
Exploring Enhanced Hydrolytic Dehydrogenation of Ammonia Borane with Porous Graphene-Supported Platinum Catalysts


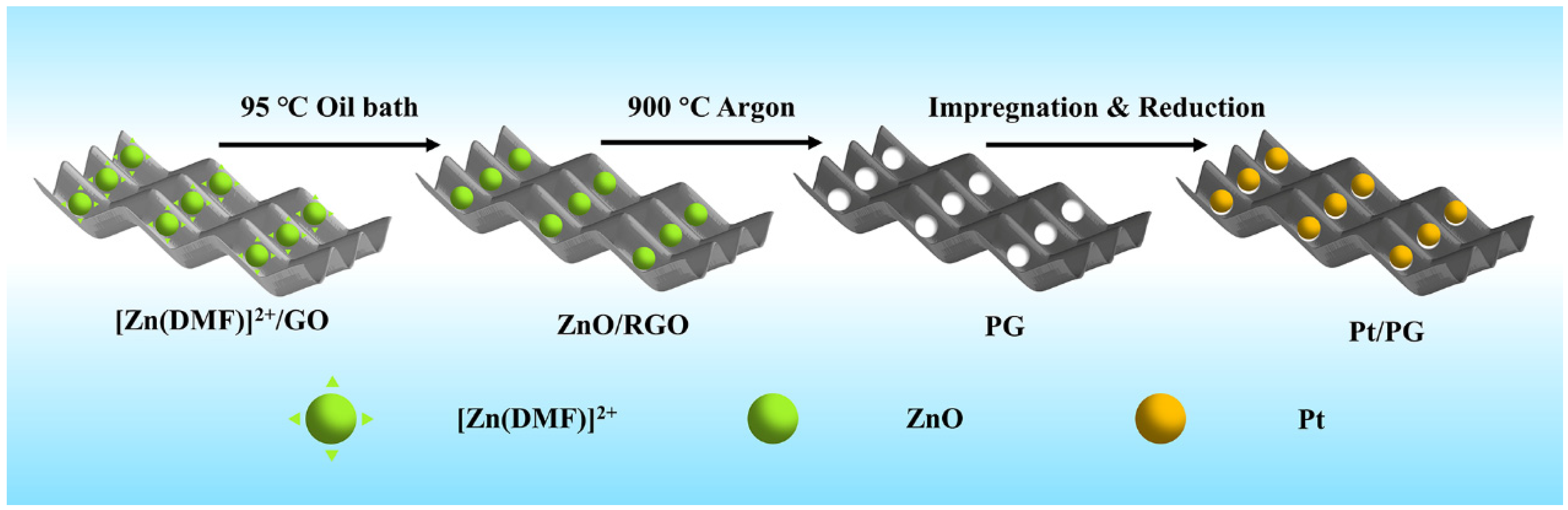{kind=link}
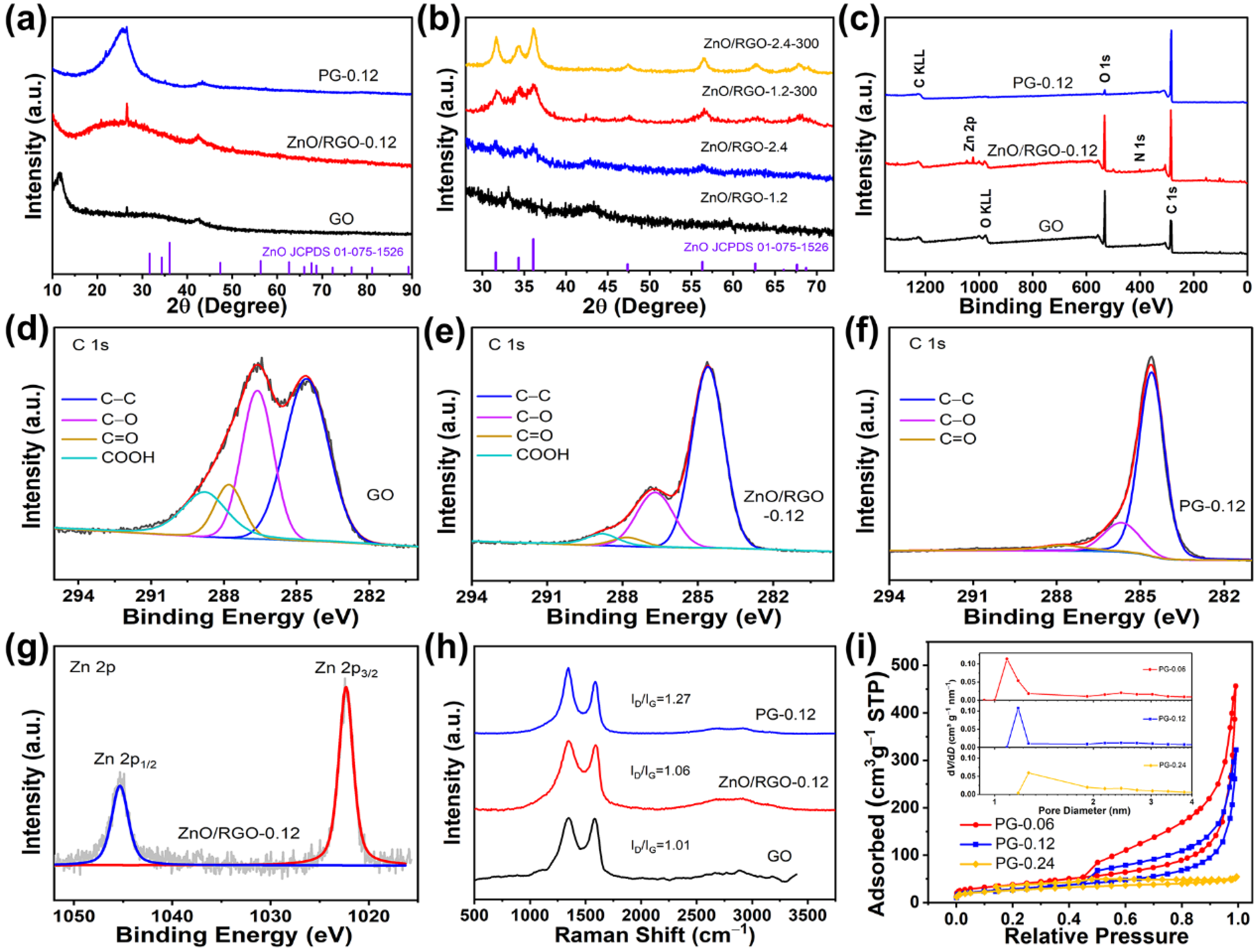{kind=link}
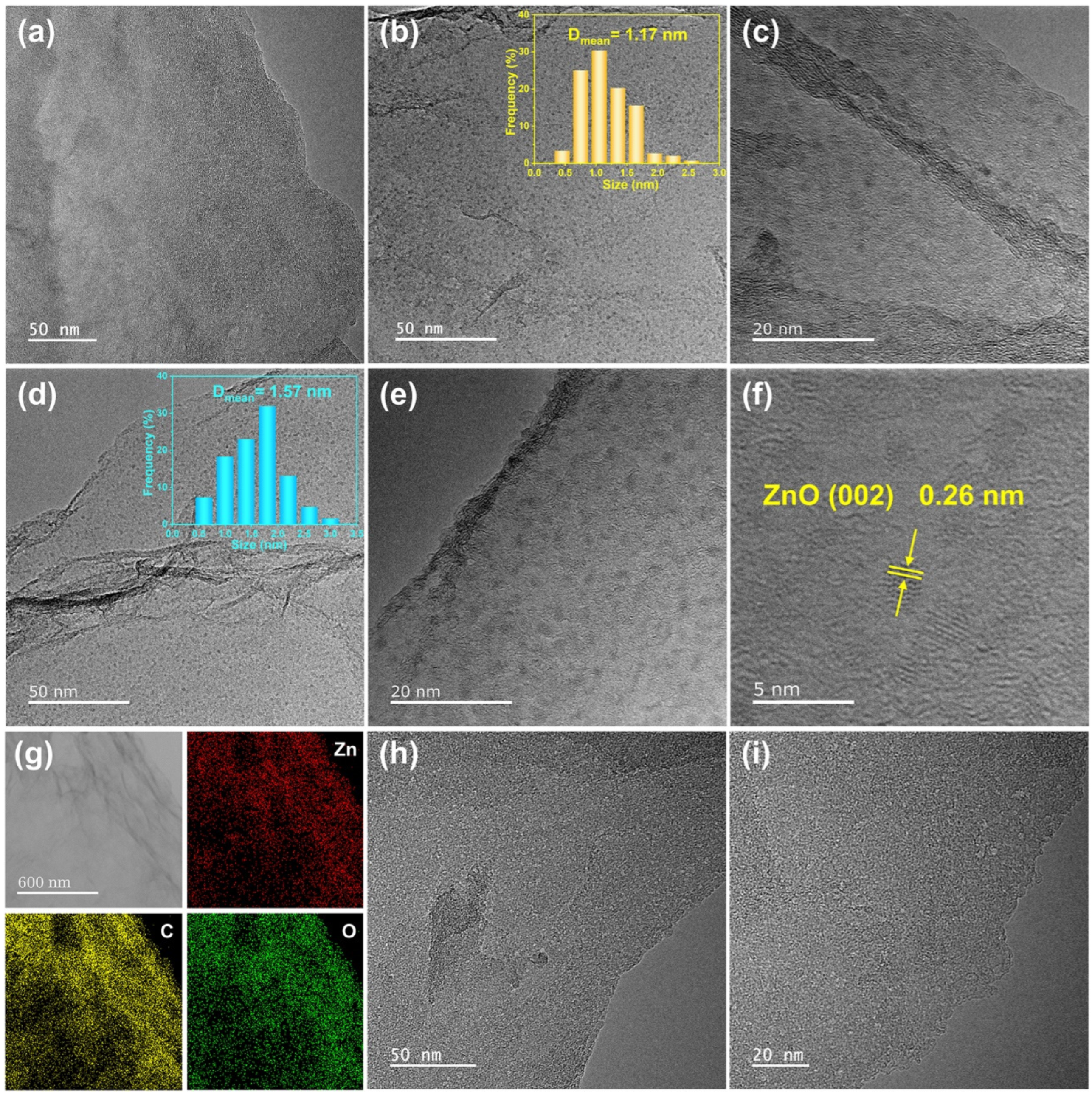{kind=link}
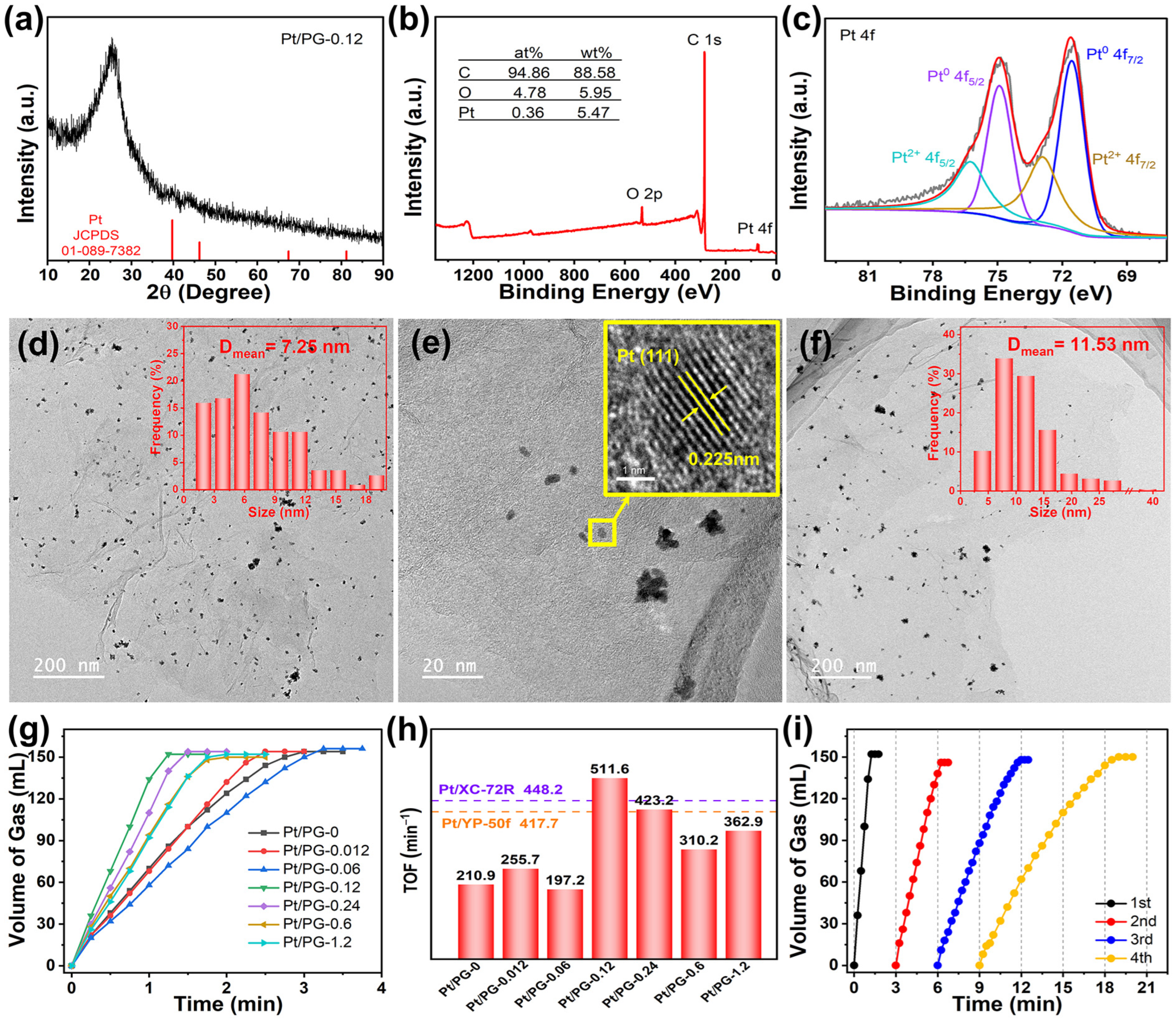{kind=link}
{kind=link}
Abstract
1. Introduction
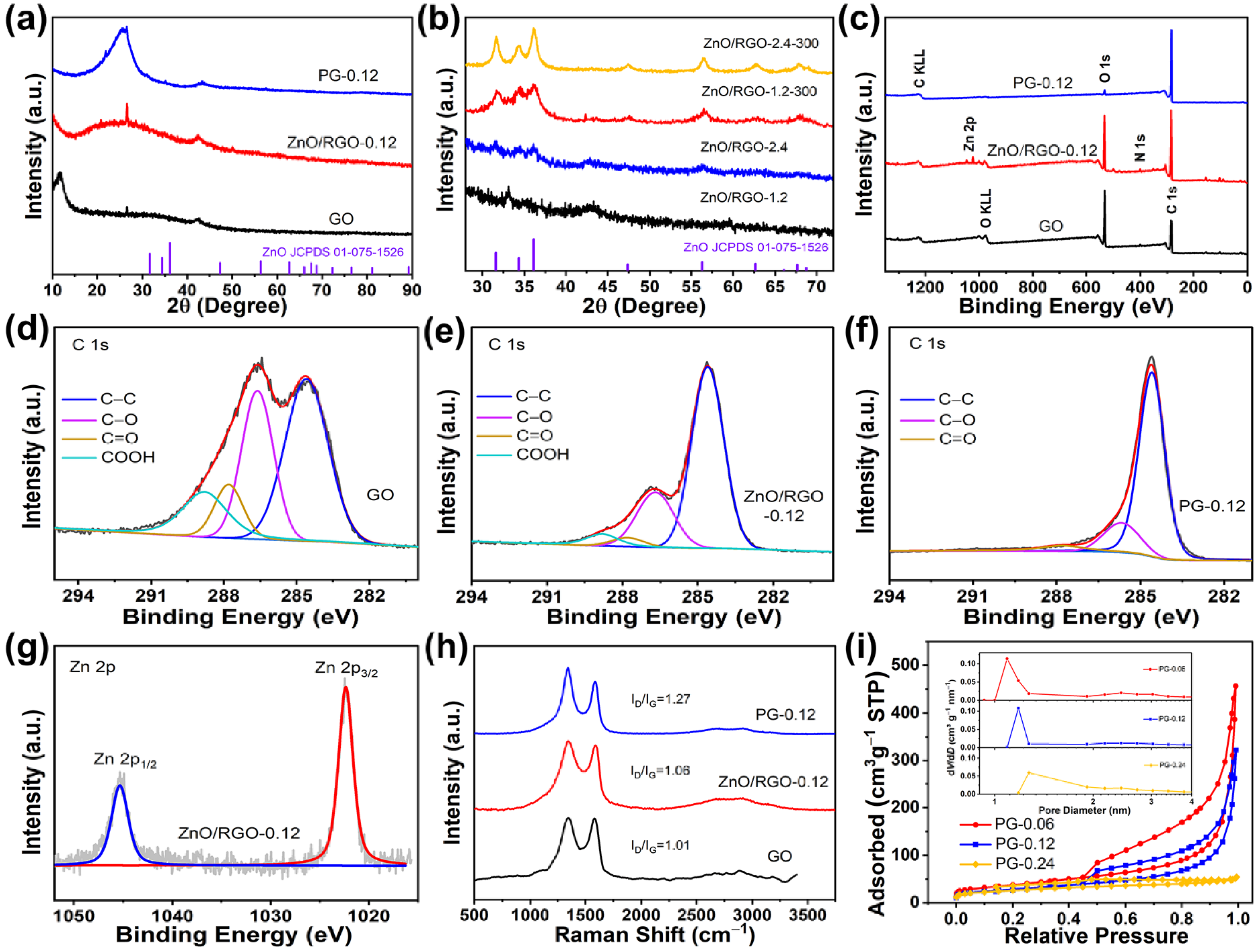
2. Results and Discussion
3. Experimental
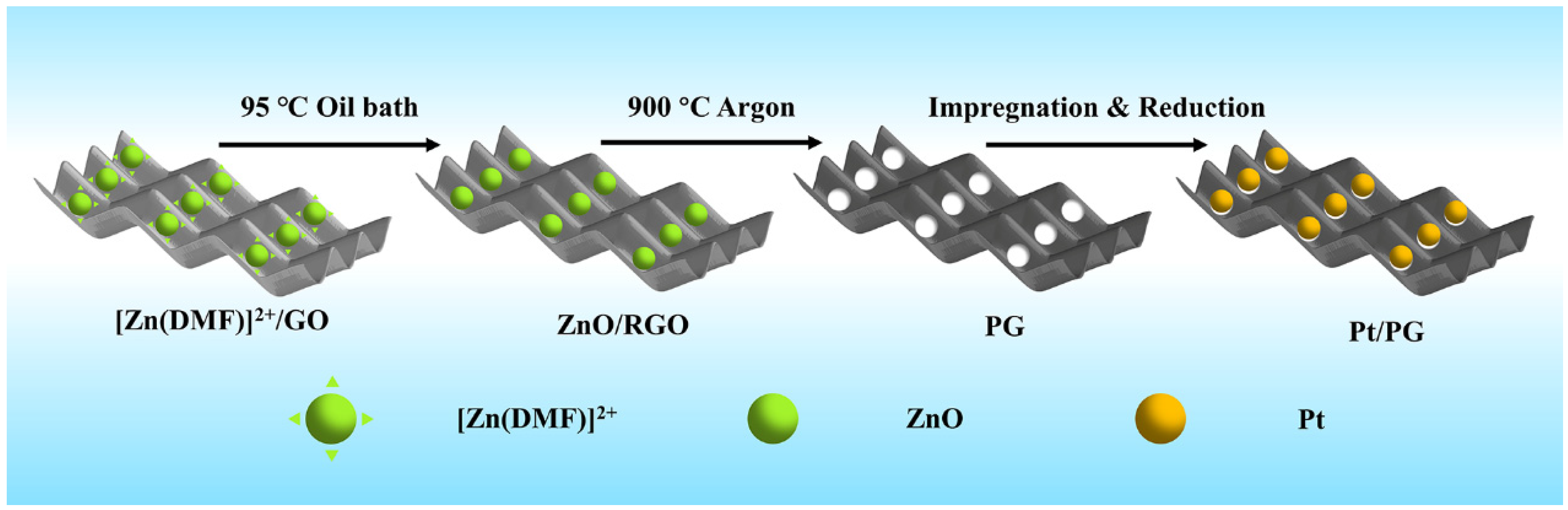
3.1. Materials Preparation
3.1.1. Preparation of GO
3.1.2. Preparation of PG
3.1.3. Preparation of Pt/PG
3.2. Characterizations
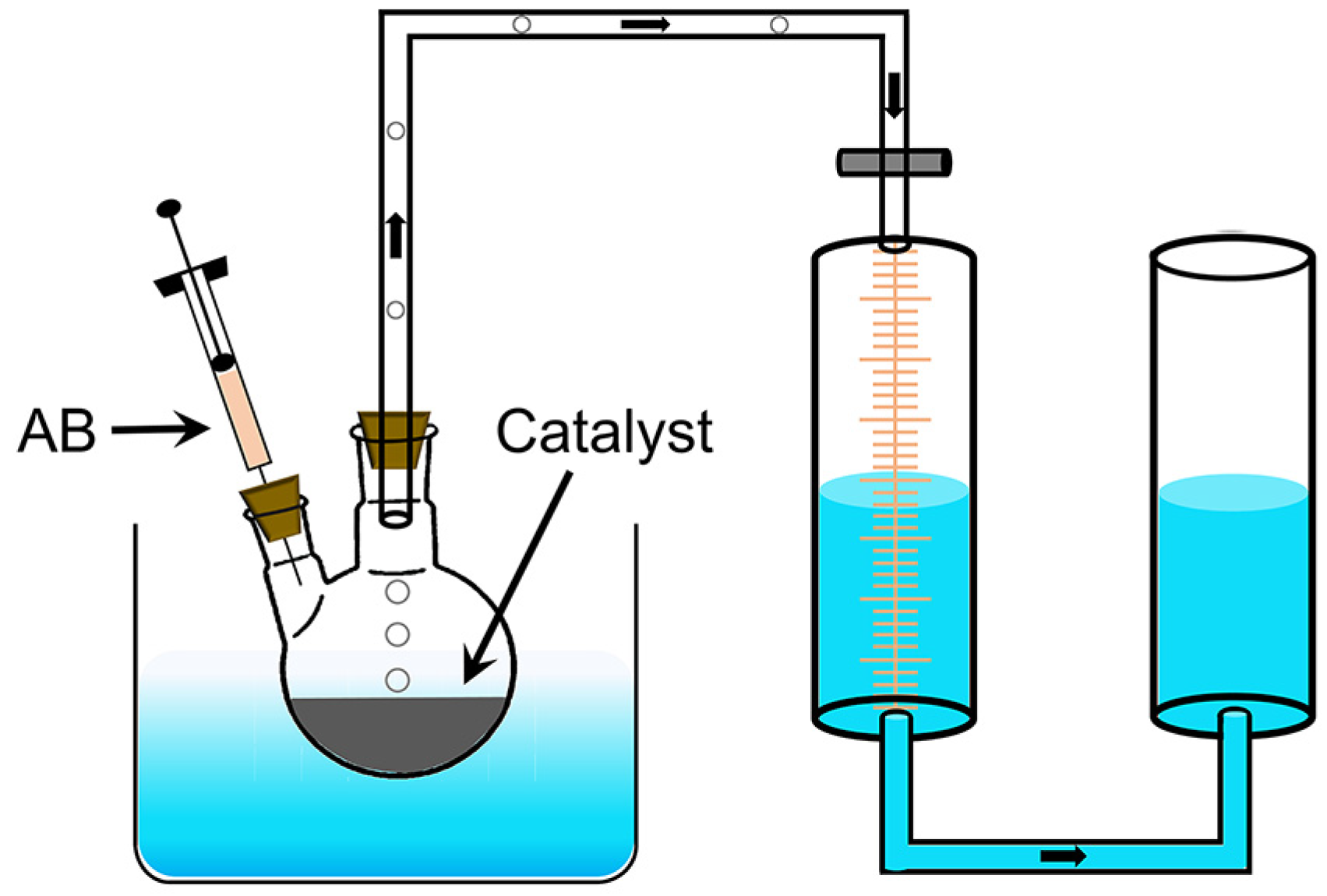
3.3. Catalytic Performance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durbin, D.J.; Malardier-Jugroot, C. Review of hydrogen storage techniques for on board vehicle applications. Int. J. Hydrogen Energy 2013, 38, 14595–14617. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, G.Z. Half-electrolysis of water with the aid of a supercapacitor electrode. ACS Appl. Energy Mater. 2023, 6, 6104–6110. [Google Scholar] [CrossRef] [PubMed]
- Komova, O.V.; Simagina, V.I.; Butenko, V.R.; Odegova, G.V.; Bulavchenko, O.A.; Nikolaeva, O.A.; Ozerova, A.M.; Lipatnikova, I.L.; Tayban, E.S.; Mukha, S.A.; et al. Dehydrogenation of ammonia borane recrystallized by different techniques. Renew. Energy 2022, 184, 460–472. [Google Scholar] [CrossRef]
- Li, D.-H.; Li, Q.-M.; Qi, S.-L.; Qin, H.-C.; Liang, X.-Q.; Li, L. Theoretical study of hydrogen production from ammonia borane catalyzed by metal and non-metal diatom-doped cobalt phosphide. Molecules 2022, 27, 8206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, Q.-L.; Tsumori, N.; Xu, Q. Immobilizing highly catalytically active noble metal nanoparticles on reduced graphene oxide: A non-noble metal sacrificial approach. J. Am. Chem. Soc. 2015, 137, 106–109. [Google Scholar] [CrossRef]
- Qiao, H.; Chen, Y. Rich amino grafted graphene oxide supported AgPd nanocatalysts for hydrogen generation from formic acid. Mater. Lett. 2024, 355, 135533. [Google Scholar] [CrossRef]
- Ibrahim, A.; Paskevicius, M.; Buckley, C.E. Chemical compression and transport of hydrogen using sodium borohydride. Sustain. Energy Fuels 2023, 7, 1196–1203. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, N.; Xu, Q.; Yu, J. Nanopore-supported metal nanocatalysts for efficient hydrogen generation from liquid-phase chemical hydrogen storage materials. Adv. Mater. 2020, 32, 2001818. [Google Scholar] [CrossRef]
- Lang, C.; Jia, Y.; Yao, X. Recent advances in liquid-phase chemical hydrogen storage. Energy Storage Mater. 2020, 26, 290–312. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Liu, J.; Wang, S.; Song, Y.; Zhang, J. Design strategies for shape-controlled nanocatalysts for efficient dehydrogenation of ammonia borane: A review. J. Alloys Compd. 2023, 961, 171001. [Google Scholar] [CrossRef]
- Wei, Q.; Qiu, S.; Yin, C.; Liu, J.; Xia, Y.; Wen, X.; Zou, Y.; Xu, F.; Sun, L.; Chu, H. Nitrogen-doped carbon encapsulated Ru-decorated Co2P supported on graphene oxide as efficient catalysts for hydrogen generation from ammonia borane. J. Alloys Compd. 2022, 921, 166207. [Google Scholar] [CrossRef]
- Li, G.-L.; Kumar Tripathi, A.; Chan, H.; Chen, S.-T.; Chang, J.-T.; Nakagawa, T.; Wang, C.-Y. Recyclable dehydrogenation/regeneration of ammonia borane nanoconfined in amino-functionalized ZIF-8 with 3-amino-1,2,4-triazole. ACS Sustain. Chem. Eng. 2023, 11, 6143–6152. [Google Scholar] [CrossRef]
- Özhava, D.; Çiğdem, Y.; Ertürk, S. Methanolysis of ammonia borane catalyzed by magnetically isolable rhodium(0) nanoparticles. Int. J. Hydrogen Energy 2023, 48, 22942–22953. [Google Scholar] [CrossRef]
- Gong, B.; Wu, H.; Sheng, L.; Zhang, W.; Wu, X. Hydrolysis of ammonia borane on a single Pt atom supported by N-doped graphene. ACS Appl. Mater. Interfaces 2022, 14, 13231–13239. [Google Scholar] [CrossRef]
- Duan, J.; Liu, X.; Bian, L.; Fan, Y.; Liu, B. Controllable synthesis of MoC and Mo2C to boost hydrogen generation from ammonia borane hydrolysis. ACS Appl. Energy Mater. 2023, 6, 1753–1762. [Google Scholar] [CrossRef]
- Duan, Y.; Guo, P.; Sui, D.; Deng, D.; Lu, T.; Yang, Y. Investigation on M@CuOx/C (M=Ru, Rh, Pd and Pt) catalysts prepared by galvanic reduction for hydrogen evolution from ammonia borane. Int. J. Hydrogen Energy 2022, 47, 36098–36109. [Google Scholar] [CrossRef]
- Rzelewska-Piekut, M.; Wolańczyk, Z.; Nowicki, M.; Regel-Rosocka, M. Precipitation of Pt, Pd, Rh, and Ru nanoparticles with non-precious metals from model and real multicomponent solutions. Molecules 2023, 28, 5188. [Google Scholar] [CrossRef]
- Meng, Y.; Sun, Q.; Zhang, T.; Zhang, J.; Dong, Z.; Ma, Y.; Wu, Z.; Wang, H.; Bao, X.; Sun, Q.; et al. Cobalt-promoted noble-metal catalysts for efficient hydrogen generation from ammonia borane hydrolysis. J. Am. Chem. Soc. 2023, 145, 5486–5495. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Z.; Chen, G.Z. Nano-scale engineering of heterojunction for alkaline water electrolysis. Materials 2024, 17, 199. [Google Scholar] [CrossRef]
- Cao, F.; Ding, R.; Rui, Z.; Wang, X.; Meng, Z.; Zhang, B.; Dong, W.; Li, J.; Liu, J.; Jiang, X. Advances in low Pt loading membrane electrode assembly for proton exchange membrane fuel cells. Molecules 2023, 28, 773. [Google Scholar] [CrossRef]
- Guan, S.; Liu, Y.; Zhang, H.; Shen, R.; Wen, H.; Kang, N.; Zhou, J.; Liu, B.; Fan, Y.; Jiang, J.; et al. Recent advances and perspectives on supported catalysts for heterogeneous hydrogen production from ammonia borane. Adv. Sci. 2023, 10, 2300726. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Peigney, A.; Laurent, C.; Flahaut, E.; Bacsa, R.R.; Rousset, A. Specific surface area of carbon nanotubes and bundles of carbon nanotubes. Carbon 2001, 39, 507–514. [Google Scholar] [CrossRef]
- Chen, Y. Diverse structural constructions of graphene-based composites for supercapacitors and metal-ion batteries. FlatChem 2022, 36, 100453. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, G.; Zhang, F. Multiple roles of graphene in heterogeneous catalysis. Chem. Soc. Rev. 2015, 44, 3023–3035. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Stoller, M.D.; Ganesh, K.J.; Cai, W.; Ferreira, P.J.; Pirkle, A.; Wallace, R.M.; Cychosz, K.A.; Thommes, M.; et al. Carbon-based supercapacitors produced by activation of graphene. Science 2011, 332, 1537–1541. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, Z.; Zhong, X.; Huang, X.; Weiss, N.O.; Huang, Y.; Duan, X. Holey graphene frameworks for highly efficient capacitive energy storage. Nat. Commun. 2014, 5, 4554. [Google Scholar] [CrossRef]
- Zhou, D.; Cui, Y.; Xiao, P.-W.; Jiang, M.-Y.; Han, B.-H. A general and scalable synthesis approach to porous graphene. Nat. Commun. 2014, 5, 4716. [Google Scholar] [CrossRef]
- Kim, H.-K.; Bak, S.-M.; Lee, S.W.; Kim, M.-S.; Park, B.; Lee, S.C.; Choi, Y.J.; Jun, S.C.; Han, J.T.; Nam, K.-W.; et al. Scalable fabrication of micron-scale graphene nanomeshes for high-performance supercapacitor applications. Energy Environ. Sci. 2016, 9, 1270–1281. [Google Scholar] [CrossRef]
- Wan, J.; Huang, L.; Wu, J.; Xiong, L.; Hu, Z.; Yu, H.; Li, T.; Zhou, J. Microwave combustion for rapidly synthesizing pore-size-controllable porous graphene. Adv. Funct. Mater. 2018, 28, 1800382. [Google Scholar] [CrossRef]
- Savaram, K.; Li, M.; Tajima, K.; Takai, K.; Hayashi, T.; Hall, G.; Garfunkel, E.; Osipov, V.; He, H. Dry microwave heating enables scalable fabrication of pristine holey graphene nanoplatelets and their catalysis in reductive hydrogen atom transfer reactions. Carbon 2018, 139, 861–871. [Google Scholar] [CrossRef]
- Lin, Y.; Han, X.; Campbell, C.J.; Kim, J.-W.; Zhao, B.; Luo, W.; Dai, J.; Hu, L.; Connell, J.W. Holey graphene nanomanufacturing: Structure, composition, and electrochemical properties. Adv. Funct. Mater. 2015, 25, 2920–2927. [Google Scholar] [CrossRef]
- Aguirre, M.E.; Rodríguez, H.B.; San Román, E.; Feldhoff, A.; Grela, M.A. Ag@ZnO core–shell nanoparticles formed by the timely reduction of Ag+ ions and zinc acetate hydrolysis in N,N-dimethylformamide: Mechanism of growth and photocatalytic properties. J. Phys. Chem. C 2011, 115, 24967–24974. [Google Scholar] [CrossRef]
- Wei, D.; Chen, L.; Tian, L.; Ramakrishna, S.; Ji, D. Zn single atoms/clusters/nanoparticles embedded in the hybrid carbon aerogels for high-performance ORR electrocatalysis. Inorg. Chem. 2023, 62, 16547–16553. [Google Scholar] [CrossRef]
- Liu, Y.-p.; Li, Y.-b.; Huang, D.-j.; Zhang, H.; Chu, K. ZnO quantum dots coupled with graphene toward electrocatalytic N2 reduction: Experimental and DFT investigations. Chem. Eur. J. 2019, 25, 11933–11939. [Google Scholar] [CrossRef]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of graphite oxide revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Zhang, D.; Yu, P.; Ma, Y. High performance supercapacitors based on reduced graphene oxide in aqueous and ionic liquid electrolytes. Carbon 2011, 49, 573–580. [Google Scholar] [CrossRef]
- Wen, X.; Deng, J.; Hu, S.; Chen, Y. High yield and high volumetric capacitance activated carbons by one-step homogeneous activation of diaphragma juglandis fructus for supercapacitors. ChemistrySelect 2023, 8, e202302002. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Yu, P.; Ma, Y. Stable dispersions of graphene and highly conducting graphene films: A new approach to creating colloids of graphene monolayers. Chem. Commun. 2009, 30, 4527–4529. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Zhang, L.; Chi, Y.; Li, Z.; Sun, X.; Gu, H.; Zhang, H.; Chen, Y.; Chen, G.Z. Effects of pore widening vs oxygenation on capacitance of activated carbon in aqueous sodium sulfate electrolyte. J. Electrochem. Soc. 2020, 167, 040524. [Google Scholar] [CrossRef]
- Chen, W.; Ji, J.; Feng, X.; Duan, X.; Qian, G.; Li, P.; Zhou, X.; Chen, D.; Yuan, W. Mechanistic insight into size-dependent activity and durability in Pt/CNT catalyzed hydrolytic dehydrogenation of ammonia borane. J. Am. Chem. Soc. 2014, 136, 16736–16739. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, X.; Kitta, M.; Xu, Q. Monodispersed Pt nanoparticles on reduced graphene oxide by a non-noble metal sacrificial approach for hydrolytic dehydrogenation of ammonia borane. Nano Res. 2017, 10, 3811–3816. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Sun, X.; Chen, Y. Exploring Enhanced Hydrolytic Dehydrogenation of Ammonia Borane with Porous Graphene-Supported Platinum Catalysts. Molecules 2024, 29, 1761. https://doi.org/10.3390/molecules29081761
Xu Z, Sun X, Chen Y. Exploring Enhanced Hydrolytic Dehydrogenation of Ammonia Borane with Porous Graphene-Supported Platinum Catalysts. Molecules. 2024; 29(8):1761. https://doi.org/10.3390/molecules29081761
Chicago/Turabian StyleXu, Zhenbo, Xiaolei Sun, and Yao Chen. 2024. "Exploring Enhanced Hydrolytic Dehydrogenation of Ammonia Borane with Porous Graphene-Supported Platinum Catalysts" Molecules 29, no. 8: 1761. https://doi.org/10.3390/molecules29081761
APA StyleXu, Z., Sun, X., & Chen, Y. (2024). Exploring Enhanced Hydrolytic Dehydrogenation of Ammonia Borane with Porous Graphene-Supported Platinum Catalysts. Molecules, 29(8), 1761. https://doi.org/10.3390/molecules29081761