Author Contributions
Conceptualization, Y.-Y.T.; methodology, Y.-Y.T.; software, J.-B.T.; validation, Y.-Y.T.; formal analysis, Y.T.; investigation, Y.L.; resources, Y.-Y.T. and J.-B.T.; data curation, Y.-Y.T.; writing—original draft preparation, Y.-Y.T. and Y.L.; writing—review and editing, Y.-Y.T. and Y.L.; visualization, J.-B.T. and Y.L.; supervision, J.-B.T.; project administration, Y.-Y.T. and Y.T.; funding acquisition, Y.-Y.T. and J.-B.T. All authors have read and agreed to the published version of the manuscript.
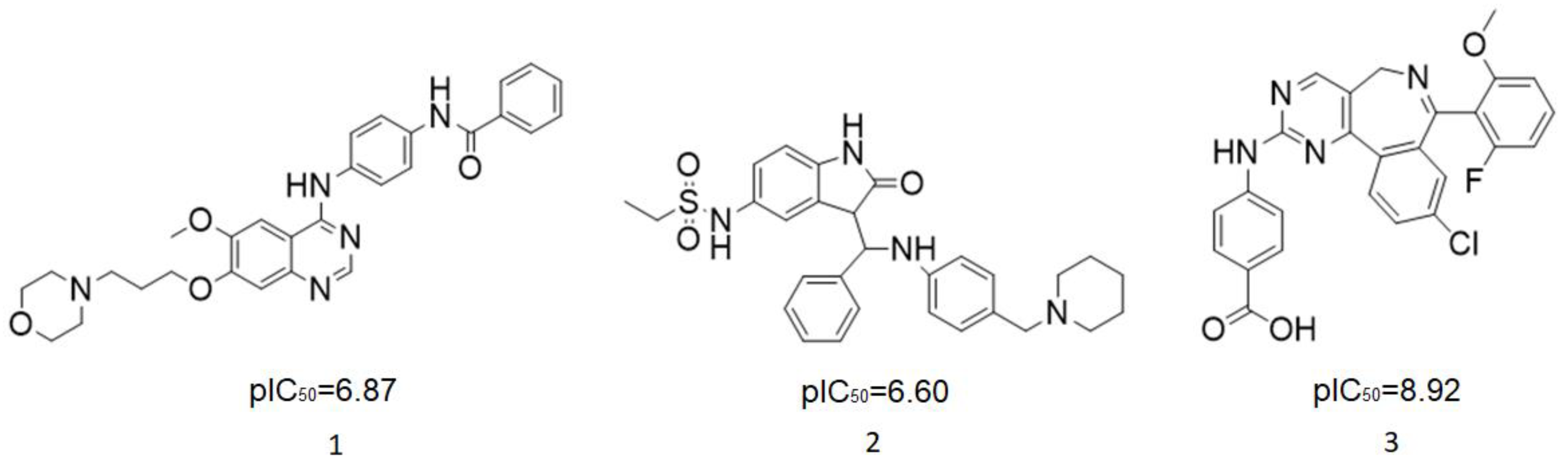
Figure 1.
Several Aurora kinase inhibitors that have entered clinical trials.
Figure 1.
Several Aurora kinase inhibitors that have entered clinical trials.
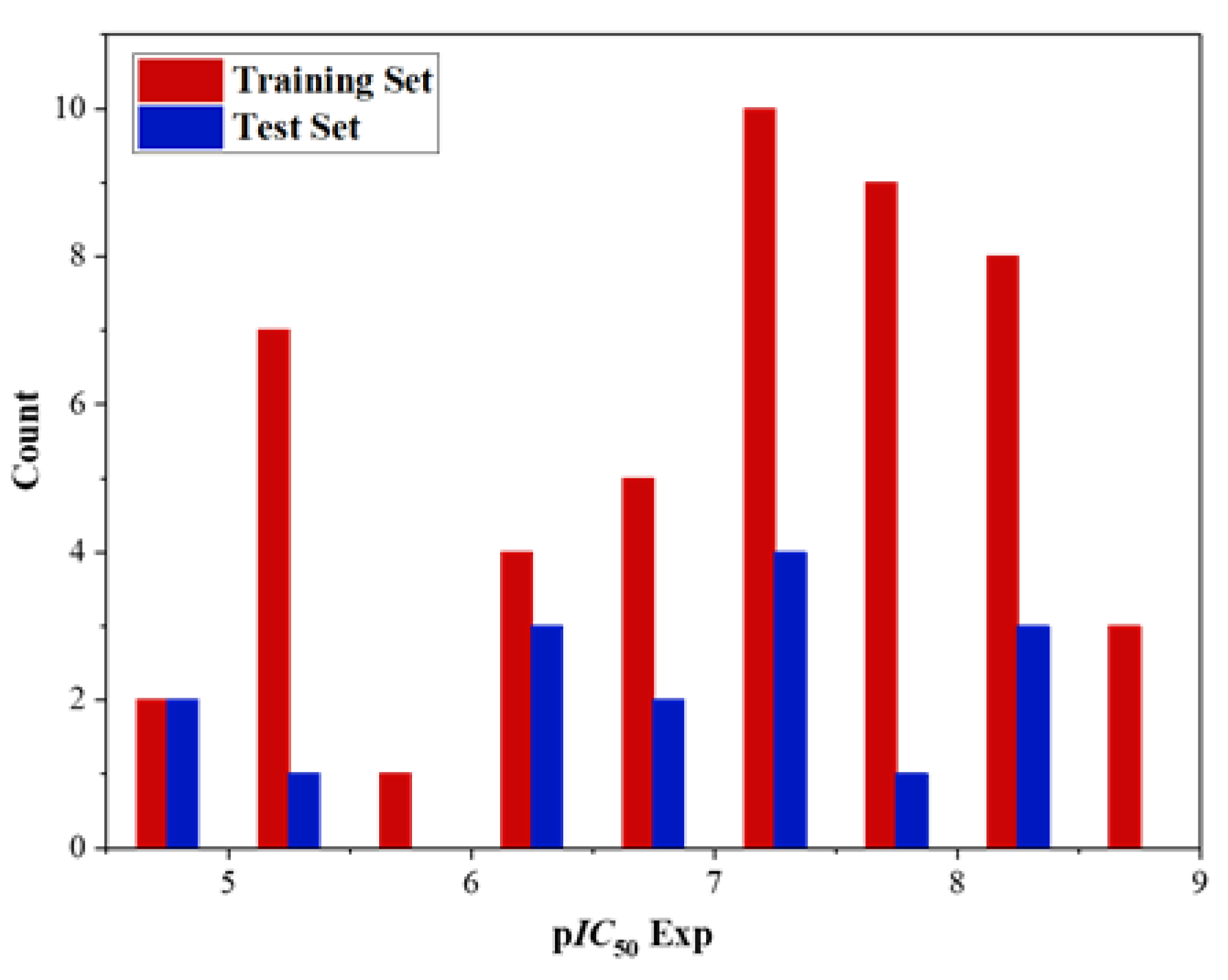
Figure 2.
Distribution of experimental inhibitory activities (pIC50) for the training and test sets compounds in the QSAR models.
Figure 2.
Distribution of experimental inhibitory activities (pIC50) for the training and test sets compounds in the QSAR models.
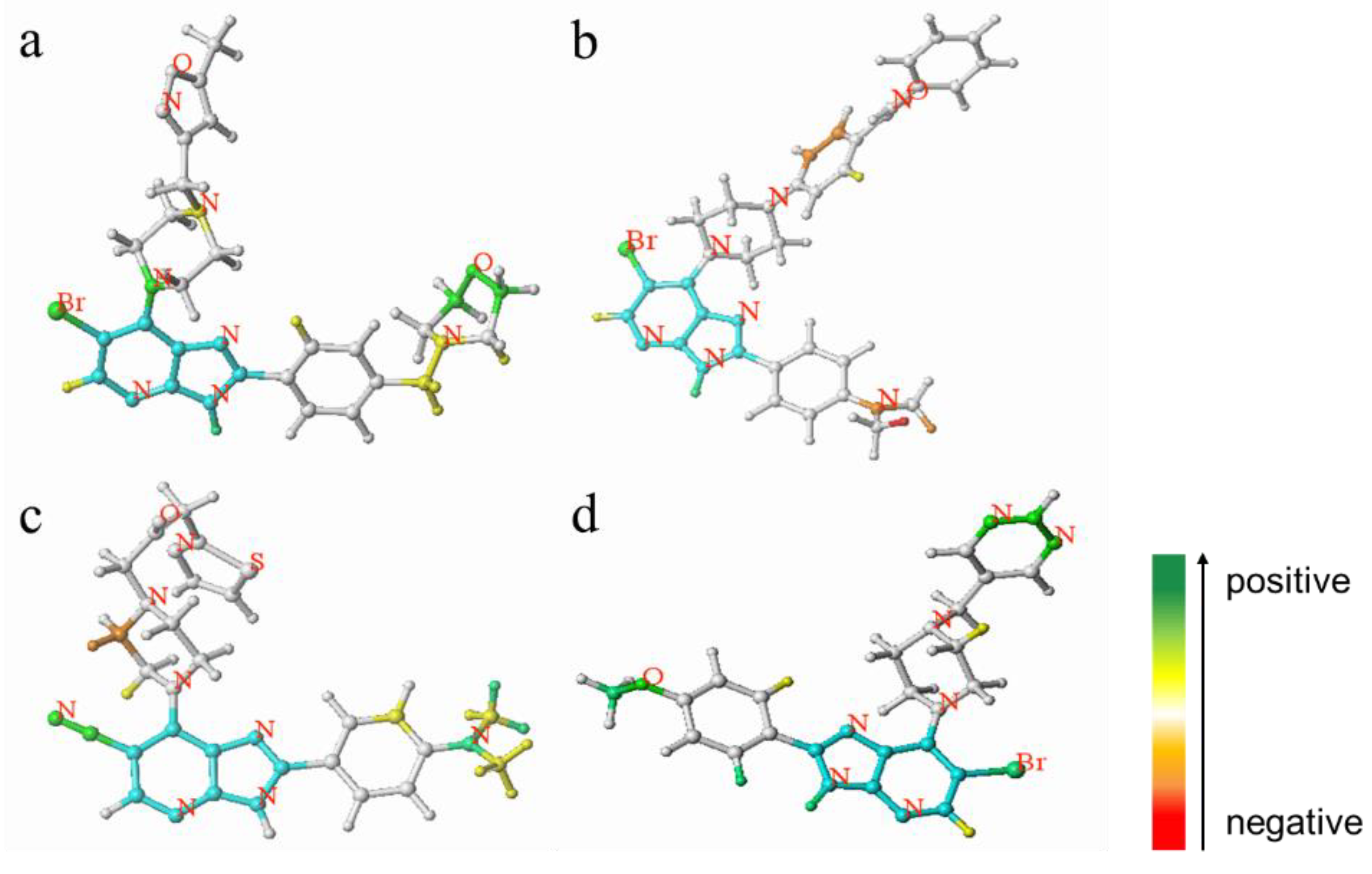
Figure 3.
Atomic contribution maps of compound 52 (a), compound 32 (b), compound 21 (c) and compound 40 (d). (Cyan indicates common backbone, green or yellow indicates positive contribution, orange and red indicate negative contribution, and white indicates intermediate contribution to biological activity).
Figure 3.
Atomic contribution maps of compound 52 (a), compound 32 (b), compound 21 (c) and compound 40 (d). (Cyan indicates common backbone, green or yellow indicates positive contribution, orange and red indicate negative contribution, and white indicates intermediate contribution to biological activity).
Figure 4.
Three-dimensional (3D) (a)spatial field and (b)static electric field action diagrams for the CoMFA model with compound 52 as the template molecule.
Figure 4.
Three-dimensional (3D) (a)spatial field and (b)static electric field action diagrams for the CoMFA model with compound 52 as the template molecule.
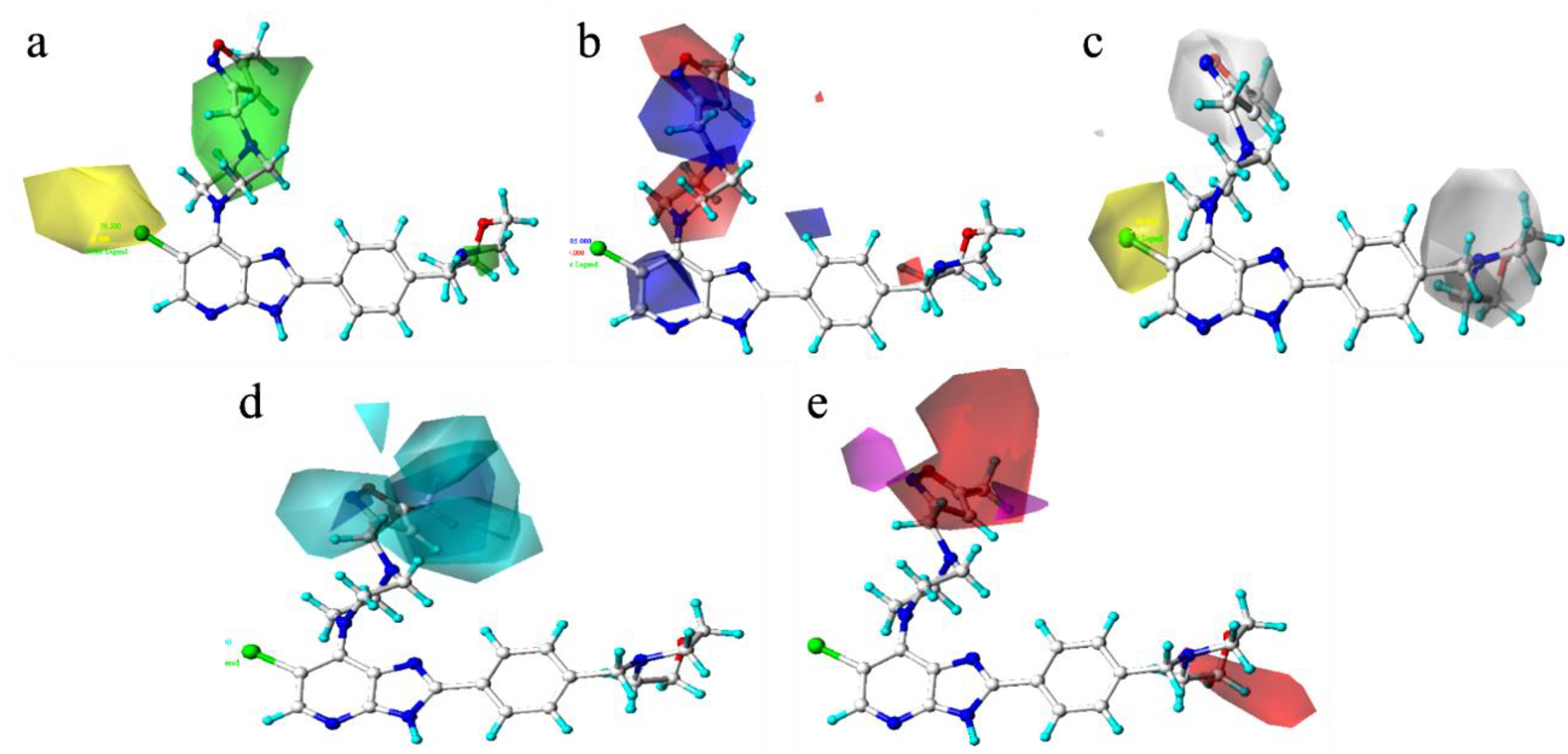
Figure 5.
Three-dimensional (3D) contours of the CoMSIA model with compound 52 as template ((a): spatial force, (b): electrostatic force, (c): hydrophobic force, (d): hydrogen bond donor force, (e): hydrogen bond acceptor force).
Figure 5.
Three-dimensional (3D) contours of the CoMSIA model with compound 52 as template ((a): spatial force, (b): electrostatic force, (c): hydrophobic force, (d): hydrogen bond donor force, (e): hydrogen bond acceptor force).
Figure 6.
Schematic representation of segmentation methods and fragments using compound 52 as a template molecule.
Figure 6.
Schematic representation of segmentation methods and fragments using compound 52 as a template molecule.
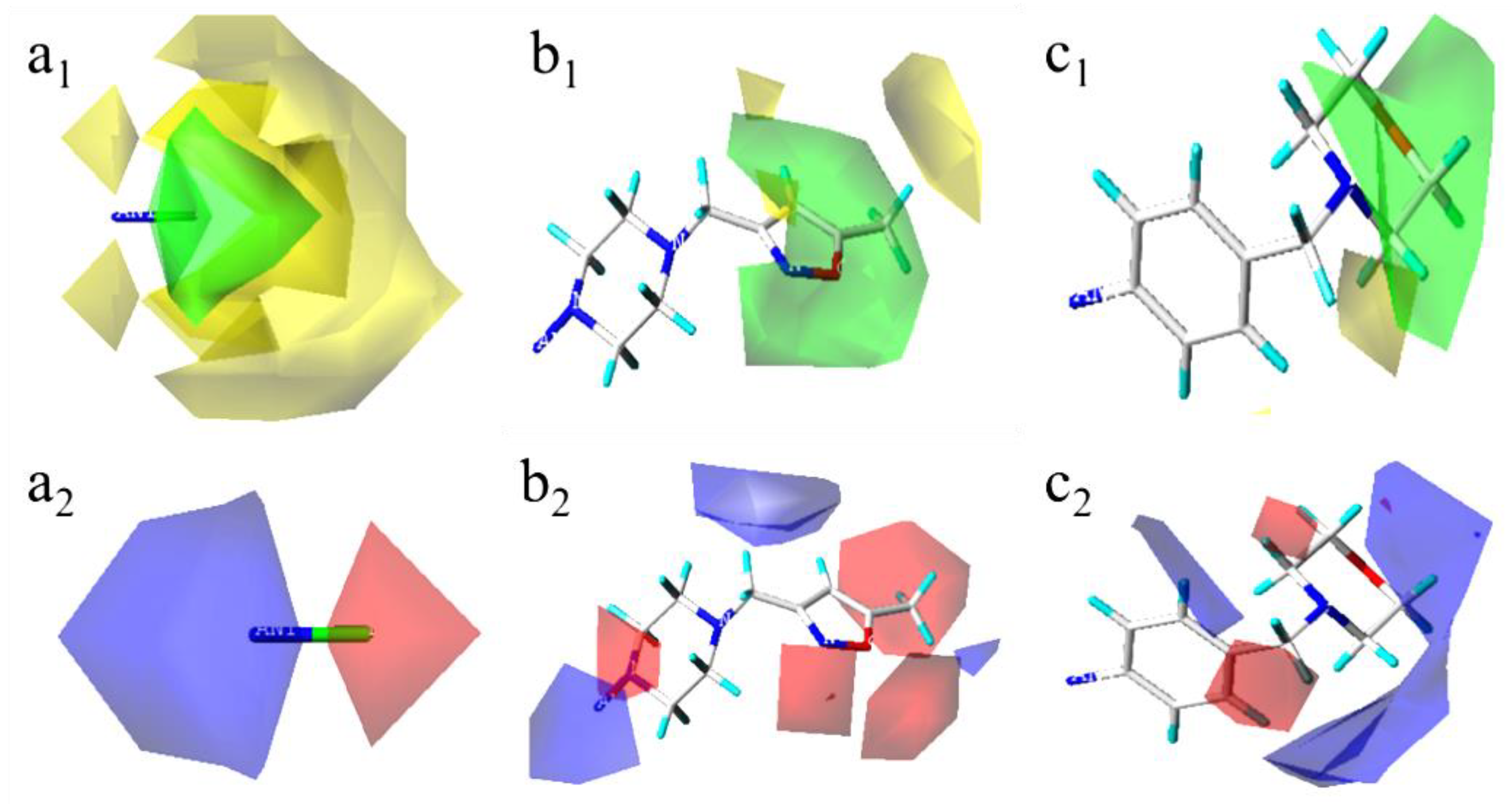
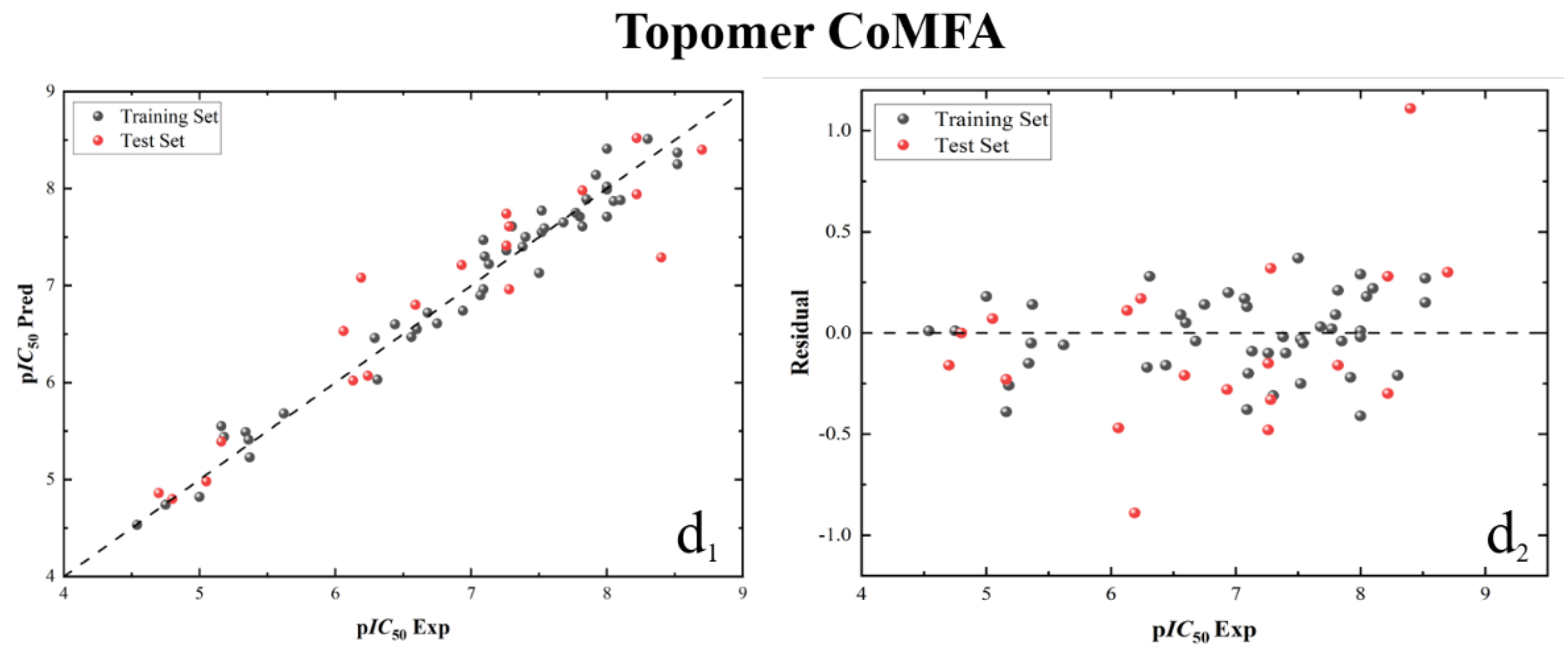
Figure 7.
Three-dimensional (3D) contour map of the Topomer CoMFA model based on template 52. (a1) the spatial contour map of Ra group; (a2) the electrostatic contour map of Ra group; (b1) the spatial contour map of Rb group; (b2) the electrostatic contour map of Rb group; (c1) the spatial contour map of Rc group; (c2) the electrostatic contour map of Rc group.
Figure 7.
Three-dimensional (3D) contour map of the Topomer CoMFA model based on template 52. (a1) the spatial contour map of Ra group; (a2) the electrostatic contour map of Ra group; (b1) the spatial contour map of Rb group; (b2) the electrostatic contour map of Rb group; (c1) the spatial contour map of Rc group; (c2) the electrostatic contour map of Rc group.
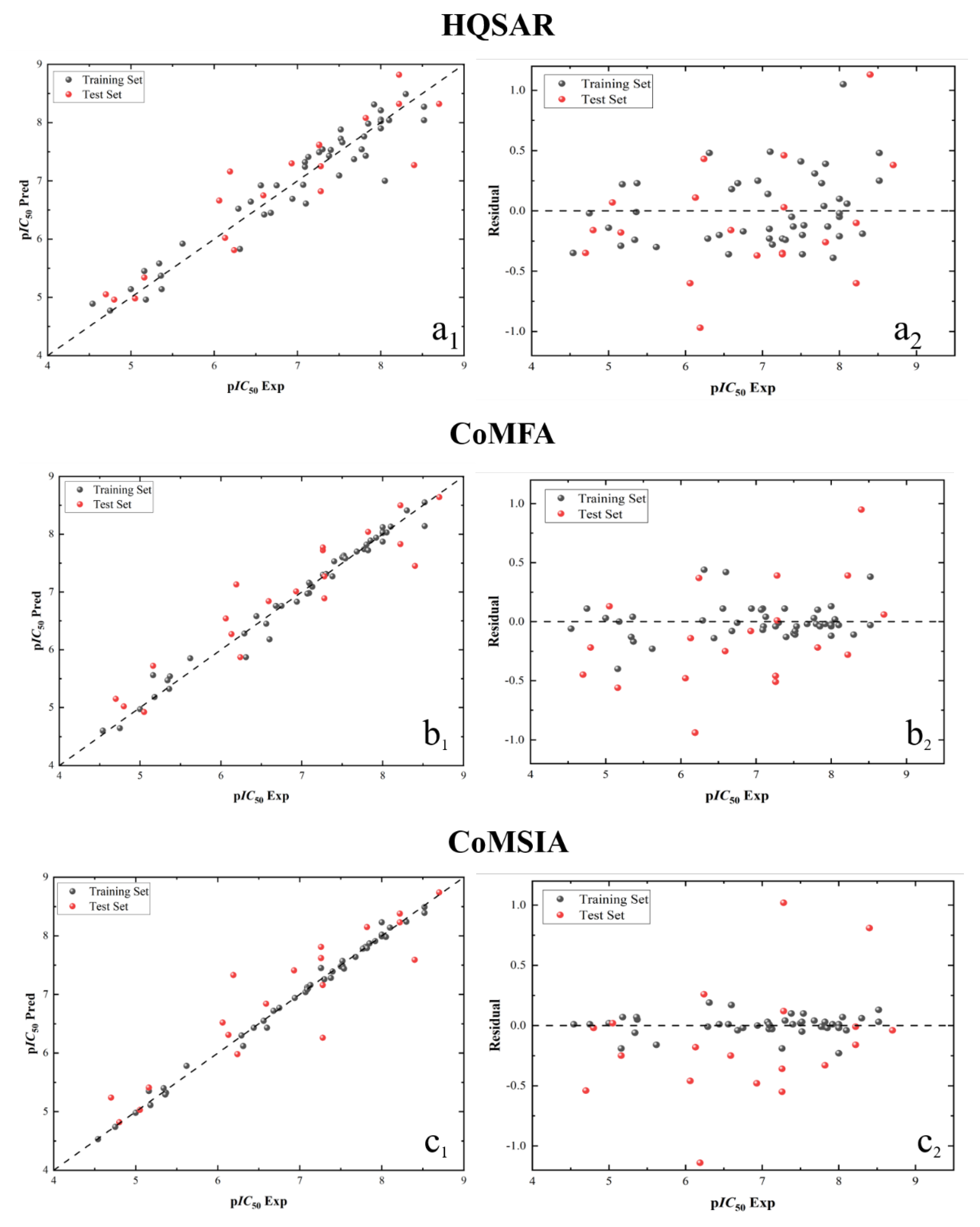
Figure 8.
Linear regression analysis diagram and residual analysis diagram of HQSAR (a1,a2), CoMFA (b1,b2), CoMSIA (c1,c2) and Topomer CoMFA (d1,d2) model results.
Figure 8.
Linear regression analysis diagram and residual analysis diagram of HQSAR (a1,a2), CoMFA (b1,b2), CoMSIA (c1,c2) and Topomer CoMFA (d1,d2) model results.
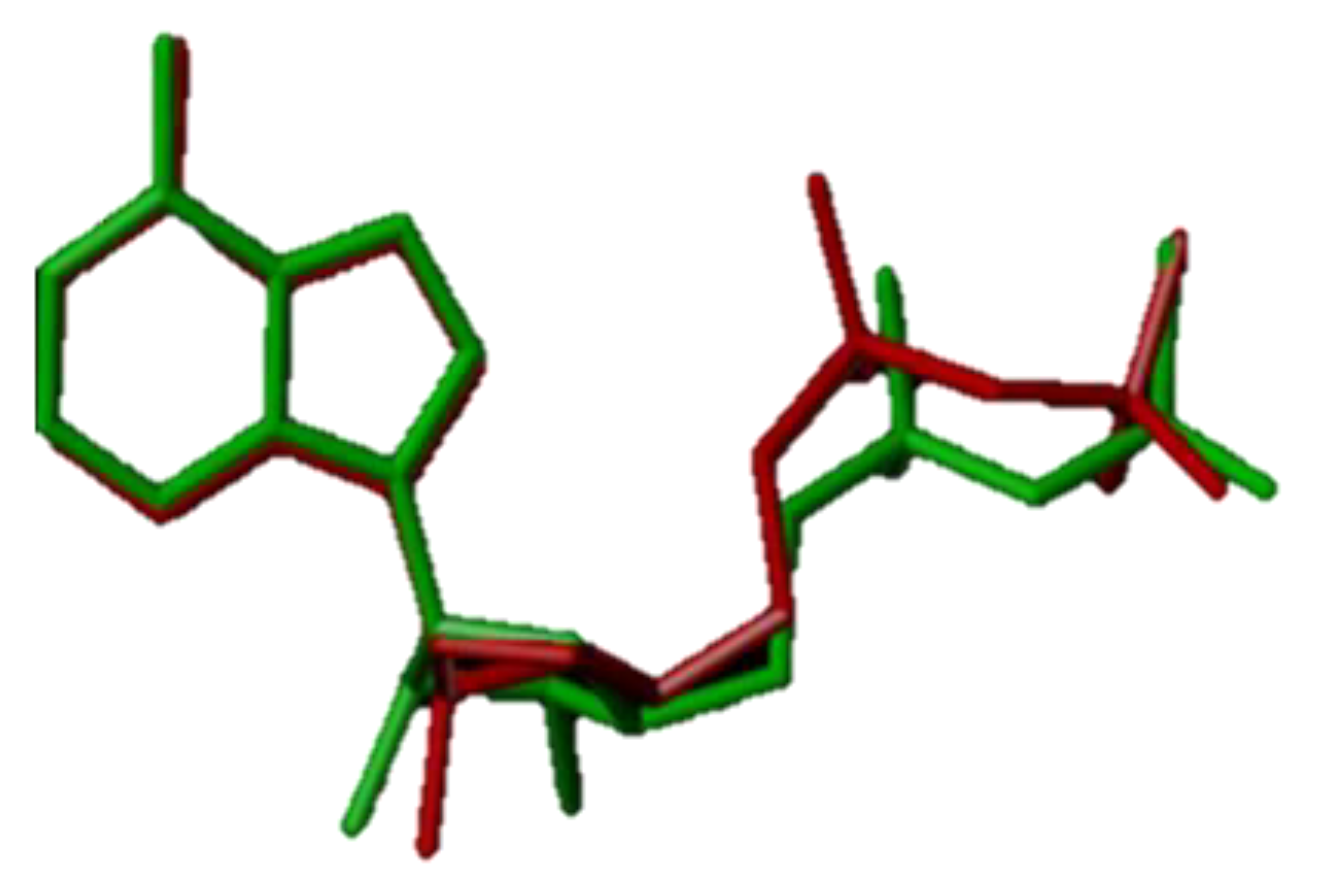
Figure 9.
Conformational superposition of docked ligand and extracted primary ligand (ADP). (The red bars in the figure represent the docked ligand and the green bars are the original ligand).
Figure 9.
Conformational superposition of docked ligand and extracted primary ligand (ADP). (The red bars in the figure represent the docked ligand and the green bars are the original ligand).
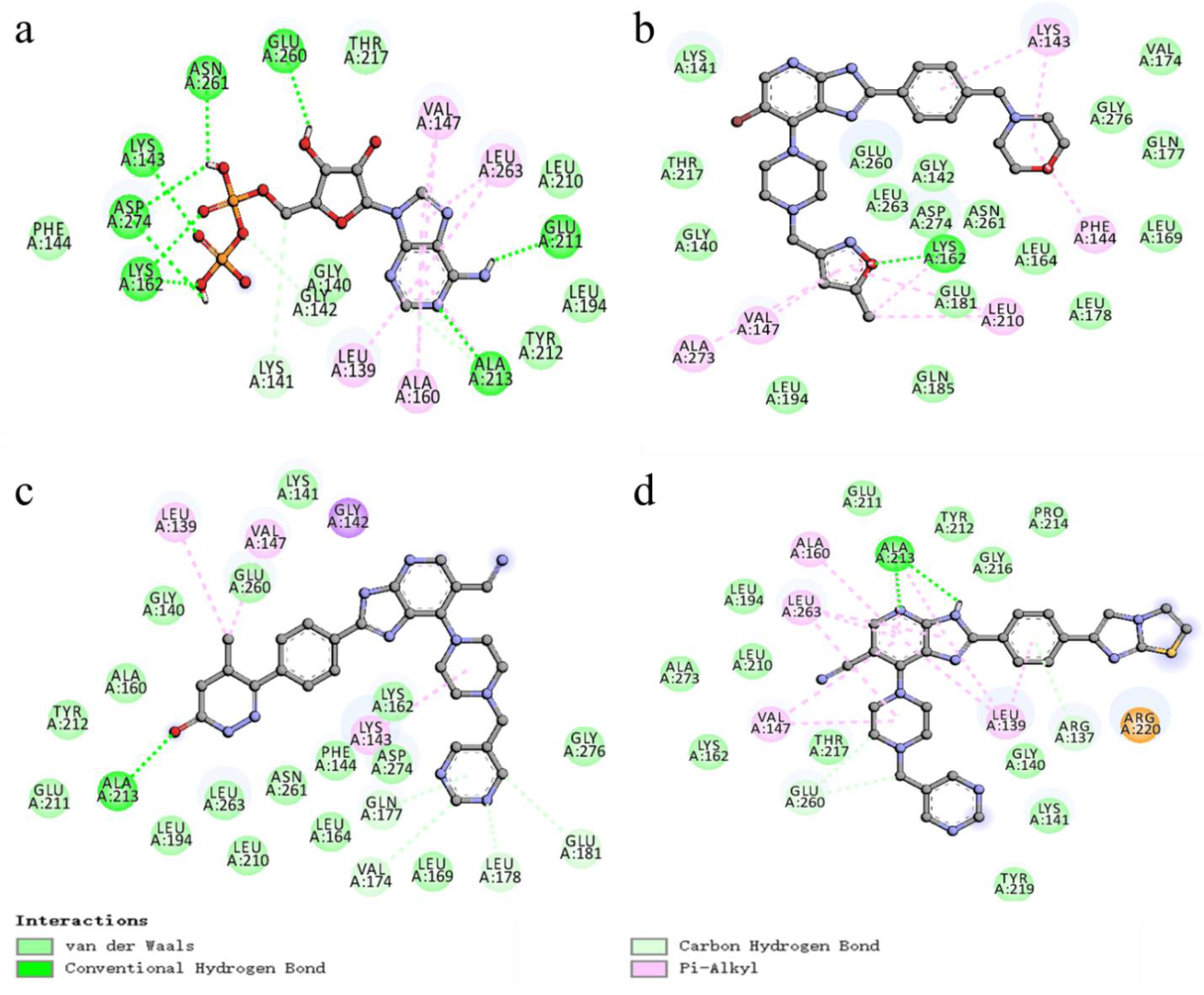
Figure 10.
Two-dimensional (2D) view of the binding conformation and interactions within the protein active site (PDB: 1MQ4). (a) original ligand ADP (b) compound 52 (c) compound N1 (d) compound N2.
Figure 10.
Two-dimensional (2D) view of the binding conformation and interactions within the protein active site (PDB: 1MQ4). (a) original ligand ADP (b) compound 52 (c) compound N1 (d) compound N2.
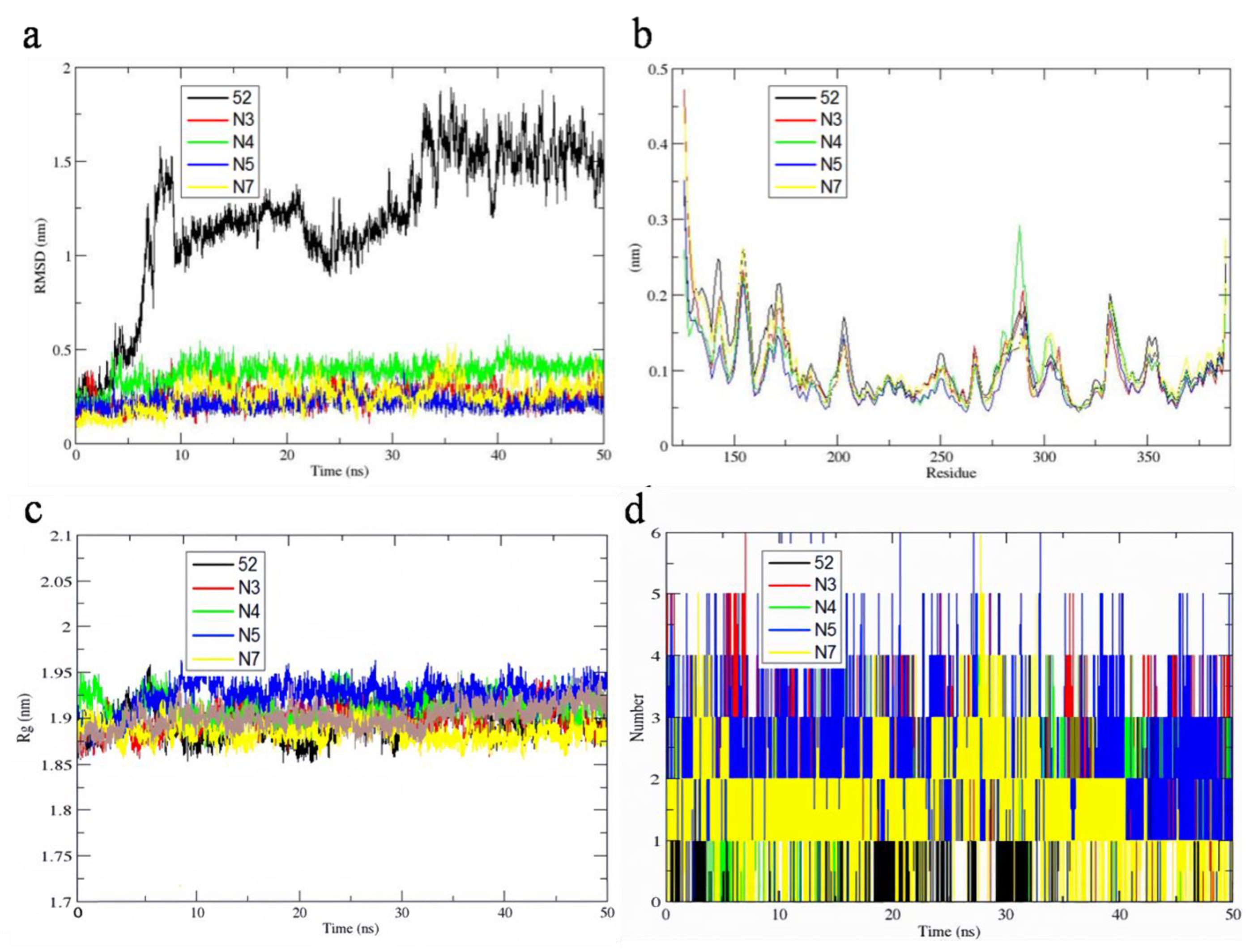
Figure 11.
RMSD values of ligands at the active site of 1MQ4 during 50 ns MD simulation (a). The RMSF values of chain A in five protein-ligand complexes (b). Radius of gyration values in five protein-ligand complexes (c). The hydrogen-bond numbers between 1MQ4 and the ligands (d).
Figure 11.
RMSD values of ligands at the active site of 1MQ4 during 50 ns MD simulation (a). The RMSF values of chain A in five protein-ligand complexes (b). Radius of gyration values in five protein-ligand complexes (c). The hydrogen-bond numbers between 1MQ4 and the ligands (d).
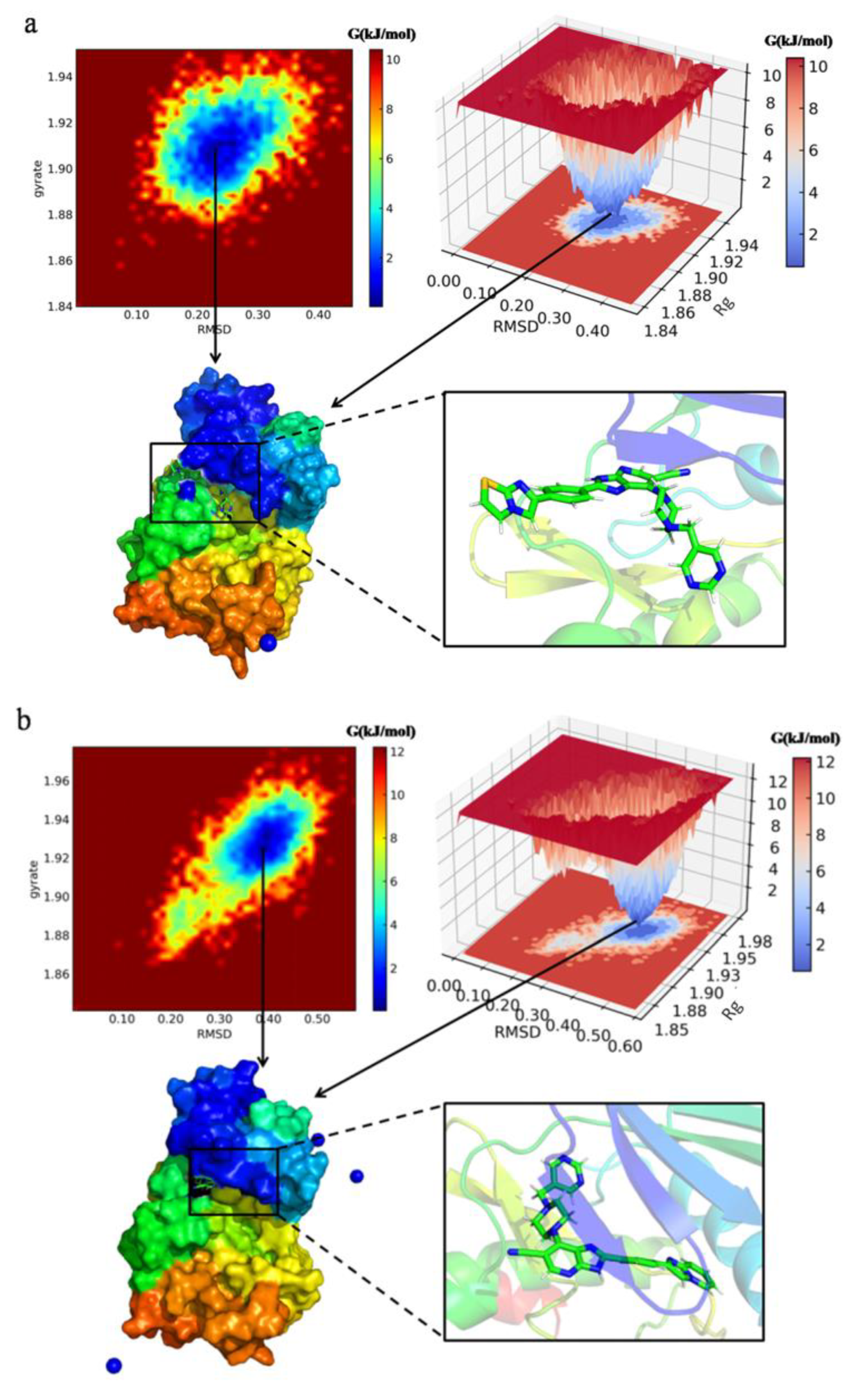
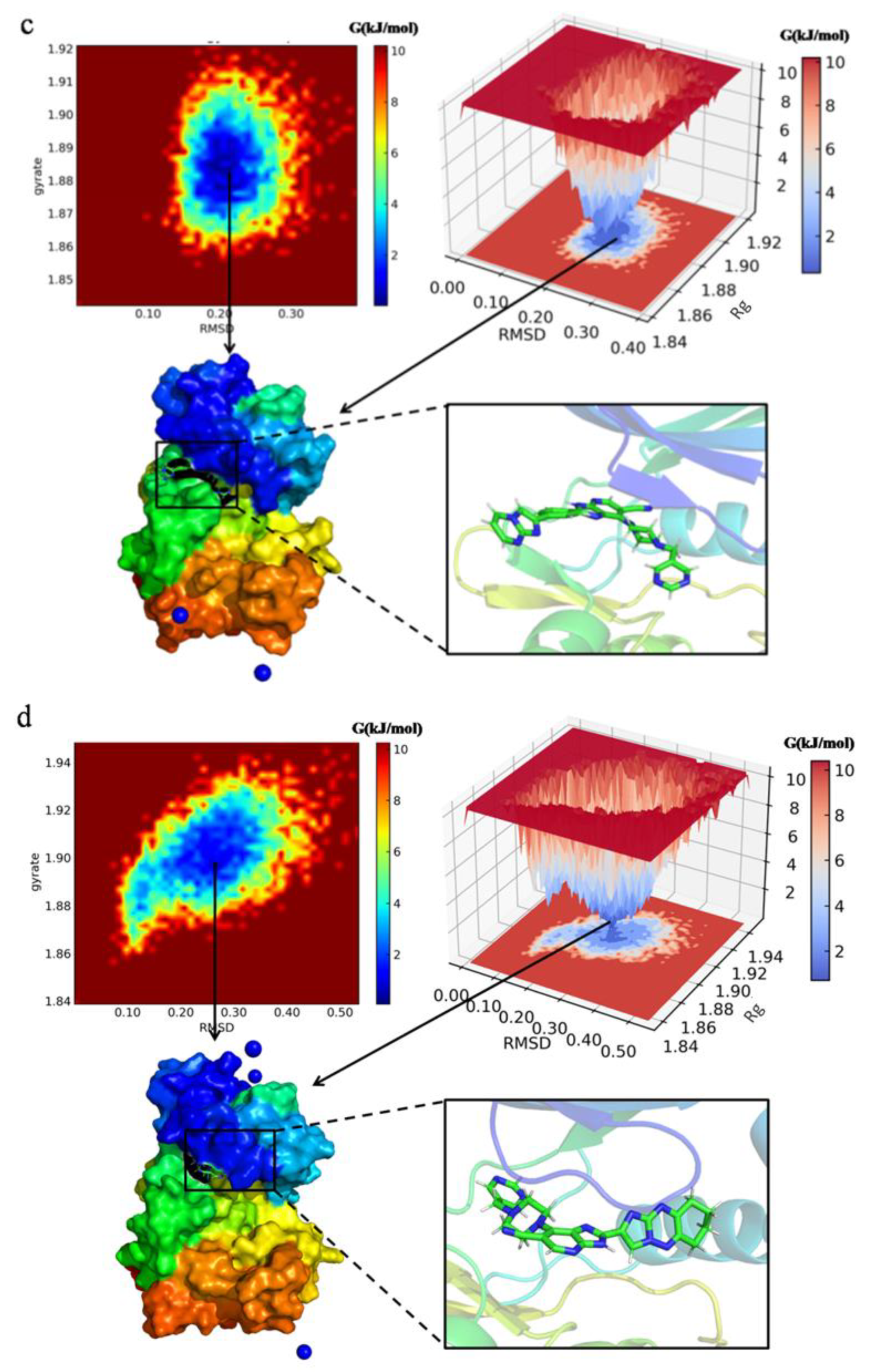
Figure 12.
Free energy landscape (FEL) plotted between RMSD and Rg coordinates, snapshot from lowest energy conformational state of the newly designed compounds ((a): N3, (b): N4, (c): N5, (d): N7) in complex with protein 1MQ4. The colored scale plot shows the free energy profile (kJ/mol), the red color region in plot represents the metastable state, and the dark blue region represents the lowest energy state.
Figure 12.
Free energy landscape (FEL) plotted between RMSD and Rg coordinates, snapshot from lowest energy conformational state of the newly designed compounds ((a): N3, (b): N4, (c): N5, (d): N7) in complex with protein 1MQ4. The colored scale plot shows the free energy profile (kJ/mol), the red color region in plot represents the metastable state, and the dark blue region represents the lowest energy state.
Table 1.
Compound structures and their activity values.
Table 1.
Compound structures and their activity values.
![Molecules 29 01772 i001]() |
|---|
| NO. | R1 | R2 | Experimental pIC50 |
|---|
| 1 | H | H | 5.37 |
| 2 * | Cl | H | 6.24 |
| 3 | Br | H | 6.31 |
| 4 | CF3 | H | 6.13 |
| 5 | Me | H | 5.16 |
| 6 * | 4-Methoxy-phenyl | H | 4.70 |
| 7 | Benzo[1,3]dioxole-5-yl | H | 4.75 |
| 8 | 3-Hydroxy-phenyl | H | 5.05 |
| 9 | Cl | Cl | 6.60 |
| 10 * | H | Me | 5.16 |
| 11 | H | Cl | 5.62 |
| 12 | H | m-Dimethylamino | 5.00 |
| 13 | H | p-Methoxy | 5.18 |
| 14 * | H | m-Methoxy | 4.80 |
| 15 | H | o-Methoxy | 4.54 |
| 16 | H | p-Pyrrolidin-1-yl | 5.34 |
| 17 | H | p-Pyrid-2-yl | 5.36 |
| 18 * | H | 4-Dimethylamino-phenyl | 6.06 |
| 19 | Cl | 4-Dimethylamino-phenyl | 7.38 |
| 20 | Br | 4-Dimethylamino-phenyl | 7.26 |
| 21 | CN | 4-Dimethylamino-phenyl | 7.30 |
| 22 * | Cyclopropyl | 4-Methoxy-phenyl | 7.28 |
| 23 | Br | H | 6.29 |
| 24 | Br | 4-Dimethylaminomethyl-phenyl | 7.80 |
| 25 | Br | Ph | 7.09 |
| 26 * | Cl | 4-Methoxy-phenyl | 7.28 |
| 27 | Cl | 4-Morpholin-4-ylmethyl-phenyl | 8.52 |
| 28 | Br | H | 6.44 |
| 29 | Cl | Phenylcarbamoylmethyl | 6.94 |
| 30 * | Br | Pyridin-3-ylcarbamoylmethyl | 6.93 |
| 31 | Cl | (3-Chloro-phenylcarbamoyl)-methyl | 7.13 |
| 32 | Br | Phenylcarbamoyl | 6.56 |
| 33 | Br | Benzenesulfonyl | 6.75 |
| 34 * | Br | Ph | 6.59 |
| 35 | Br | Isobutyl | 6.68 |
| 36 | Br | 1-Pyridin-4-yl-ethyl | 7.07 |
| 37 | Cl | 1-Phenyl-ethyl | 7.10 |
| 38 * | Br | Pyridin-4-ylmethyl | 7.26 |
| 39 | Br | Pyridin-3-ylmethyl | 8.05 |
| 40 | Br | Pyrimidin-5-ylmethyl | 8.10 |
| 41 | Br | 4-Chloro-benzyl | 7.40 |
| 42 * | Br | Cyclopropylmethyl | 6.19 |
| 43 | Br | 5-Methyl-isoxazol-3-ylmethyl | 7.77 |
| 44 | Br | 2-Ethyl-oxazol-4-ylmethyl | 7.09 |
| 45 | Br | 1-Methyl-4H-imidazol-2-ylmethyl | 7.50 |
| 46 * | Br | Thiazol-4-ylmethyl | 8.40 |
| 47 | Cl | Pyridin-4-ylmethyl | 7.68 |
| 48 | Br | Pyridin-4-ylmethyl | 7.82 |
| 49 | Br | Pyridin-3-ylmethyl | 8.30 |
| 50 * | Br | Pyrimidin-5-ylmethyl | 8.22 |
| 51 | Br | 4-Chloro-benzyl | 7.92 |
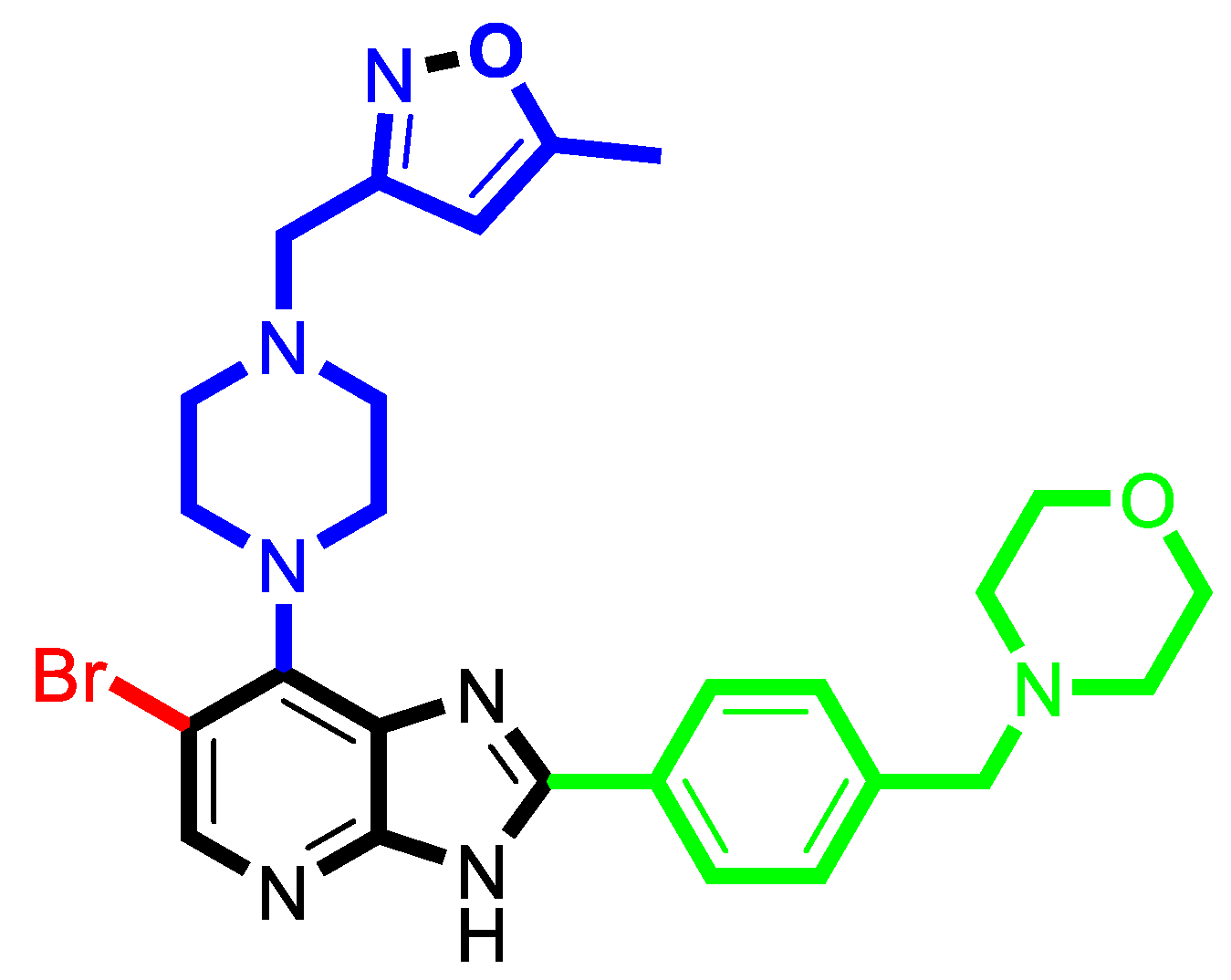
| 52 | Br | 5-Methyl-isoxazol-3-ylmethyl | 8.70 |
| 53 | Br | 1-Methyl-4H-imidazol-2-ylmethyl | 7.52 |
| 54 * | Br | Thiazol-4-ylmethyl | 8.22 |
| 55 | Br | Piperazin-1-ylmethyl | 8.00 |
| 56 | Br | Dimethylaminomethyl | 7.85 |
| 57 | Br | Aminomethyl | 8.00 |
| 58 * | Cl | Aminomethyl | 7.26 |
| 59 | Br | 2-Hydroxy-ethoxy | 7.52 |
| 60 | Cl | 2-Hydroxy-ethoxy | 7.54 |
| 61 | Br | Piperazin-1-yl | 8.00 |
| 62 * | Br | 4-Methyl-piperazin-1-yl | 7.82 |
| 63 | Cl | 4-Methyl-piperazin-1-yl | 8.00 |
| 64 | Br | Pyrazol-1-ylmethyl | 8.52 |
| 65 | Cl | Pyrazol-1-ylmethyl | 8.00 |
Table 2.
HQSAR model results using different fragment characteristic parameters for fixed holographic length (4–7).
Table 2.
HQSAR model results using different fragment characteristic parameters for fixed holographic length (4–7).
| Model | Fragment Distinction | Fragment Size | N | HL | q2 | r2 | SEE | SEEcv |
|---|
| 1-01 | A | 4–7 | 6 | 83 | 0.87 | 0.957 | 0.247 | 0.432 |
| 1-02 | B | 4–7 | 6 | 151 | 0.812 | 0.933 | 0.310 | 0.519 |
| 1-03 | C | 4–7 | 5 | 307 | 0.870 | 0.964 | 0.226 | 0.427 |
| 1-04 | A/B | 4–7 | 4 | 71 | 0.844 | 0.937 | 0.293 | 0.463 |
| 1-05 | A/C | 4–7 | 6 | 257 | 0.861 | 0.967 | 0.217 | 0.446 |
| 1-06 | A/H | 4–7 | 2 | 401 | 0.800 | 0.844 | 0.453 | 0.512 |
| 1-07 | A/CH | 4–7 | 6 | 151 | 0.872 | 0.961 | 0.237 | 0.428 |
| 1-08 | A/DA | 4–7 | 4 | 199 | 0.859 | 0.948 | 0.267 | 0.440 |
| 1-09 | CH/DA | 4–7 | 5 | 199 | 0.860 | 0.955 | 0.251 | 0.443 |
| 1-10 | A/B/C | 4–7 | 5 | 257 | 0.861 | 0.960 | 0.237 | 0.442 |
| 1-11 | A/B/H | 4–7 | 2 | 401 | 0.796 | 0.839 | 0.459 | 0.517 |
| 1-12 | A/B/CH | 4–7 | 6 | 307 | 0.864 | 0.968 | 0.214 | 0.441 |
| 1-13 | A/B/DA | 4–7 | 3 | 61 | 0.841 | 0.926 | 0.314 | 0.462 |
| 1-14 | A/C/H | 4–7 | 2 | 53 | 0.794 | 0.832 | 0.470 | 0.519 |
| 1-15 | A/C/CH | 4–7 | 6 | 257 | 0.865 | 0.968 | 0.215 | 0.440 |
| 1-16 | A/C/DA | 4–7 | 3 | 199 | 0.850 | 0.921 | 0.325 | 0.448 |
| 1-17 | A/H/CH | 4–7 | 2 | 151 | 0.803 | 0.846 | 0.449 | 0.508 |
| 1-18 | A/H/DA | 4–7 | 3 | 151 | 0.832 | 0.907 | 0.353 | 0.474 |
| 1-19 | A/B/C/H | 4–7 | 4 | 97 | 0.805 | 0.900 | 0.371 | 0.516 |
| 1-20 | A/B/C/CH | 4–7 | 6 | 97 | 0.860 | 0.965 | 0.223 | 0.448 |
| 1-21 | A/B/C/DA | 4–7 | 3 | 71 | 0.839 | 0.916 | 0.335 | 0.464 |
| 1-22 | A/B/H/CH | 4–7 | 2 | 401 | 0.801 | 0.844 | 0.453 | 0.510 |
| 1-23 | A/B/H/DA | 4–7 | 3 | 59 | 0.833 | 0.894 | 0.377 | 0.473 |
| 1-24 | A/B/CH/DA | 4–7 | 3 | 151 | 0.839 | 0.922 | 0.324 | 0.465 |
| 1-25 | A/C/CH/DA | 4–7 | 2 | 199 | 0.849 | 0.898 | 0.366 | 0.445 |
| 1-26 | A/H/CH/DA | 4–7 | 3 | 151 | 0.835 | 0.904 | 0.358 | 0.470 |
| 1-27 | A/B/C/H/CH | 4–7 | 4 | 97 | 0.814 | 0.901 | 0.369 | 0.505 |
| 1-28 | A/B/C/H/DA | 4–7 | 5 | 97 | 0.863 | 0.946 | 0.276 | 0.438 |
| 1-29 | A/B/C/CH/DA | 4–7 | 6 | 59 | 0.863 | 0.962 | 0.235 | 0.440 |
| 1-30 | A/B/H/CH/DA | 4–7 | 3 | 401 | 0.828 | 0.901 | 0.365 | 0.480 |
| 1-31 | A/C/H/CH/DA | 4–7 | 3 | 199 | 0.834 | 0.902 | 0.362 | 0.472 |
| 1-32 | A/B/C/H/CH/DA | 4–7 | 5 | 97 | 0.832 | 0.941 | 0.287 | 0.485 |
Table 3.
HQSAR model analysis with the same fragment type (A/CH) and different fragment length.
Table 3.
HQSAR model analysis with the same fragment type (A/CH) and different fragment length.
| Model | Fragment Distinction | Fragment Size | N | HL | q2 | r2 | SEE | SEEcv |
|---|
| 1-07 | A/CH | 4–7 | 6 | 151 | 0.872 | 0.961 | 0.237 | 0.428 |
| 2-01 | A/CH | 1–3 | 6 | 83 | 0.772 | 0.882 | 0.412 | 0.572 |
| 2-02 | A/CH | 1–4 | 6 | 257 | 0.885 | 0.952 | 0.262 | 0.406 |
| 2-03 | A/CH | 2–4 | 5 | 257 | 0.892 | 0.948 | 0.271 | 0.389 |
| 2-04 | A/CH | 2–5 | 5 | 401 | 0.874 | 0.945 | 0.277 | 0.420 |
| 2-05 | A/CH | 3–6 | 6 | 307 | 0.884 | 0.959 | 0.243 | 0.409 |
| 2-06 | A/CH | 5–8 | 4 | 151 | 0.855 | 0.933 | 0.302 | 0.446 |
| 2-07 | A/CH | 6–9 | 6 | 199 | 0.881 | 0.967 | 0.219 | 0.414 |
| 2-08 | A/CH | 7–10 | 6 | 151 | 0.859 | 0.956 | 0.251 | 0.450 |
| 2-09 | A/CH | 8–11 | 6 | 353 | 0.868 | 0.966 | 0.222 | 0.436 |
| 2-10 | A/CH | 9–12 | 5 | 401 | 0.858 | 0.956 | 0.248 | 0.446 |
Table 4.
Statistical Parameters of CoMFA and CoMSIA Models.
Table 4.
Statistical Parameters of CoMFA and CoMSIA Models.
| Model | Field | q2 | r2 | N | F | SEE | Contribution |
|---|
| CoMFA | S/E | 0.866 | 0.983 | 6 | 403.587 | 0.156 | 0.486/0.514 |
| 3-01 | S | 0.847 | 0.922 | 4 | 130.757 | 0.326 | 1 |
| 3-02 | E | 0.804 | 0.965 | 6 | 193.892 | 0.224 | 1 |
| 3-03 | H | 0.729 | 0.931 | 5 | 116.254 | 0.311 | 1 |
| 3-04 | D | 0.198 | 0.982 | 8 | 2.454 | 0.982 | 1 |
| 3-05 | A | 0.490 | 0.728 | 3 | 40.052 | 0.604 | 1 |
| 3-06 | S/E | 0.864 | 0.965 | 5 | 234.646 | 0.223 | 0.372/0.628 |
| 3-07 | S/H | 0.833 | 0.953 | 4 | 222.533 | 0.254 | 0.487/0.513 |
| 3-08 | S/D | 0.841 | 0.939 | 5 | 132.909 | 0.292 | 0.828/0.172 |
| 3-09 | S/A | 0.846 | 0.939 | 5 | 131.767 | 0.293 | 0.535/0.465 |
| 3-10 | E/H | 0.831 | 0.982 | 6 | 380.156 | 0.161 | 0.610/0.390 |
| 3-11 | E/D | 0.819 | 0.977 | 6 | 301.906 | 0.18 | 0.840/0.160 |
| 3-12 | E/A | 0.815 | 0.952 | 5 | 169.461 | 0.26 | 0.672/0.328 |
| 3-13 | H/D | 0.722 | 0.974 | 8 | 190.455 | 0.196 | 0.838/0.162 |
| 3-14 | H/A | 0.810 | 0.986 | 8 | 361.979 | 0.143 | 0.538/0.462 |
| 3-15 | D/A | 0.482 | 0.774 | 3 | 51.450 | 0.55 | 0.222/0.778 |
| 3-16 | S/E/H | 0.867 | 0.977 | 5 | 364.504 | 0.18 | 0.263/0.458/0.279 |
| 3-17 | S/E/D | 0.855 | 0.974 | 5 | 319.939 | 0.192 | 0.321/0.545/0.134 |
| 3-18 | S/E/A | 0.859 | 0.970 | 6 | 227.267 | 0.207 | 0.276/0.453/0.271 |
| 3-19 | S/H/D | 0.833 | 0.985 | 9 | 283.698 | 0.153 | 0.424/0.450/0.127 |
| 3-20 | S/H/A | 0.852 | 0.966 | 5 | 241.583 | 0.22 | 0.332/0.321/0.346 |
| 3-21 | S/D/A | 0.863 | 0.958 | 7 | 132.490 | 0.25 | 0.489/0.125/0.386 |
| 3-22 | E/H/D | 0.832 | 0.994 | 8 | 857.566 | 0.093 | 0.538/0.341/0.122 |
| 3-23 | E/H/A | 0.839 | 0.982 | 6 | 381.917 | 0.161 | 0.443/0.290/0.266 |
| 3-24 | H/D/A | 0.839 | 0.989 | 8 | 466.395 | 0.126 | 0.476/0.104/0.421 |
| 3-25 | S/E/H/D | 0.860 | 0.986 | 5 | 589.859 | 0.142 | 0.236/0.398/0.256/0.110 |
| 3-26 | S/E/H/A | 0.867 | 0.984 | 6 | 427.260 | 0.152 | 0.208/0.346/0.231/0.215 |
| 3-27 | S/E/D/A | 0.871 | 0.985 | 9 | 283.928 | 0.152 | 0.241/0.419/0.098/0.242 |
| 3-28 | S/H/D/A | 0.865 | 0.991 | 8 | 525.498 | 0.119 | 0.286/0.311/0.112/0.291 |
| 3-29 | E/H/D/A | 0.860 | 0.994 | 8 | 802.661 | 0.097 | 0.403/0.273/0.095/0.229 |
| 3-30 | S/E/H/D/A | 0.877 | 0.995 | 9 | 802.161 | 0.091 | 0.183/0.316/0.221/0.089/0.192 |
Table 5.
Results of the Topomer CoMFA model.
Table 5.
Results of the Topomer CoMFA model.
| N | q2 | r2 | q2 Stderr | r2 Stderr | F | SEE |
|---|
| 6 | 0.905 | 0.971 | 0.36 | 0.20 | 369.402 | 0.199 |
Table 6.
Comparison of predicted activity value and residual value of HQSAR, CoMFA, CoMSIA and Topomer CoMFA models.
Table 6.
Comparison of predicted activity value and residual value of HQSAR, CoMFA, CoMSIA and Topomer CoMFA models.
| NO. | pIC50 Exp | HQSAR | COMFA | CoMSIA | Tomoper |
|---|
| pIC50 Pred | Residual | pIC50 Pred | Residual | pIC50 Pred | Residual | pIC50 Pred | Residual |
|---|
| 01 | 5.37 | 5.14 | 0.23 | 5.54 | −0.17 | 5.32 | 0.05 | 5.23 | 0.14 |
| 02 * | 6.24 | 5.81 | 0.43 | 5.87 | 0.37 | 5.98 | 0.27 | 6.07 | 0.17 |
| 03 | 6.31 | 5.83 | 0.48 | 5.87 | 0.44 | 6.12 | 0.19 | 6.03 | 0.28 |
| 04 * | 6.13 | 6.02 | 0.11 | 6.27 | −0.14 | 6.31 | −0.18 | 6.02 | 0.11 |
| 05 | 5.16 | 5.45 | −0.29 | 5.56 | −0.40 | 5.35 | −0.19 | 5.55 | −0.39 |
| 06 * | 4.70 | 5.05 | −0.35 | 5.15 | −0.45 | 5.24 | −0.54 | 4.86 | −0.16 |
| 07 | 4.75 | 4.77 | −0.03 | 4.64 | 0.11 | 4.74 | 0.01 | 4.74 | 0.00 |
| 08 * | 5.05 | 4.98 | 0.07 | 4.92 | 0.12 | 5.03 | 0.02 | 4.98 | 0.07 |
| 09 | 6.60 | 6.42 | 0.18 | 6.18 | 0.42 | 6.43 | 0.17 | 6.55 | 0.05 |
| 10 * | 5.16 | 5.34 | −0.19 | 5.72 | −0.56 | 5.41 | −0.26 | 5.39 | −0.23 |
| 11 | 5.62 | 5.92 | −0.30 | 5.85 | −0.23 | 5.78 | −0.16 | 5.68 | −0.06 |
| 12 | 5.00 | 5.14 | −0.14 | 4.97 | 0.03 | 4.98 | 0.02 | 4.82 | 0.18 |
| 13 | 5.18 | 4.96 | 0.22 | 5.18 | 0.00 | 5.11 | 0.07 | 5.44 | −0.26 |
| 14 * | 4.80 | 4.96 | −0.16 | 5.02 | −0.22 | 4.82 | −0.02 | 4.80 | 0.00 |
| 15 | 4.54 | 4.89 | −0.35 | 4.60 | −0.06 | 4.53 | 0.01 | 4.53 | 0.01 |
| 16 | 5.34 | 5.58 | −0.24 | 5.47 | −0.13 | 5.40 | −0.06 | 5.49 | −0.15 |
| 17 | 5.36 | 5.37 | −0.01 | 5.32 | 0.04 | 5.29 | 0.06 | 5.41 | −0.05 |
| 18 * | 6.06 | 6.66 | −0.60 | 6.54 | −0.48 | 6.52 | −0.46 | 6.53 | −0.47 |
| 19 | 7.38 | 7.43 | −0.05 | 7.27 | 0.11 | 7.28 | 0.09 | 7.40 | −0.02 |
| 20 | 7.26 | 7.49 | −0.23 | 7.30 | −0.04 | 7.45 | −0.19 | 7.36 | −0.10 |
| 21 | 7.30 | 7.54 | −0.24 | 7.31 | −0.01 | 7.26 | 0.04 | 7.61 | −0.31 |
| 22 * | 7.28 | 6.82 | 0.46 | 6.89 | 0.39 | 6.26 | 1.02 | 6.96 | 0.32 |
| 23 | 6.29 | 6.52 | −0.23 | 6.28 | 0.01 | 6.30 | −0.01 | 6.46 | −0.17 |
| 24 | 7.80 | 7.76 | 0.04 | 7.82 | −0.03 | 7.81 | −0.01 | 7.71 | 0.09 |
| 25 | 7.09 | 7.24 | −0.14 | 6.98 | 0.11 | 7.12 | −0.03 | 6.96 | 0.13 |
| 26 * | 7.28 | 7.25 | 0.03 | 7.27 | 0.01 | 7.16 | 0.12 | 7.61 | −0.33 |
| 27 | 8.52 | 8.04 | 0.49 | 8.55 | −0.03 | 8.49 | 0.04 | 8.25 | 0.27 |
| 28 | 6.44 | 6.64 | −0.21 | 6.58 | −0.14 | 6.43 | 0.01 | 6.60 | −0.16 |
| 29 | 6.94 | 6.69 | 0.25 | 6.83 | 0.11 | 6.94 | 0.00 | 6.74 | 0.20 |
| 30 * | 6.93 | 7.30 | −0.37 | 7.01 | −0.08 | 7.41 | −0.48 | 7.21 | −0.28 |
| 31 | 7.13 | 7.41 | −0.29 | 7.09 | 0.04 | 7.16 | −0.03 | 7.22 | −0.09 |
| 32 | 6.56 | 6.92 | −0.36 | 6.45 | 0.11 | 6.55 | 0.00 | 6.47 | 0.09 |
| 33 | 6.75 | 6.92 | −0.17 | 6.76 | −0.01 | 6.77 | −0.02 | 6.61 | 0.14 |
| 34 * | 6.59 | 6.75 | −0.16 | 6.84 | −0.25 | 6.84 | −0.25 | 6.80 | −0.21 |
| 35 | 6.68 | 6.45 | 0.23 | 6.76 | −0.08 | 6.72 | −0.04 | 6.72 | −0.04 |
| 36 | 7.07 | 6.93 | 0.14 | 6.97 | 0.11 | 7.04 | 0.03 | 6.90 | 0.17 |
| 37 | 7.10 | 6.61 | 0.49 | 7.14 | −0.04 | 7.10 | 0.00 | 7.30 | −0.20 |
| 38 * | 7.26 | 7.61 | −0.35 | 7.72 | −0.46 | 7.81 | −0.55 | 7.41 | −0.15 |
| 39 | 8.05 | 7.00 | 1.05 | 8.03 | 0.01 | 7.98 | 0.06 | 7.87 | 0.18 |
| 40 | 8.10 | 8.04 | 0.06 | 8.13 | −0.04 | 8.14 | −0.04 | 7.88 | 0.22 |
| 41 | 7.40 | 7.53 | −0.13 | 7.53 | −0.13 | 7.39 | 0.01 | 7.50 | −0.10 |
| 42 * | 6.19 | 7.16 | −0.98 | 7.13 | −0.94 | 7.33 | −1.14 | 7.08 | −0.89 |
| 43 | 7.77 | 7.54 | 0.23 | 7.74 | 0.03 | 7.78 | −0.01 | 7.75 | 0.02 |
| 44 | 7.09 | 7.32 | −0.23 | 7.16 | −0.07 | 7.08 | 0.02 | 7.47 | −0.38 |
| 45 | 7.50 | 7.09 | 0.40 | 7.60 | −0.11 | 7.48 | 0.02 | 7.13 | 0.37 |
| 46 * | 8.40 | 7.27 | 1.13 | 7.45 | 0.95 | 7.59 | 0.81 | 7.29 | 1.11 |
| 47 | 7.68 | 7.37 | 0.31 | 7.70 | −0.03 | 7.64 | 0.04 | 7.65 | 0.03 |
| 48 | 7.82 | 7.43 | 0.39 | 7.72 | 0.11 | 7.79 | 0.04 | 7.61 | 0.21 |
| 49 | 8.30 | 8.49 | −0.19 | 8.41 | −0.11 | 8.24 | 0.06 | 8.51 | −0.21 |
| 50 * | 8.22 | 8.82 | −0.60 | 8.50 | −0.28 | 8.38 | −0.16 | 8.52 | −0.30 |
| 51 | 7.92 | 8.31 | −0.39 | 7.94 | −0.02 | 7.91 | 0.02 | 8.14 | −0.22 |
| 52 * | 8.70 | 8.32 | 0.38 | 8.64 | 0.06 | 8.74 | −0.04 | 8.40 | 0.30 |
| 53 | 7.52 | 7.88 | −0.35 | 7.63 | −0.10 | 7.49 | 0.03 | 7.77 | −0.25 |
| 54 * | 8.22 | 8.32 | −0.10 | 7.83 | 0.39 | 8.23 | −0.01 | 7.94 | 0.28 |
| 55 | 8.00 | 8.05 | −0.05 | 8.04 | −0.04 | 8.02 | −0.02 | 7.99 | 0.01 |
| 56 | 7.85 | 7.98 | −0.13 | 7.89 | −0.04 | 7.87 | −0.02 | 7.89 | −0.04 |
| 57 | 8.00 | 7.90 | 0.10 | 7.87 | 0.13 | 7.99 | 0.01 | 7.71 | 0.29 |
| 58 * | 7.26 | 7.62 | −0.36 | 7.77 | −0.51 | 7.62 | −0.36 | 7.74 | −0.48 |
| 59 | 7.52 | 7.72 | −0.20 | 7.60 | −0.07 | 7.57 | −0.04 | 7.55 | −0.03 |
| 60 | 7.54 | 7.66 | −0.12 | 7.58 | −0.04 | 7.44 | 0.09 | 7.59 | −0.05 |
| 61 | 8.00 | 8.05 | −0.05 | 8.02 | −0.02 | 8.02 | −0.02 | 7.99 | 0.01 |
| 62 * | 7.82 | 8.08 | −0.25 | 8.04 | −0.22 | 8.15 | −0.33 | 7.98 | −0.16 |
| 63 | 8.00 | 8.02 | −0.02 | 8.03 | −0.03 | 8.02 | −0.02 | 8.02 | −0.02 |
| 64 | 8.52 | 8.27 | 0.25 | 8.14 | 0.38 | 8.39 | 0.14 | 8.37 | 0.15 |
| 65 | 8.00 | 8.21 | −0.21 | 8.12 | −0.12 | 8.23 | −0.23 | 8.41 | −0.41 |
Table 7.
External verification of the QSAR Model.
Table 7.
External verification of the QSAR Model.
| Parameters | Criterion | HQSAR | COMFA | CoMSIA | Topomer |
|---|
| | 0.814 | 0.829 | 0.758 | 0.855 |
| k | 0.85 < k < 1.15 | 0.977 | 0.980 | 0.978 | 0.984 |
| k′ | 0.85 < k′ < 1.15 | 1.018 | 1.015 | 1.017 | 1.012 |
| | 0.828 | 0.838 | 0.803 | 0.860 |
| | 0.807 | 0.779 | 0.770 | 0.836 |
| | 0.610 | 0.609 | 0.559 | 0.647 |
| | 0.584 | 0.539 | 0.523 | 0.615 |
| | 0.026 | 0.070 | 0.036 | 0.033 |
| RMSE | RMSE → 0 | 0.501 | 0.480 | 0.530 | 0.443 |
| MAE | MAE → 0 | 0.543 | 0.547 | 0.565 | 0.462 |
| CCC | CCC > 0.85 | 0.902 | 0.902 | 0.888 | 0.922 |
Table 8.
Molecular structure of the new compounds and activity value prediction using the Topomer CoMFA model.
Table 9.
Molecular docking results and scoring function.
Table 9.
Molecular docking results and scoring function.
| Molecule | pIC50 Pred | Contribution Value | Scoring Function |
|---|
| Ra | Rb | Rc | Total-Score | Crash | Polar |
|---|
| Template | 8.70 | 0.80 | 1.54 | 1.77 | 6.3210 | −1.7537 | 1.2747 |
| N1 | 9.27 | 1.08 | 1.68 | 2.20 | 8.1293 | −2.0803 | 0.5836 |
| N2 | 9.23 | 1.08 | 1.68 | 2.14 | 5.4437 | −1.6456 | 1.1618 |
| N3 | 9.18 | 1.08 | 1.68 | 2.10 | 7.8842 | −1.4843 | 2.2739 |
| N4 | 9.14 | 1.08 | 1.68 | 2.05 | 7.2484 | −2.5719 | 2.4922 |
| N5 | 9.11 | 1.08 | 1.68 | 2.02 | 7.1447 | −1.5927 | 1.1425 |
| N6 | 9.04 | 1.08 | 1.68 | 1.95 | 4.6770 | −2.6022 | 1.1778 |
| N7 | 9.02 | 1.08 | 1.68 | 1.93 | 7.5526 | −2.0592 | 2.3005 |
| N8 | 8.98 | 1.08 | 1.68 | 1.92 | 5.8277 | −3.2544 | 1.9236 |
| N9 | 9.00 | 1.08 | 1.68 | 1.90 | 5.8477 | −3.1327 | 1.4617 |
| N10 | 8.96 | 1.08 | 1.68 | 1.89 | 6.4953 | −1.6457 | 1.9826 |
Table 10.
The binding energies for newly designed compounds (N3, N4, N5, N7) combined with PCAF protein (5TPX) by MM/PBSA study.
Table 10.
The binding energies for newly designed compounds (N3, N4, N5, N7) combined with PCAF protein (5TPX) by MM/PBSA study.
| Energy Terms (kcal/mol) | 52 | N3 | N4 | N5 | N7 |
|---|
| −50.813 ± 4.865 | −172.076 ± 1.610 | −193.759 ± 1.693 | −191.519 ± 1.474 | −174.278 ± 1.446 |
| −61.006 ± 2.977 | −56.241 ± 4.117 | −47.689 ± 1.434 | −86.251 ± 3.671 | −52.961 ± 1.484 |
| 90.704 ± 6.468 | 178.769 ± −4.801 | 208.975 ± 2.246 | 237.124 ± 4.049 | 195.046 ± 5.573 |
| −5.824 ± 0.532 | −18.960 ± 0.157 | −22.428 ± 0.122 | −21.198 ± 0.172 | −20.861 ± 0.172 |
| −26.939 ± 5.302 | −68.515 ± 2.218 | −54.869 ± 1.705 | −62.317 ± 2.418 | −52.857 ± 4.201 |
Table 11.
ADMET prediction results.
Table 11.
ADMET prediction results.
| Molecular | Absorption | Distribution | Metabolism | Excretion | Toxicity |
|---|
| HIA | F | PPB | BBB | CYP450 3A4 | CYP450 2D6 | CYP450 2C9 | T1/2 | CL | AMES | Carcinogenicity |
|---|
| N1 | 99.43% | 58.57% | 0.975 | 0.986 | Inhibitor | Non | Inhibitor | 0.784 | 6.378 | Non | Non |
| N2 | 98.61% | 58.57% | 0.955 | 0.822 | Inhibitor | Non | Inhibitor | 0.733 | 9.318 | Non | Non |
| N3 | 96.67% | 61.43% | 0.903 | 0.996 | Inhibitor | Non | Inhibitor | 0.489 | 8.638 | Non | Non |
| N4 | 95.80% | 50.00% | 1.001 | 0.992 | Inhibitor | Non | Inhibitor | 0.709 | 8.848 | Non | Non |
| N5 | 96.75% | 55.71% | 0.997 | 0.991 | Inhibitor | Non | Inhibitor | 0.442 | 7.315 | Non | Non |
| N6 | 92.07% | 58.57% | 1.042 | 0.989 | Inhibitor | Non | Inhibitor | 0.408 | 6.781 | Non | Non |
| N7 | 97.73% | 57.14% | 0.817 | 0.994 | Inhibitor | Non | Inhibitor | 0.249 | 3.946 | Non | Non |
| N8 | 93.23% | 55.71% | 0.883 | 0.990 | Inhibitor | Non | Inhibitor | 0.594 | 4.794 | Non | Non |
| N9 | 98.91% | 58.57% | 0.866 | 1.000 | Inhibitor | Non | Inhibitor | 0.158 | 6.421 | Non | Non |
| N10 | 92.65% | 58.57% | 0.929 | 0.988 | Inhibitor | Non | Inhibitor | 0.301 | 5.548 | Non | Non |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}































