
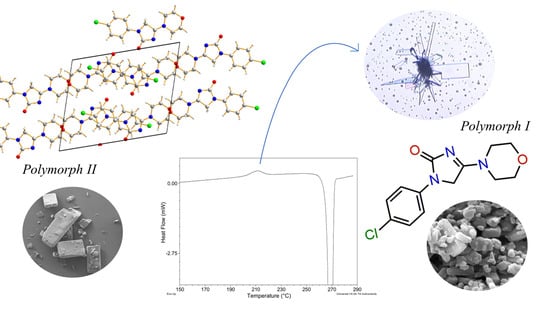
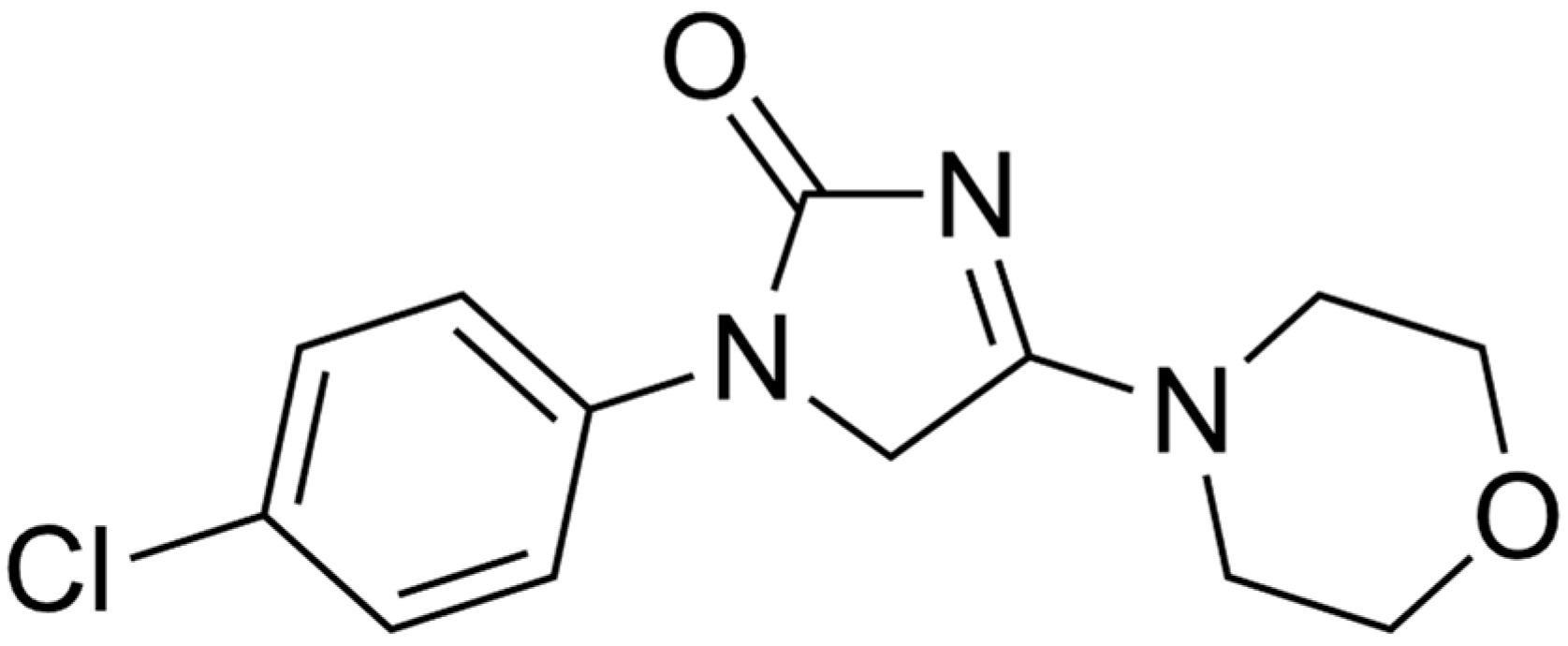
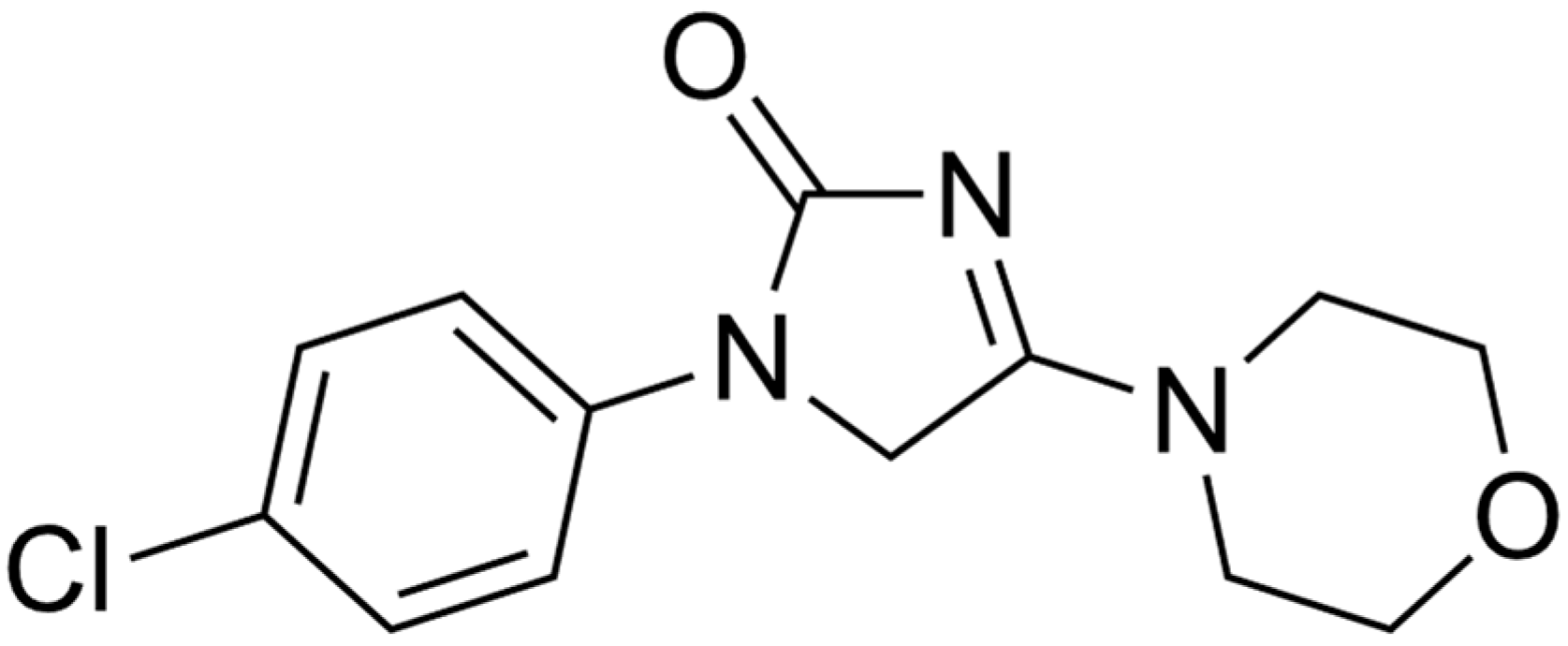
New Findings on the Crystal Polymorphism of Imepitoin

 ,
,  ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
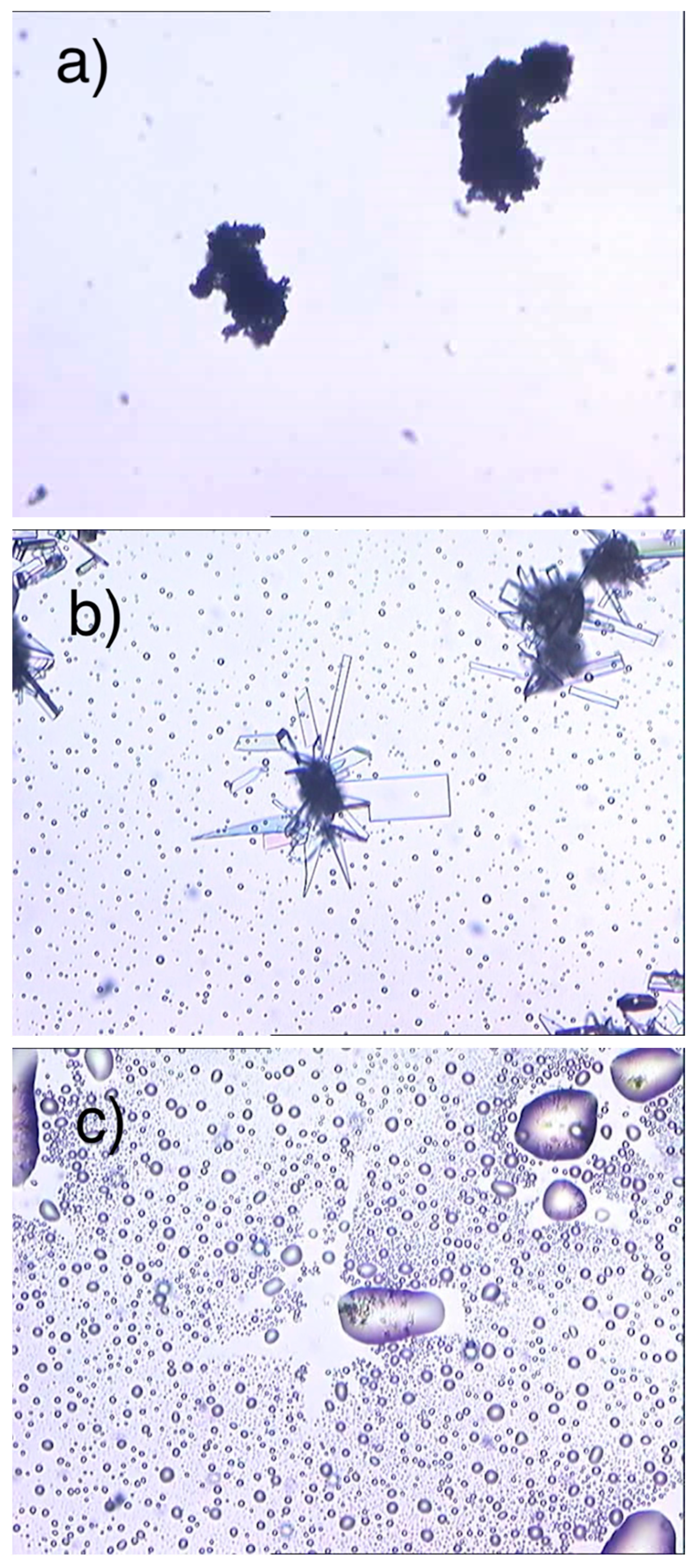
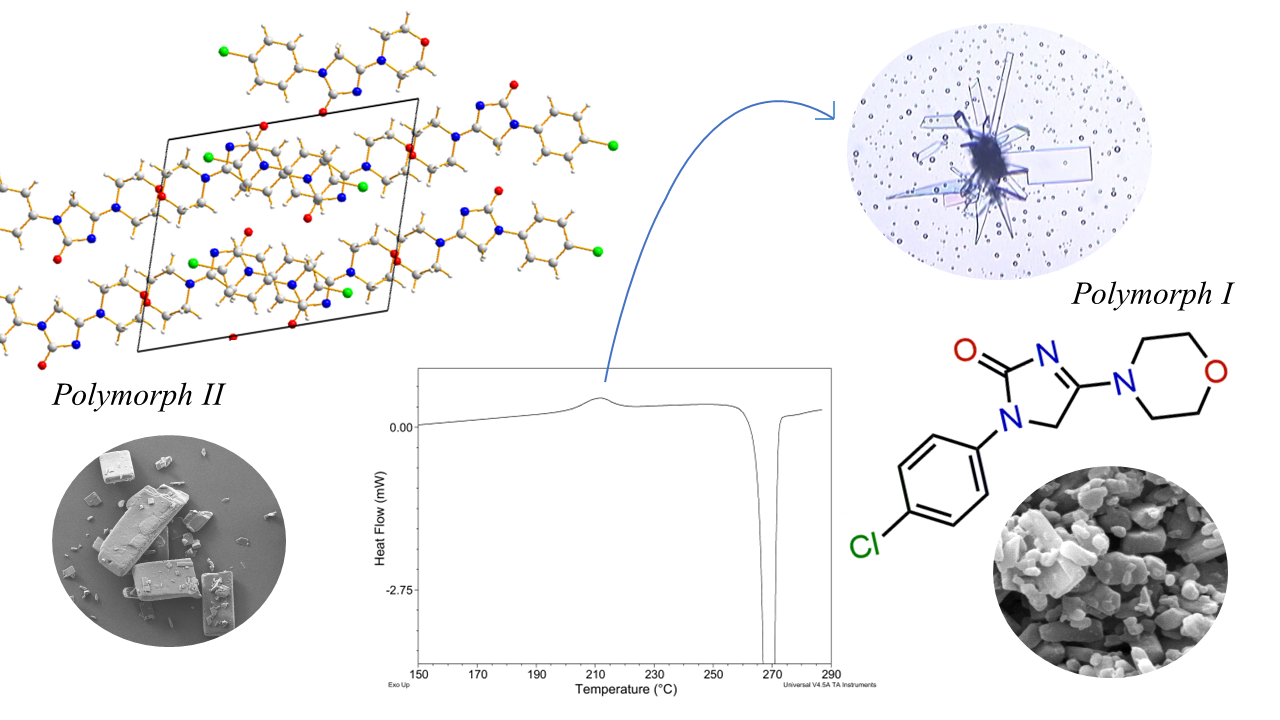
2.1. SEM Measurements
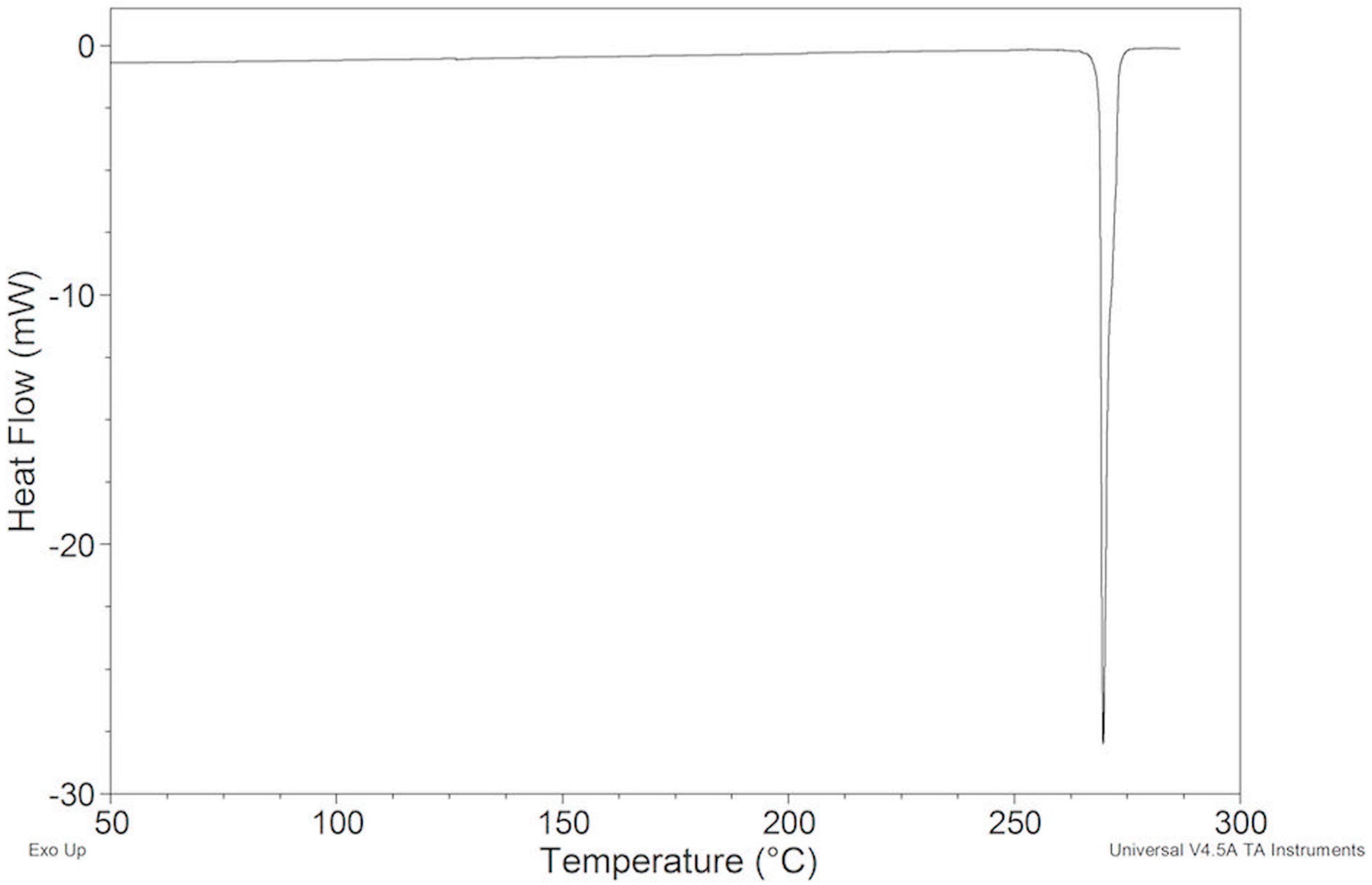
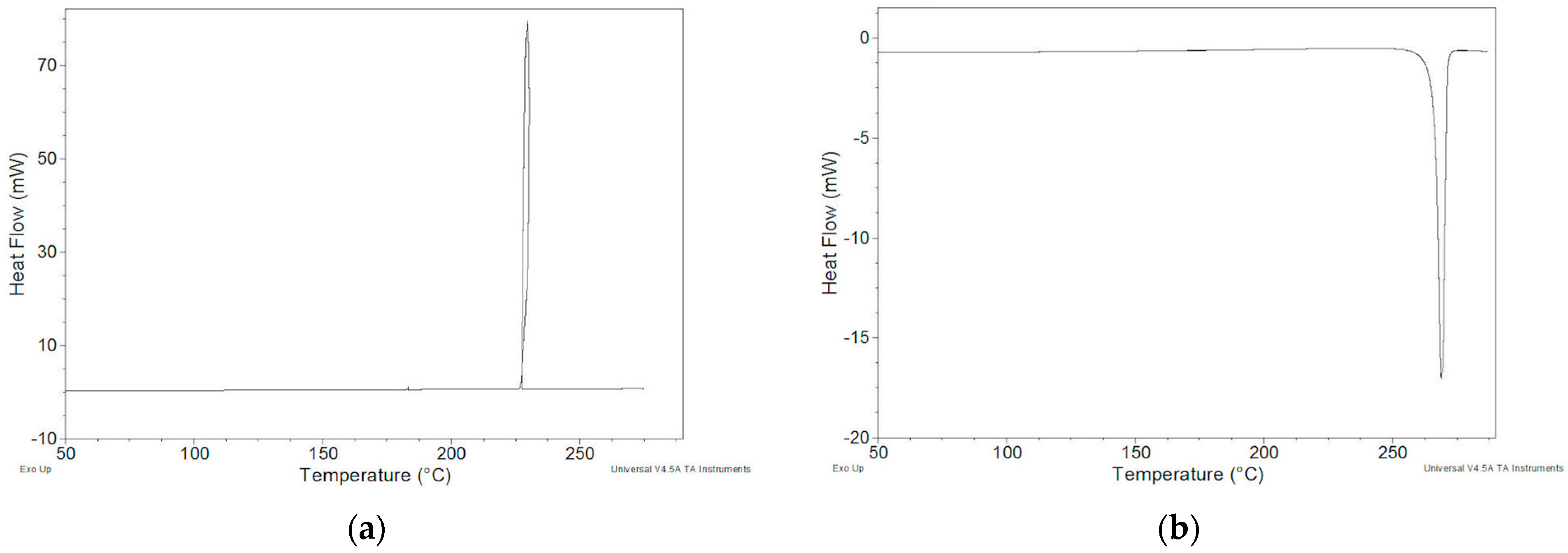
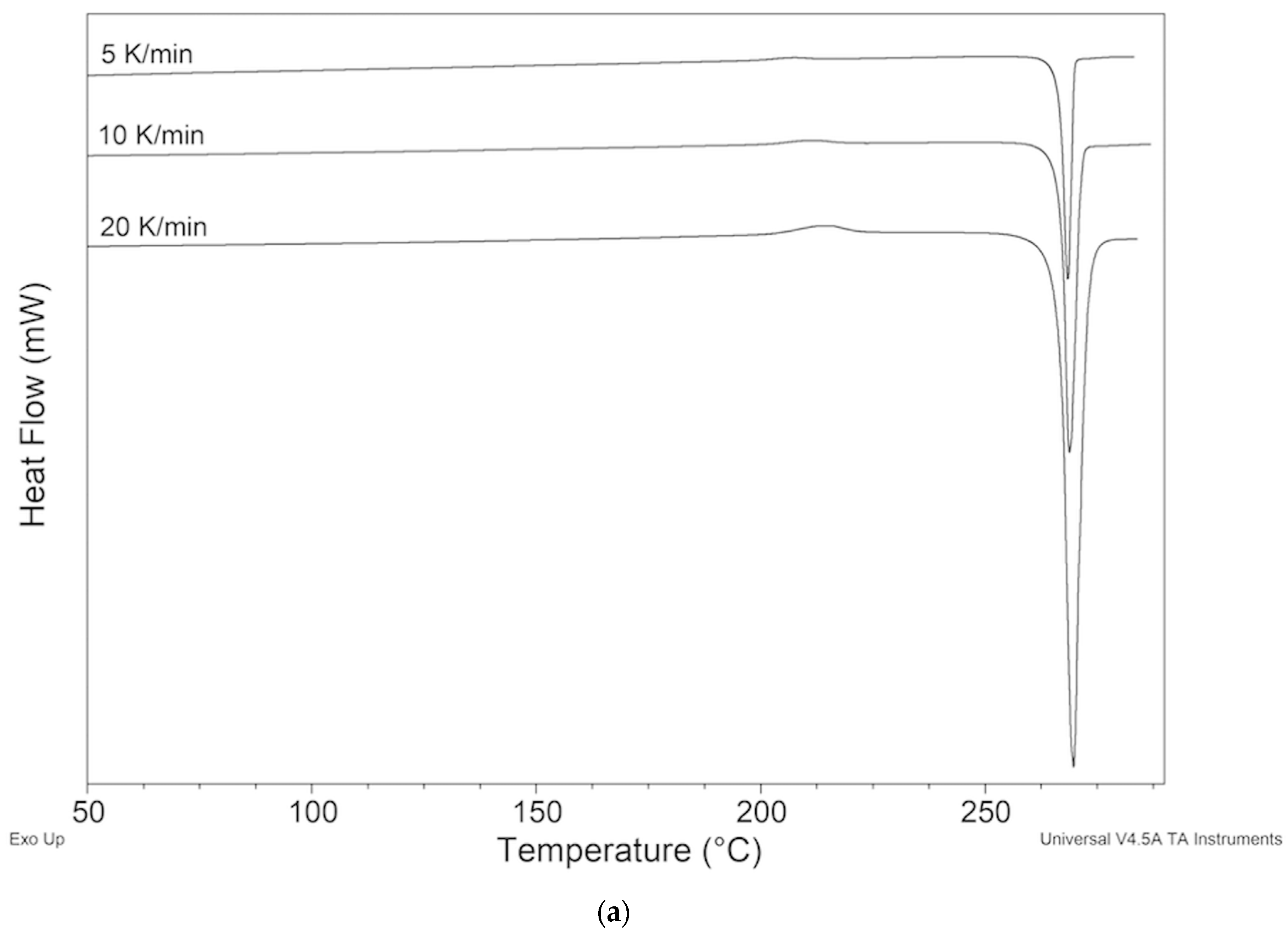
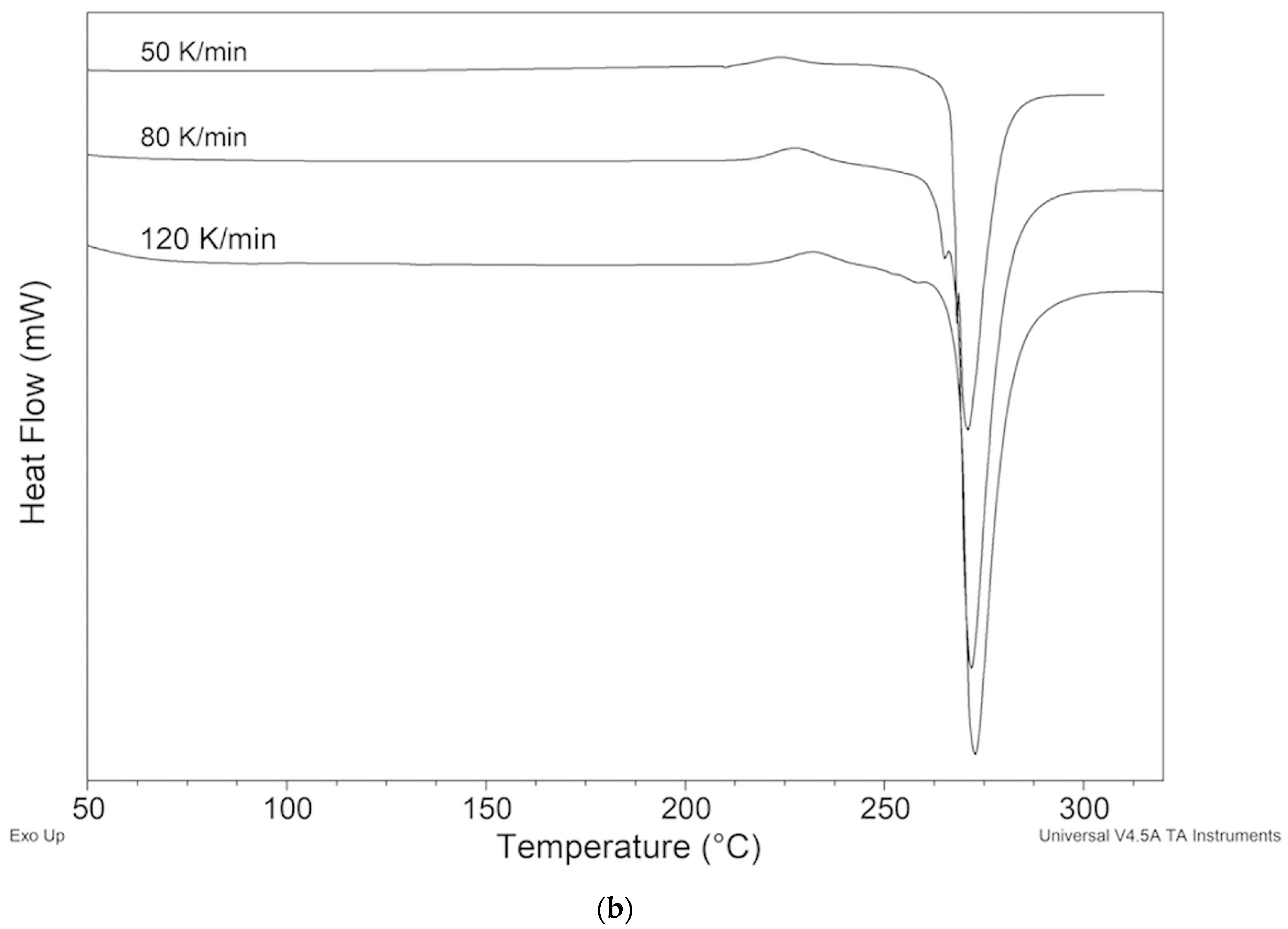
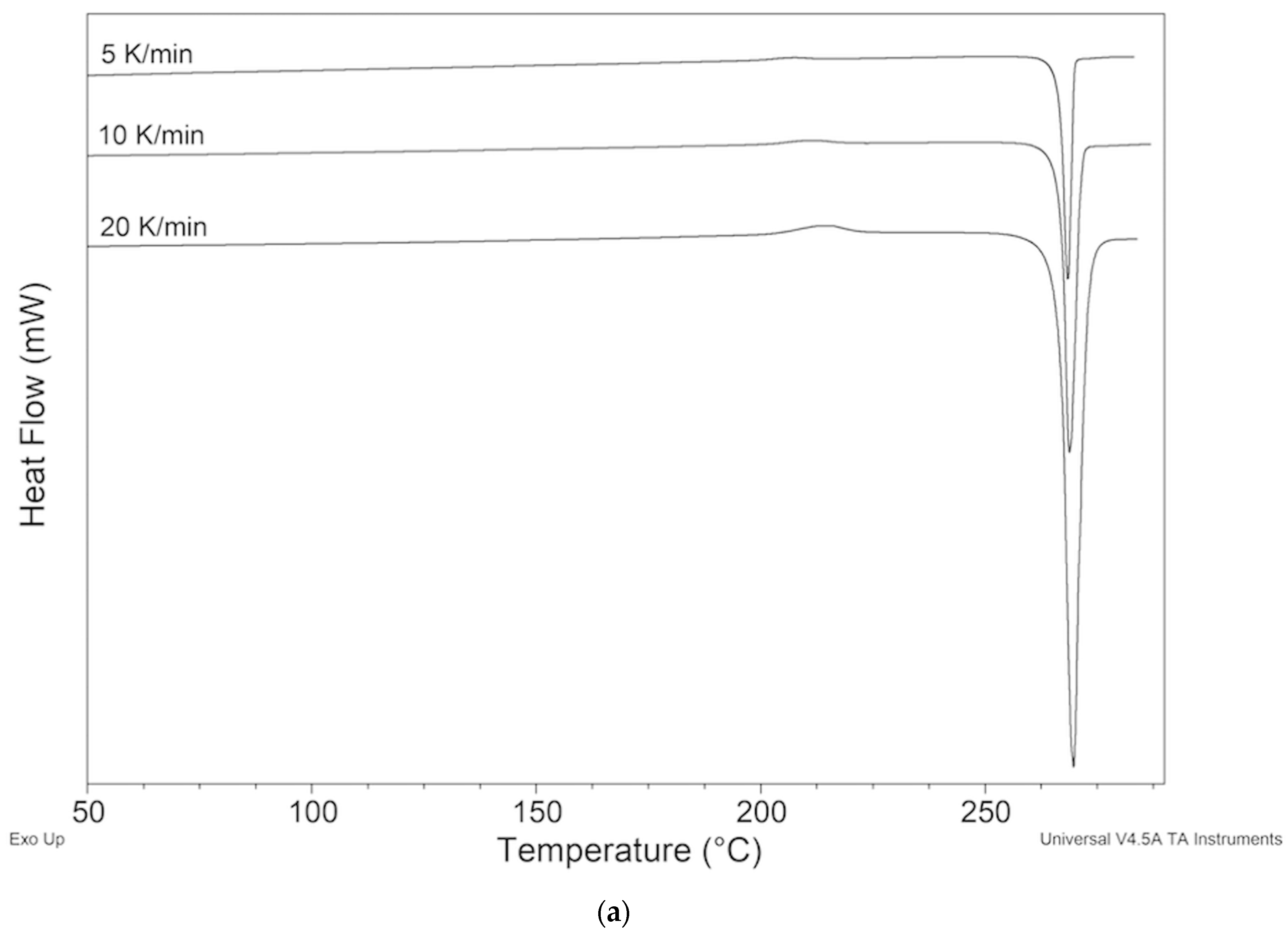
2.2. Thermal Behavior
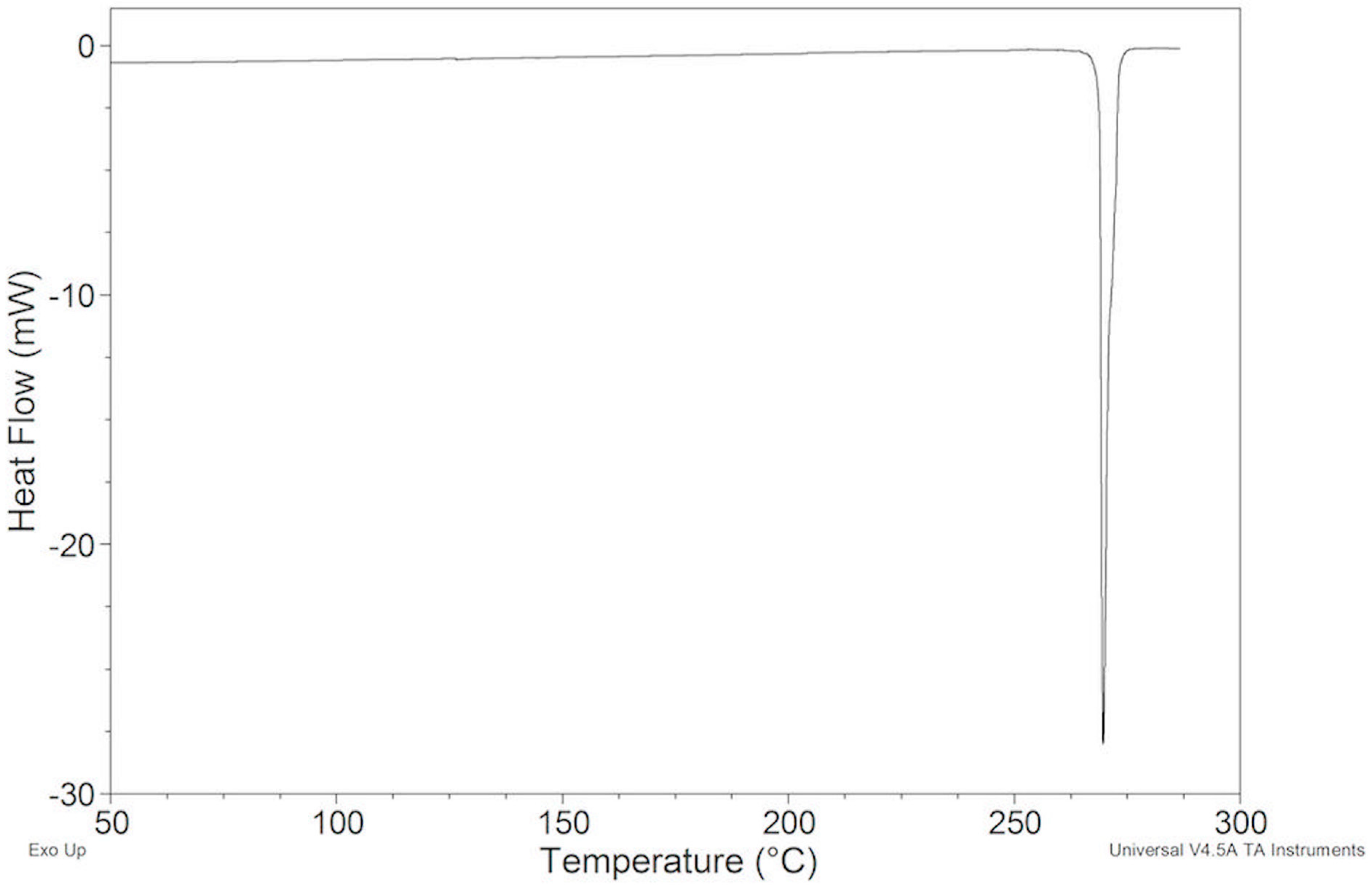
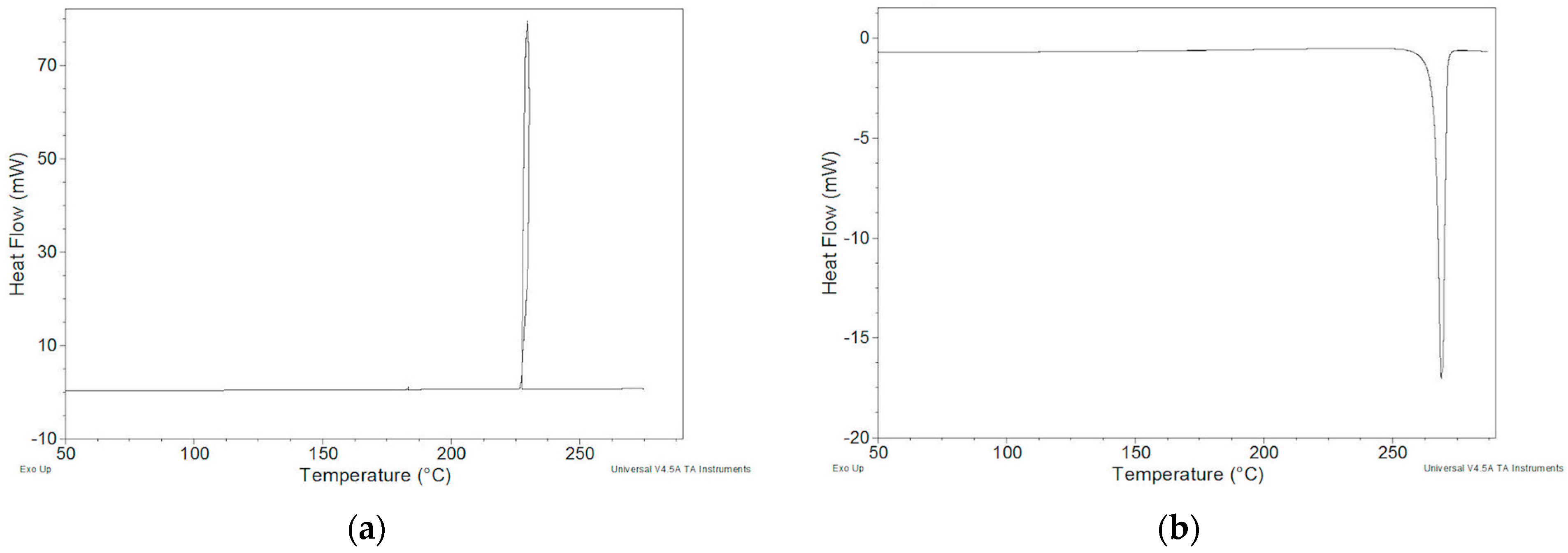
2.2.1. ImeTQ Sample
2.2.2. ImeGR
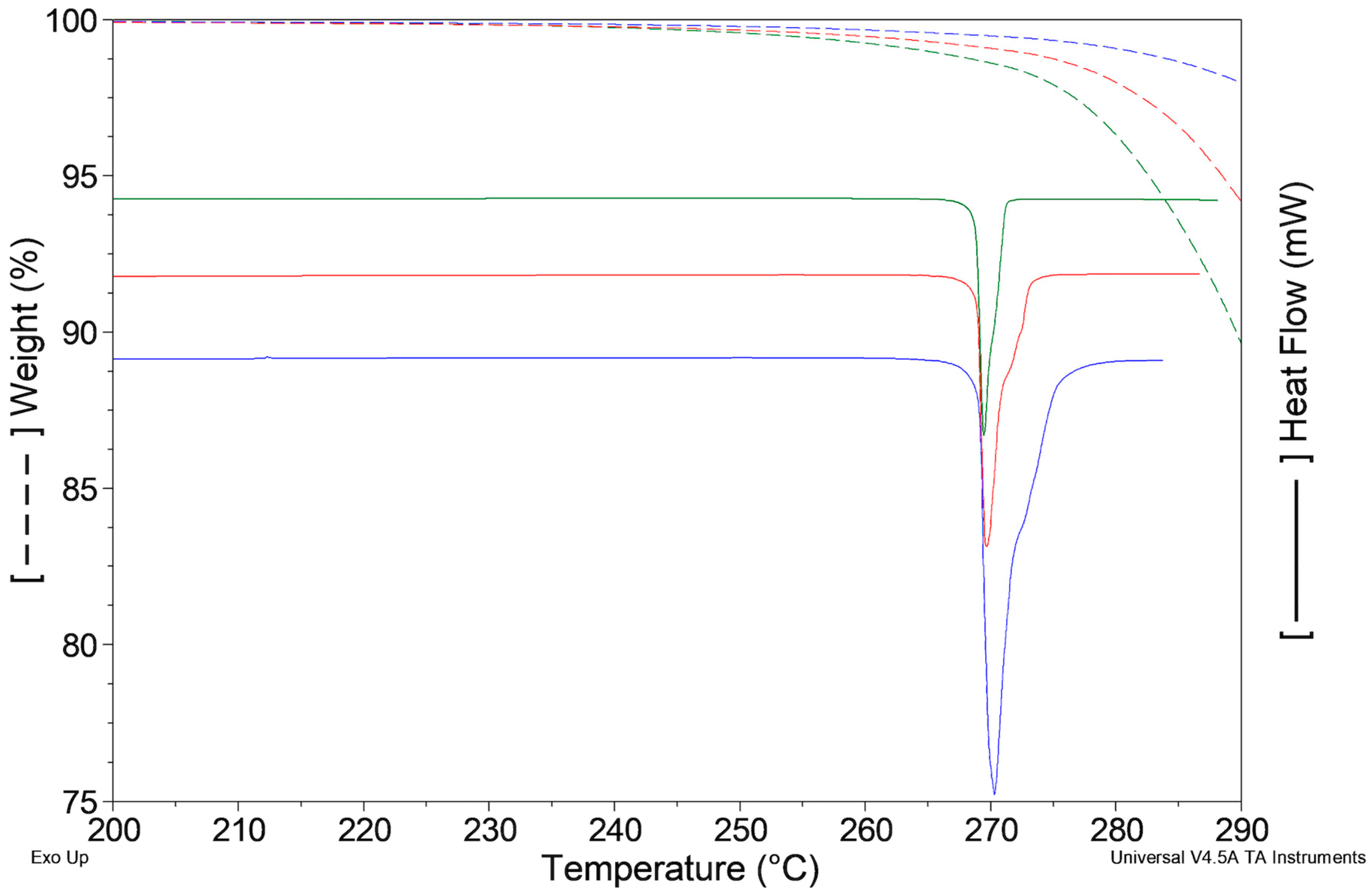
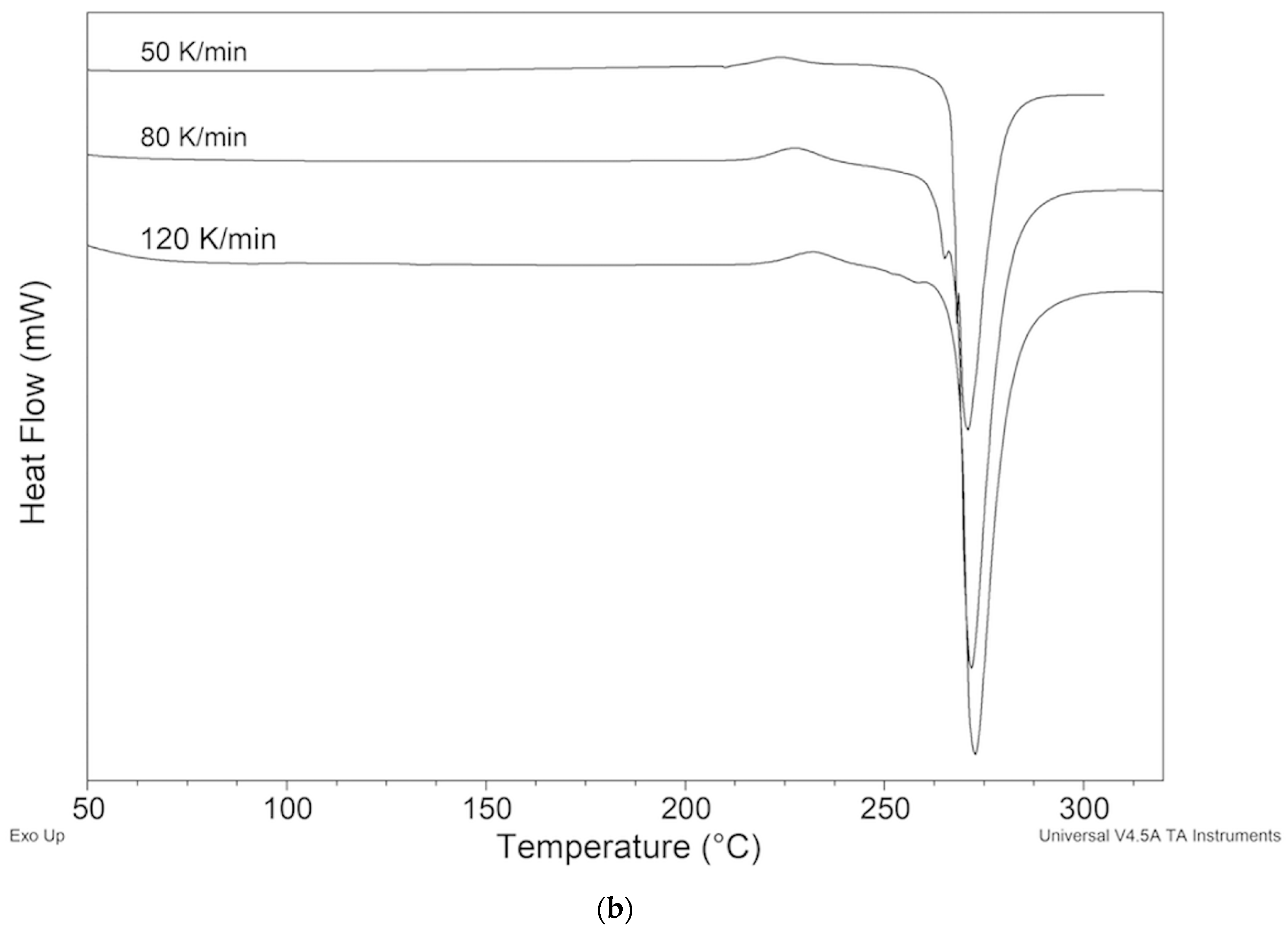
2.2.3. Samples Recrystallized by Different Solvents
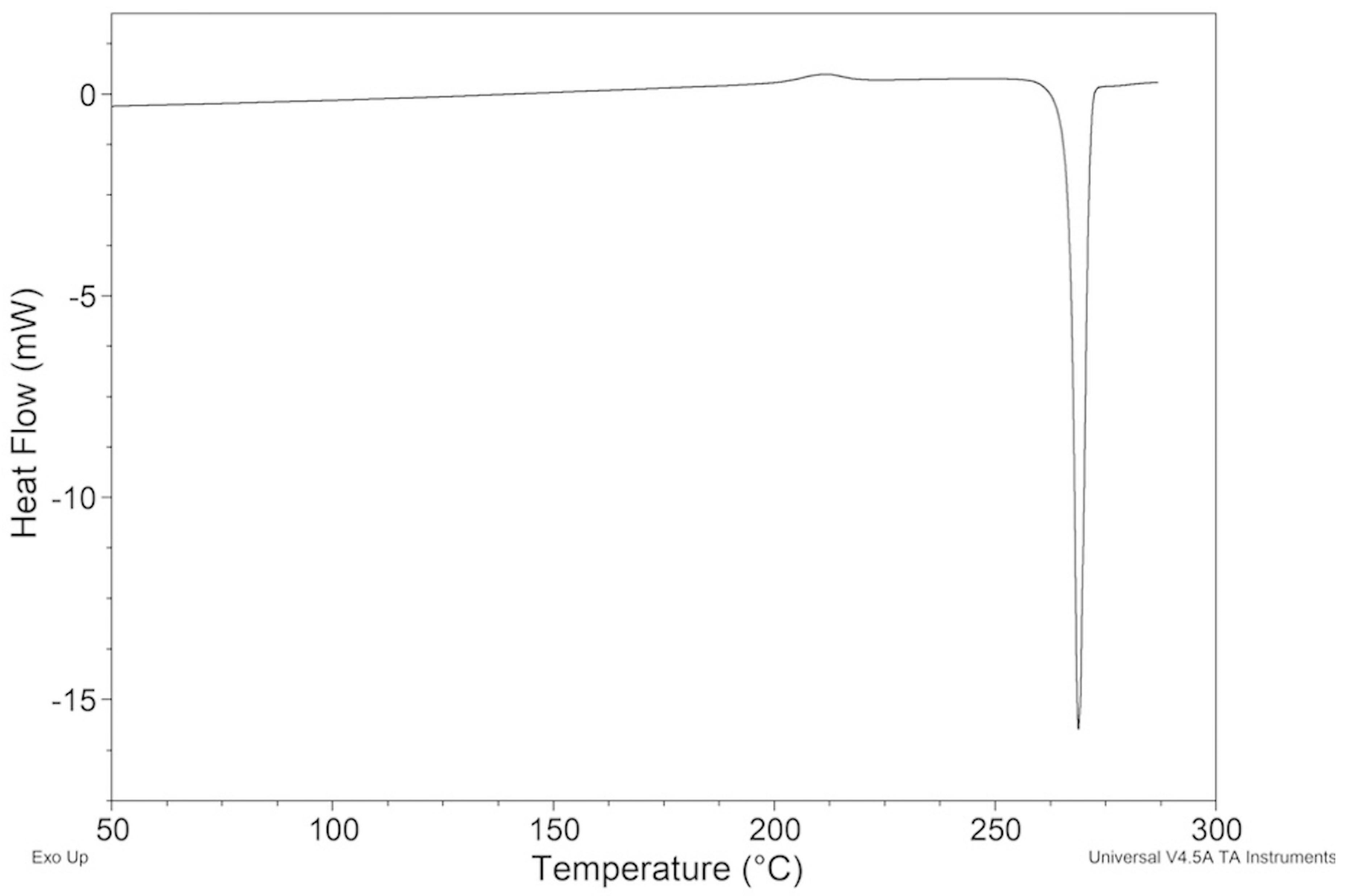
2.2.4. Samples Crystallized from Xylene
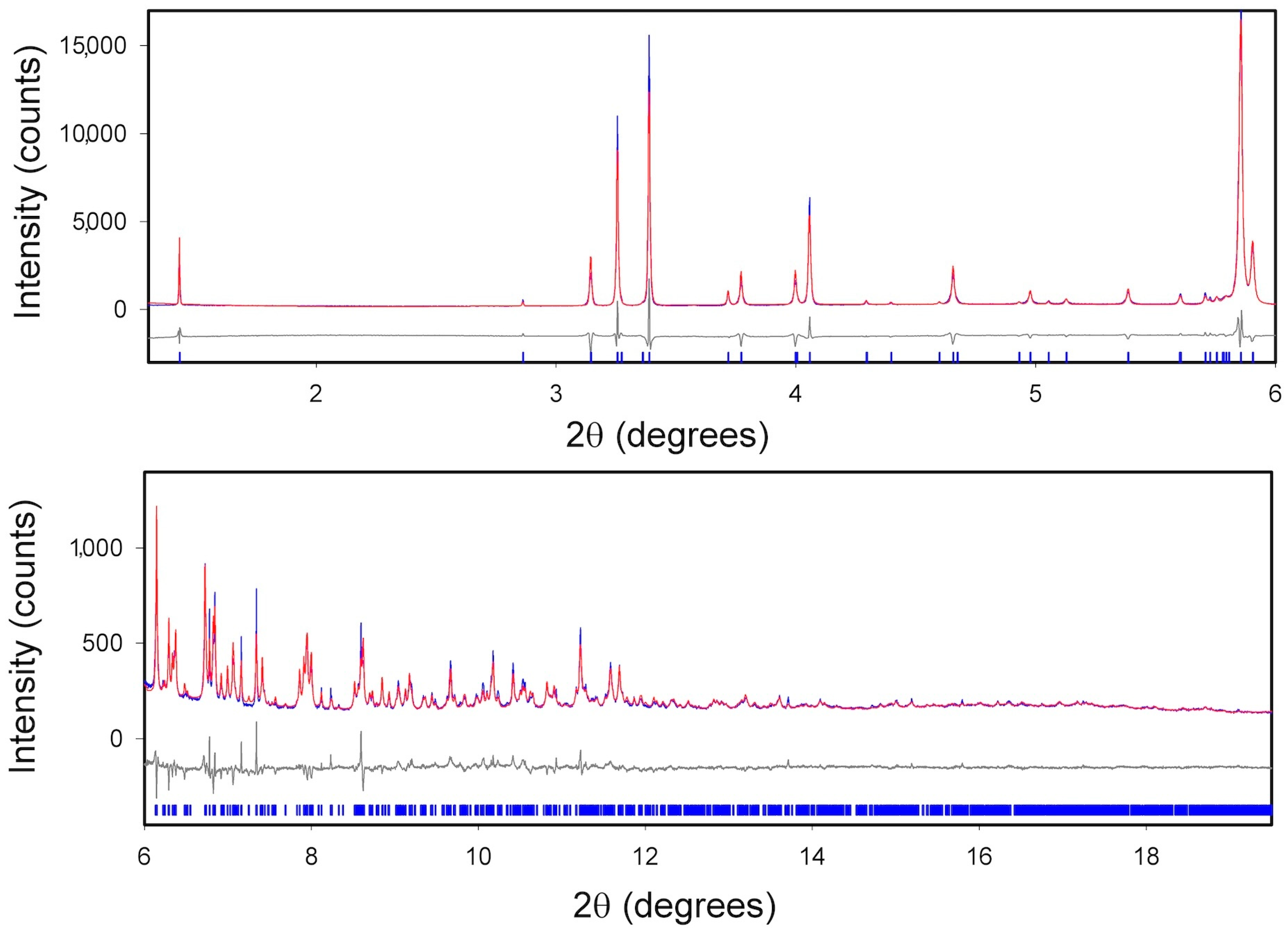
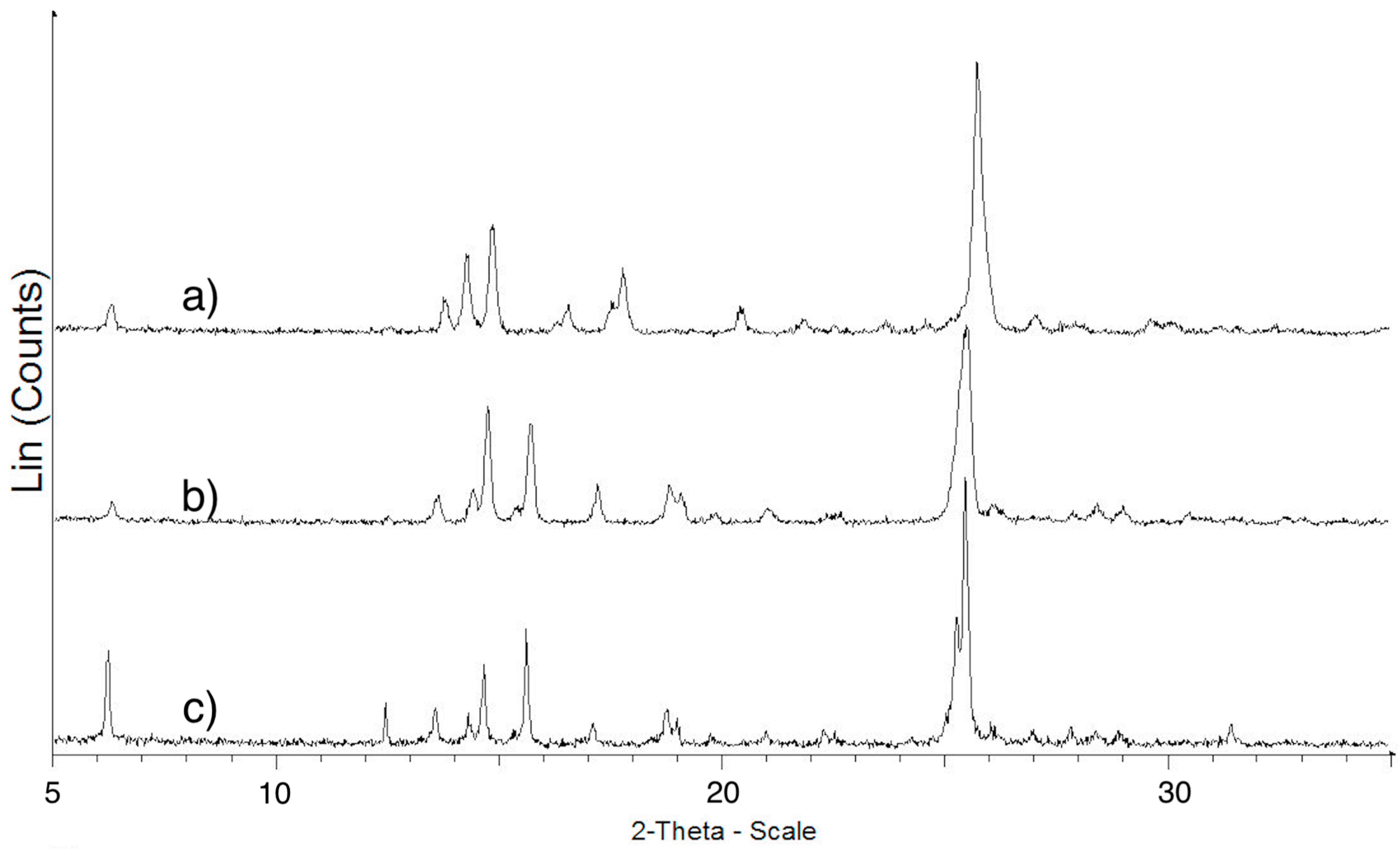
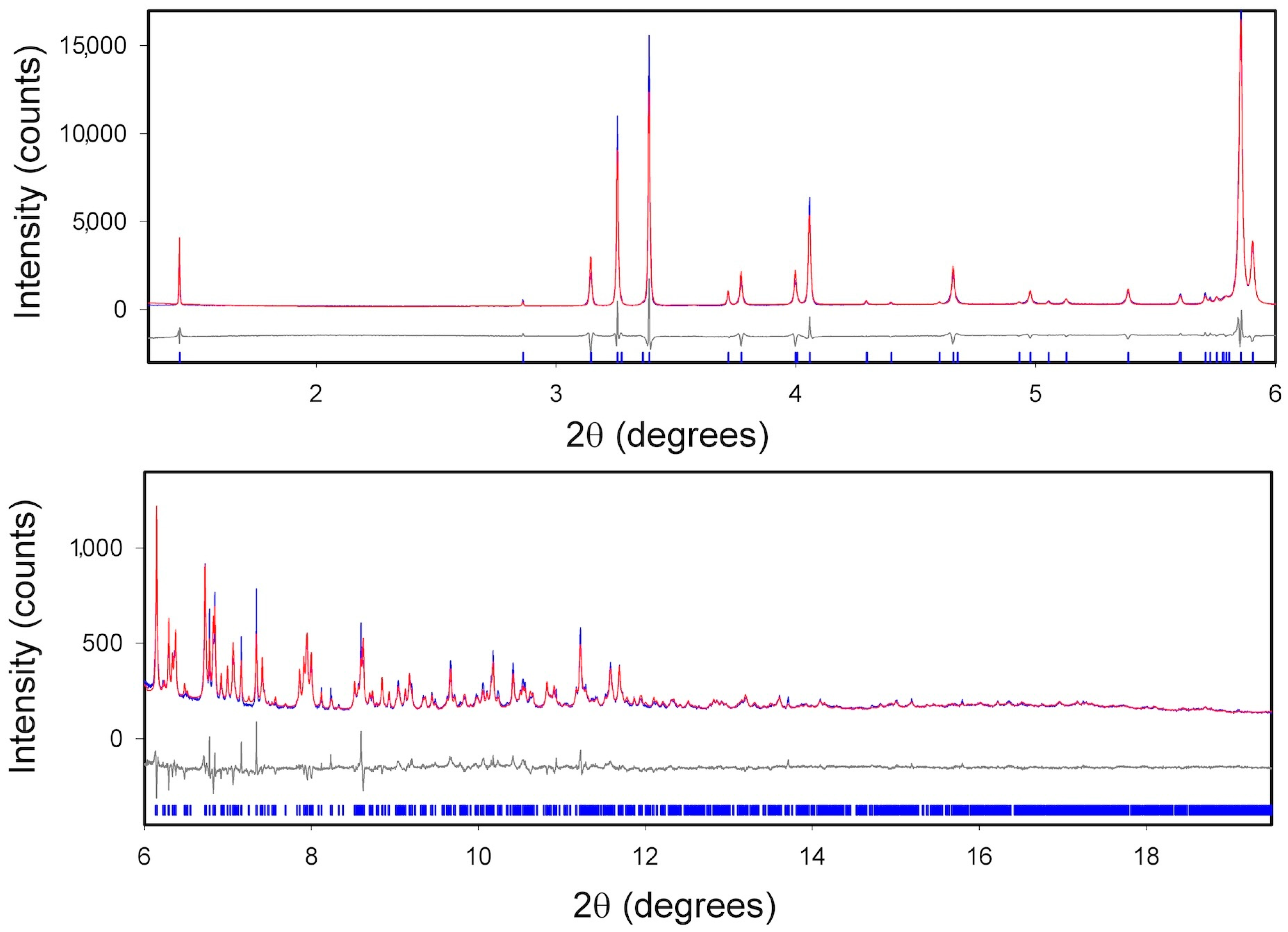
2.3. XRPD Measurements
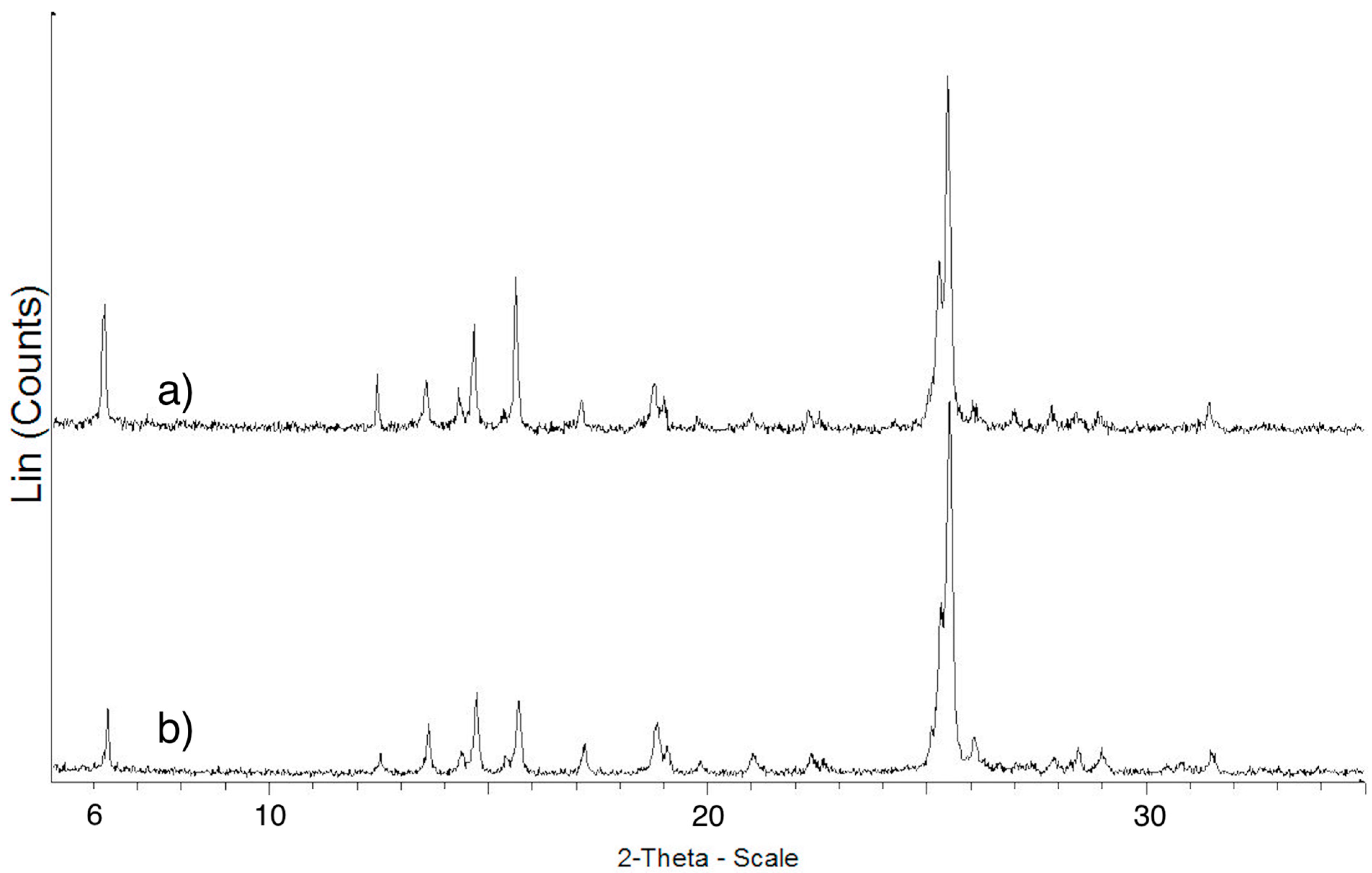
2.3.1. ImeTQ
2.3.2. ImeGR
2.3.3. Samples Crystallized from Various Solvents
2.3.4. ImeXyl
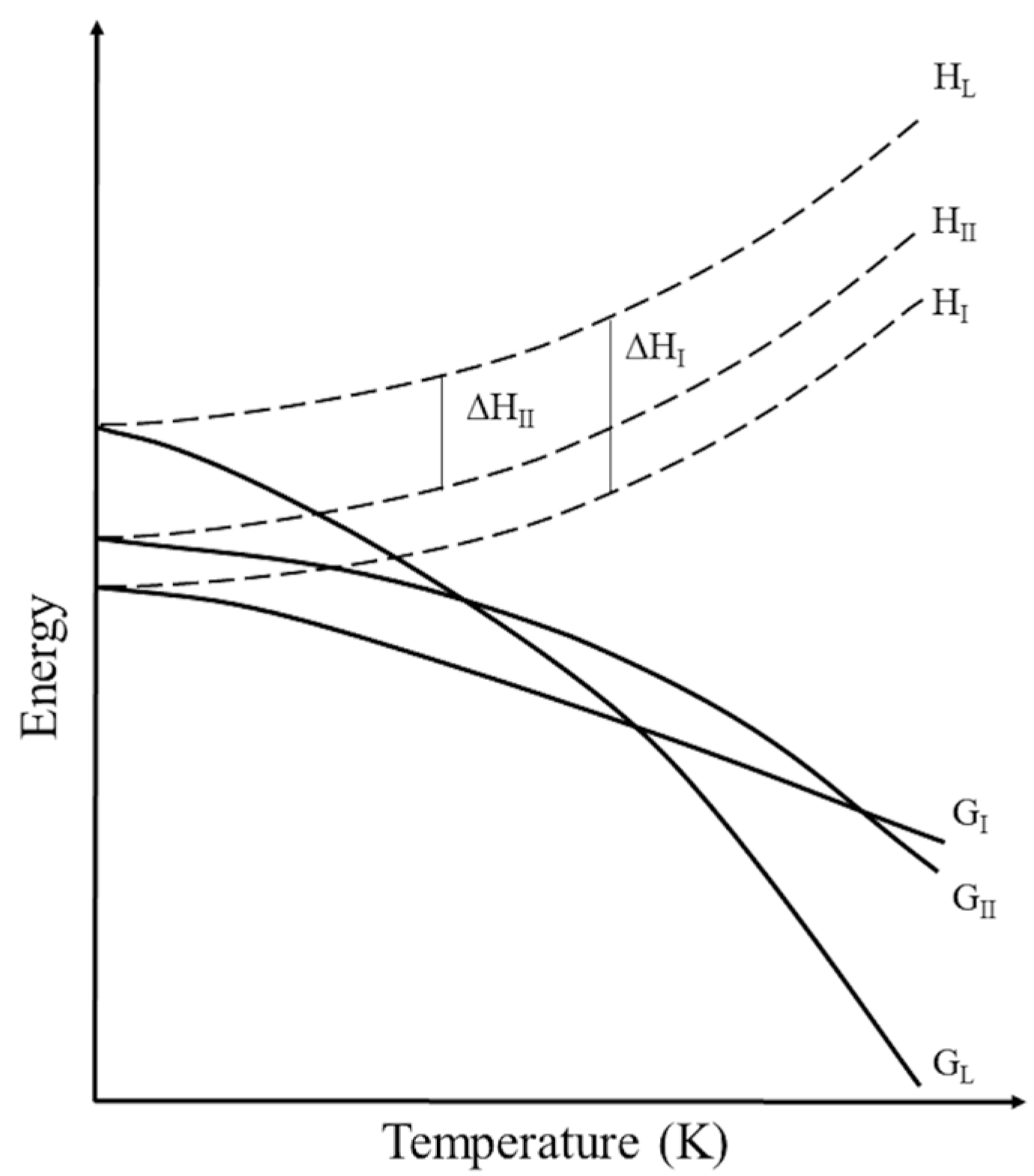
2.4. Nature of the Polymorphic Transition of ImeXyl
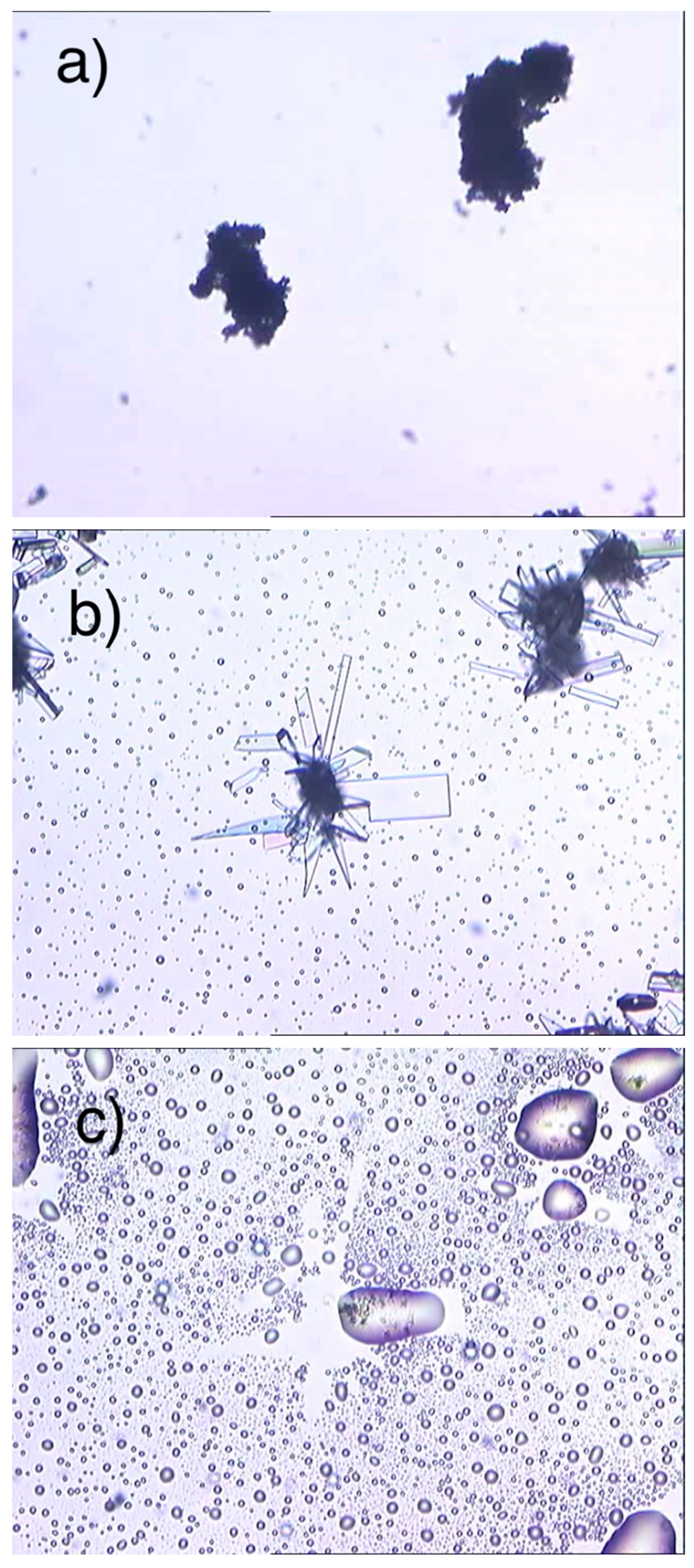
2.5. Measurements HSM
ImeXyl
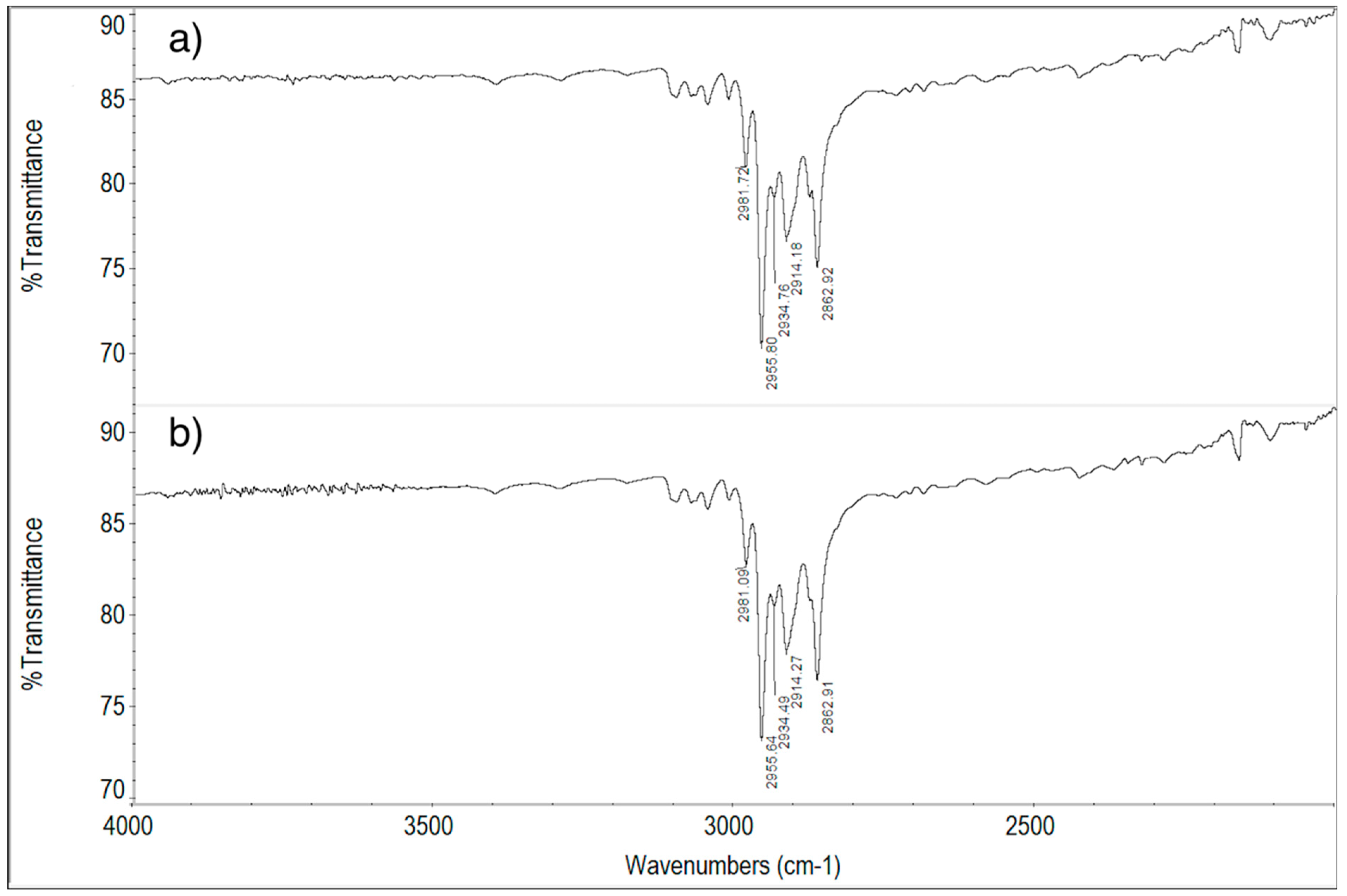
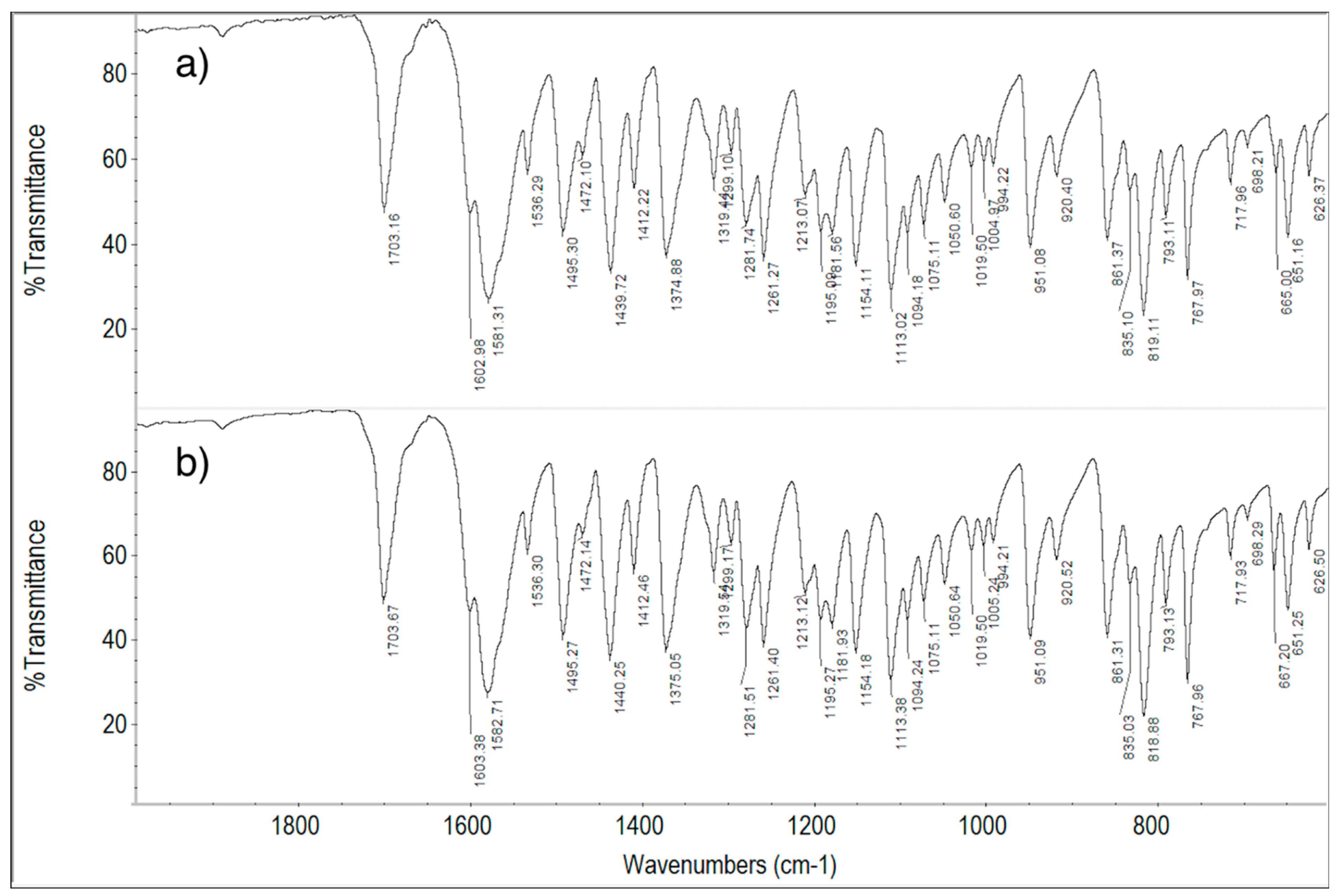
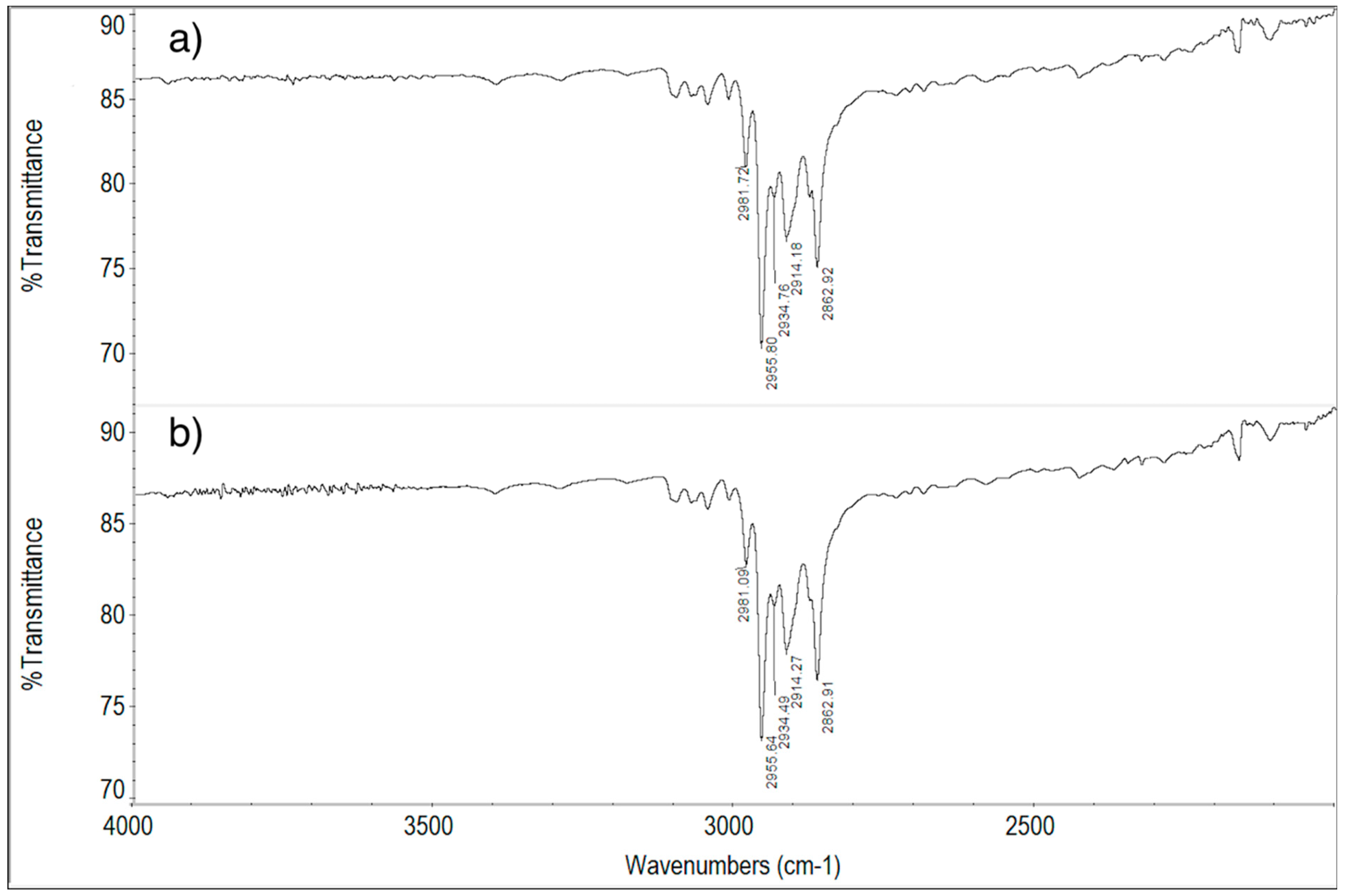
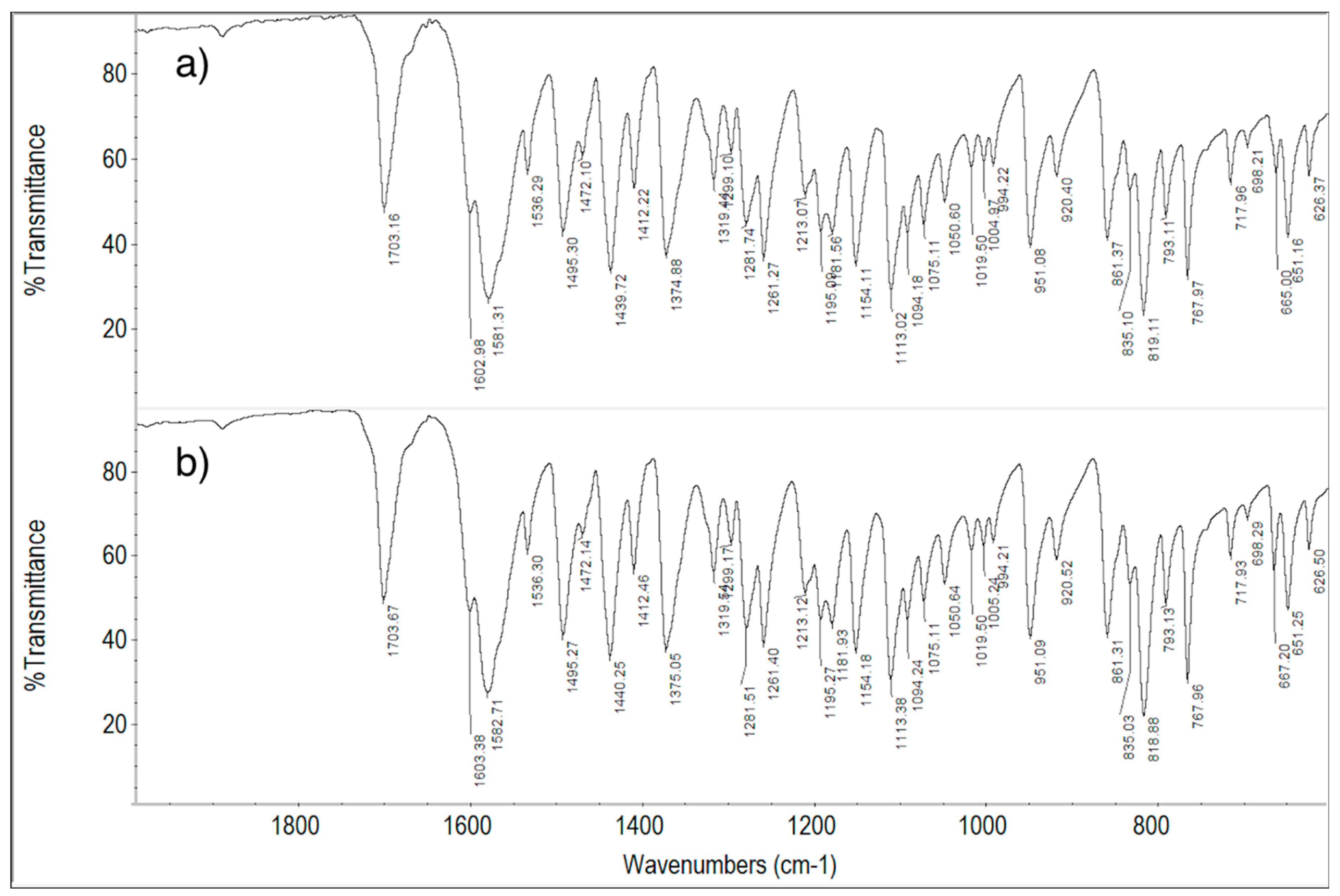
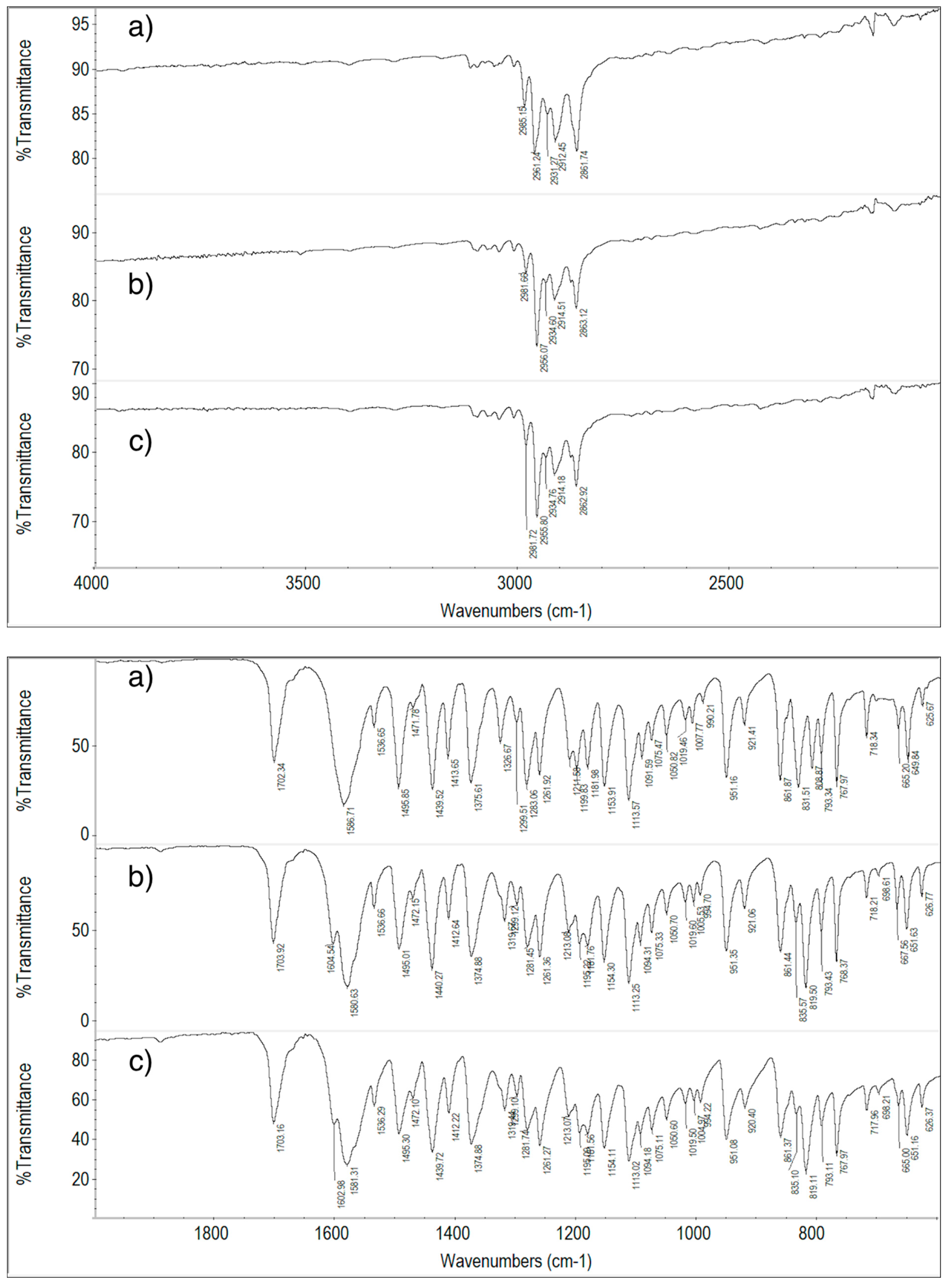
2.6. FT-IR Spectroscopy Measurements
2.6.1. ImeTQ and ImeGR
2.6.2. ImeXyl
2.7. Raman Measurements
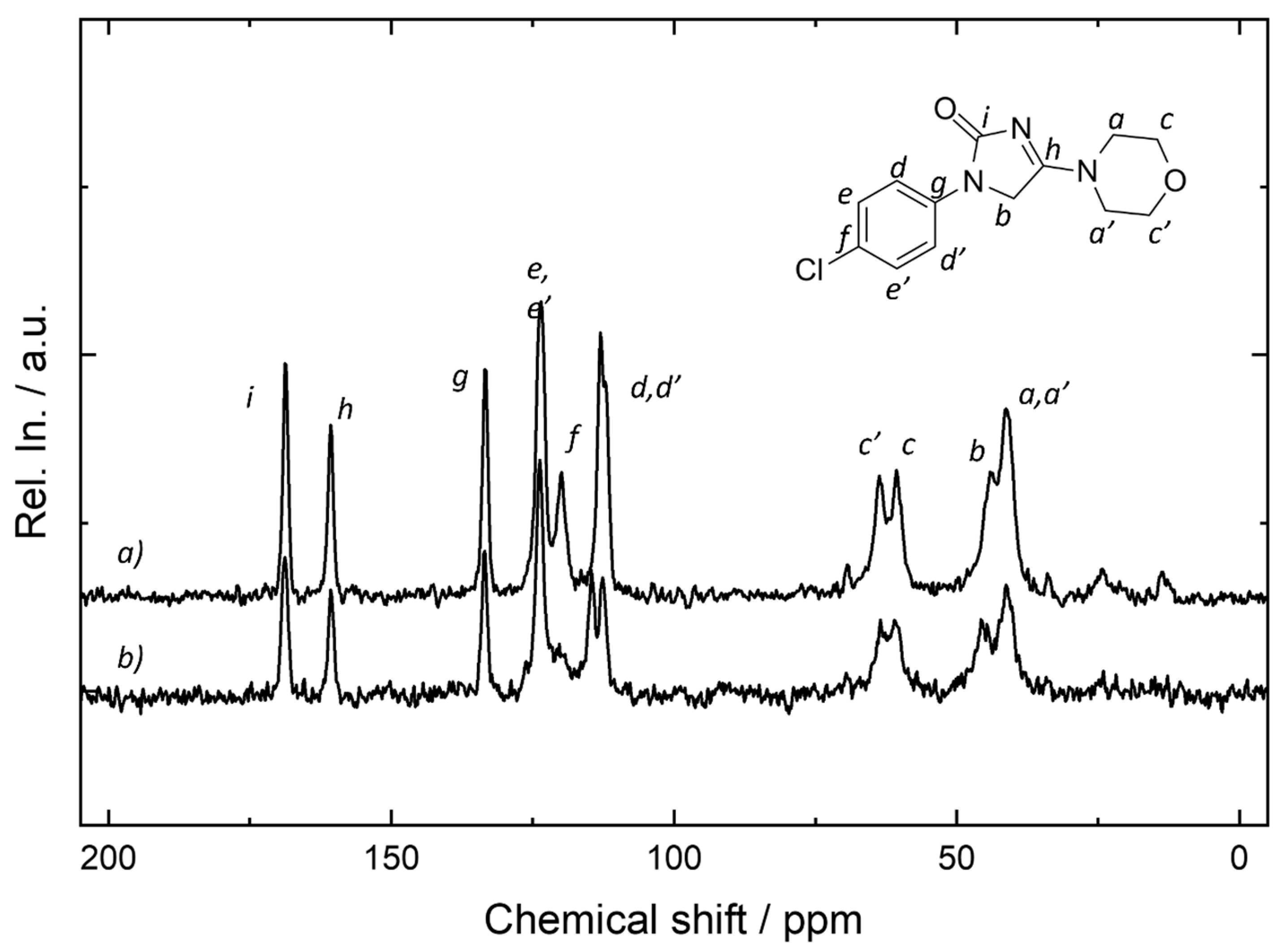
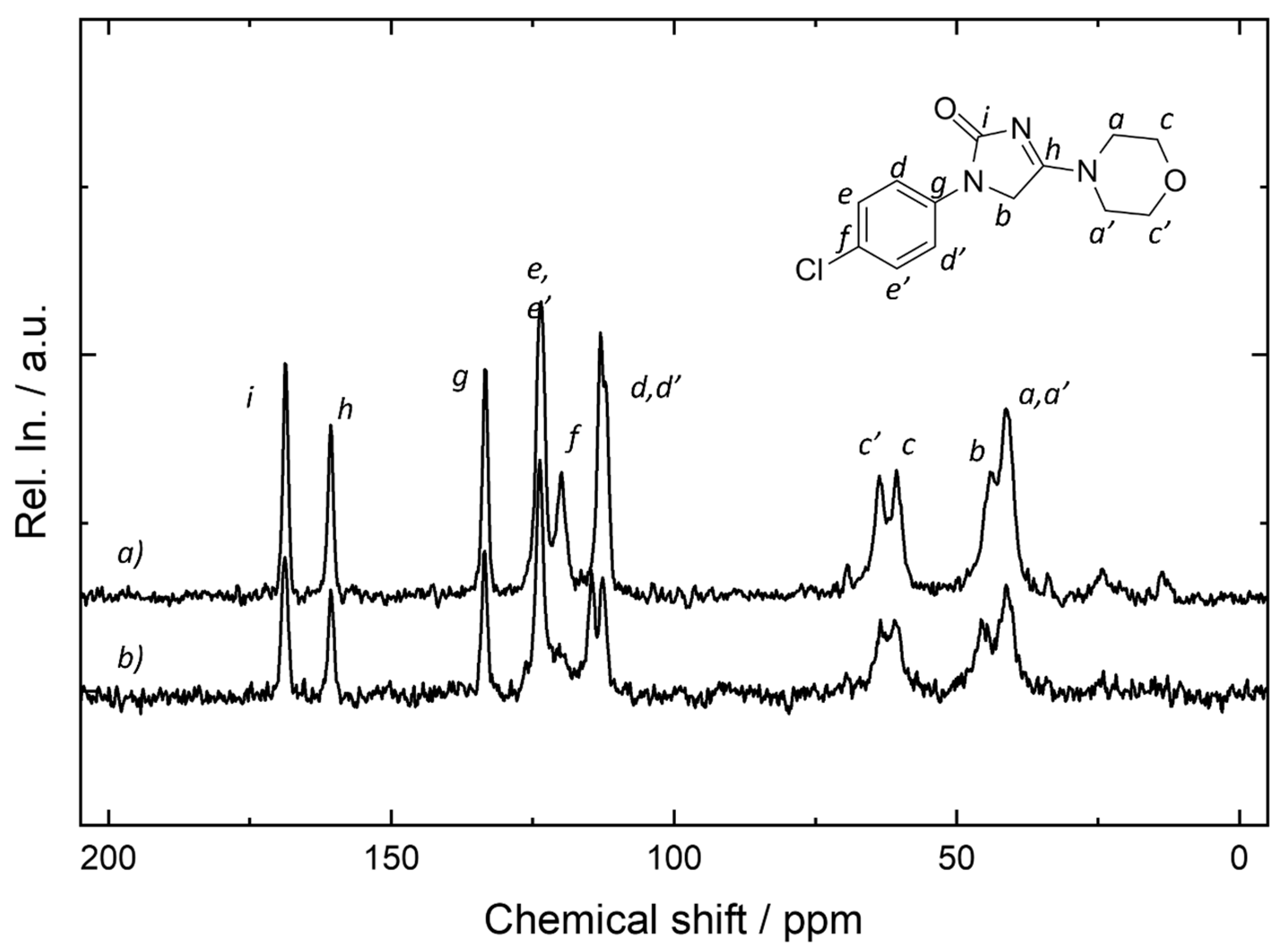
2.8. NMR Measurements
2.9. Kinetic Stability of the Polymorph II
3. Materials and Methods
3.1. Materials
3.2. Grinding
3.3. Solvent Crystallization
3.4. Sonication, Quenching, and Kneading
3.5. SEM Measurements
3.6. Thermal Measurements
3.7. XRPD Measurements
3.8. HSM Measurements
3.9. FT-IR Measurements
3.10. Raman Measurements
3.11. NMR Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernstein, J. Polymorphism—A perspective. Cryst. Growth Des. 2010, 11, 632–650. [Google Scholar] [CrossRef]
- Rietveld, I.B.; Barrio, M.; Lloveras, P.; Céolin, R.; Tamarit, J.L. Polymorphism of spironolactone: An unprecedented case of monotropy turning to enantiotropy with a huge difference in the melting temperatures. Int. J. Pharm. 2018, 552, 193–205. [Google Scholar] [CrossRef]
- Rai, S.K.; Gunnam, A.; Beran, G.J.O.; Kaduk, J.A.; Nangia, A.K. Polymorphs, solvatomorphs, hydrate, and perhydrate of dabrafenib. Cryst. Growth Des. 2023, 23, 1179–1188. [Google Scholar] [CrossRef]
- de Moraes, M.G.F.; Barreto, A.G.; Secchi, A.R.; de Souza, M.B.; da Cunha Lage, P.L.; Myerson, A.S. Polymorphism of praziquantel: Role of cooling crystallization in access to solid forms and discovery of new polymorphs. Cryst. Growth Des. 2023, 23, 1247–1258. [Google Scholar] [CrossRef]
- Silva, J.F.C.; Pereira Silva, P.S.; Ramos Silva, M.; Fantechi, E.; Chelazzi, L.; Ciattini, S.; Eusébio, M.E.S.; Rosado, M.T.S. Amorphous solid forms of ranolazine and tryptophan and their relaxation to metastable polymorphs. Cryst. Growth Des. 2023, 23, 6679–6691. [Google Scholar] [CrossRef] [PubMed]
- António, C.T.; Évora, A.O.L.; Bernardes, C.E.S.; Diogo, H.P.; de Araujo, G.L.B.; Smith, P.; Byrn, S.R.; Fausto, R.; Minas da Piedade, M.E. Polymorphism in erlotinib hydrochloride: New insights into relative stability, thermal behavior, and structural differences of forms A and B. Cryst. Growth Des. 2023, 23, 7374–7384. [Google Scholar] [CrossRef]
- Griesser, U.J.; Weigand, D.; Rollinger, J.M.; Haddow, M.; Gstrein, E. The crystal polymorphs of metazachlor: Identification and thermodynamic stability. J. Therm. Anal. Calorim. 2004, 77, 511–522. [Google Scholar] [CrossRef]
- Rietveld, I.B.; Céolin, R. Phenomenology of crystalline polymorphism: Overall monotropic behavior of the cardiotonic agent FK664 forms A and B. J. Therm. Anal. Calorim. 2015, 120, 1079–1087. [Google Scholar] [CrossRef]
- McCrone, W.C. Polymorphism. In Physics and Chemistry of the Organic Solid State; Fox, D., Labes, M., Weissberger, A., Eds.; Interscience: New York, NY, USA, 1965; Volume 2, pp. 726–767. [Google Scholar]
- Li, F.; Chen, S.; Hu, H.; Liang, C.; Sun, S.; Jin, C.; Chen, F. Crystallization selectivity of ribavirin solution and amorphous phase. Molecules 2023, 28, 6320. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, R.; Thakur, T.S. Crystal polymorphism in pharmaceutical science. Compr. Supramol. Chem. II 2017, 5, 283–309. [Google Scholar]
- Bruni, G.; Pardi, F.; Capsoni, D.; Berbenni, V.; Bini, M.; Valle, G.; Milanese, C.; Girella, A.; Marini, A. Febantel: Looking for new polymorphs. J. Therm. Anal. Calorim. 2017, 130, 1605–1612. [Google Scholar] [CrossRef]
- Bruni, G.; Gozzo, F.; Capsoni, D.; Bini, M.; Macchi, P.; Simoncic, P.; Berbenni, V.; Milanese, C.; Girella, A.; Ferrari, S.; et al. Thermal, spectroscopic, and ab initio structural characterization of caprofen polymorphs. J. Pharm. Sci. 2011, 100, 2321–2332. [Google Scholar] [CrossRef] [PubMed]
- Bruni, G.; Milanese, C.; Berbenni, V.; Sartor, F.; Villa, M.; Marini, A. Crystalline and amorphous phases of a new drug. J. Therm. Anal. Calorim. 2010, 102, 297–303. [Google Scholar] [CrossRef]
- Han, R.; Qu, H.; Li, Y.; Liu, Y.; Tao, T.; Tang, W.; Li, Z.; Gong, J. Development strategies of polymorphs and solvates for enhancing powder properties: A case study of thiothiamine. Cryst. Growth Des. 2023, 23, 7482–7495. [Google Scholar] [CrossRef]
- Michalak, O.; Łaszcz, M.; Jatczak, K.; Witkowska, A.; Bujak, I.; Groman, A.; Cybulski, M. New polymorphic forms of pemetrexed diacid and their use for the preparation of pharmaceutically pure amorphous and hemipentahydrate forms of pemetrexed disodium. Molecules 2015, 20, 13814–13829. [Google Scholar] [CrossRef] [PubMed]
- Masciocchi, N.; Zampieri, M.; Abbinante, V.M.; Zambra, M.; Barreca, G. Thermal and Structural Characterization of Two Crystalline Polymorphs of Tafamidis Free Acid. Molecules 2023, 27, 7411. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.Y.; Gao, X.; Niu, Y.J.; Shi, X.L.; Zhou, W. Polymorphic characterization, pharmacokinetics, and anti-inflammatory activity of ginsenoside compound K polymorphs. Molecules 2021, 26, 1983. [Google Scholar] [CrossRef] [PubMed]
- Rundfeldt, C.; Dost, R.; Loscher, W.; Tipold, A.; Unverferth, K.; Lankau, H.J. Use of Dihydroimidazolones for the Treatment of Dogs. U.S. Patent US 9,469,611 B2, 18 October 2016. [Google Scholar]
- Rostock, A.; Tober, C.; Dost, R.; Rundfeldt, C.; Bartsch, R.; Egerland, U.; Stark, B.; Schupke, H.; Kronbach, T.; Lankau, H.J.; et al. AWD 131-138. ADD-233089. Anxiolytic, anticonvulsant. Drugs Future 1998, 23, 253–255. [Google Scholar]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res. 2013, 103, 2–30. [Google Scholar] [CrossRef] [PubMed]
- Tipold, A.; Keefe, T.J.; Lösker, W.; Rundfeldt, C.; De Vries, F. Clinical efficacy and safety of imepitoin in comparison with phenobarbital for the control of idiopathic epilepsy in dogs. J. Vet. Pharmacol. Ther. 2014, 38, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Hoffmann, K.; Twele, F.; Potschka, H.; Töllner, K. The novel antiepileptic drug imepitoin compares favorably to other GABA-mimetic drugs in a seizure threshold model in mice and dogs. Pharmacol. Res. 2013, 77, 39–46. [Google Scholar] [CrossRef]
- Kaduk, J.A.; Gindhart, A.M.; Gates-Rector, S.; Blanton, T.N. Crystal structure of imepitoin, C13H14ClN3O2. Powder Diffr. 2022, 37, 206–210. [Google Scholar] [CrossRef]
- Pindelska, E.; Sokal, A.; Kolodziejski, W. Pharmaceutical cocrystals, salts and polymorphs: Advanced characterization techniques. Adv. Drug Deliv. Rev. 2017, 117, 111–146. [Google Scholar] [CrossRef] [PubMed]
- Guranda, D.T.; Gil’deeva, G.N. Drugs synthesis methods and manufacturing technology. Pharm. Chem. J. 2010, 44, 254–260. [Google Scholar] [CrossRef]
- Willart, J.F.; Durand, M.; Briggner, L.E.; Marx, A.; Danède, F.; Descamps, M. Solid-state amorphization of linaprazan by mechanical milling and evidence of polymorphism. J. Pharm. Sci. 2013, 102, 2214–2220. [Google Scholar] [CrossRef]
- Hasa, D.; Jones, W. Screening for new pharmaceutical solid forms using mechanochemistry: A practical guide. Adv. Drug Deliv. Rev. 2017, 117, 147–161. [Google Scholar] [CrossRef]
- Kaur Bhangu, S.; Ashokkumar, M.; Lee, J. Ultrasound assisted crystallization of paracetamol: Crystal size distribution and polymorph control. Cryst. Growth Des. 2016, 16, 1934–1941. [Google Scholar] [CrossRef]
- Zhang, S.; Lee, T.W.Y.; Chow, A.H.L. Crystallization of itraconazole polymorphs from melt. Cryst. Growth Des. 2016, 16, 3791–3801. [Google Scholar] [CrossRef]
- Zhou, Y.; Lv, C.; Liu, X.; Gao, J.; Gao, Y.; Wang, T.; Huang, X. An overview on polymorph preparation methods of active pharmaceutical ingredients. Cryst. Growth Des. 2024, 24, 584–600. [Google Scholar] [CrossRef]
- Lee, E.H. A practical guide to pharmaceutical polymorph screening & selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar]
- Coelho, A.A. Indexing of powder diffraction patterns by iterative use of singular value decomposition. J. Appl. Cryst. 2003, 36, 86–95. [Google Scholar] [CrossRef]
- Altomare, A.; Campi, G.; Cuocci, C.; Eriksson, L.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. Advances in powder diffraction pattern indexing: N-TREOR09. J. Appl. Crystallogr. 2009, 42, 768–775. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Burger, A.; Ramberger, R. On the polymorphism of pharmaceuticals and other molecular crystals. Mikrochimica Acta 1979, II, 259–271. [Google Scholar] [CrossRef]
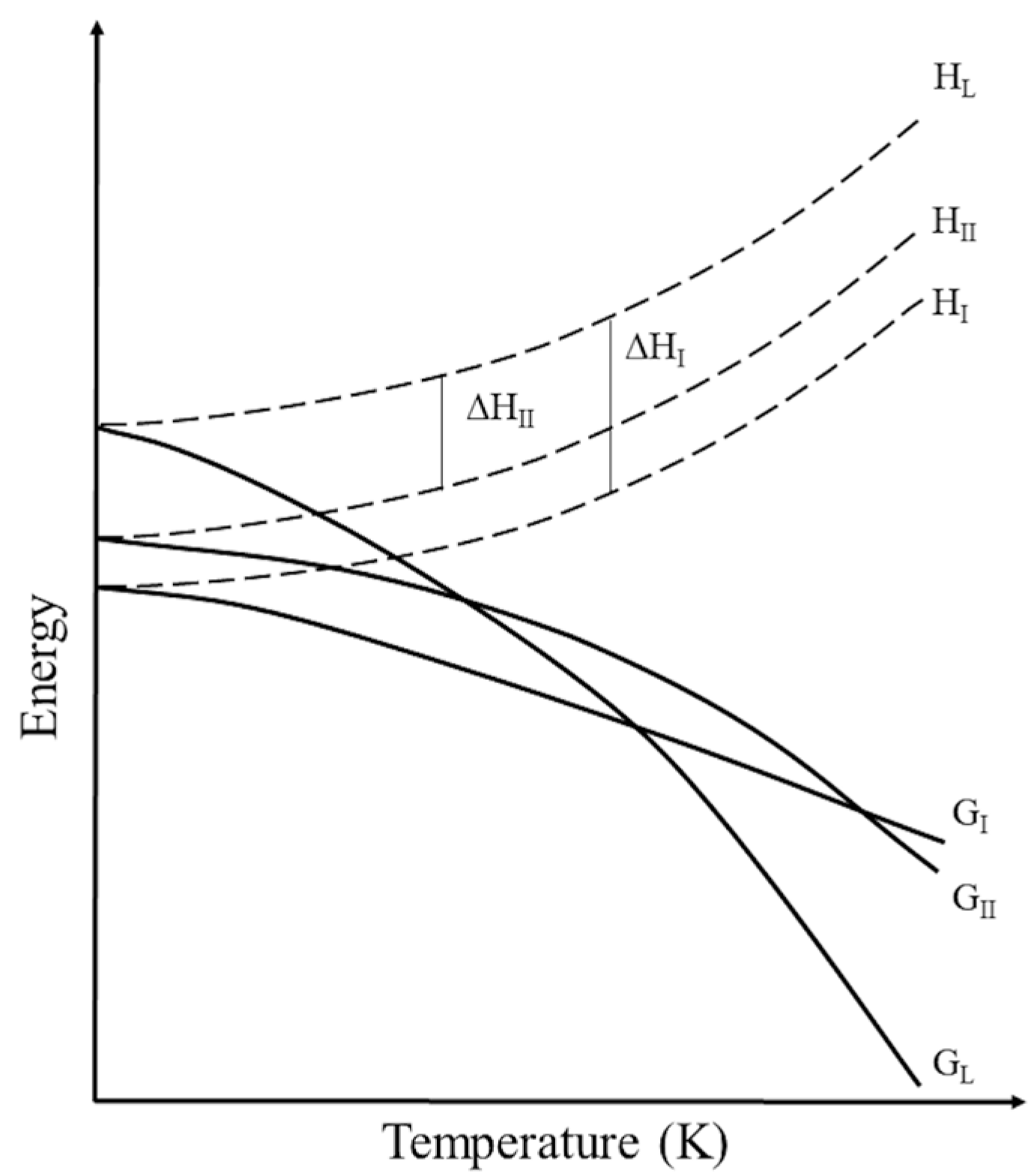
- Perlovich, G.; Surov, A. Polymorphism of monotropic forms: Relationships between thermochemical and structural characteristics. Acta Cryst. B 2020, 76, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Inferring thermodynamic stability relationship of polymorphs from melting data. J. Pharm. Sci. 1995, 84, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Tuschel, D. Raman Spectroscopy and Polymorphism. Spectroscopy 2019, 34, 11. [Google Scholar]
- Zhao, L.B.; Huang, Y.F.; Liu, X.M.; Anema, J.R.; Wu, D.Y.; Ren, B.; Tian, Z.Q. A DFT study on photoinduced surface catalytic coupling reactions on nanostructured silver: Selective formation of azobenzene derivatives from para-substituted nitrobenzene and aniline. Phys. Chem. Chem. Phys. 2012, 14, 12919–12929. [Google Scholar] [CrossRef] [PubMed]
- Jun, B.H.; Kim, G.; Noh, M.S.; Kang, H.; Kim, Y.K.; Cho, M.H.; Jeong, D.H.; Lee, Y.S. Surface-enhanced Raman scattering-active nanostructures and strategies for bioassays. Nanomedicine 2011, 6, 1463–1480. [Google Scholar] [CrossRef] [PubMed]
- Markham, L.M.; Mayne, L.C.; Hudson, B.S.; Zgierski, M.Z. Resonance Raman Studies of Imidazole, Imidazolium, and Their Derivatives: The Effect of Deuterium Substitution. J. Phys. Chem. 1993, 97, 10319–10325. [Google Scholar] [CrossRef]
- Parveen, S.; Al-Alshaikh, M.A.; Panicker, C.Y.; El-Emam, A.A.; Narayana, B.; Saliyan, V.V.; Sarojini, B.K.; van Alsenoy, C. Vibrational and structural observations and molecular docking study on 1-{3-(4-chlorophenyl)-5-[4-(propan-2-yl)phenyl]-4,5-dihydro-1H-pyrazol-1-yl}-ethanone. J. Mol. Struct. 2016, 1112, 136–146. [Google Scholar] [CrossRef]
- Fitch, A.; Dejoie, C.; Covacci, E.; Confalonieri, G.; Grendal, O.; Claustre, L.; Guillou, P.; Kieffer, J.; de Nolf, W.; Petitdemange, S.; et al. ID22—The high-resolution powder-diffraction beamline at ESRF. J. Synchrotron Radiat. 2023, 30, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Dejoie, C.; Coduri, M.; Petitdemange, S.; Giacobbe, C.; Covacci, E.; Grimaldi, O.; Autran, P.-O.; Mogodi, M.W.; Sisak Jund, D.; Fitch, A.N. Combining a nine-crystal multi-analyser stage with a two-dimensional detector for high-resolution powder X-ray diffraction. J. Appl. Cryst. 2018, 51, 1721–1733. [Google Scholar] [CrossRef]
- TOPAS4.2; Bruker: Karlsruhe, Germany, 2009.
- ACD/ChemSketch; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2003.
- Stewart, J.J.P. MOPAC2016, Version 18.305L; Stewart Computational Chemistry: Colorado Springs, CO, USA, 2016. Available online: http://OpenMOPAC.net/(accessed on 7 April 2024).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
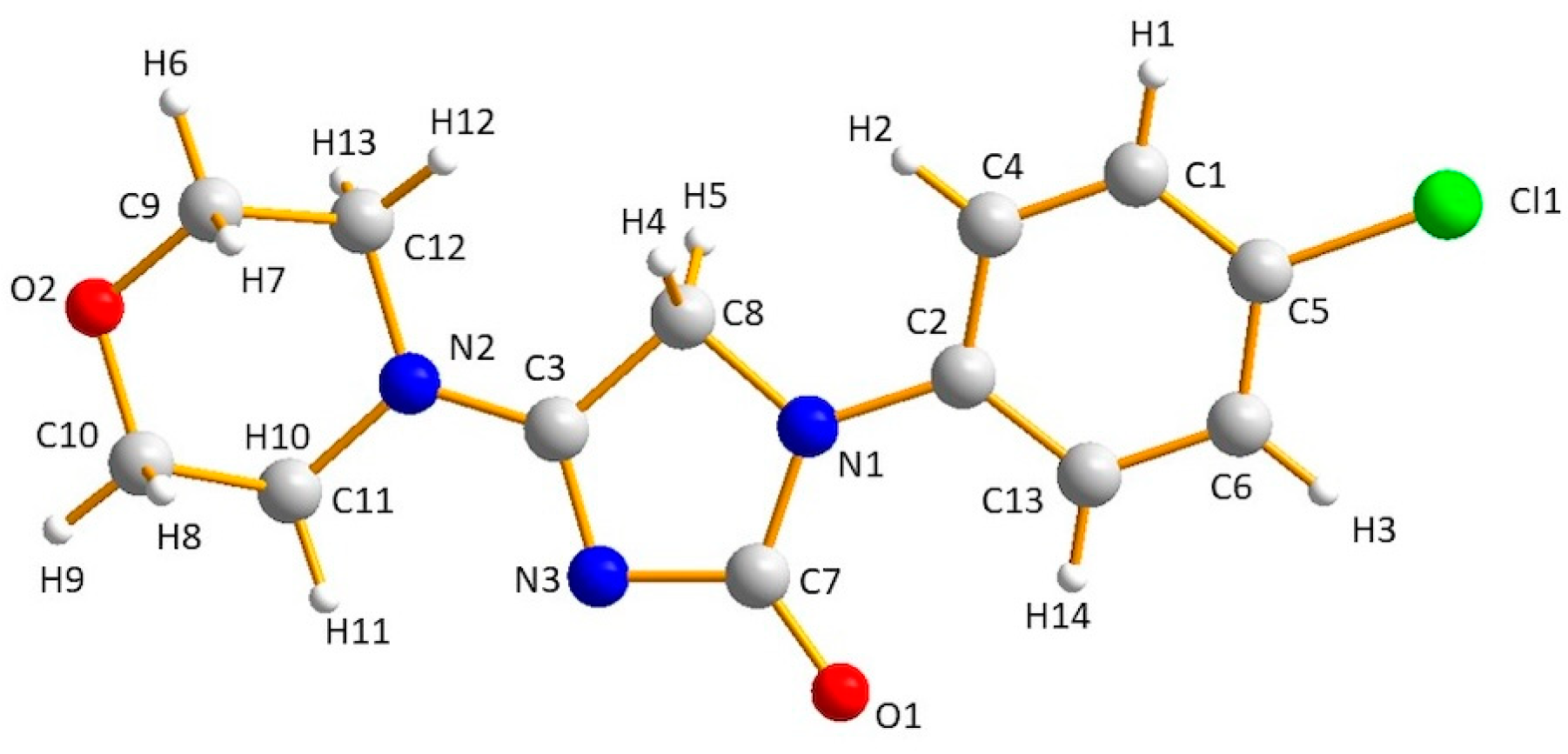
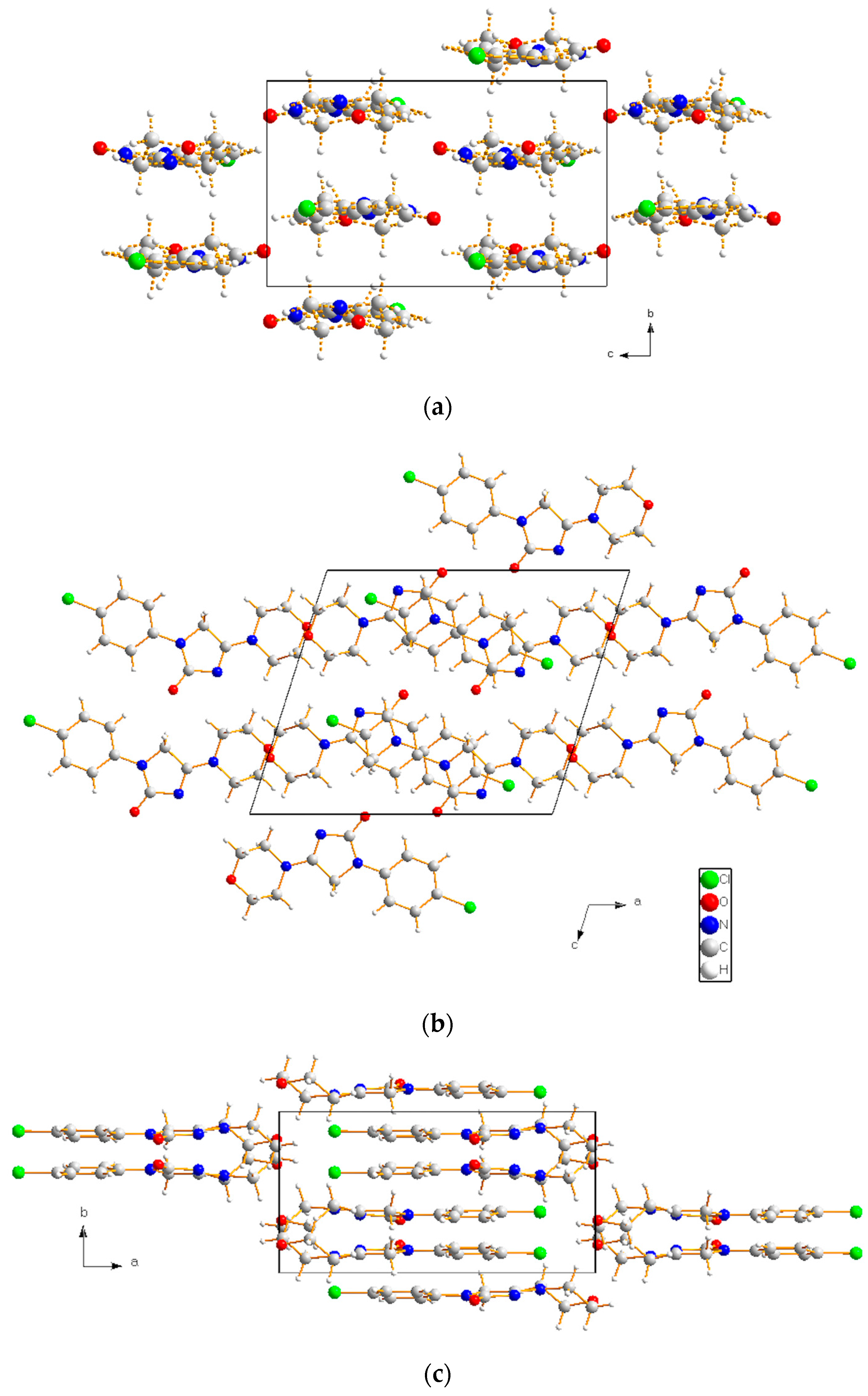
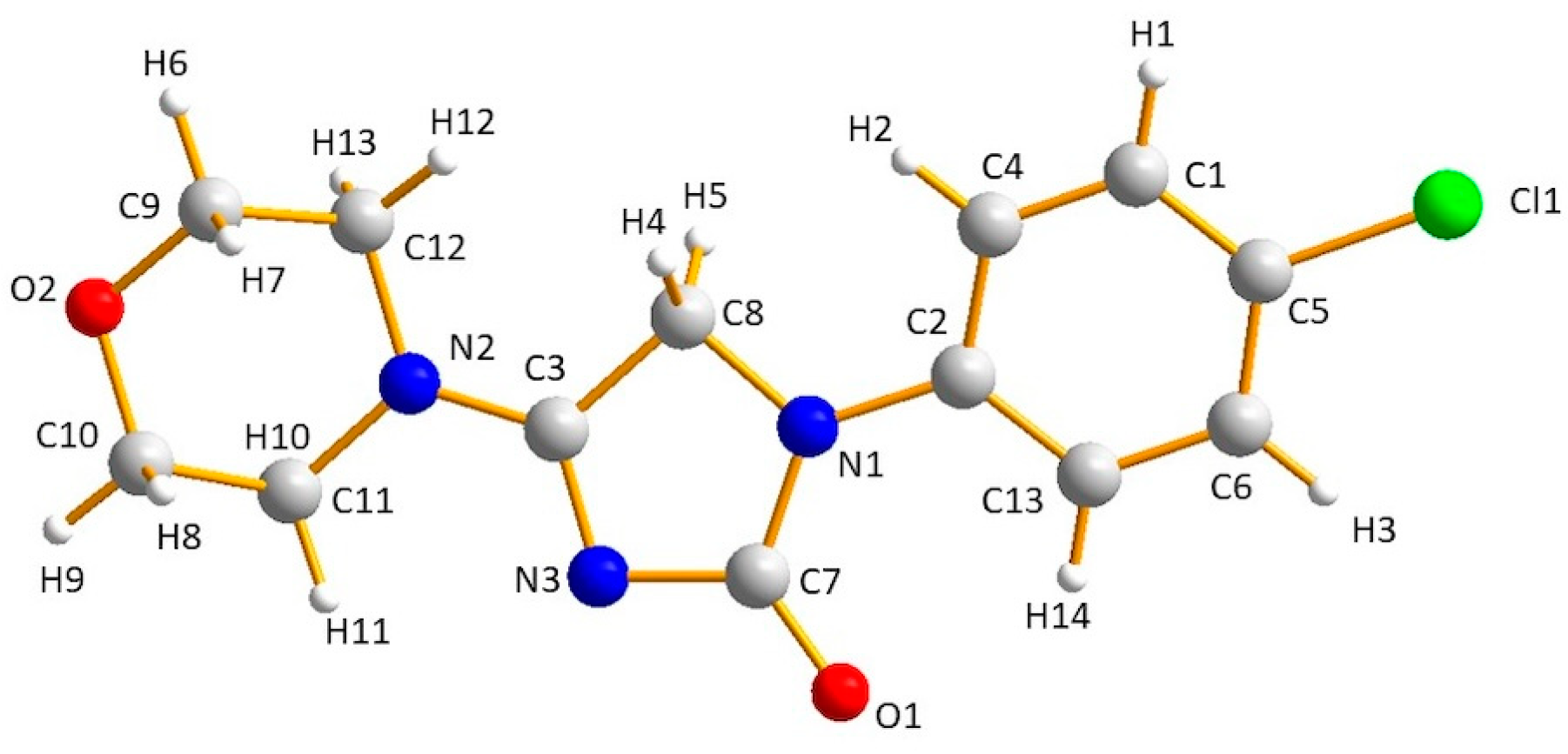
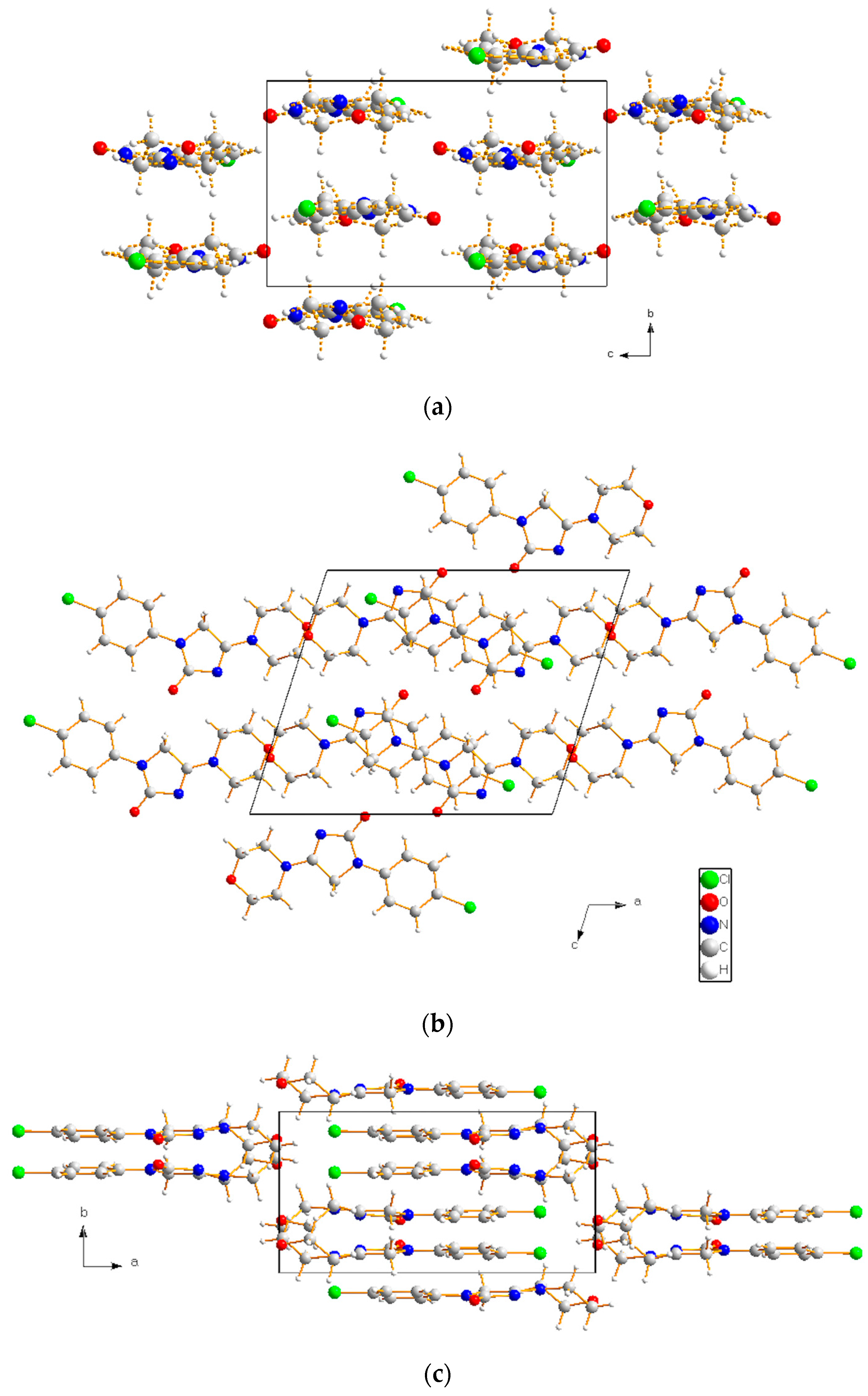
| H-Bond | A···H (Å) (II) | A···H (Å) (I) |
|---|---|---|
| O2···H6 (or H7, H8, H9) O2···H8 (or H13) O2···H10 | 1.96–1.99 * 2.66–2.70 2.82 | 2.09–2.19 * 2.62 2.96 |
| O1···H14 O1···H4 O1···H12 | 2.24 * 2.35 2.78 | 2.10 * 2.03 2.77 |
| N3···H11 N3···H6 N3···H4 N3···H12 | 2.41 * 2.93 3.04 * 3.04 | 2.39 * 2.87 3.23 * 3.12 |
| Cl1···H1 (or H3) Cl1···H10 Cl1···H8 | 2.79–2.80 * 3.02 3.10 | 2.80–2.87 * 2.85 3.01 |
| Vibration | Wavenumbers, cm−1 |
|---|---|
| C=O stretching | 1703.4 |
| C=N and aromatic ring stretching | 1602.9 1581.8 |
| Aromatic ring stretching | 1495.1 |
| Aromatic ring in-plane bending | 1281.7 1261.3 1154.1 |
| Combined C-Cl and ring stretching | 1113.0 |
| CH out-of-plane bending (2 adjacent H) | 819.3 |
| C-Cl stretching | 667.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruni, G.; Capsoni, D.; Pellegrini, A.; Altomare, A.; Coduri, M.; Ferrara, C.; Galinetto, P.; Molteni, R. New Findings on the Crystal Polymorphism of Imepitoin. Molecules 2024, 29, 1724. https://doi.org/10.3390/molecules29081724
Bruni G, Capsoni D, Pellegrini A, Altomare A, Coduri M, Ferrara C, Galinetto P, Molteni R. New Findings on the Crystal Polymorphism of Imepitoin. Molecules. 2024; 29(8):1724. https://doi.org/10.3390/molecules29081724
Chicago/Turabian StyleBruni, Giovanna, Doretta Capsoni, Anna Pellegrini, Angela Altomare, Mauro Coduri, Chiara Ferrara, Pietro Galinetto, and Renato Molteni. 2024. "New Findings on the Crystal Polymorphism of Imepitoin" Molecules 29, no. 8: 1724. https://doi.org/10.3390/molecules29081724