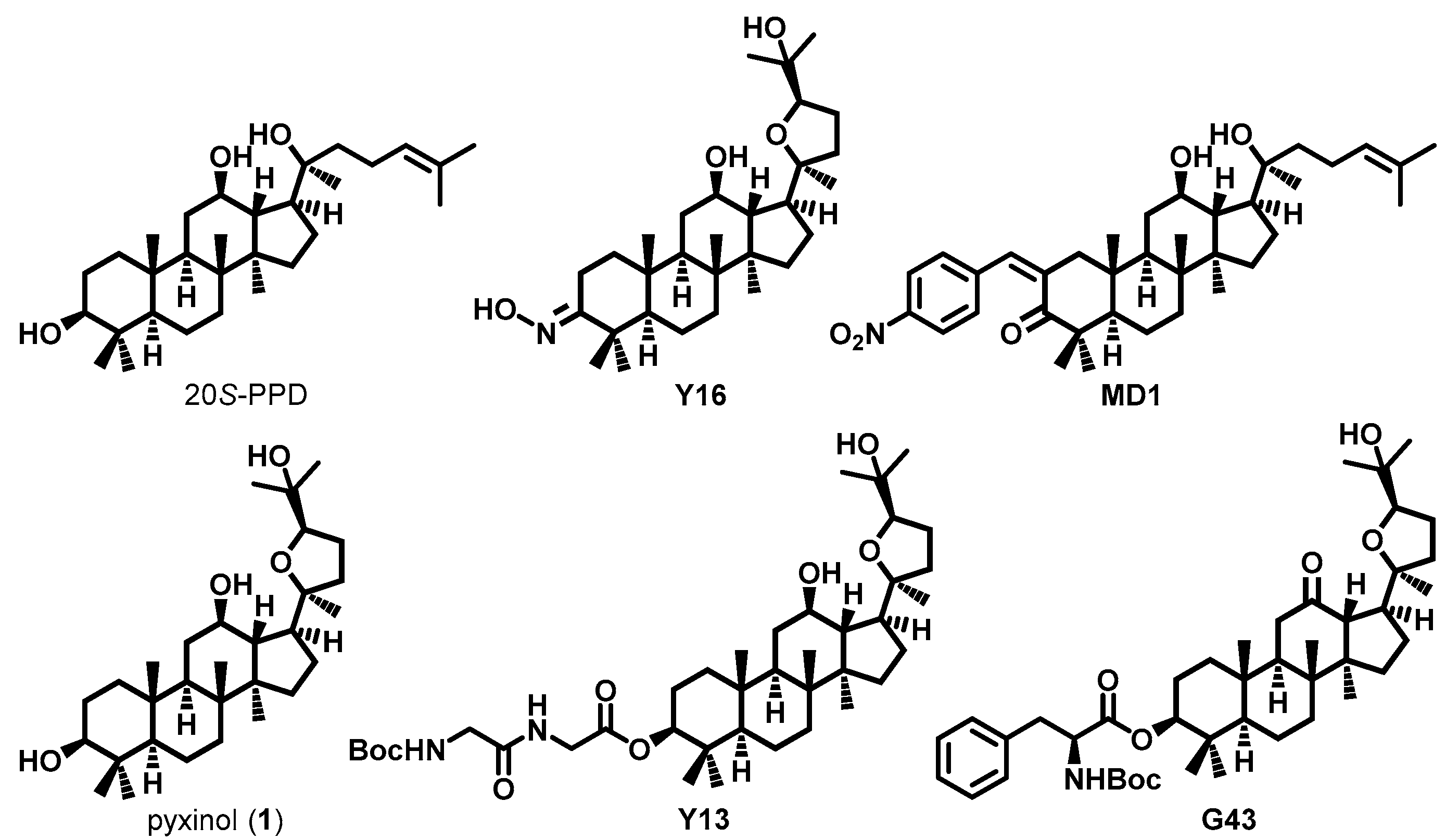
1. Introduction
2. Results and Discussion
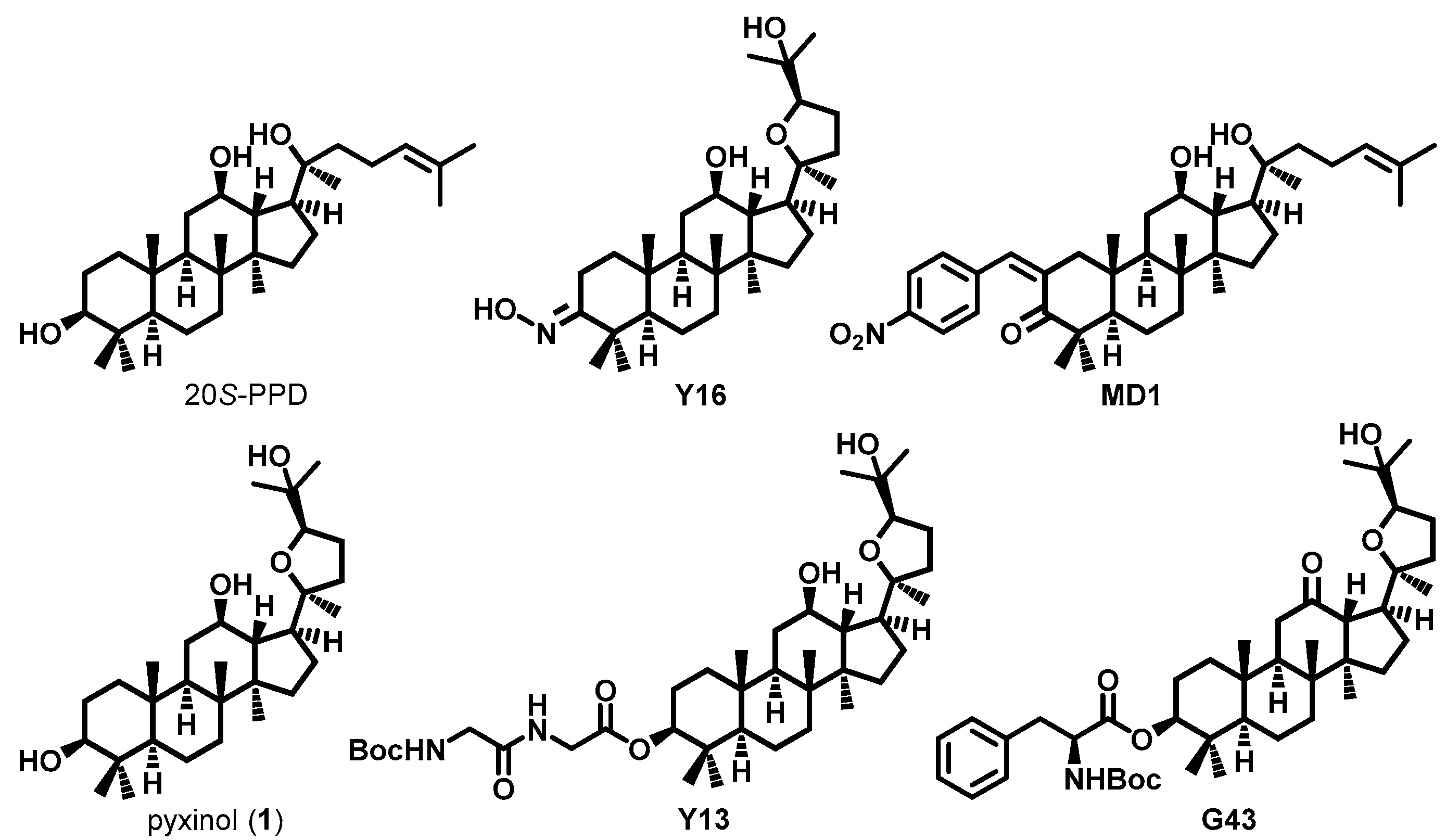
2.1. Designing Pyxinol Derivatives and Virtual Screening
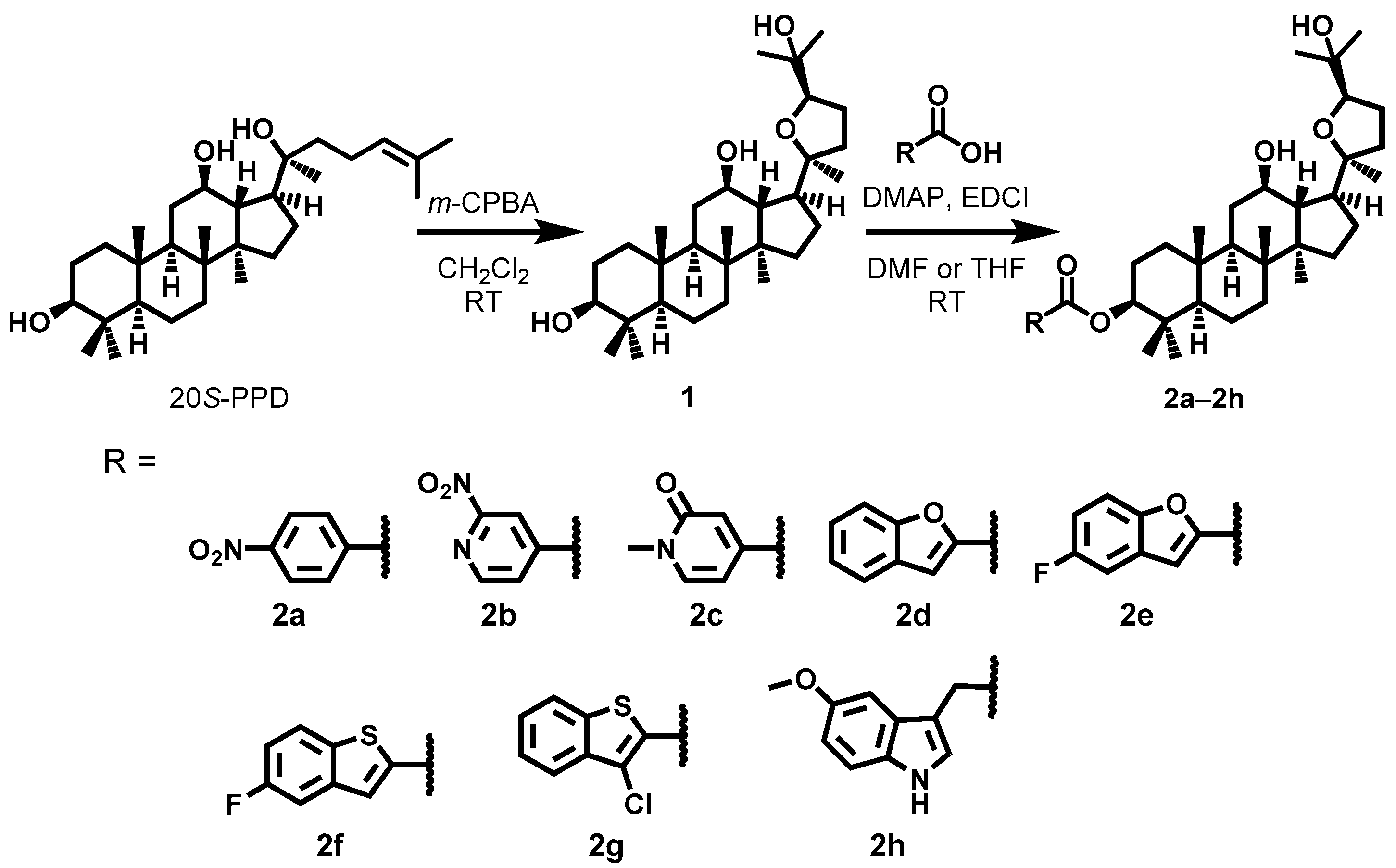
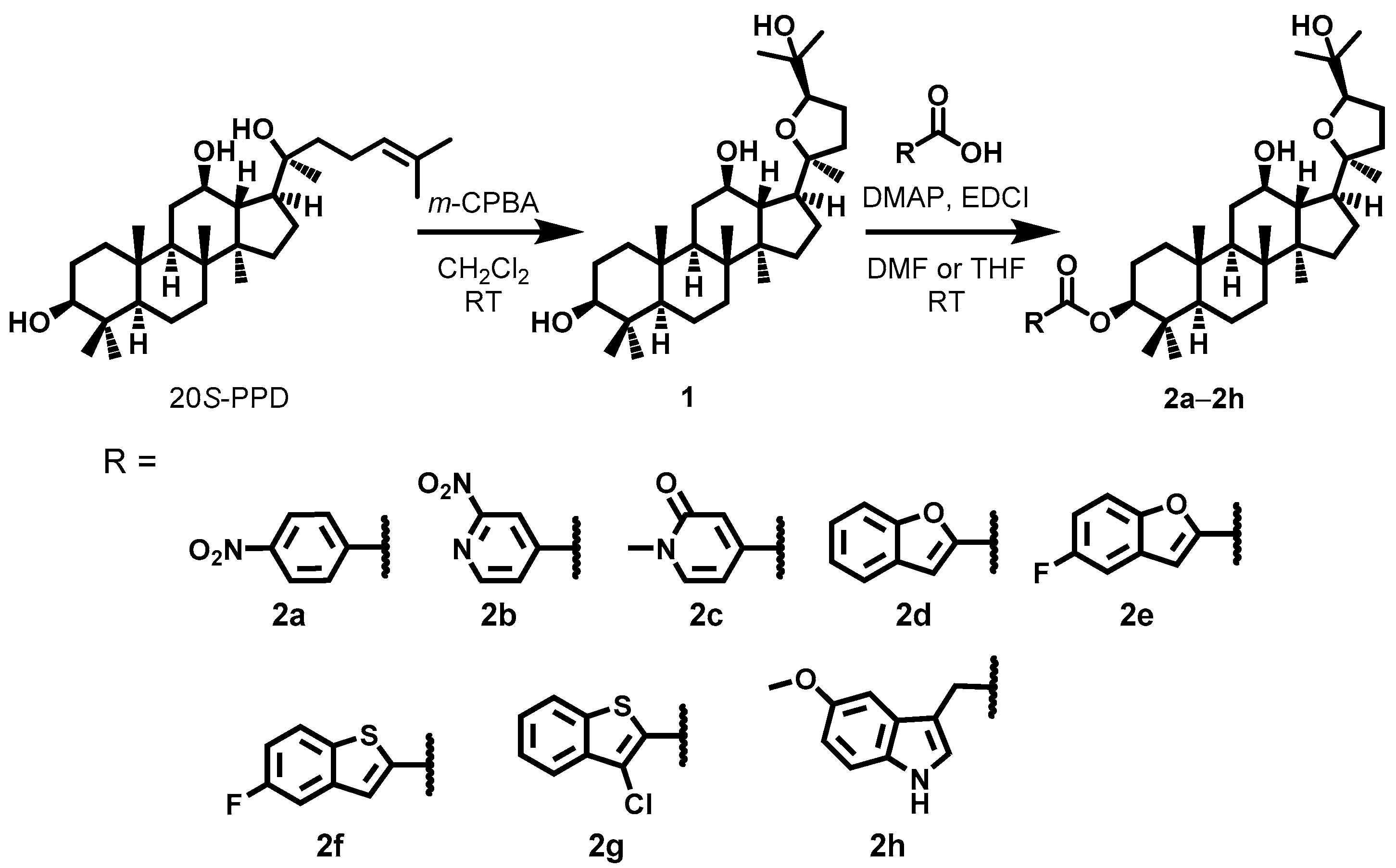
2.2. Synthesis
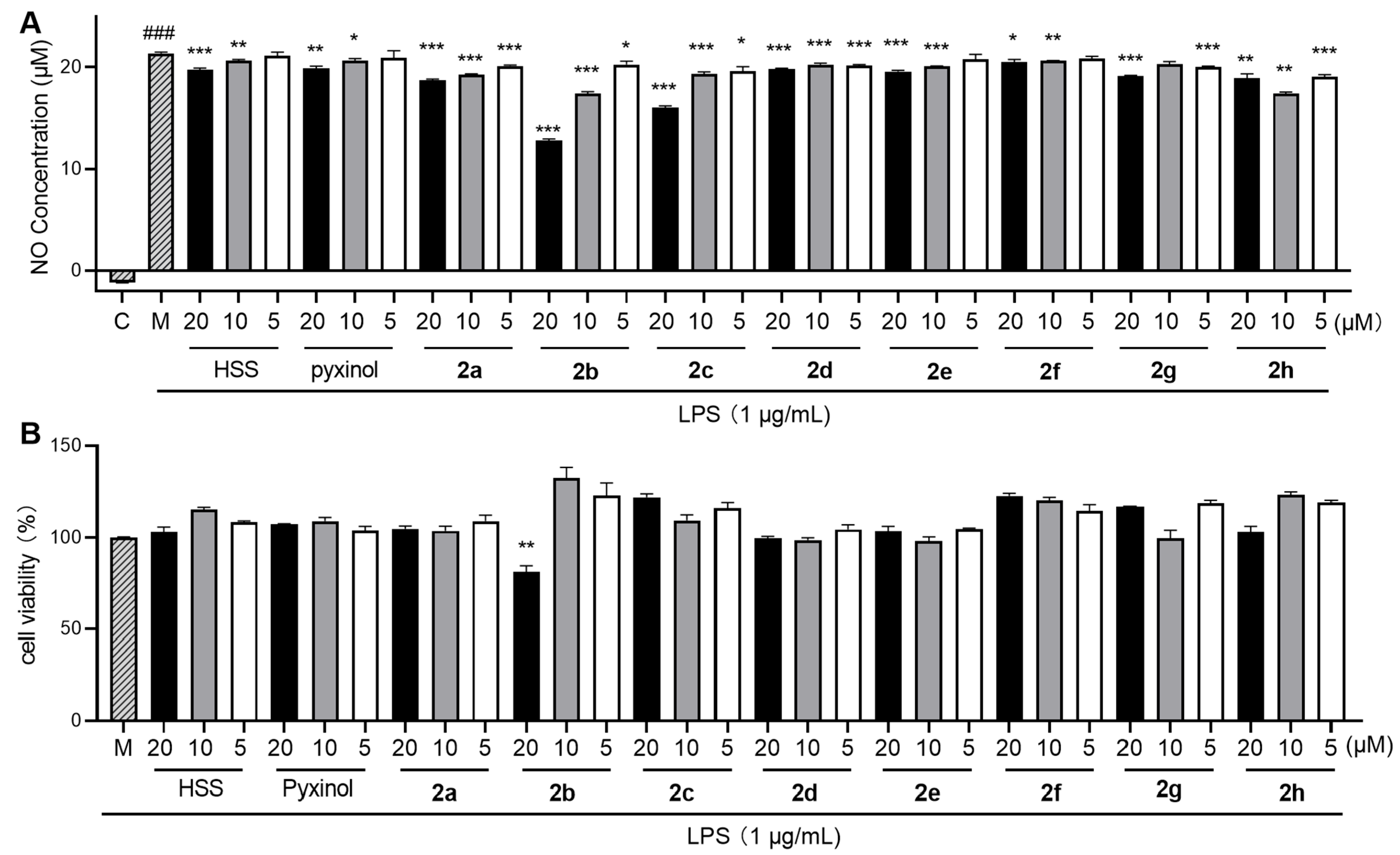
2.3. Inhibition of LPS-Triggered Nitric Oxide (NO) Release
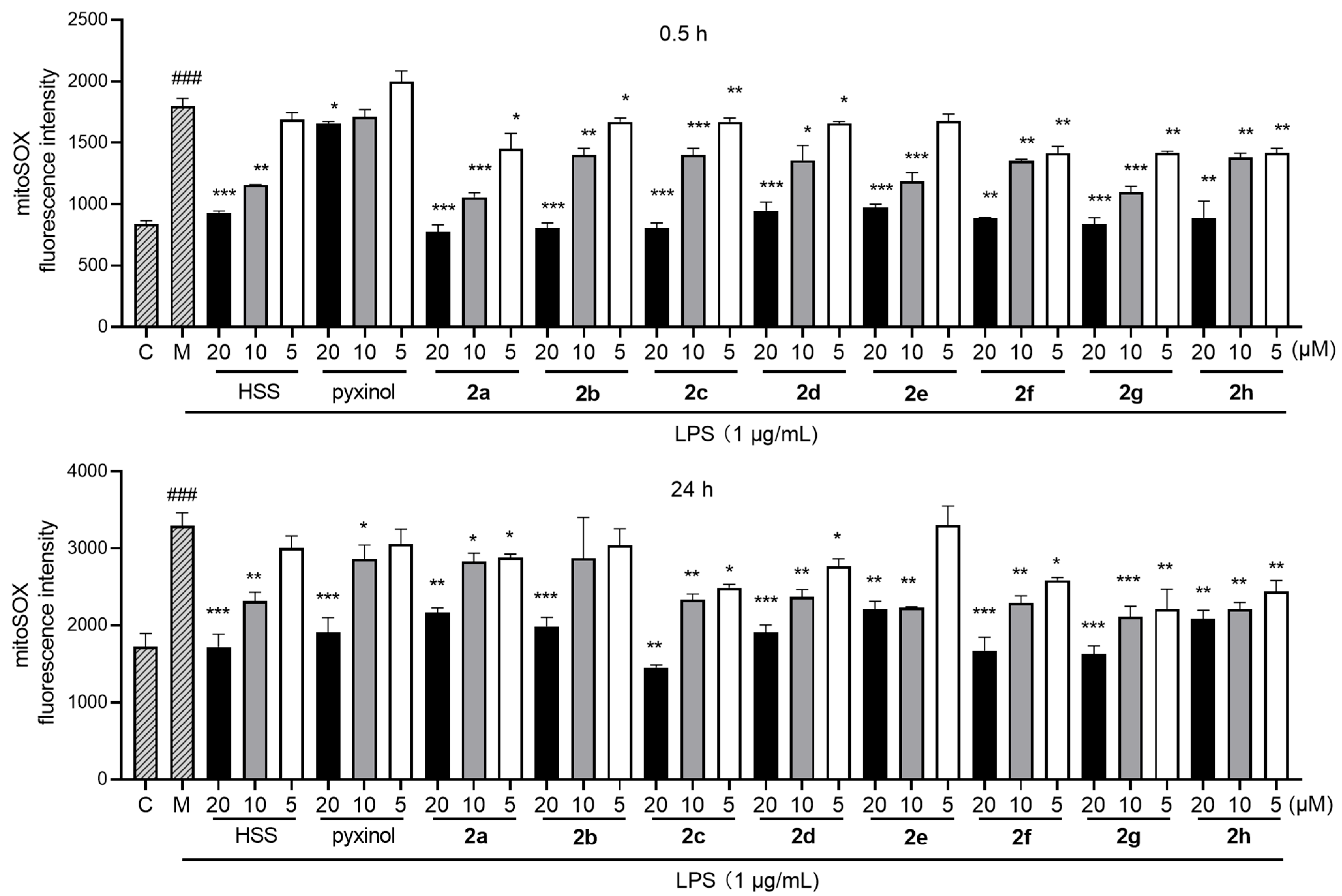
2.4. Inhibition of LPS-Triggered Generation of Mitochondrial Reactive Oxygen Species (MtROS)
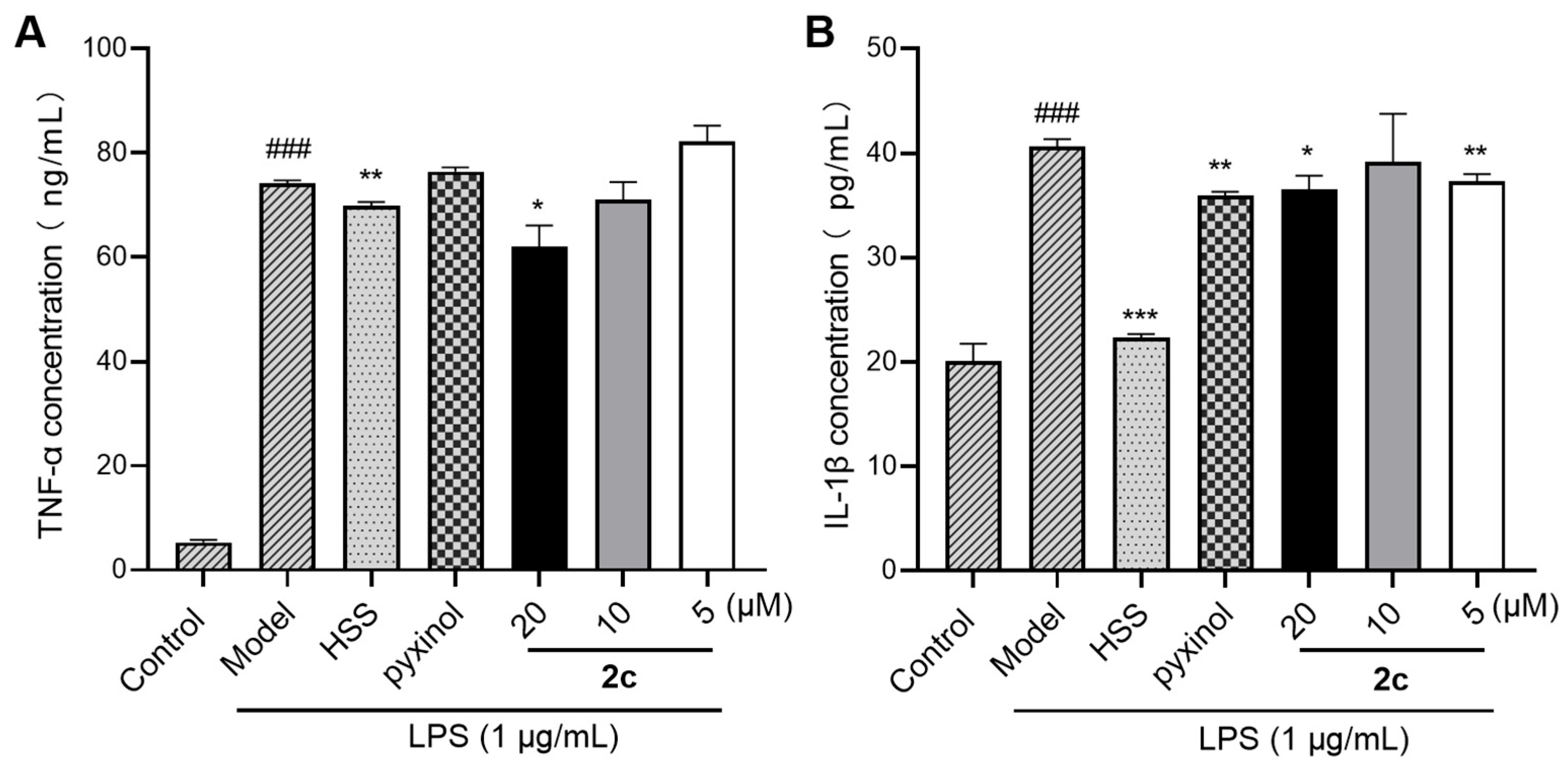
2.5. Inhibition of LPS-Triggered Cytokine Release
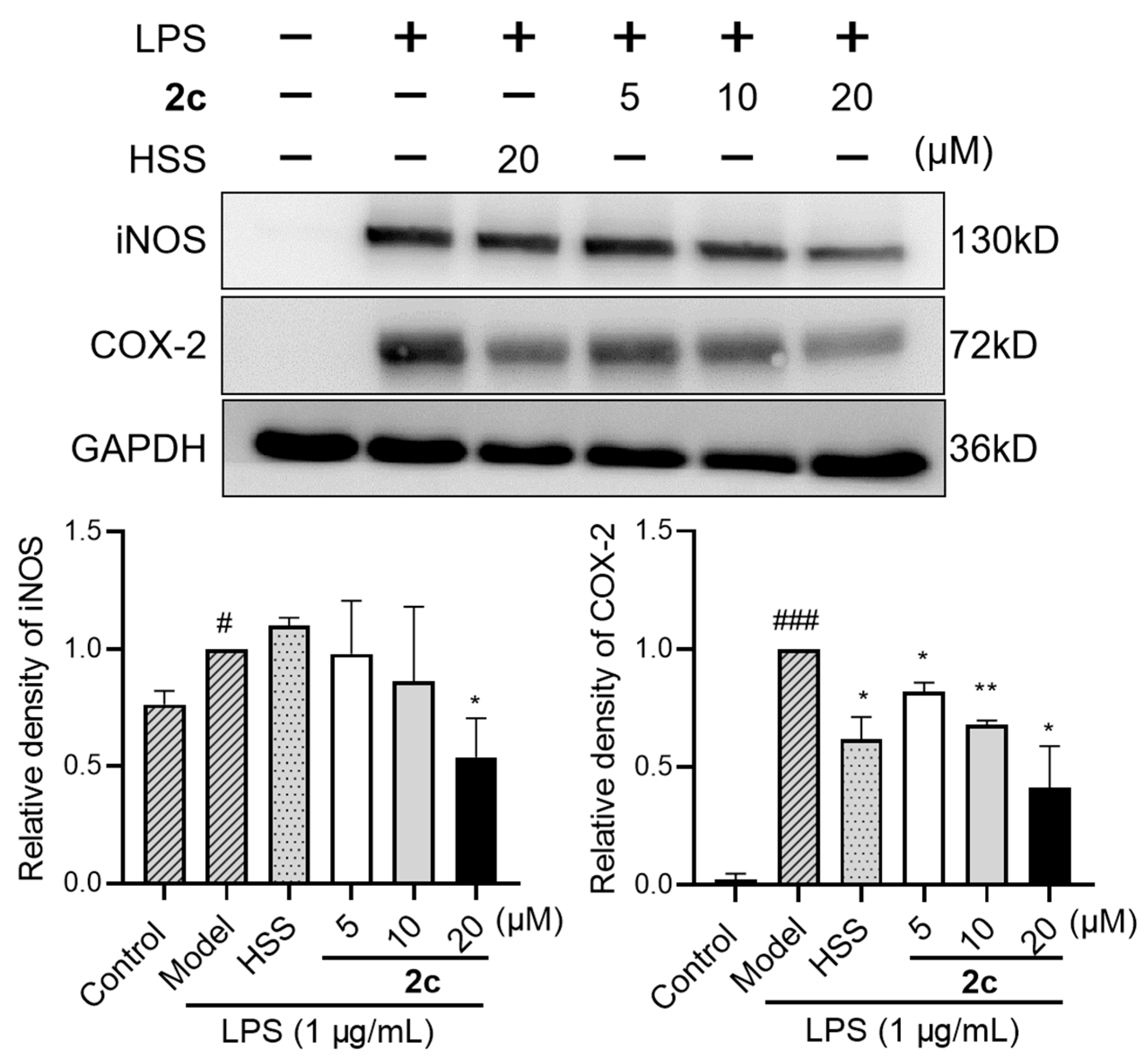
2.6. Inhibition of LPS-Triggered Generation of Inducible Nitric Oxide Synthase (iNOS) and Cyclooxygenase-2 (COX-2)
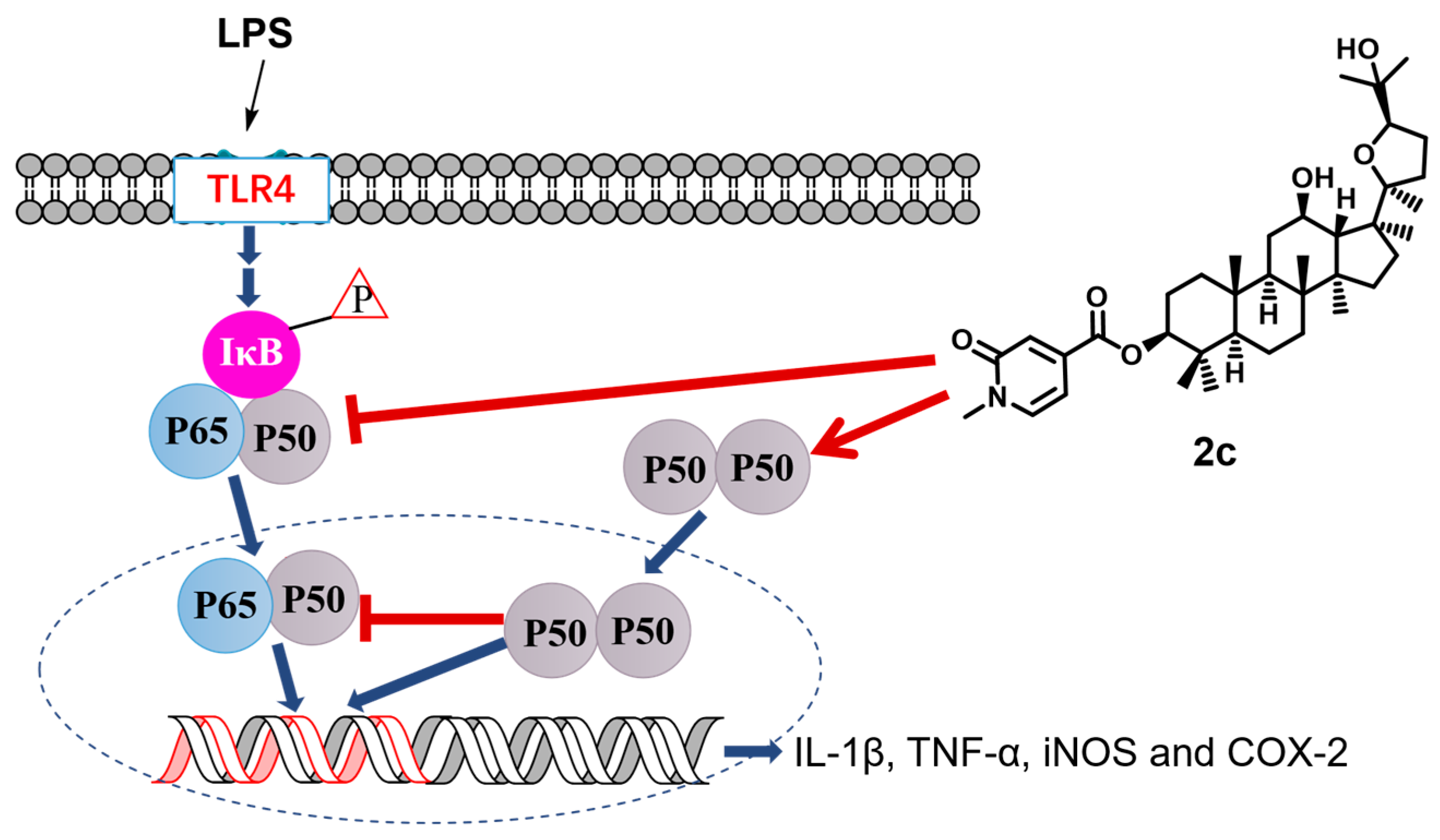
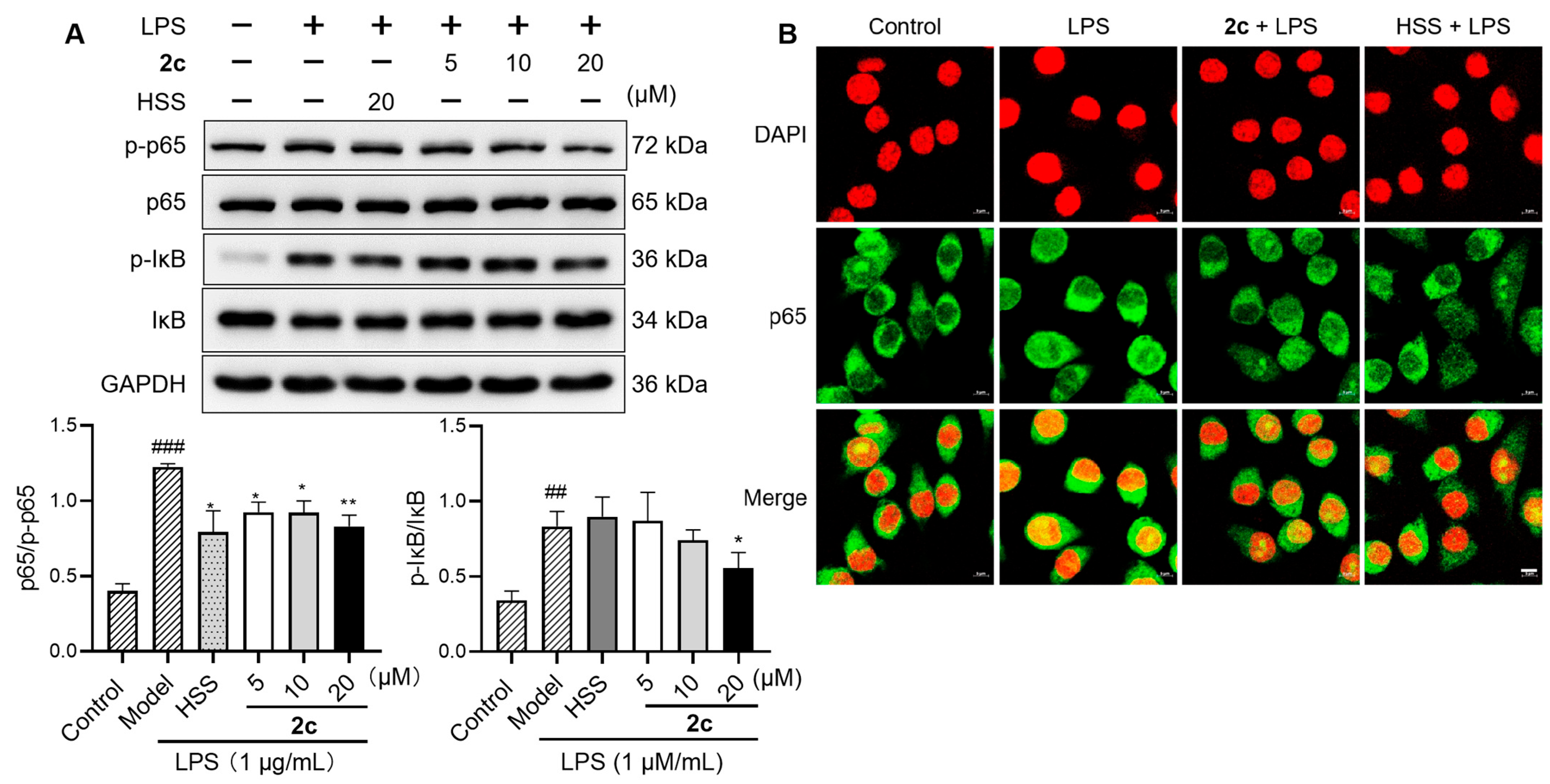
2.7. Inhibition of LPS-Triggered Activation of NF-κB
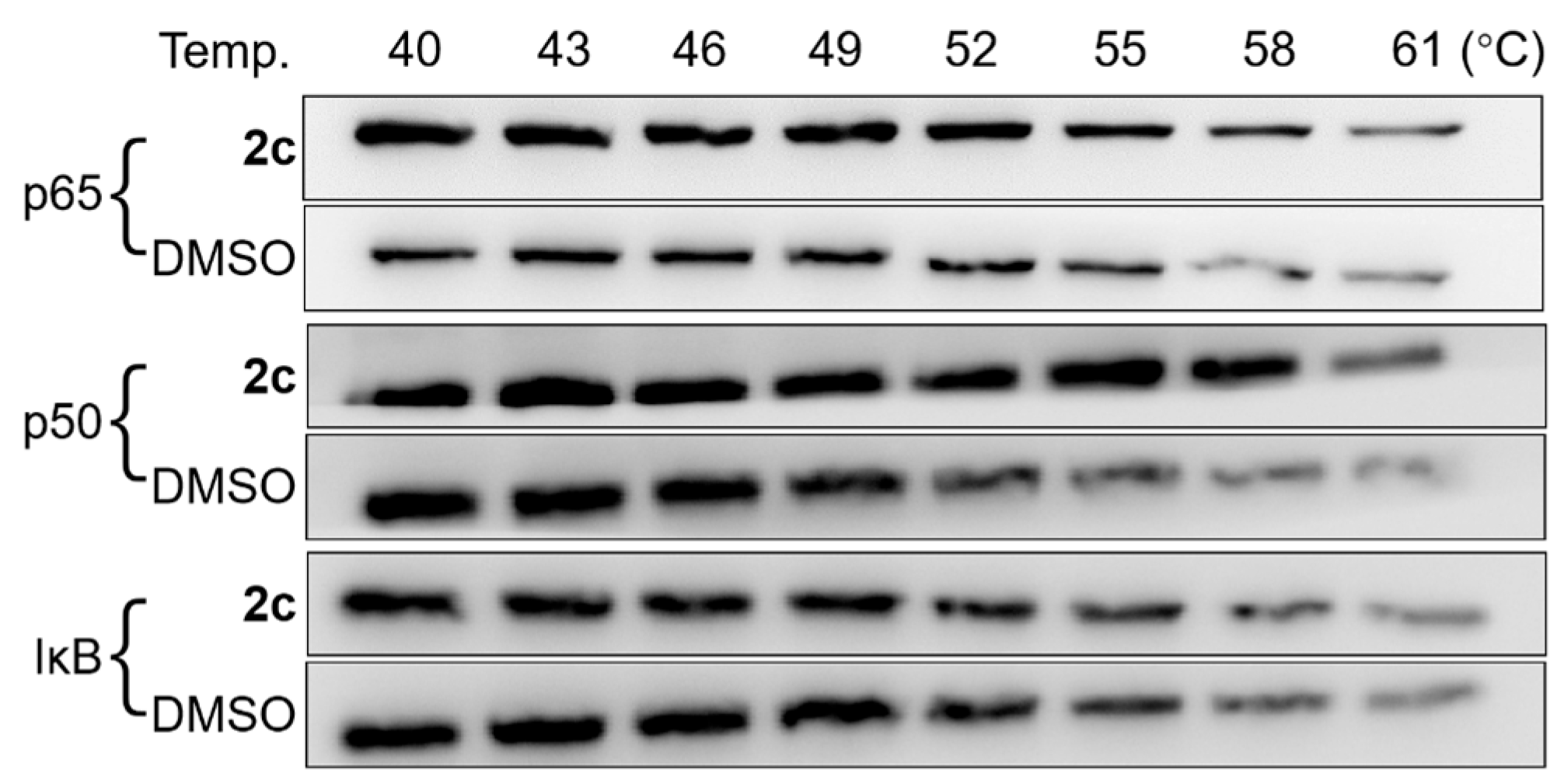
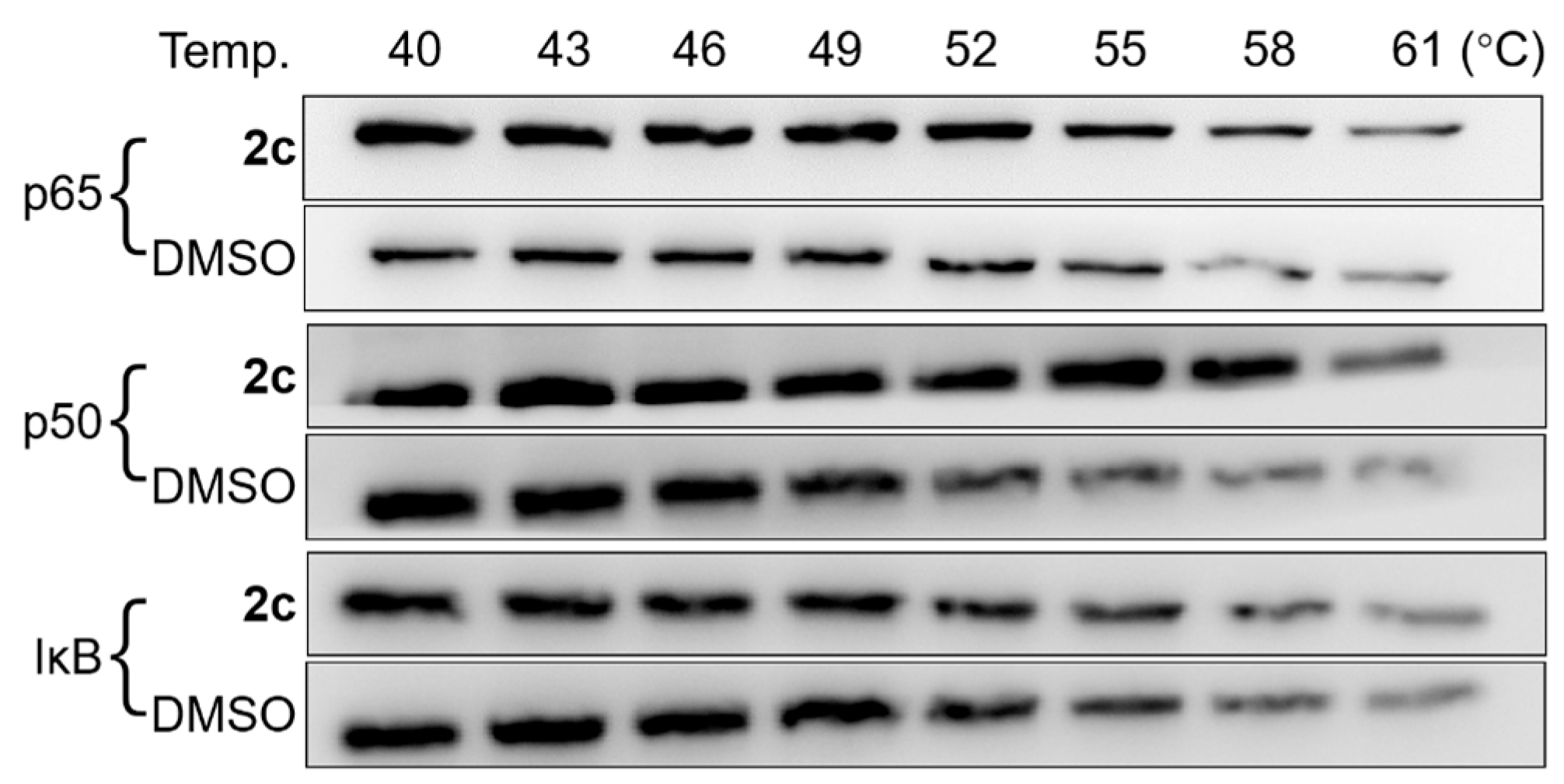
2.8. CETSA
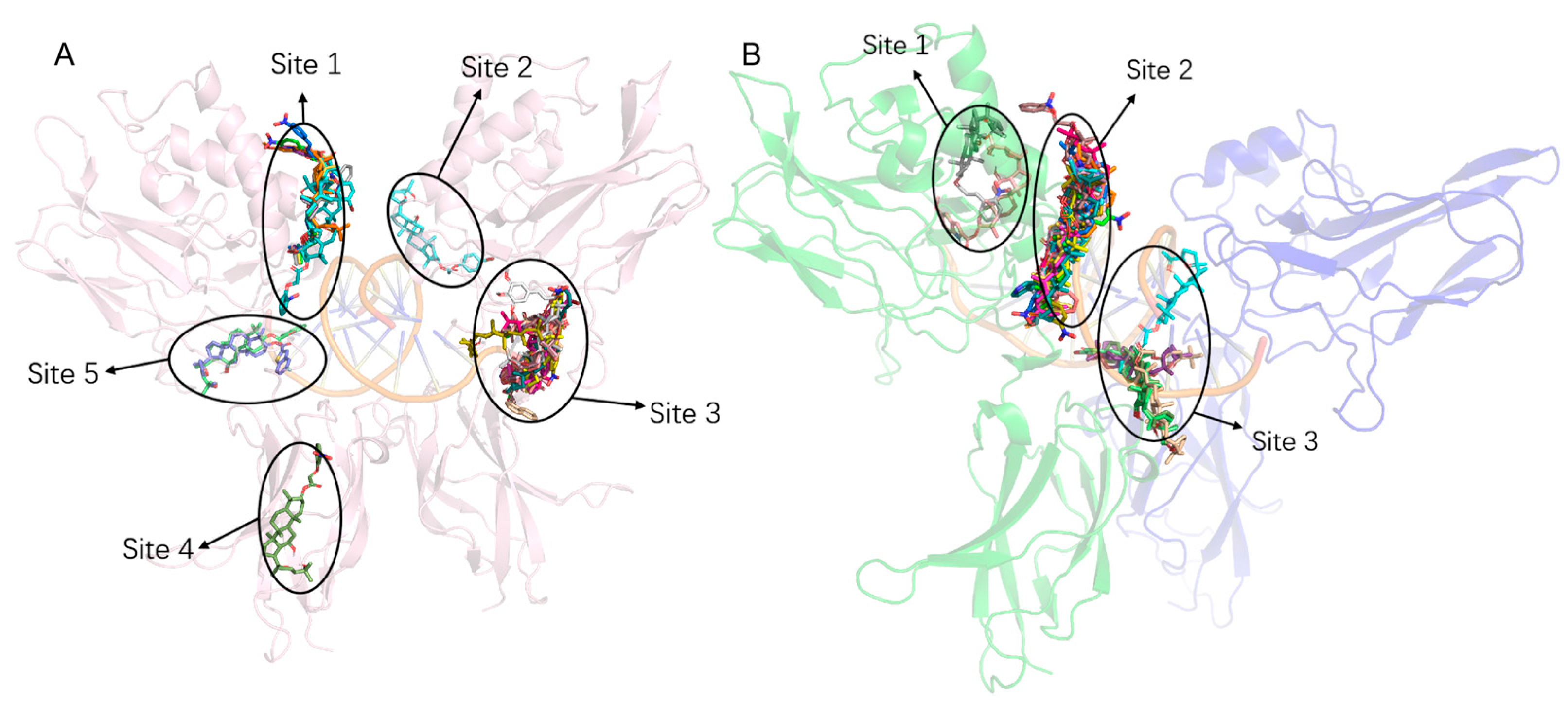
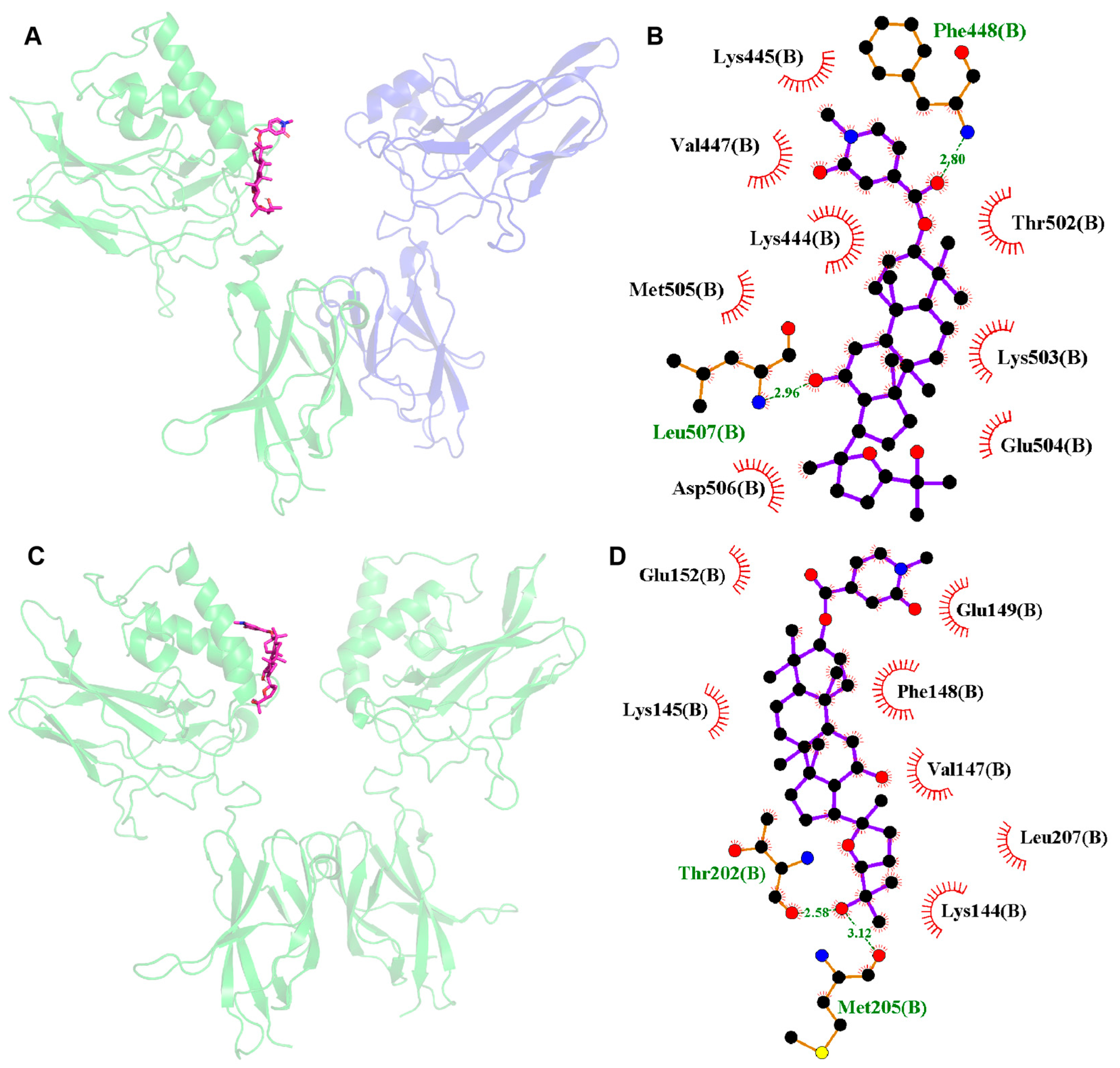
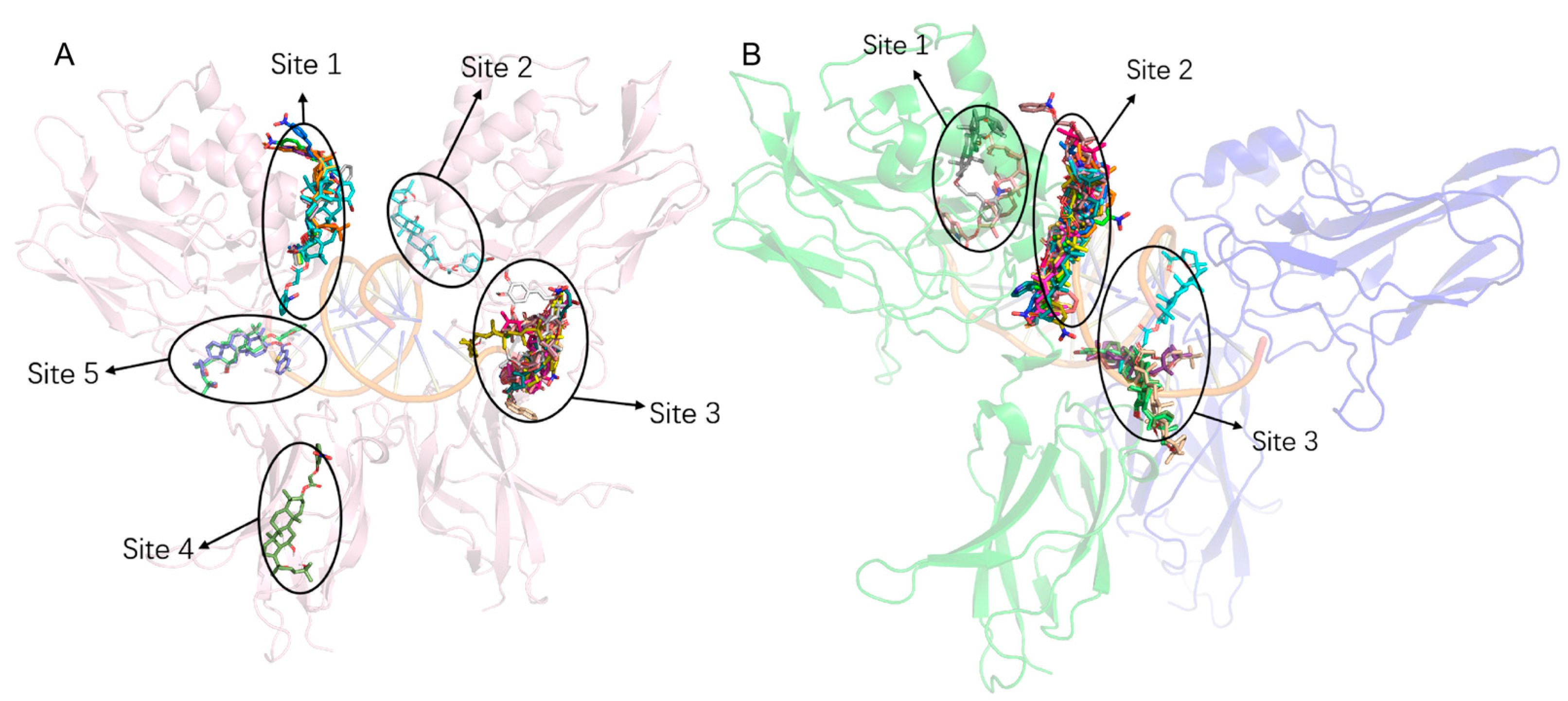
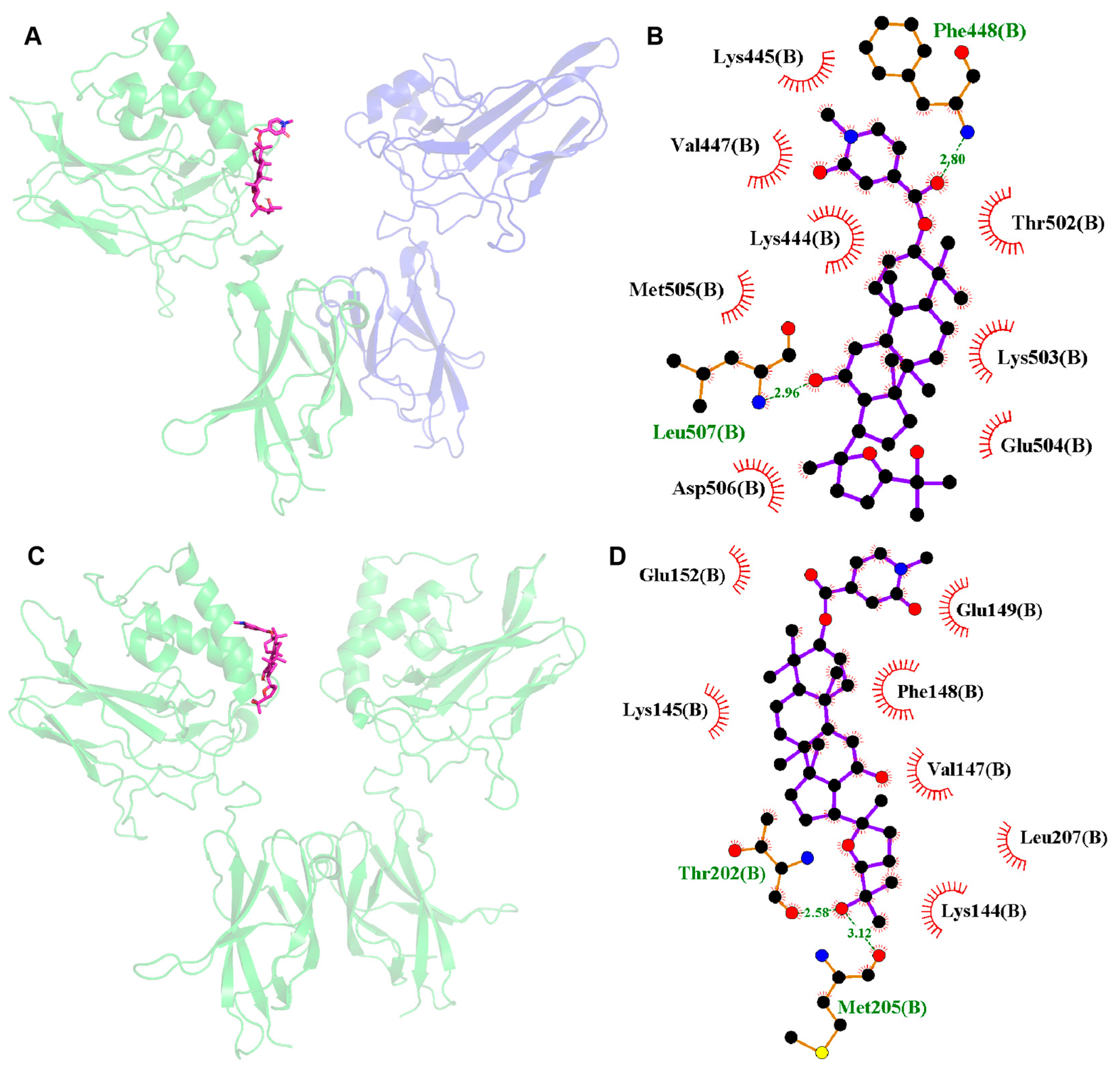
2.9. Molecular Docking
3. Materials and Methods
3.1. Virtual Screening and Molecular Docking Analysis
3.2. Chemistry
3.3. Synthesis of 2a–2h
- Compound 2a, white solid, yield: 75%; m.p.: 167–168 °C; +28.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.29 (d, J = 9.1 Hz, 2H), 8.19 (d, J = 8.8 Hz, 2H), 4.76 (dd, J = 10.2, 6.0 Hz, 1H), 3.86 (dd, J = 8.9, 6.7 Hz, 1H), 3.54 (td, J = 10.4, 4.7 Hz, 1H), 2.21 (td, J = 10.0, 3.1 Hz, 1H), 2.10–0.94 (m, 21H), 1.28 (s, 3H), 1.28 (s, 3H), 1.10 (s, 3H), 1.02 (s, 3H), 1.01 (s, 3H), 0.94 (s, 3H), and 0.93 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 164.4, 150.5, 136.4, 130.7 (2C), 123.6 (2C), 86.6, 85.5, 82.9, 71.0, 70.2, 56.2, 52.1, 50.5, 49.5, 48.0, 39.9, 38.7, 38.4, 37.2, 34.8, 32.7, 31.5, 31.3, 28.7, 28.2, 28.0, 27.7, 26.2, 25.1, 23.8, 18.3, 18.3, 16.8, 16.5, and 15.5; HRMS (ESI, positive): m/z [M+Na]+ calculated for C37H55N1O7Na+ 648.3871, found 648.3872.
- Compound 2b, white solid, yield: 60%; m.p.: 220–222 °C; +33.7 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.81 (d, J = 4.7 Hz, 1H), 8.74 (s, 1H), 8.22 (dd, J = 4.8, 1.2 Hz, 1H), 4.82 (dd, J = 9.1, 7.1 Hz, 1H), 3.86 (dd, J = 8.9, 6.7 Hz, 1H), 3.54 (td, J = 10.5, 4.6 Hz, 1H), 2.21 (td, J = 10.1, 3.1 Hz, 1H), 2.08–0.91 (m, 21H), 1.28 (s, 3H), 1.28 (s, 3H), 1.10 (s, 3H), 1.03 (s, 3H), 1.01 (s, 3H), 0.95 (s, 3H), 0.93 (s, 3H), and 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 162.6, 157.4, 149.9, 142.7, 128.4, 117.6, 86.5, 85.4, 84.0, 70.9, 70.1, 56.1, 52.0, 50.4, 49.4, 47.9, 39.8, 38.6, 38.3, 37.1, 34.7, 32.6, 31.4, 31.2, 28.6, 28.2, 27.9, 27.6, 26.1, 25.0, 23.7, 18.2 (2C), 16.7, 16.4, and 15.4; HRMS (ESI, positive): m/z [M+Na]+ calculated for C36H54N2O7Na+ 649.3823, found 649.3823.
- Compound 2c, colorless syrup, yield: 80%; +37.6 (c 1.0, CH3OH); 1H NMR (400 MHz, CDCl3): δ 7.35 (d, J = 6.9 Hz, 1H), 7.20 (s, 1H), 6.66 (dd, J = 7.0, 1.5 Hz, 1H), 4.66 (dd, J = 10.9, 5.1 Hz, 1H), 3.85 (dd, J = 8.7, 6.7 Hz, 1H), 3.58 (s, 3H), 3.53 (td, J = 10.6, 4.4 Hz, 1H), 2.20 (td, J = 10.0, 4.2 Hz, 1H), 2.08–0.77 (m, 21H), 1.28 (s, 3H), 1.27 (s, 3H), 1.10 (s, 3H), 1.00 (s, 3H), 0.96 (s, 3H), 0.91 (s, 6H), and 0.89 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 164.2, 162.9, 141.4, 138.5, 122.1, 104.6, 86.5, 85.4, 82.9, 70.9, 70.1, 56.1, 52.0, 50.4, 49.4, 47.9, 39.8, 38.6, 38.2, 37.8, 37.1, 34.7, 32.6, 31.3, 31.2, 28.6, 28.1, 27.9, 27.6, 26.1, 25.0, 23.5, 18.1 (2C), 16.6, 16.4, and 15.4; HRMS (ESI, positive): m/z [M+Na]+ calculated for C37H57N1O6Na+ 634.4078, found 634.4062.
- Compound 2d, pale yellow syrup, yield: 62%; +28.4 (c 0.5, CHCl3);1H NMR (400 MHz, CDCl3): δ 7.67 (dt, J = 8.0, 0.9 Hz, 1H), 7.59 (dd, J = 8.4, 0.7 Hz, 1H), 7.48 (d, J = 0.8 Hz, 1H), 7.44 (ddd, J = 8.5, 7.1, 1.4 Hz,1H), 7.30 (ddd, J = 8.0, 7.1, 0.8 Hz, 1H), 4.78 (dd, J = 9.1, 7.1 Hz, 1H), 4.31 (t, J = 6.6 Hz, 1H), 3.85 (dd, J = 8.8, 6.6 Hz, 1H), 3.54 (td, J = 10.4, 4.7 Hz, 1H), 2.20 (td, J = 10.1, 3.2 Hz, 1H), 2.07–0.88 (m, 21H), 1.28 (s, 3H), 1.27 (s, 3H), 1.10 (s, 3H), 1.01 (s, 3H), 1.01 (s, 3H), 0.94 (s, 6H), and 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 159.5, 155.7, 146.0, 128.8, 127.4, 123.7, 122.7, 113.3, 112.4, 86.5, 85.4, 82.1, 70.9, 70.1, 56.1, 52.0, 50.4, 49.4, 48.0, 39.8, 38.6, 38.3, 37.1, 34.8, 32.6, 31.3, 31.2, 28.6, 28.1, 27.9, 27.6, 26.1, 25.0, 23.8, 18.2, 18.1, 16.6, 16.4, and 15.4; HRMS (ESI, positive): m/z [M+Na]+ calculated for C39H56O6Na+ 643.3969, found 643.3953.
- Compound 2e, white solid, yield: 65%; m.p.: 116–117 °C; +26.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.53 (dd, J = 9.1, 4.1 Hz, 1H), 7.44 (s, 1H), 7.32 (dd, J = 8.2, 2.5 Hz, 1H), 7.17 (td, J = 9.1, 2.5 Hz, 1H), 4.78 (dd, J = 8.7, 7.6 Hz, 1H), 3.85 (dd, J = 8.8, 6.9 Hz, 1H), 3.54 (td, J = 10.4, 4.4 Hz, 1H), 2.20 (td, J = 10.1, 3.3 Hz, 1H), 2.08–0.85 (m, 21H), 1.28 (s, 3H), 1.27 (s, 3H), 1.10 (s, 3H), 1.01 (s, 6H), 0.93 (s, 6H), and 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 159.4 (d, J = 240.8 Hz, 1C), 159.1, 152.0, 147.6, 127.7 (d, J = 11.6 Hz, 1C), 115.7 (d, J = 26.0 Hz, 1C), 113.3 (d, J = 15.4 Hz, 1C), 113.2, 107.7 (d, J = 25.0 Hz, 1C), 86.5, 85.4, 82.4, 70.9, 70.1, 56.1, 52.0, 50.4, 49.4, 48.0, 39.8, 38.6, 38.3, 37.1, 34.7, 32.6, 31.3, 31.2, 28.6, 28.1, 27.9, 27.6, 26.1, 25.0, 23.8, 18.2, 18.1, 16.6, 16.4, and 15.4; HRMS (ESI, positive): m/z [M+Na]+ calculated for C39H55F1O6Na+ 661.3875, found 661.3861.
- Compound 2f, white solid, yield: 60%; m.p.: 186–188 °C; +30.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.97 (s, 1H), 7.79 (dd, J = 8.9, 4.8 Hz, 1H), 7.53 (dd, J = 9.1, 2.5 Hz, 1H), 7.21 (td, J = 8.9, 2.6 Hz, 1H), 4.71 (dd, J = 10.9, 5.4 Hz, 1H), 3.85 (dd, J = 8.8, 6.6 Hz, 1H), 3.54 (td, J = 10.4, 4.7 Hz, 1H), 2.20 (td, J = 10.1, 3.1 Hz, 1H), 2.10–0.88 (m, 21H), 1.28 (s, 3H), 1.27 (s, 3H), 1.10 (s, 3H), 1.01 (s, 6H), 0.94 (s, 3H), 0.94 (s, 3H), and 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 162.2, 160.8 (d, J = 243.7 Hz, 1C), 139.6 (d, J = 9.6 Hz, 1C), 137.6, 136.8, 129.4 (d, J = 3.9 Hz, 1C), 124.0 (d, J = 9.6 Hz, 1C), 116.0 (d, J = 26.0 Hz, 1C), 110.5 (d, J = 22.2 Hz, 1C), 86.5, 85.4, 82.6, 70.9, 70.1, 56.1, 52.0, 50.4, 49.4, 48.0, 39.8, 38.6, 38.3, 37.1, 34.7, 32.6, 31.4, 31.2, 28.6, 28.1, 27.9, 27.6, 26.1, 25.0, 23.7, 18.2, 18.2, 16.6, 16.4, and 15.4; HRMS (ESI, positive): m/z [M+Na]+ calculated for C39H55F1S1O5Na+ 677.3646, found 677.3632.
- Compound 2g, white solid, yield: 60%; m.p.: 248–249 °C; +46.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.97 (dd, J = 6.6, 1.4 Hz, 1H), 7.81 (dd, J = 6.7, 1.5 Hz, 1H), 7.53 (td, J = 7.2, 1.6 Hz, 1H), 7.49 (td, J = 7.2, 1.4 Hz, 1H), 4.74 (dd, J = 11.4, 5.1 Hz, 1H), 3.85 (dd, J = 8.8, 6.9 Hz, 1H), 3.54 (td, J = 10.5, 4.5 Hz, 1H), 2.20 (td, J = 10.1, 3.1 Hz, 1H), 2.10–0.90 (m, 21H), 1.28 (s, 3H), 1.27 (s, 3H), 1.10 (s, 3H), 1.03 (s, 3H), 1.01 (s, 3H), 0.96 (s, 3H), 0.94 (s, 3H), and 0.93 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 161.2, 138.7, 137.3, 128.1, 126.9, 125.5, 123.9, 122.8 (2C), 86.6, 85.5, 83.1, 71.0, 70.2, 56.2, 52.1, 50.5, 49.5, 48.1, 39.9, 38.7, 38.3, 37.2, 34.9, 32.7, 31.5, 31.3, 28.7, 28.2, 28.0, 27.7, 26.2, 25.1, 23.9, 18.3, 18.3, 16.8, 16.5, and 15.5; HRMS (ESI, positive): m/z [M+Na]+ calculated for C39H55Cl1S1O5Na+ 693.3351, found 693.3342.
- Compound 2h, pale yellow syrup, yield: 50%; +28.1 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.24 (d, J = 8.8 Hz, 1H), 7.15 (d, J = 1.9 Hz, 1H), 7.05 (d, J = 2.5 Hz, 1H), 6.85 (dd, J = 8.8, 2.5 Hz, 1H), 4.49 (dd, J = 11.0, 5.2 Hz, 1H), 3.85 (s, 3H), 3.84 (dd, J = 8.8, 6.6 Hz, 1H), 3.73 (d, J = 0.5 Hz, 2H), 3.50 (td, J = 10.6, 4.6 Hz, 1H), 2.18 (td, J = 10.1, 3.1 Hz, 1H), 2.08–0.77 (m, 21H), 1.27 (s, 3H), 1.26 (s, 3H), 1.09 (s, 3H), 0.97 (s, 3H), 0.88 (s, 3H), 0.85 (s, 3H), 0.79 (s, 3H), and 0.75 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 171.8, 154.1, 131.2, 127.7, 123.6, 112.6, 111.8, 108.7, 100.6, 86.5, 85.4, 81.1, 70.9, 70.1, 56.0, 55.9, 52.0, 50.4, 49.4, 48.0, 39.8, 38.6, 37.9, 37.1, 34.7, 32.6, 31.8, 31.3, 31.2, 28.6, 27.9, 27.9, 27.6, 26.1, 25.0, 23.7, 18.1 (2C), 16.4, 16.4, and 15.4. HRMS (ESI, positive): m/z [M+Na]+ calculated for C41H61N1O6Na+ 686.4391, found 686.4383.
3.4. Cell Culture and NO Release Assay
3.5. Cell Viability
3.6. MitoSOX Assay
3.7. ELISA
3.8. Western Blotting
3.9. Immunofluorescence Staining
3.10. CETSA
3.11. Statistical Analyses
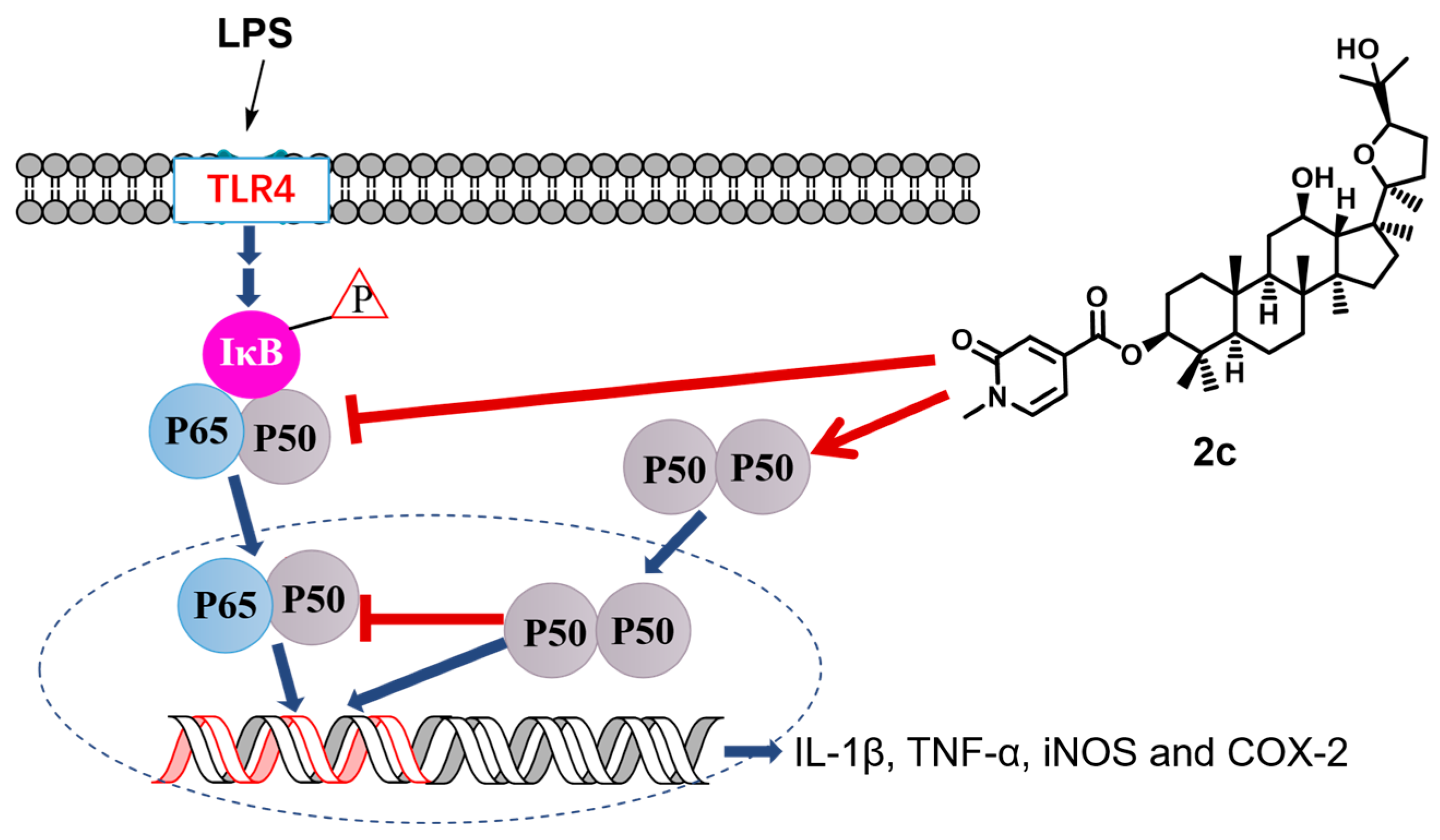
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Poll, T.; Shankar-Hari, M.; Wiersinga, W.J. The immunology of sepsis. Immunity 2021, 54, 2450–2464. [Google Scholar] [CrossRef] [PubMed]
- Płóciennikowska, A.; Hromada-Judycka, A.; Borzęcka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Ma, X.Y.; Wang, H.; Ding, B.; Zhong, H.; Ghosh, S.; Lengyel, P. The interferon-inducible p202a protein modulates NF-κB activity by inhibiting the binding to DNA of p50/p65 heterodimers and p65 homodimers while enhancing the binding of p50 homodimers. J. Biol. Chem. 2003, 278, 23008–23019. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.N.; Kundu, J.K.; Cha, Y.N.; Surh, Y.J. Resolvin D1 stimulates efferocytosis through p50/p50-mediated suppression of tumor necrosis factor-α expression. J. Cell Sci. 2013, 126, 4037–4047. [Google Scholar] [CrossRef]
- Kim, J.H.; Yi, Y.S.; Kim, M.Y.; Cho, J.Y. Role of ginsenosides, the main active components of Panax ginseng, in inflammatory responses and diseases. J. Ginseng Res. 2017, 41, 435–443. [Google Scholar] [CrossRef]
- Hyun, S.H.; Bhilare, K.D.; In, G.; Park, C.K.; Kim, J.H. Effects of Panax ginseng and ginsenosides on oxidative stress and cardiovascular diseases: Pharmacological and therapeutic roles. J. Ginseng Res. 2022, 46, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, H.; Liu, W.; Cao, H.; Hu, X.; Gao, X.; Xu, F.; Li, Z.; Hua, H.; Li, D. Dammarane-type leads panaxadiol and protopanaxadiol for drug discovery: Biological activity and structural modification. Eur. J. Med. Chem. 2020, 189, 112087. [Google Scholar] [CrossRef] [PubMed]
- Karra, A.G.; Konstantinou, M.; Tzortziou, M.; Tsialtas, I.; Kalousi, F.D.; Garagounis, C.; Hayes, J.M.; Psarra, A.G. Potential dissociative glucocorticoid receptor activity for protopanaxadiol and protopanaxatriol. Int. J. Mol. Sci. 2019, 20, 94. [Google Scholar] [CrossRef]
- Yang, G.; Mi, X.; Wang, Y.; Li, S.; Yu, L.; Huang, X.; Tan, S.; Yu, H. Fusion of Michael-acceptors enhances the anti-inflammatory activity of ginsenosides as potential modulators of the NLRP3 signaling pathway. Bioorg. Chem. 2023, 134, 106467. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Li, D.; Zhong, D. Identification of 20(S)-protopanaxadiol metabolites in human liver microsomes and human hepatocytes. Drug Metab. Dispos. 2011, 39, 472–483. [Google Scholar] [CrossRef]
- Wang, W.; Wu, X.; Wang, L.; Meng, Q.; Liu, W. Stereoselective property of 20 (S)-protopanaxadiol ocotillol type epimers affects its absorption and also the inhibition of P-glycoprotein. PLoS ONE 2014, 9, e98887. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Wu, X.; Xu, L.; Meng, Q.; Liu, W. Stereoselective formation and metabolism of 20(S)-protopanaxadiol ocotillol type epimers in vivo and in vitro. Chirality 2015, 27, 170–176. [Google Scholar] [CrossRef]
- Feng, R.Q.; Liu, J.; Wang, Z.H.; Zhang, J.W.; Cates, C.; Rousselle, T.; Meng, Q.G.; Li, J. The structure-activity relationship of ginsenosides on hypoxia-reoxygenation induced apoptosis of cardiomyocytes. Biochem. Biophys. Res. Commun. 2017, 494, 556–568. [Google Scholar] [CrossRef]
- Wang, T.; Meng, Q.; Zhang, J.; Bi, Y.; Jiang, N. Study on the structure-function relationship of 20(S)-panaxadiol and its epimeric derivatives in myocardial injury induced by isoproterenol. Fitoterapia 2010, 81, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Q.; Yang, Y.T.; Yang, Q.; Li, Y.; Jiang, Y.T.; Fu, F.H.; Wang, H.B. Novel fluorescent pyxinol-based probes: Design, synthesis and biological evaluation. Chin. J. Org. Chem 2017, 37, 2109–2114. [Google Scholar] [CrossRef]
- Liu, J.L.; Liu, Y.H.; Yu, H.; Zhang, Y.; Hsu, A.C.Y.; Zhang, M.M.; Gou, Y.W.; Sun, W.; Wang, F.; Li, P.Y.; et al. Design, synthesis and biological evaluation of novel pyxinol derivatives with anti-heart failure activity. Biomed. Pharmacother. 2021, 133, 111050. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.C.; Wang, K.Y.; Xu, S.; Kong, L.T.; Bi, Y.; Li, X.P. Recent advances in the semisynthesis, modifications and biological activities of ocotillol-type triterpenoids. Molecules 2020, 25, 5562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Cao, Y.C.; Wang, K.Y.; Shi, Z.Y.; Wang, R.D.; Meng, Q.G.; Bi, Y. Design, synthesis, and antibacterial evaluation of novel ocotillol derivatives and their synergistic effects with conventional antibiotics. Molecules 2021, 26, 5969. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, C.; Zhang, H.; Bi, Y.; Chen, X.; Tian, H.; Xie, X.; Meng, Q.; Lewis, P.J.; Xu, J. Synthesis and biological evaluation of novel ocotillol-type triterpenoid derivatives as antibacterial agents. Eur. J. Med. Chem. 2013, 68, 444–453. [Google Scholar] [CrossRef]
- Ren, Q.W.; Yang, G.Q.; Guo, M.Q.; Guo, J.W.; Li, Y.; Lu, J.; Yang, Q.; Tang, H.H.; Fang, X.J.; Sun, Y.X.; et al. Design, synthesis, and discovery of ocotillol-type amide derivatives as orally available modulators of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2019, 161, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Zhang, H.; Zhang, G.N.; Wang, Y.J.; Kathawala, R.J.; Si, R.; Patel, B.A.; Xu, J.; Chen, Z.S. Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents. Oncotarget 2015, 6, 24277–24290. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, M.; Liu, S.; Zou, Z.; Ren, R.; Zhang, C.; Xie, H.; Sun, J.; Qi, Y.; Qu, Q.; et al. Pyxinol bearing amino acid residues: Easily achievable and promising modulators of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2021, 216, 113317. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zhang, D.D.; Ma, G.S.; Su, Z.Y.; Liu, M.M.; Wang, R.; Meng, Q.G.; Bi, Y.; Wang, H.B. Design, synthesis, and biological evaluation of ocotillol derivatives fused with 2-aminothiazole via A-ring as modulators of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2022, 243, 114784. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, S.; Zhang, C.; Yu, L.; Zou, Z.; Wang, C.; Gao, M.; Li, S.; Ma, Y.; Xu, R.; et al. Discovery of pyxinol amide derivatives bearing amino acid residues as nonsubstrate allosteric inhibitors of P-glycoprotein-mediated multidrug resistance. J. Med. Chem. 2023, 66, 8628–8642. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, H.; Fu, S.Z.; Tan, L.Y.; Liu, J.L.; Zhou, B.S.; Li, L.; Liu, Y.H.; Wang, C.X.; Li, P.Y.; et al. Synthesis and anti-hepatocarcinoma effect of amino acid derivatives of pyxinol and ocotillol. Molecules 2021, 26, 780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Zeng, X.M.; Zhang, S.Y.; Zhou, Y.Y.; Zhou, Z.W. Novel ocotillol-derived lactone derivatives: Design, synthesis, bioactive evaluation, SARs and preliminary antibacterial mechanism. Mol. Divers. 2022, 26, 2103–2120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mi, X.; Du, Y.; Li, S.; Yu, L.; Gao, M.; Yang, X.; Song, Z.; Yu, H.; Yang, G. Design, synthesis, and anti-inflammatory activities of 12-dehydropyxinol derivatives. Molecules 2023, 28, 1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Gao, M.; Sun, Y.; Wang, C.; Fang, X.; Gao, H.; Diao, W.; Yu, H. Design, synthesis and anti-inflammatory activity of 3-amino acid derivatives of ocotillol-type sapogenins. Eur. J. Med. Chem. 2020, 202, 112507. [Google Scholar] [CrossRef]
- Sun, Y.; Fang, X.; Gao, M.; Wang, C.; Gao, H.; Bi, W.; Tang, H.; Cui, Y.; Zhang, L.; Fan, H.; et al. Synthesis and structure–activity relationship of pyxinol derivatives as novel anti-inflammatory agents. ACS Med. Chem. Lett. 2020, 11, 457–463. [Google Scholar] [CrossRef]
- Pan, C.H.; Shan, H.J.; Wu, T.Y.; Liu, W.; Lin, Y.W.; Xia, W.Y.; Wang, F.; Zhou, Z.B.; Yu, X.W. 20(S)-Protopanaxadiol inhibits titanium particle-induced inflammatory osteolysis and RANKL-mediated osteoclastogenesis via MAPK and NF-kappa B signaling pathways. Front. Pharmacol. 2019, 9, 1538. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, J.; Rhee, M.H.; Yu, T.; Baek, K.-S.; Sung, N.Y.; Kim, Y.; Yoon, K.; Kim, J.H.; Kwak, Y.-S.; et al. Molecular mechanism of protopanaxadiol saponin fraction-mediated anti-inflammatory actions. J. Ginseng Res. 2015, 39, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.W.; Rey, F.A.; Sodeoka, M.; Verdine, G.L.; Harrison, S.C. Structure of the NF-kappa B p50 homodimer bound to DNA. Nature 1995, 373, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kawatkar, A.; Schefter, M.; Hermansson, N.O.; Snijder, A.; Dekker, N.; Brown, D.G.; Lundbäck, T.; Zhang, A.X.; Castaldi, M.P. CETSA beyond soluble targets: A broad application to multipass transmembrane proteins. ACS Chem. Biol. 2019, 14, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef] [PubMed]
- Neha, K.; Wakode, S. Contemporary advances of cyclic molecules proposed for inflammation. Eur. J. Med. Chem. 2021, 221, 113493. [Google Scholar] [CrossRef]
- Yang, G.Q.; Li, Y.; Yang, Q.; Yue, X.; Yao, L.; Jiang, Y.T. Simple and efficient synthesis of pseudoginsenoside HQ. Chin. J. Org. Chem 2017, 37, 1530–1536. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, G.; Cao, D.; Wang, F.; Zhang, F.; Shao, H.; Jiao, W. New monoterpene glycoside paeoniflorin derivatives as NO and IL-1β inhibitors: Synthesis and biological evaluation. Molecules 2023, 28, 6922. [Google Scholar] [CrossRef]
- Fock, E.M.; Parnova, R.G. Protective Effect of mitochondria-targeted antioxidants against inflammatory response to lipopolysaccharide challenge: A review. Pharmaceutics 2021, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-W.; Wang, F.; Turk, A.; Park, J.-S.; Ma, H.; Ma, Y.; Noh, H.-R.; Sui, G.; Shin, D.-S.; Lee, M.-K.; et al. Zaluzanin C alleviates inflammation and lipid accumulation in Kupffer cells and hepatocytes by regulating mitochondrial ROS. Molecules 2023, 28, 7484. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Liu, M.; Xu, H.; Chuan, J.; Yang, Z. Lipopolysaccharide activated NF-kB signaling by regulating HTRA1 expression in human retinal pigment epithelial cells. Molecules 2023, 28, 22 36. [Google Scholar] [CrossRef]
- Malek, S.; Huxford, T.; Ghosh, G. Ikappa Balpha functions through direct contacts with the nuclear localization signals and the DNA binding sequences of NF-kappaB. J. Biol. Chem. 1998, 273, 25427–25435. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xie, H.; Wang, C.; Zhang, C.; Yu, L.; Zhang, L.; Liu, X.; Xu, R.; Song, Z.; Liu, R.; et al. Design, synthesis, and discovery of Eudistomin Y derivatives as lysosome-targeted antiproliferation agents. Eur. J. Med. Chem. 2023, 250, 115193. [Google Scholar] [CrossRef]
- Fan, H.Y.; Wang, X.K.; Li, X.; Ji, K.; Du, S.H.; Liu, Y.; Kong, L.L.; Xu, J.C.; Yang, G.Q.; Chen, D.Q.; et al. Curcumin, as a pleiotropic agent, improves doxorubicin-induced nephrotic syndrome in rats. J. Ethnopharmacol. 2020, 250, 112502. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔGbinding (kcal/mol) | |
|---|---|---|
| 1NFK | 1VKX | |
| 2a | −8.49 | −7.71 |
| 2b | −9.96 | −9.38 |
| 2c | −8.22 | −7.98 |
| 2d | −7.90 | −7.44 |
| 2e | −7.27 | −7.49 |
| 2f | −6.90 | −7.42 |
| 2g | −8.23 | −7.56 |
| 2h | −6.83 | −7.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, S.; Zou, Z.; Luan, X.; Chen, C.; Li, S.; Zhang, Z.; Quan, M.; Li, X.; Zhu, W.; Yang, G. Synthesis, Anti-Inflammatory Activities, and Molecular Docking Study of Novel Pyxinol Derivatives as Inhibitors of NF-κB Activation. Molecules 2024, 29, 1711. https://doi.org/10.3390/molecules29081711
Tan S, Zou Z, Luan X, Chen C, Li S, Zhang Z, Quan M, Li X, Zhu W, Yang G. Synthesis, Anti-Inflammatory Activities, and Molecular Docking Study of Novel Pyxinol Derivatives as Inhibitors of NF-κB Activation. Molecules. 2024; 29(8):1711. https://doi.org/10.3390/molecules29081711
Chicago/Turabian StyleTan, Shuai, Zongji Zou, Xuwen Luan, Cheng Chen, Shuang Li, Zhen Zhang, Mengran Quan, Xiang Li, Wei Zhu, and Gangqiang Yang. 2024. "Synthesis, Anti-Inflammatory Activities, and Molecular Docking Study of Novel Pyxinol Derivatives as Inhibitors of NF-κB Activation" Molecules 29, no. 8: 1711. https://doi.org/10.3390/molecules29081711
APA StyleTan, S., Zou, Z., Luan, X., Chen, C., Li, S., Zhang, Z., Quan, M., Li, X., Zhu, W., & Yang, G. (2024). Synthesis, Anti-Inflammatory Activities, and Molecular Docking Study of Novel Pyxinol Derivatives as Inhibitors of NF-κB Activation. Molecules, 29(8), 1711. https://doi.org/10.3390/molecules29081711