


Electronic Effects in a Green Protocol for (Hetero)Aryl-S Coupling
 , , , , ,
, , , , ,
Abstract
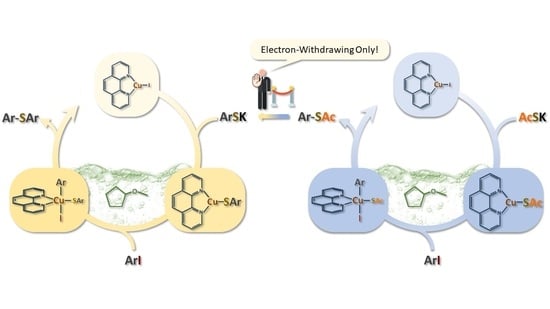
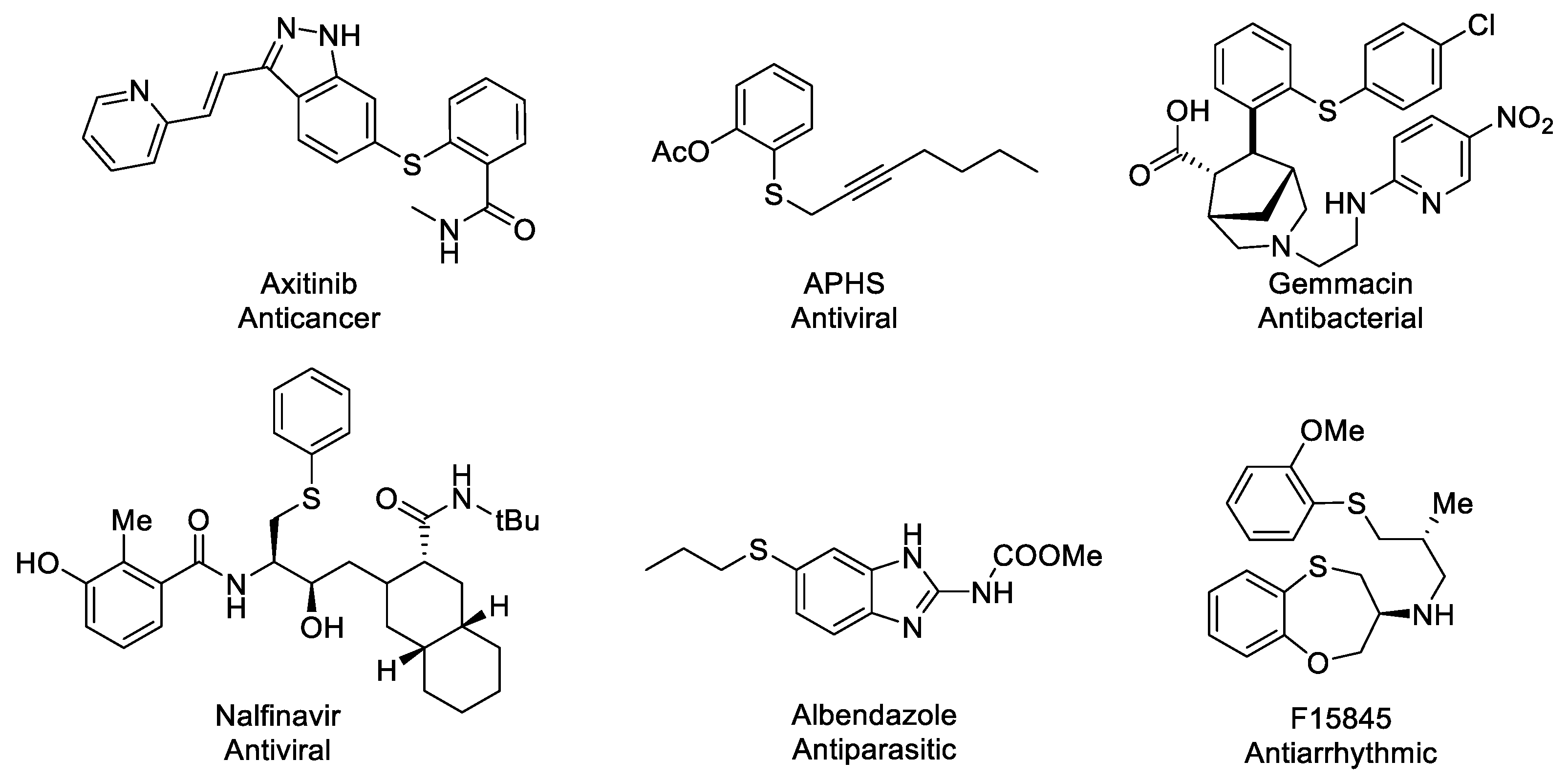
1. Introduction
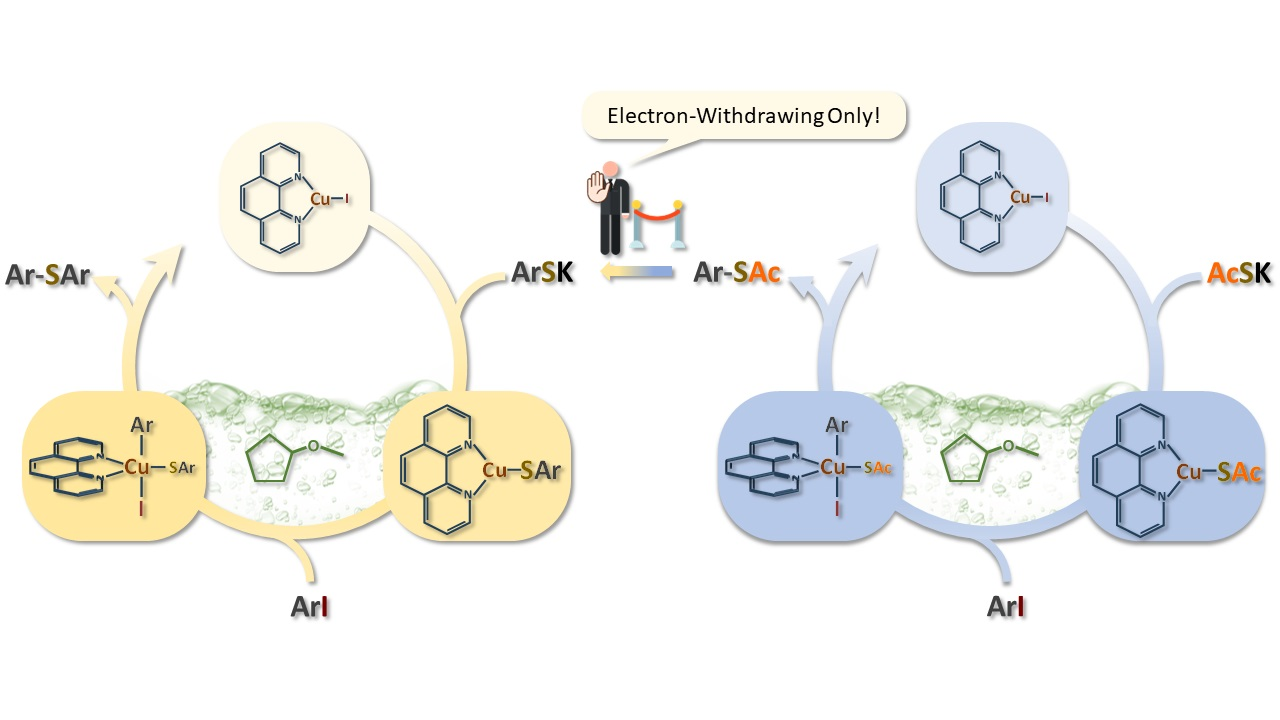
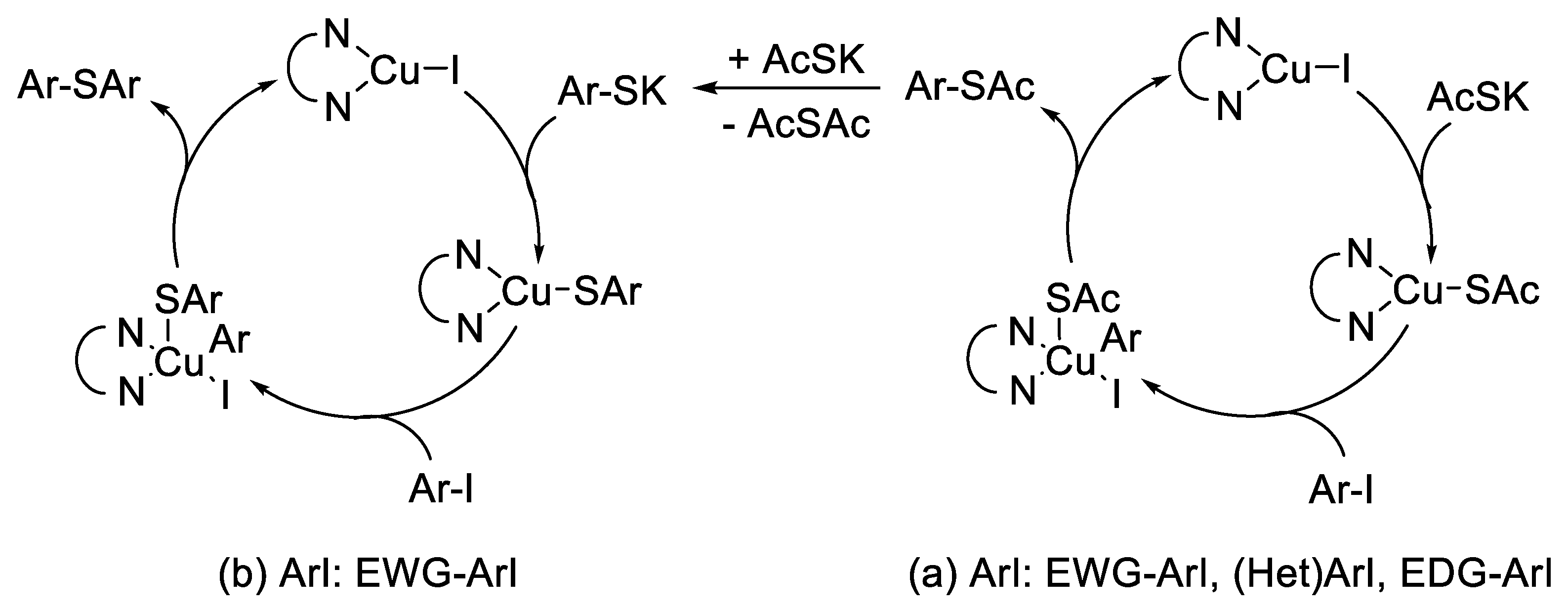
2. Results and Discussion
3. Materials and Methods
3.1. Materials
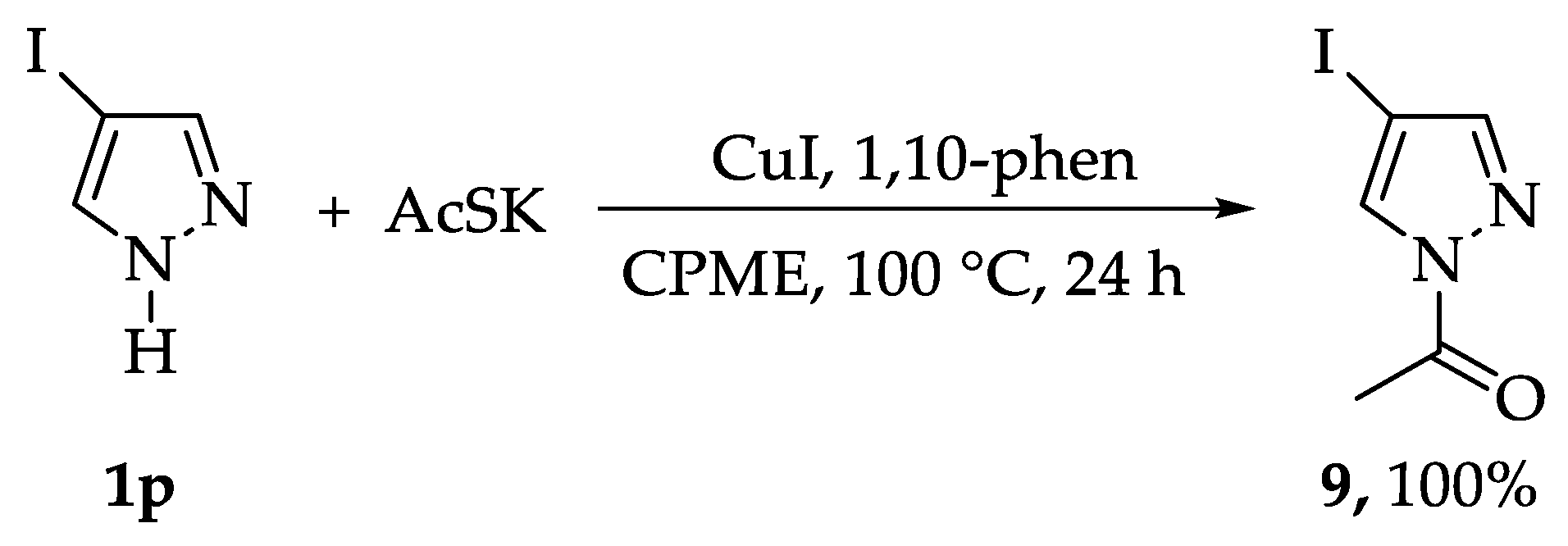
3.2. General Procedure for the Synthesis of the Substrates 4a–4o, 5, and 9
3.3. General Procedure for the Synthesis of Substrates 4a, 4f, and 4h–4j under Microwave Irradiation
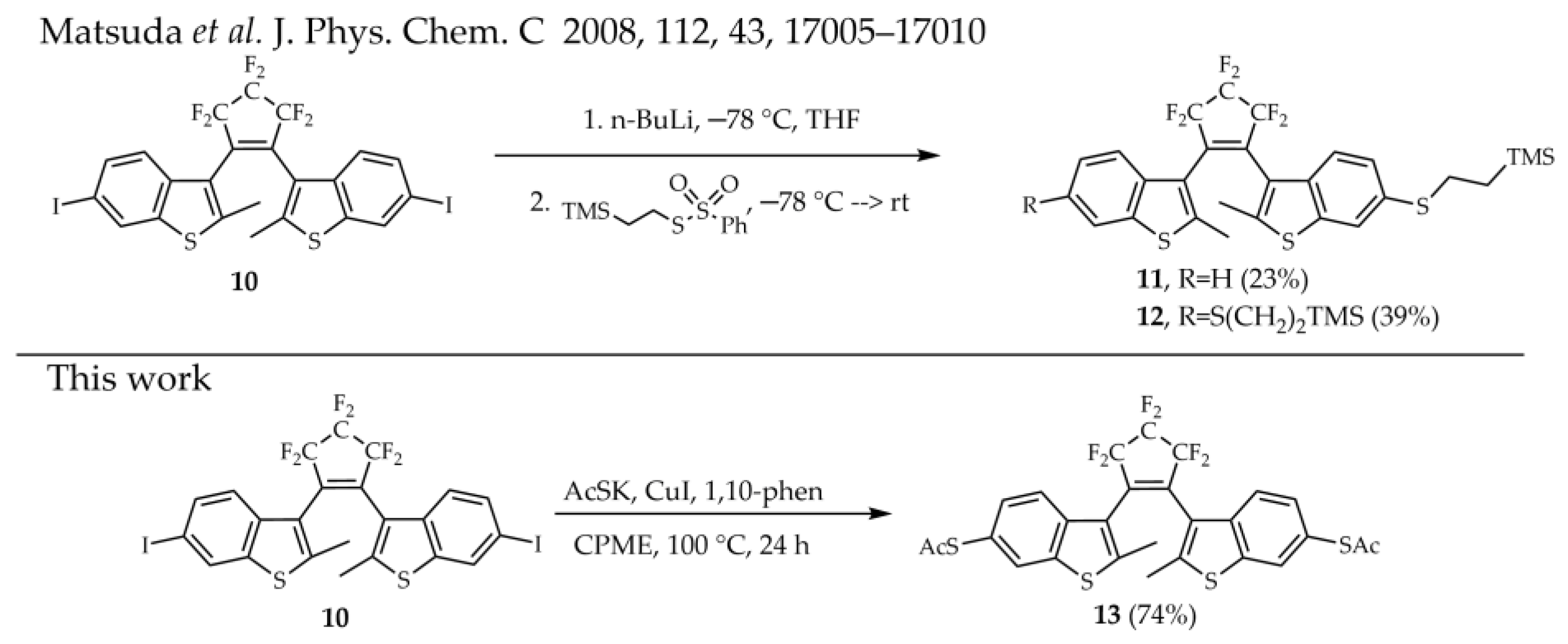
3.4. Synthesis of 3,3′-(Perfluorocyclopent-1-ene-1,2-diyl)bis(2-methyl-6-(acetylthio)-benzo[b]thiophene) 13
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perry, C.M.; Frampton, J.E.; McCormack, P.L.; Siddiqui, M.A.A.; Cvetković, R.S. Nelfinavir. Drugs 2005, 65, 2209–2244. [Google Scholar] [CrossRef]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Dubey, A.V.; Gharat, S.B.; Vijay Kumar, A. Glycerol as a Recyclable Solvent for Copper-Mediated Ligand-Free C-S Cross-Coupling Reaction: Application to Synthesis of Gemmacin Precursor. ChemistrySelect 2017, 2, 4852–4856. [Google Scholar] [CrossRef]
- Fang, X.; Li, J.; Wang, C.-J. Organocatalytic Asymmetric Sulfa-Michael Addition of Thiols to α,β-Unsaturated Hexafluoroisopropyl Esters: Expeditious Access to (R)-Thiazesim. Org. Lett. 2013, 15, 3448–3451. [Google Scholar] [CrossRef] [PubMed]
- Saint, D.A. Persistent (Current) in the Face of Adversity … A New Class of Cardiac Anti-Ischaemic Compounds on the Horizon? Br. J. Pharmacol. 2009, 156, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Zwettler, N.; Engbæk, J.; Lundsgaard, R.; Paranowska, I.; Nielsen, T.; Clyens, S.; Christiansen, J.; Andersen, M.Ø. Chemical Surface Functionalization of Bulk Poly (p-Phenylene Sulfide) Yields a Stable Sulfonic Acid Catalyst. React. Funct. Polym. 2015, 88, 47–54. [Google Scholar] [CrossRef]
- Bai, Y.; Li, Z.; Cheng, B.; Zhang, M.; Su, K. Higher UV-Shielding Ability and Lower Photocatalytic Activity of TiO2@SiO2/APTES and Its Excellent Performance in Enhancing the Photostability of Poly(p-Phenylene Sulfide). RSC Adv. 2017, 7, 21758–21767. [Google Scholar] [CrossRef]
- Gao, Y.; Su, K.; Wang, X.; Li, Z. A Metal-Nano GO Frameworks/PPS Membrane with Super Water Flux and High Dyes Interception. J. Membr. Sci. 2019, 574, 55–64. [Google Scholar] [CrossRef]
- Gao, Y.; Su, K.; Li, Z.; Cheng, B. Graphene Oxide Hybrid Poly(p-Phenylene Sulfide) Nanofiltration Membrane Intercalated by Bis(Triethoxysilyl) Ethane. Chem. Eng. J. 2018, 352, 10–19. [Google Scholar] [CrossRef]
- Antoine, R.; Broyer, M.; Dugourd, P. Metal Nanoclusters: From Fundamental Aspects to Electronic Properties and Optical Applications. Sci. Technol. Adv. Mater. 2023, 24, 2222546. [Google Scholar] [CrossRef] [PubMed]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef] [PubMed]
- Jevric, M.; Broman, S.L.; Nielsen, M.B. Palladium-Mediated Strategies for Functionalizing the Dihydroazulene Photoswitch: Paving the Way for Its Exploitation in Molecular Electronics. J. Org. Chem. 2013, 78, 4348–4356. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jiang, X. Transfer of Sulfur: From Simple to Diverse. Chem. Asian J. 2013, 8, 2546–2563. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-F.; Liu, Y.-C.; Badsara, S.S. Transition-Metal-Catalyzed C-S Bond Coupling Reaction. Chem. Asian J. 2014, 9, 706–722. [Google Scholar] [CrossRef] [PubMed]
- Geiger, V.J.; Oechsner, R.M.; Gehrtz, P.H.; Fleischer, I. Recent Metal-Catalyzed Methods for Thioether Synthesis. Synthesis 2022, 53, 5139–5167. [Google Scholar] [CrossRef]
- Behera, P.K.; Choudhury, P.; Behera, P.; Swain, A.; Pradhan, A.K.; Rout, L. Transition Metal Catalysed C-S Cross-Coupling Reactions at Room Temperature. ChemistrySelect 2022, 7, e202202919. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C–S, C–Se, and C–Te Bond Formations via Cross-Coupling and Atom-Economic Addition Reactions. Achievements and Challenges. Chem. Rev. 2022, 122, 16110–16293. [Google Scholar] [CrossRef] [PubMed]
- Soria-Castro, S.M. Potassium Thioacetate. Synlett 2012, 23, 2997–2998. [Google Scholar] [CrossRef][Green Version]
- Soria-Castro, S.M.; Peñéñory, A.B. Efficient Cu-Catalyzed Base-Free C–S Coupling under Conventional and Microwave Heating. A Simple Access to S-Heterocycles and Sulfides. Beilstein J. Org. Chem. 2013, 9, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Soria-Castro, S.M.; Andrada, D.M.; Caminos, D.A.; Argüello, J.E.; Robert, M.; Peñéñory, A.B. Mechanistic Insight into the Cu-Catalyzed C–S Cross-Coupling of Thioacetate with Aryl Halides: A Joint Experimental–Computational Study. J. Org. Chem. 2017, 82, 11464–11473. [Google Scholar] [CrossRef]
- Thomas, A.M.; Sherin, D.R.; Asha, S.; Manojkumar, T.K.; Anilkumar, G. Exploration of the Mechanism and Scope of the CuI/DABCO Catalysed CS Coupling Reaction. Polyhedron 2020, 176, 114269. [Google Scholar] [CrossRef]
- Cyclopentyl Methyl Ether (CPME)|Specialty Solvents. Available online: https://www.zeon.co.jp/en/business/enterprise/special/solvent-cpme/ (accessed on 13 March 2023).
- Watanabe, K.; Yamagiwa, N.; Torisawa, Y. Cyclopentyl Methyl Ether as a New and Alternative Process Solvent. Org. Process Res. Dev. 2007, 11, 251–258. [Google Scholar] [CrossRef]
- Azzena, U.; Carraro, M.; Pisano, L.; Monticelli, S.; Bartolotta, R.; Pace, V. Cyclopentyl Methyl Ether: An Elective Ecofriendly Ethereal Solvent in Classical and Modern Organic Chemistry. ChemSusChem 2019, 12, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K. The Toxicological Assessment of Cyclopentyl Methyl Ether (CPME) as a Green Solvent. Molecules 2013, 18, 3183–3194. [Google Scholar] [CrossRef] [PubMed]
- Bates, C.G.; Gujadhur, R.K.; Venkataraman, D. A General Method for the Formation of Aryl−Sulfur Bonds Using Copper(I) Catalysts. Org. Lett. 2002, 4, 2803–2806. [Google Scholar] [CrossRef] [PubMed]
- Park, N.; Park, K.; Jang, M.; Lee, S. One-Pot Synthesis of Symmetrical and Unsymmetrical Aryl Sulfides by Pd-Catalyzed Couplings of Aryl Halides and Thioacetates. J. Org. Chem. 2011, 76, 4371–4378. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y. Cu(I)-Catalysed Coupling of Arylsulfinic Salts with Aryl Bromides. J. Chem. Res. 2014, 38, 447–449. [Google Scholar] [CrossRef]
- Sperotto, E.; van Klink, G.P.M.; de Vries, J.G.; van Koten, G. Ligand-Free Copper-Catalyzed C−S Coupling of Aryl Iodides and Thiols. J. Org. Chem. 2008, 73, 5625–5628. [Google Scholar] [CrossRef] [PubMed]
- Kwong, F.Y.; Buchwald, S.L. A General, Efficient, and Inexpensive Catalyst System for the Coupling of Aryl Iodides and Thiols. Org. Lett. 2002, 4, 3517–3520. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Chen, H.-H. 1,1,1-Tris(Hydroxymethyl)Ethane as a New, Efficient, and Versatile Tripod Ligand for Copper-Catalyzed Cross-Coupling Reactions of Aryl Iodides with Amides, Thiols, and Phenols. Org. Lett. 2006, 8, 5609–5612. [Google Scholar] [CrossRef]
- Rajarathnam, D.; Babu, J.; Nadar, P.A. Enhanced reactivity in the ammonolysis of phenyl thiolacetates in aqueous medium. Int. J. Chem. Kinet. 2002, 34, 18–26. [Google Scholar] [CrossRef]
- Shen, G.; Lu, Q.; Wang, Z.; Sun, W.; Zhang, Y.; Huang, X.; Sun, M.; Wang, Z. Environmentally Friendly and Recyclable CuCl2-Mediated C–S Bond Coupling Strategy Using DMEDA as Ligand, Base, and Solvent. Synthesis 2022, 54, 184–198. [Google Scholar] [CrossRef]
- Zou, Y.; Huang, Q.; Huang, T.; Ni, Q.; Zhang, E.; Xu, T.; Yuan, M.; Li, J. CuI/1,10-Phen/PEG Promoted Decarboxylation of 2,3-Diarylacrylic Acids: Synthesis of Stilbenes under Neutral and Microwave Conditions with an in Situ Generated Recyclable Catalyst. Org. Biomol. Chem. 2013, 11, 6967–6974. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Backes, B.J. Efficient Preparation of S-Aryl Thioacetates from Aryl Halides and Potassium Thioacetate. Tetrahedron Lett. 2007, 48, 3033–3037. [Google Scholar] [CrossRef]
- Kahrs, C.; Schmidtmann, M.; Wickleder, M.S.; Christoffers, J. Pyridine and Pyrimidine Functionalized Benzene Sulfonic and Disulfonic Acids as New Linker Compounds for Coordination Polymers. Eur. J. Org. Chem. 2018, 2018, 6499–6506. [Google Scholar] [CrossRef]
- Matsuda, K.; Yamaguchi, H.; Sakano, T.; Ikeda, M.; Tanifuji, N.; Irie, M. Conductance Photoswitching of Diarylethene−Gold Nanoparticle Network Induced by Photochromic Reaction. J. Phys. Chem. C 2008, 112, 17005–17010. [Google Scholar] [CrossRef]
- Sakano, T.; Yamaguchi, H.; Tanifuji, N.; Irie, M.; Matsuda, K. Percolation-Type Photoswitching Behavior in Conductance of Diarylethene–Silver Nanoparticle Networks. Chem. Lett. 2008, 37, 634–635. [Google Scholar] [CrossRef]
- Matsuda, K.; Irie, M. Photochromism of Diarylethenes with Two Nitronyl Nitroxides: Photoswitching of an Intramolecular Magnetic Interaction. Chem. Eur. J. 2001, 7, 3466–3473. [Google Scholar] [CrossRef]
- Barbero, M.; Degani, I.; Dughera, S.; Fochi, R. An Improved, General Procedure to S-Aryl Thiol Esters: A New Synthetic Application of Dry Arenediazonium o-Benzenedisulfonimides. Synthesis 2003, 2003, 1225–1230. [Google Scholar] [CrossRef]
- Yang, F.; He, G.C.; Sun, S.-H.; Song, T.-T.; Min, X.-T.; Ji, D.-W.; Guo, S.-Y.; Chen, Q.-A. Selective C–S Bond Constructions Using Inorganic Sulfurs via Photoinduced Electron Donor–Acceptor Activation. J. Org. Chem. 2022, 87, 14241–14249. [Google Scholar] [CrossRef]
- Pijper, T.C.; Robertus, J.; Browne, W.R.; Feringa, B.L. Mild Ti-mediated transformation of t-butyl thio-ethers into thio-acetates. Org. Biomol. Chem. 2014, 13, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-Z.; Wei, W.-L.; Niu, Y.-L.; Li, X.; Wang, M.; Gao, W.-C. Homocouplings of Sodium Arenesulfinates: Selective Access to Symmetric Diaryl Sulfides and Diaryl Disulfides. Molecules 2022, 27, 6232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Y.; Gao, T.; Yan, P.; Zou, X.; Li, H. Diastereoselective self-assembly of a triple-stranded europium helicate with light modulated chiroptical properties. Dalton Trans. 2021, 50, 4604–4612. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry 1 | Ar-X | Ligand | Product | Yield (%) 2 |
| 1 | Ph-I (1) | 1,10-phen | 4 | 98 |
| 2 | Ph-I (1) | 1,10-phen | 4 | 30 3 |
| 3 | Ph-I (1) | 1,10-phen | 4 | 56 4 |
| 4 | Ph-Br (2) | 1,10-phen | - | - |
| 5 | m-I2C6H4 (3) | 1,10-phen | 5 | 87 5 |
| 6 | Ph-I (1) | DABCO | 4 | 15 |
| 7 | Ph-Br (2) | DABCO | - | - |
| 8 | Ph-I (1) | 2,2′-Bipy | 4 | 10 |
| 9 | Ph-Br (2) | 2,2′-Bipy | - | - |
 | ||||
|---|---|---|---|---|
| Entry 1 | Ph-X(1) | Conversion | Product | Entry 1 |
| (%) 2 | 4(%) 2 | 6(%) 2 | ||
| 1 |  1a | 100 | 100 (98) | - |
| 100 3 | 100 3 | - | ||
| 2 |  1b | 94 | 100 (90) | - |
| 3 |  1c | 74 | 100 (71) | - |
| 4 |  1d | 40 | 100 (36) | - |
| 5 |  1e | 97 | 100 (94) | - |
| 6 |  1f | 90 | 100 (87) | - |
| 88 3 | 100 3 | - | ||
| 7 |  1g | 100 | 93 | 7 |
| 8 |  1h | 100 | 92 (87) | 8 |
| 100 3 | 79 3 | 21 3 | ||
| 9 |  1i | 100 | 90 (87) | 10 |
| 100 3 | 85 3 | 15 3 | ||
| 10 |  1j | 100 | 73 | 27 |
| 100 3 | 70 3 | 303 | ||
| 11 |  1k | 100 | 81 (76) | 19 |
| 12 |  1l | 66 | 100 (62) | - |
| 13 |  1m | 88 | 100 (88) | - |
| 14 |  1n | 80 | 100 (72) | - |
| 15 |  1o | 60 | 100 (57) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carraro, M.; Are, C.; Azzena, U.; De Luca, L.; Gaspa, S.; Satta, G.; Holzer, W.; Pace, V.; Pisano, L. Electronic Effects in a Green Protocol for (Hetero)Aryl-S Coupling. Molecules 2024, 29, 1714. https://doi.org/10.3390/molecules29081714
Carraro M, Are C, Azzena U, De Luca L, Gaspa S, Satta G, Holzer W, Pace V, Pisano L. Electronic Effects in a Green Protocol for (Hetero)Aryl-S Coupling. Molecules. 2024; 29(8):1714. https://doi.org/10.3390/molecules29081714
Chicago/Turabian StyleCarraro, Massimo, Camillo Are, Ugo Azzena, Lidia De Luca, Silvia Gaspa, Giuseppe Satta, Wolfgang Holzer, Vittorio Pace, and Luisa Pisano. 2024. "Electronic Effects in a Green Protocol for (Hetero)Aryl-S Coupling" Molecules 29, no. 8: 1714. https://doi.org/10.3390/molecules29081714
APA StyleCarraro, M., Are, C., Azzena, U., De Luca, L., Gaspa, S., Satta, G., Holzer, W., Pace, V., & Pisano, L. (2024). Electronic Effects in a Green Protocol for (Hetero)Aryl-S Coupling. Molecules, 29(8), 1714. https://doi.org/10.3390/molecules29081714