3,4,5,6-Tetra-
O-acetyl-2-deoxy-2-(
N-ethylacetamido)-
l-glucononitrile
(11) [
53]. Colorless needles, m.p. 103–105 °C, [α]
D20 −49.0°; [α]
57820 −51.4°; [α]
54620 −58.5°, [α]
43620 −100.6°, [α]
36520 −160.5° (
c 1.0, chloroform); IR (KBr) ṽ
max/cm
−1 2239 (C≡N), 1755, 1649 (C=O), 1274, 1262, 1206 (C-O-C), 1057, 1029 (C-O); Raman ṽ
max/cm
−1 2239 (C≡N), 1736 (C=O);
1H NMR (500 MHz, CDCl
3) δ 5.57 (m, 2H, H-3, H-4), 5.48 (d,
J2,3 = 9.0 Hz, 1H, H-2), 5.11 (ddd,
J5,6 = 3.0 Hz,
J5,6′ = 5.0 Hz,
J4,5 = 8.5 Hz, H-5), 4.27 (dd,
J5,6 = 3.0 Hz,
J6,6′ = 12.5 Hz, 1H, H-6), 4.07 (dd,
J5,6′ = 5.0 Hz,
J6,6′ = 12.5 Hz, 1H, H-6′), 3.41 (m, 2H, CH
2), 2.20, 2.12, 2.08, 2.07, 2.06, 2.05 (s, 15H, CH
3), 1.35 (t,
J = 7.0 Hz, 3H, CH
3);
13C{
1H} NMR (125 MHz, CDCl
3) δ 170.88, 170.53, 169.69, 169.46, 169.36 (C=O), 114.78 (C≡N), 68.20, 68.04, 67.85 (C-3, C-4, C-5), 61.56 (C-6), 45.34 (C-2), 42.86 (CH
2), 21.09, 20.73, 20.65, 20.52, 20.33, 14.48 (CH
3). HRMS-(ESI-TOF)
m/
z [M + H]
+ calcd. for C
18H
27N
2O
9: 415.1717. Found: 415.1706.
m/
z [M + NH
4]
+ calcd. for C
18H
30N
3O
9: 432.1982. Found: 432.1969.
m/
z [M
2 + Na]
+ calcd. for C
36H
52N
4O
18Na: 851.3174. Found: 851.3133.
3,4,5,6-Tetra-
O-acetyl-2-deoxy-2-(
N-
n-propylacetamido)-
l-glucononitrile (
12) [
53]. Colorless needles, m.p. 115–116 °C, [α]
D20 −48.0°; [α]
57820 −48.9°; [α]
54620 −55.5°, [α]
43620 −95.6°, [α]
36520 −154.2° (
c 0.4, chloroform); IR (KBr) ṽ
max/cm
−1 2233 (C≡N), 1750 (C=O ester), 1655 (C=O amide), 1415, 1360, 1258 and 1210 (C-O-C), 1057, 1030 (C-O);
1H NMR (400 MHz, CDCl
3) δ 5.60 (t, 2H, H-3, H-4), 5.34 (d,
J2,3 = 8.8 Hz, 1H, H-2), 5.14 (m,
J5,6 = 3.2 Hz,
J5,6′ = 4.8 Hz, H-5), 4.27 (dd,
J5,6 = 3.0 Hz,
J6,6′ = 12.0 Hz, 1H, H-6), 4.07 (dd,
J5,6′ = 3.0 Hz,
J6,6′ = 12.0 Hz, 1H, H-6′), 3.25 (m, 2H, CH
2), 2.21, 2.22, 2.08, 2.07, 2.05 (s, 15H, CH
3), 0.97 (t,
J = 7.2 Hz, 3H, CH
3);
13C{
1H} NMR (100 MHz, CDCl
3) δ 170.94, 170.54, 169.72, 169.44, 169.32 (C=O), 114.70 (C≡N), 68.15, 68.08, 67.87 (C-3, C-4, C-5), 61.57 (C-6), 45.94 (C-2), 50.21, 22.54 (CH
2), 21.11, 20.74, 20.65, 20.52, 20.35, 11.19 (CH
3).
3,4,5,6,7-Penta-
O-acetyl-2-deoxy-2-(
N-ethylacetamido)-
d-
glycero-
l-
gluco-heptononitrile (
13) [
51]. Colorless needles, m.p. 120–122 °C, [α]
D20 −11.8°; [α]
57820 −11.8°; [α]
54620 −15.0°, [α]
43620 −26.0° (
c 0.5, chloroform); IR (KBr) ṽ
max/cm
−1 2255 (C≡N), 1749, 1662 (C=O), 1275, 1203 (C-O-C), 1067, 1036 (C-O); Raman ṽ
max/cm
−1 2254 (C≡N), 1743, 1661 (C=O);
1H-NMR (500 MHz, CDCl
3) δ 5.57 (dd,
J3,4 = 1.5 Hz,
J2,3 = 10.0 Hz, 1H, H-3), 5.44 (dd,
J3,4 = 1.5 Hz,
J4,5 = 10.0 Hz, 1H, H-4), 5.38 (d,
J2,3 = 10.0 Hz, 1H, H-2), 5.30 (dd,
J5,6 = 2.0 Hz,
J4,5 = 7.5 Hz, 1H, H-5), 5.23 (ddd,
J5,6 = 1.5 Hz,
J6,7 = 5.0 Hz,
J6,7′ = 7.0 Hz, H-6), 4.28 (dd,
J6,7 = 5.5 Hz,
J7,7′ = 12.0 Hz, 1H, H-7), 3.85 (dd,
J6,7′ = 7.0 Hz,
J7,7′ = 11.5 Hz, 1H, H-7′), 3.39 (m, 2H, CH
2), 2.21, 2.15, 2.14, 2.12, 2.06, 2.04 (s, 18H, CH
3), 1.35 (t,
J = 7.0 Hz, 3H, CH
3);
13C{
1H} NMR (100 MHz, CDCl
3) δ 170.70, 170.40, 170.32, 169.74, 169.61, 169.44 (C=O), 114.81 (C≡N), 67.76, 67.40, 67.36, 66.84 (C-3, C-4, C-5, C-6), 61.85 (C-7), 45.16 (C-2), 42.94 (CH
2), 21.06, 20.73, 20.61, 20.57, 20.49, 20.38, 14.51 (CH
3). HRMS-(ESI-TOF)
m/
z [M + H]
+ calcd. for C
21H
31N
2O
11: 487.1928. Found: 487.1904.
m/
z [M + NH
4]
+ calcd. for C
21H
34N
3O
11: 504.2193. Found: 504.2158.
3,4,5,6,7-Penta-
O-acetyl-2-deoxy-2-(
N-phenylamino)-
d-
glycero-
d-
altro-heptononitrile (
17) [
56]. White solid, m.p. 100–102 °C, [α]
D20 −43.6°; [α]
57820 −45.9°; [α]
54620 −55.2°, [α]
43620 −103.2°, [α]
36520 −183.6° (
c 0.5, chloroform); IR (KBr) ṽ
max/cm
−1 3349 (NH), 2229 (C≡N), 1771, 1747 (C=O), 1509 (NH), 1252, 1231, 1213 (C-O-C), 1061 (C-O);
1H NMR (500 MHz, CDCl
3) δ 7.26 (m, 2H, H-arom), 6.91 (t, 1H, H-arom), 6.70 (d, 2H, H-arom), 5.62 (dd,
J2,3 = 4.0 Hz,
J3,4 = 7.0 Hz, 1H, H-3), 5.46 (dd,
J4,5 = 3.5 Hz,
J3,4 = 7.0 Hz, 1H, H-4), 5.40 (dd,
J4,5 = 3.5 Hz,
J5,6 = 7.0 Hz, 1H, H-5), 5.30 (ddd,
J6,7 = 3.0 Hz,
J6,7′ = 6.0 Hz,
J5,6 = 7.0 Hz, 1H, H-6), 4.52 (dd,
J2,3 = 4.0 Hz,
J2,NH = 11.0 Hz, 1H, H-2), 4.37 (dd,
J6,7 = 3.0 Hz,
J7,7′ = 12.5 Hz, 1H, H-7), 4.11 (d,
J2,NH = 11.0 Hz, 1H, NH), 4.10 (dd,
J6,7′ = 6.0 Hz,
J7,7′ = 12.5 Hz, 1H, H-7′), 2.23, 2.12, 2.07, 2.06, 2.05 (s, 15H, CH
3);
13C{
1H} NMR (100 MHz, CDCl
3) δ 170.60, 170.01, 169.83, 169.53, 169.27, 169.11 (C=O), 143.96, 129.70, 120.91 (C-arom), 116.70 (C≡N), 114.46 (C-arom), 69.84, 69.65, 69.21 (C-3, C-4, C-5, C-6), 61.75 (C-7), 47.26 (C-2), 20.86, 20.67, 20.62 (CH
3).
3,4,5,6,7-Penta-
O-benzoyl-2-deoxy-2-(
N-ethylbenzamido)-
d-
glycero-
l-
gluco-heptononitrile (
18) [
51]. White solid, m.p. 79–81 °C, [α]
D20 −9.4°; [α]
57820 −10.0°; [α]
54620 −11.4°, [α]
43620 −12.6° (
c 0.5, chloroform) [Lit. [
51] m. p. 80 °C, [α]
D20 −23.9° (
c 0.3, pyridine)]; IR (KBr) ṽ
max/cm
−1 1730, 1655 (C=O), 1259 (C-O-C), 1090, 1068, 1024 (C-O); Raman ṽ
max/cm
−1 2250 (C≡N), 1732, 1658 (C=O);
1H NMR (500 MHz, CDCl
3) δ 8.10–7.00 (m, 30H, H-arom), 6.12 (bs, 2H, H-3, H-4), 6.04 (bs, 1H, H-5), 5.91 (s, 1H, H-6), 5.76 (bs, 1H, H-2), 4.60 (dd,
J6,7 = 4.5 Hz,
J7,7′ = 11.5 Hz, 1H, H-7), 4.48 (dd,
J6,7′ = 7.0 Hz,
J7,7′ = 11.5 Hz, 1H, H-7), 3.47 (bs, 1H, CH
2), 3.26 (bs, 1H, CH
2), 1.05 (bs, 3H, CH
3);
13C{
1H} NMR (100 MHz, CDCl
3) δ 171.90, 171.85, 165.85, 165.37, 165.23, 165.01(C=O),134.45, 133.98, 133.88, 133.77, 133.36, 133.01, 130.13, 130.08, 130.03, 1298.88, 129.69, 129.27, 128.72, 128.66, 128.47, 128.44, 128.24, 126.41 (C-arom), 114.87 (C≡N), 69.70, 69.17, 68.68 (C-3, C-4, C-5, C-6), 62.86 (C-7), 47.52 (C-2), 43.44 (CH
2), 14.01 (CH
3). HRMS-(ESI-TOF)
m/
z [M + H]
+ calcd. for C
51H
43N
2O
11: 859.2867. Found: 859.2833.
m/
z [M + NH
4]
+ calcd. for C
51H
46N
3O
11: 876.3132. Found: 876.3097.
3,4,5,6,7-Penta-
O-benzoyl-2-deoxy-2-(
N-ethylbenzamido)-
d-
glycero-
d-
galacto-heptononitrile (
20) [
51]. White solid, m. p. 166–168 °C, [α]
D20 +6.9° (
c 0.5, pyridine) [Lit. [
51] m. p. 170 °C, [α]
D24 +5.8° (
c 0.37, pyridine)]; IR (KBr) ṽ
max/cm
−1 2243 (C≡N), 1742, 1720, 1639 (C=O), 1264 (C-O-C), 1093, 1069, 1026 (C-O); Raman ṽ
max/cm
−1 2250 (C≡N), 1746, 1639 (C=O); [α]
D22 +4.2°; [α]
57822 +4.2°; [α]
54622 +4.2°; [α]
43622 +4.6° (
c 0.5, chloroform);
1H-NMR (500 MHz, CDCl
3) δ 8.06–7.88 (m, 10H, H-arom), 7.60–7.24 (m, 20H, H-arom), 6.40 (bs, 2H, H-2, H-5), 6.07 (t,
J3,4 = 5.5 Hz, 1H, H-4), 5.98 (dd,
J2,3 = 2.5 Hz,
J3,4 = 5.5 Hz,1H, H-3), 5.85 (ddd,
J6,7 = 3.0 Hz,
J6,7′ = 5.0 Hz,
J5,6 = 8.0 Hz, 1H, H-6), 4.79 (dd,
J6,7 = 3.0 Hz,
J7,7′ = 12.5 Hz, 1H, H-7), 4.48 (dd,
J6,7′ = 5.0 Hz,
J7,7′ = 12.5 Hz, 1H, H-7′), 3.61 (m, 1H, CH
2), 3.48 (m, 1H, CH
2), 1.10 (t,
J = 7.5 Hz, 3H, CH
3);
13C{
1H} NMR (125 MHz, CDCl
3) 171.95, 166.03, 165.52, 165.33, 165.27, 165.09 (C=O),134.95, 133.69, 133.62, 133.58, 133.23, 133.02, 130.11, 129.98, 129.90, 129.84, 129.42, 129.11, 128.97, 128.76, 128.63, 128.58, 128.52, 128.43, 128.30, 126.49 (C-arom), 114.80 (C≡N), 70.69, 69.43, 69.02, 68.37 (C-3, C-4, C-5, C-6), 62.61 (C-7), 47.38 (C-2), 43.50 (CH
2), 15.05 (CH
3). HRMS-(ESI-TOF)
m/
z [M + H]
+ calcd. for C
51H
43N
2O
11: 859.2822. Found: 859.2826.
m/
z [M + NH
4]
+ calcd. for C
51H
46N
3O
11: 876.3132. Found: 876.3081.
2-(Benzyloxycarbonyl)amino-2-deoxy-
d-
glycero-
d-
ido-heptononitrile (
26) [
52]. M. p. 200–202 °C (dec.), [α]
D22 −25.4° (
c 0.5, pyridine); IR (KBr) ṽ
max/cm
−1 3440, 3400–3200 (OH), 3300 (NH), 2250 (C≡N), 1695 (C=O amide I), 1535 (C=O amide II), 1285 (C-O-C), 1085, 1050, 1040 (C-O);
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 155.93 (C=O), 136.62, 128.52 (2C), 128.07, 127.94 (2C) (aromatics), 118.71 (C≡N), 72.13, 71.98, 71.38, 68.87, 66.17, 63.26 (C-7), 45.85 (C-2).
2-(
N-Benzylamino)-2-deoxy-
d-
glycero-
l-
gluco-heptononitrile (
32) [
49]. M. p. 130–132 °C (dec.), [α]
D22 −25.4° (
c 0.5, pyridine); IR (KBr) ṽ
max/cm
−1 3500–3100, 3020, 2230 (C≡N), 1610, 1500, 1430, 1090, 1050, 760, 700 (C-O);
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 138.95, 128.62, 128.49, 127.27 (C-arom), 119.76 (C≡N), 72.56, 71.30, 70.78, 69.52 (C-3, C-4, C-5, C-6), 63.71 (C-7), 51.50 (CH
2), 50.87 (C-2).
2-(
N-Benzylamino)-2-deoxy-
d-
glycero-
d-
ido-heptononitrile (
33) [
49]. M. p. 200–202 °C (dec.), [α]
D22 −81.6° (
c 0.5, pyridine); IR (KBr) ṽ
max/cm
−1 3400–3200, 2230 (C≡N), 1610, 1500, 740, 690;
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 138.95, 128.62, 128.49, 127.27 (C-arom), 119.76 (C≡N), 72.56, 71.30, 70.78, 69.52 (C-3, C-4, C-5, C-6), 63.71 (C-7), 51.50 (CH
2), 50.87 (C-2).
3,4,5,6,7-Penta-
O-acetyl-2-(
N-benzylacetamido)-2-deoxy-
d-
glycero-
l-
gluco-heptononitrile (
37). A suspension of 2-benzylamino-2-deoxy-
d-
glycero-
l-
gluco-heptononitrile (
32) [
49] (2.1 g, 7.1 mmol) in pyridine (10 mL) was treated with acetic anhydride (10 mL) and the mixture was kept for 24 h at room temperature. After this time, it was poured into ice–water, and a white solid was separated by stirring and scraping. It was filtered, washed with cold water, and dried under a vacuum on silica gel. Compound
37 was obtained as a colorless solid (3.6 g, 92%). Crystallized from ethanol, it formed colorless needles. M. p. 135–137 °C, [α]
D20 −11.2° (
c 2.0, pyridine) (Lit. [
49] for
14, m. p. 83–85 °C); IR (KBr) ṽ
max/cm
−1 2247 (C≡N), 1754, 1672 (C=O), 1250, 1215 (C-O-C), 1033 (C-O);
1H NMR (500 MHz, CDCl
3) δ 7.41 (m, 2H, H-arom), 7.34 (m, 1H, H-arom), 7.24 (d, 2H, H-arom), 5.58 (d,
J3,4 =
J4,5 = 9.5 Hz, 1H, H-4), 5.53 (d,
J2,3 =
J3,4 = 9.5 Hz, 1H, H-3), 5.38 (d,
J2,3 = 9.5 Hz, 1H, H-2), 5.32 (dd,
J5,6 = 2.0 Hz,
J4,5 = 9.5 Hz, 1H, H-5), 5.23 (t,
J6,7 = 5.5 Hz, 1H, H-6), 4.67 (d,
J = 7.5 Hz, 1H, CH
2), 4.44 (d,
J = 8.0 Hz, 1H, CH
2), 4.29 (dd,
J6,7 = 5.5 Hz,
J7,7′ = 12.0 Hz, 1H, H-7), 3.86 (dd,
J6,7′ = 7.5 Hz,
J7,7′ = 12.0 Hz, 1H, H-7′), 2.19, 2.17, 2.12, 2.11, 2.05, 1.99 (s, 18H, CH
3);
13C{
1H} NMR (125 MHz, CDCl
3) δ 171.62, 170.43, 170.37, 169.83, 169.79, 169.42 (C=O), 135.36, 129.18, 128.10, 126.00 (C-arom), 114.32 (C≡N), 67.89, 67.83, 67.45, 66.92 (C-3, C-4, C-5, C-6), 61.89 (C-7), 51.78 (CH
2), 46.81 (C-2), 21.81, 20.75, 20.63, 20.50 (CH
3). Anal. Calcd. for C
26H
32N
2O
11: C, 56.93; H, 5.88; N, 5.11. Found: C, 56.84; H, 5.90; N, 5.23. HRMS-(ESI-TOF)
m/
z [M + H]
+ calcd. for C
26H
33N
2O
11: 549.2084. Found: 549.2069.
m/
z [M + NH
4]
+ calcd. for C
26H
36N
3O
11: 566.2350. Found: 566.2330.
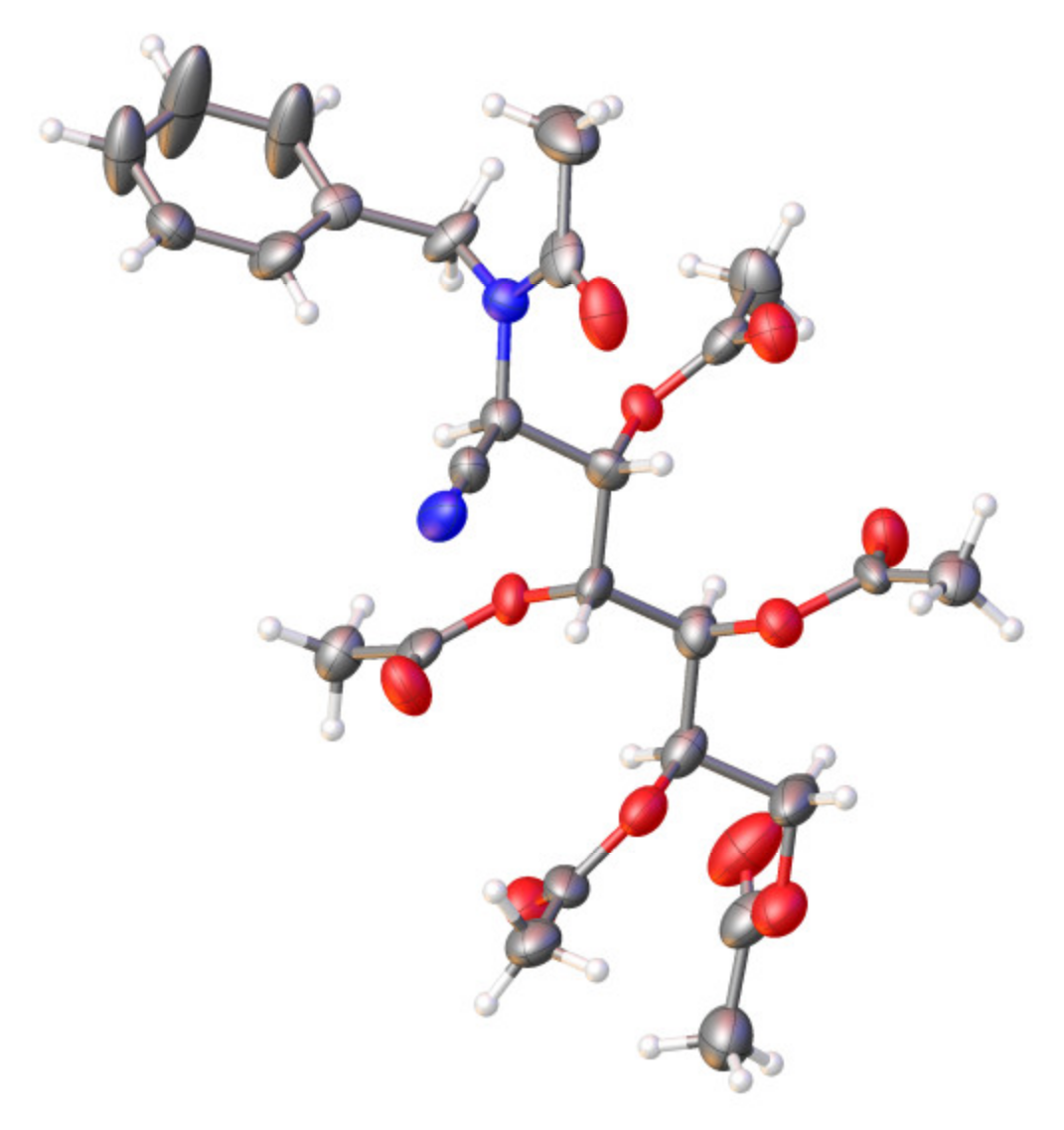
3,4,5,6,7-Penta-
O-acetyl-2-(
N-benzylacetamido)-2-deoxy-
d-
glycero-d-
ido-heptononitrile (
38). A suspension of 2-benzylamino-2-deoxy-
d-
glycero-
d-
ido-heptononitrile (
33) [
49] (0.50 g, 1.7 mmol) in pyridine (3.0 mL) was treated with acetic anhydride (2.5 mL) and the mixture was kept for 24 h at room temperature. After this time, it was poured into ice–water, and a white solid was separated by stirring and scraping. It was filtered, washed with cold water, and dried under a vacuum on silica gel. Compound
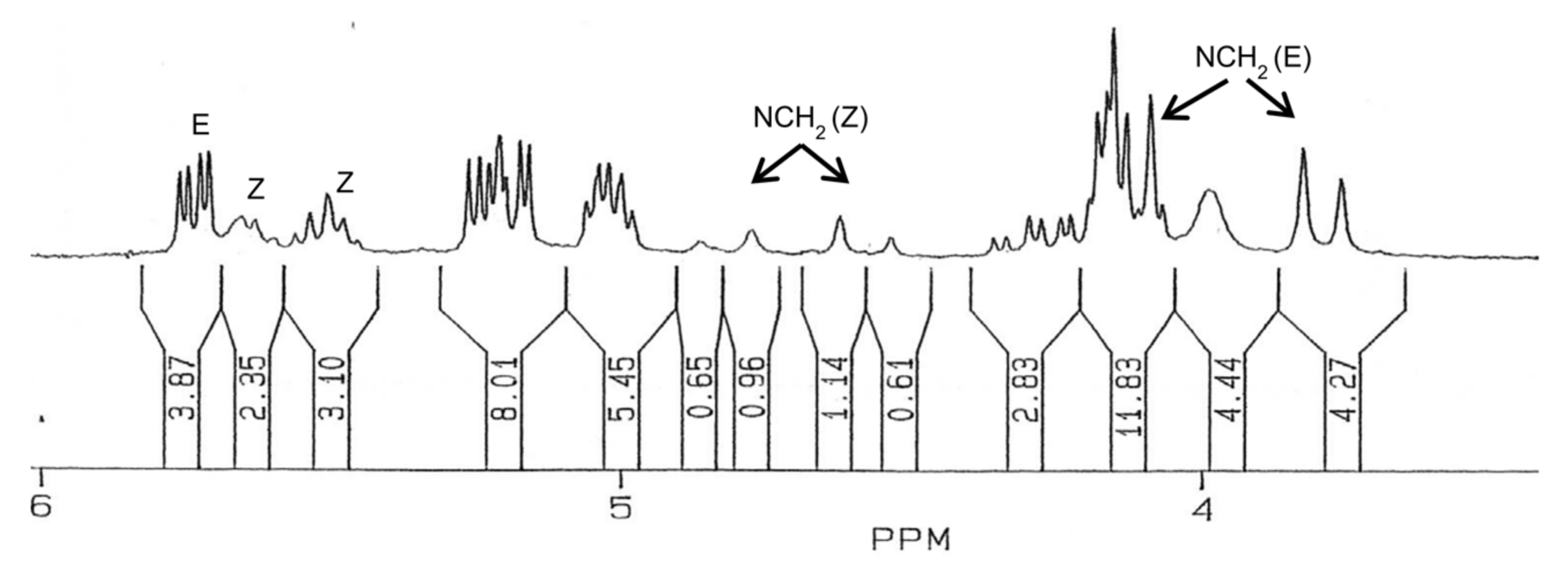
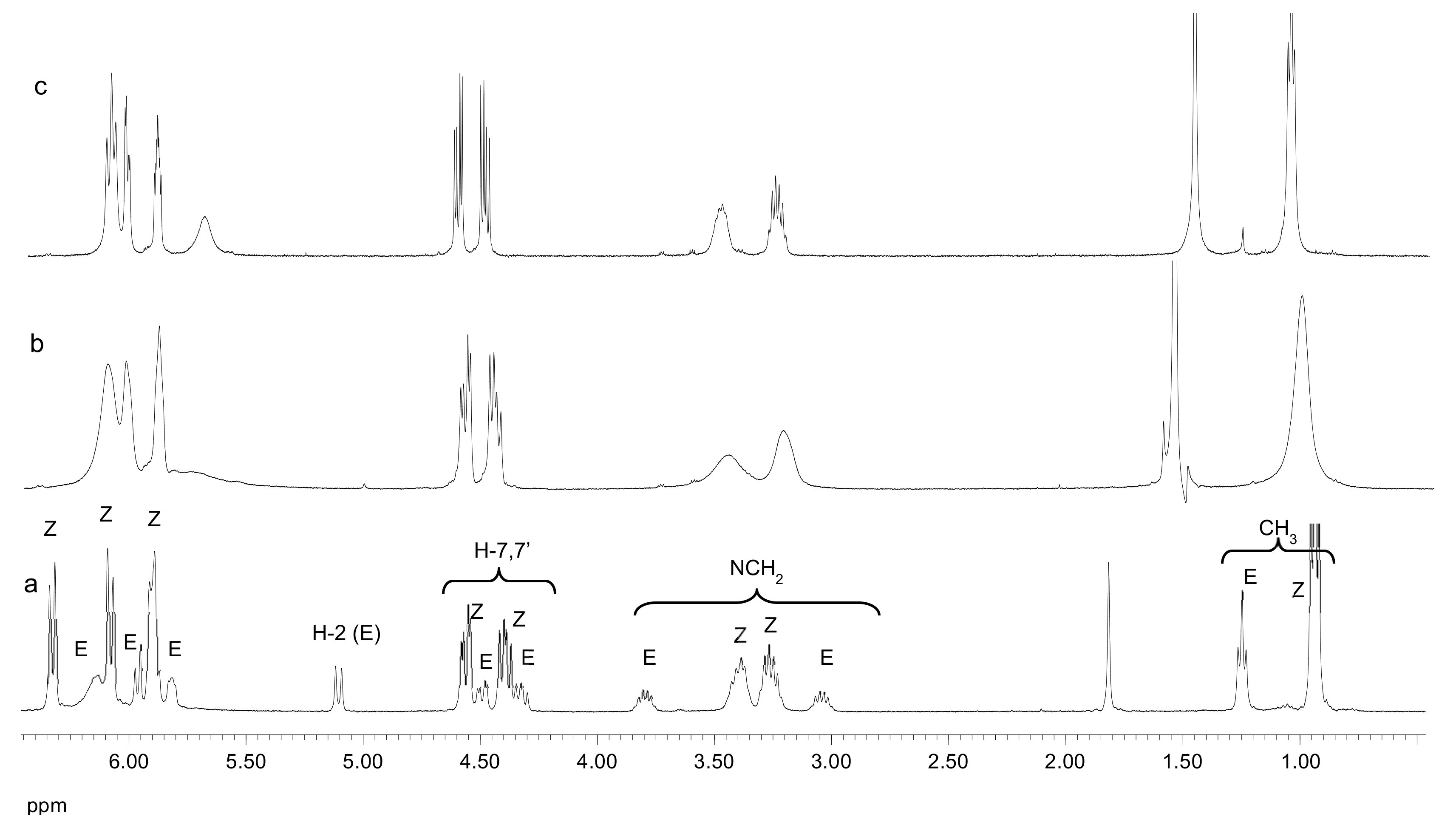
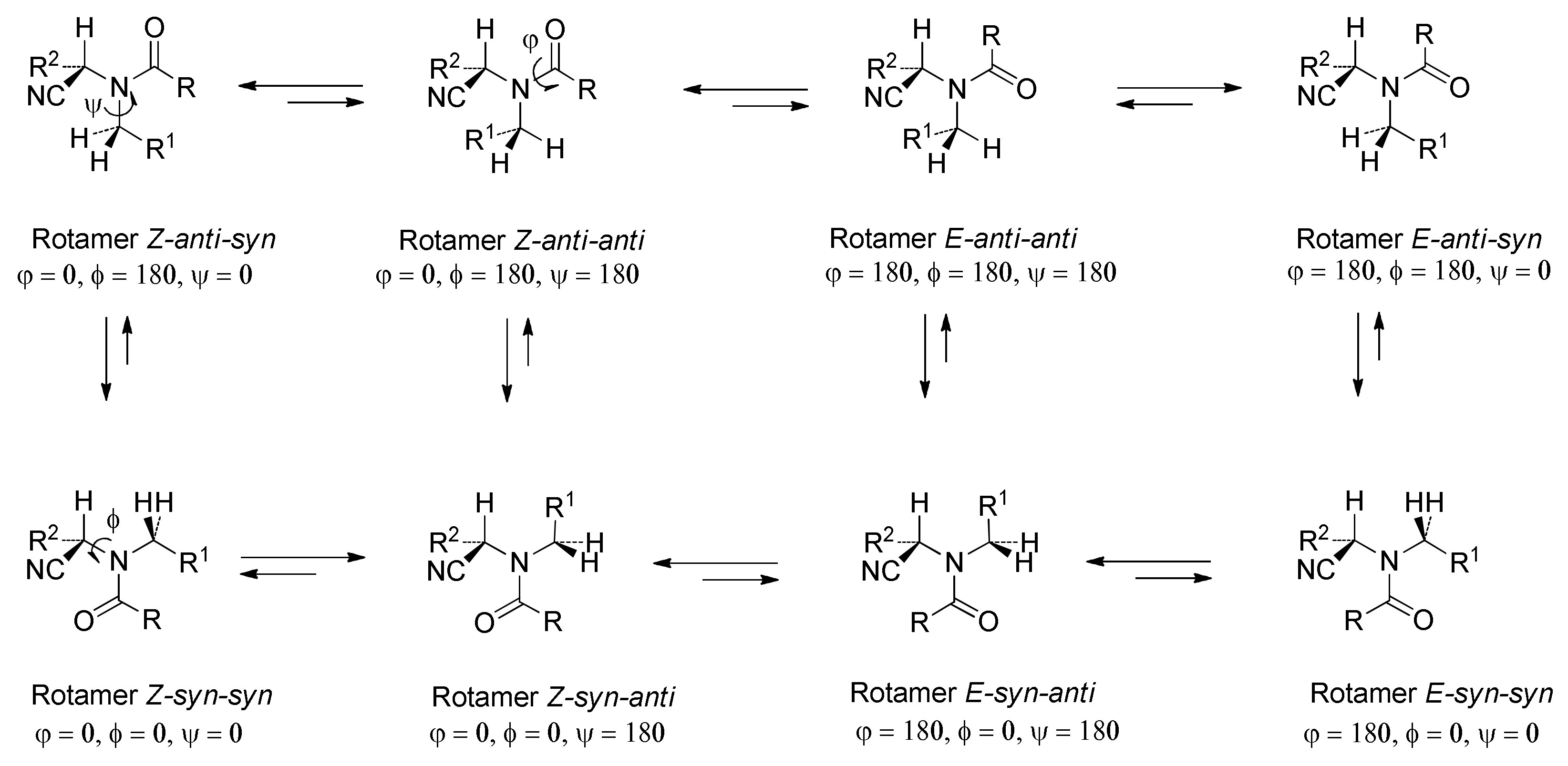
38 was obtained as an amorphous, colorless, partially melted solid (0.81 g, 87%). M. p. 42–46 °C, [α]
D20 −10.0° (
c 1.0, pyridine) (Lit. [
49] for
15, m. p. room temperature ~25 °C; [α]
D20 −9.6° (
c 1.27, pyridine)); IR (KBr) ṽ
max/cm
−1 2240 (C≡N), 1750 (C=O acetate),1670 (C=O amide), 1375, 1210 (C-O-C), 1050 (C-O);
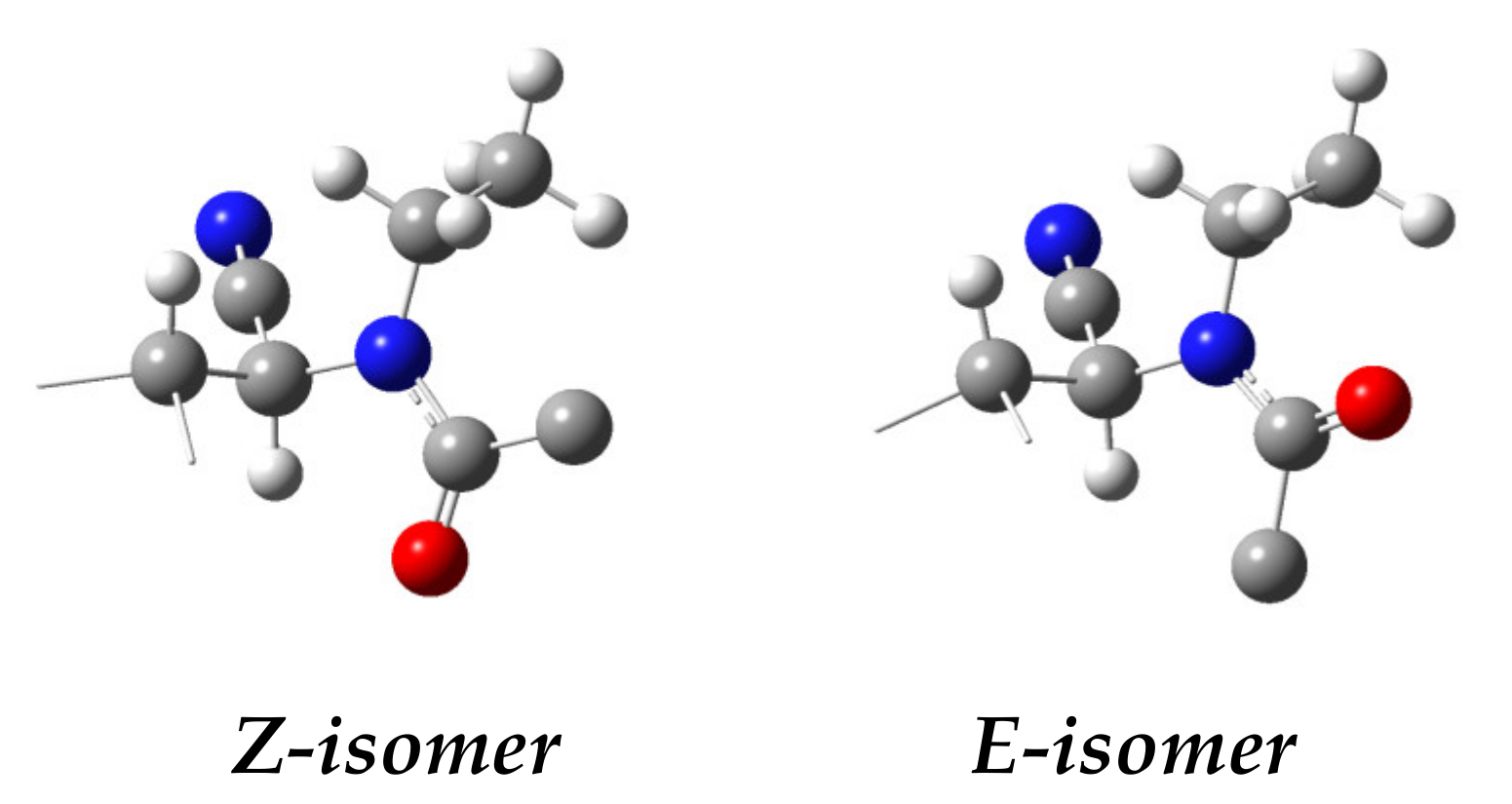
1H NMR (200 MHz, CDCl
3) δ 7.42–7.23 (m, 5H, H-arom), 5.73 (dd,
J3,4 = 7.0 Hz,
J4,5 = 3.1 Hz, 1H, H-4
E-
syn), 5.68 (m, 1H, H-3
Z-syn), 5.62 (d,
J2,3 = 5.0 Hz, 1H, H-2
Z-syn), 5.50 (m, 2H, H-4, H-5
Z-syn), 5.23 (dd,
J2,3 = 3.9 Hz,
J3,4 = 7.3 Hz, 1H, H-3
E-
syn), 5.19 (dd,
J4,5 = 3.1 Hz,
J5,6 = 7.8 Hz, 1H, H-5
E-
syn), 5.02 (m, 2H, H-6
E-
syn and
Z-syn), 4.82 (d,
J = 17.6 Hz, 1H, PhCH
2 Z-syn), 4.58 (d,
J = 17.6 Hz, 1H, PhCH
2 Z-syn), 4.32 (dd,
J6,7 = 4.2 Hz,
J7,7 = 12.3 Hz, 1H, H-7
E-
syn), 4.17 (m, 2H, H-7, H-7′
Z-syn), 4.16 (dd,
J6,7′ = 2.1 Hz,
J7,7′ = 12.3 Hz, 1H, H-7′
E-
syn), 4.08 (d,
J = 12.8 Hz, 1H, PhCH
2 E-
syn), 3.99 (d,
J2,3 = 3.9 Hz, 1H, H-2
E-
syn), 3.80 (d,
J = 13.0 Hz, 1H, PhCH
2 E-
syn), 2.15, 2.13, 2.11, 2.10, 2.08, 2.06, 2.04, 1.99 (s, 36H, CH
3);
13C{
1H} NMR (50 MHz, CDCl
3) δ 172.02 (C=O
Z-syn), 170.63 (
Z-syn), 170.33 (
E-
syn), 170.14 (
E-
syn), 169.66 (
Z-syn), 169.61 (
E-
syn), 169.42 (
Z-syn), 169.20 (
E-
syn) (C=O), 137.52 (
E-
syn), 135.49 (
Z-syn), 129.03 (
Z-syn), 128.60 (
E-
syn), 128.30 (
E-
syn), 127.91 (
Z-syn), 127.76 (
Z-syn), 126.03 (
Z-syn) (C-arom), 116.96 (C≡N
E-
syn), 114.41 (C≡N
Z-syn), 70.15 (
E-
syn), 69.27 (
Z-syn), 68.91 (
Z-syn), 68.63 (
Z-syn), 68.08 (
E-
syn), 68.00 (
Z-syn), 67.93 (
Z-syn), 67.76 (
E-
syn) (C-3, C-4, C-5, C-6), 61.22 (C-7
Z-syn), 61.11 (C-7
E-
syn), 52.08 (CH
2 Z-syn), 51.43 (CH
2 E-
syn), 49.35 (C-2
E-
syn), 46.94 (C-2
Z-syn), 21.76 (CH
3 Z-syn), 20.62, 20.54, 20.46, 20.37, 20.31 (CH
3). Anal. Calcd. for C
26H
32N
2O
11: C, 56.93; H, 5.88; N, 5.11. Found: C, 56.72; H, 5.67; N, 5.33.
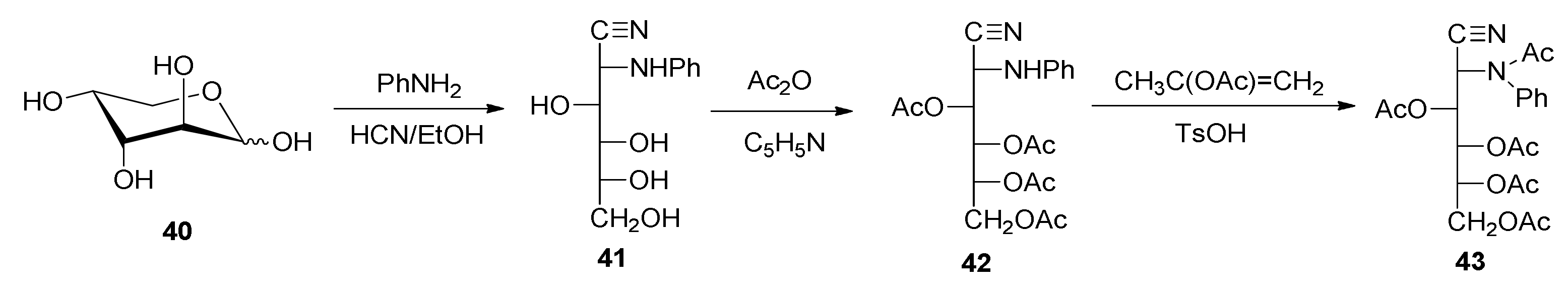
2-Deoxy-2-phenylamino-d-glucononitrile (41). To a suspension of d-arabinose (10.0 g, 66.6 mmol) in ethanol (30.0 mL) and water (3.0 mL), aniline (8.0 mL, 87.6 mmol) was added and the mixture was refluxed until dissolution. The reddish solution was concentrated in vacuo to a thick oil, which was treated with ethyl ether. The ethereal layer was decanted and the treatment was repeated twice. The residue was dissolved in hot ethanol and crystallized into N-phenyl-d-arabinopyranosylamine. The solid was dissolved by heating and then cooled rapidly to room temperature, and then anhydrous hydrogen cyanide (7–8 mL) was added. After one hour at room temperature, it was stored in a refrigerator overnight. The resulting solid was filtered, washed with cold ethanol and diethyl ether, and dried under a vacuum over silica gel (9.75 g, 58%). An additional yield of crystals was obtained from the mother liquors (2.17 g, 13%) (total yield: 11.92 g, 71%). White solid, m. p. 150–152 °C; [α]D18 +129.2°; [α]57818 +133.9°; [α]54618 +156.0° (c 0.5, pyridine); IR (KBr) ṽmax/cm−1 3500–3100 (OH, NH), 2229 (C≡N), 1603, 1499, 754, 693 (phenyl), 1142, 1103, 1053, 1034 (C-O, C-N); 13C{1H} NMR (125 MHz, DMSO-d6) δ 144.64 (C≡N), 70.78, 70.24, 68.97 (C-3, C-4, C-5), 63.36 (C-6), 52.86 (C-2).
3,4,5,6-Tetra-O-acetil-2-deoxy-2-phenylamino-d-glucononitrile (42). A suspension of 2-deoxy-2-phenylamino-d-glucononitrile (41) (0.50 g, 2.0 mmol) in pyridine (3.5 mL) was treated with acetic anhydride (2.8 mL) and the mixture was kept under agitation for 24 h at 0 °C. After this time, it was poured into ice–water and, by stirring and scraping, a white solid was separated. It was filtered, washed with cold water, and dried under a vacuum over silica gel (0.74 g, 89%). Crystallization from 96% ethanol gave rise to a white microcrystalline powder. M. p. 135–136 °C; [α]D22 +415.4°; [α]57822 +416.4°; [α]54622 +423.0°; [α]43622 +461.4° (c 0.5, chloroform); IR (KBr) ṽmax/cm−1 3347 (NH), 2239 (C≡N), 1763, 1743 (C=O), 1215 (C-O-C), 1081, 1050 (C-O); Raman ṽmax/cm−1 2236 (C≡N), 1737 (C=O); 1H NMR (500 MHz, CDCl3) δ 7.26 (m, 2H, H-arom), 6.91 (t, 1H, H-arom), 6.78 (d, 2H, H-arom), 5.57 (dd, J3,4 = 1.5 Hz, J4,5 = 9.0 Hz, 1H, H-4), 5.28 (ddd, J5,6 = 3.0 Hz, J5,6′ = 4.0 Hz, J4,5 = 9.0 Hz, 1H, H-5), 5.25 (dd, J3,4 = 2.0 Hz, J2,3 = 6.0 Hz, 1H, H-3), 4.72 (dd, J2,3 = 6.0 Hz, JNH,2 = 11.0 Hz, 1H, H-2), 4.59 (d, JNH,2 = 11.0 Hz, 1H, NH), 4.28 (dd, J5,6 = 2.5 Hz, J6,6′ = 12.5 Hz, 1H, H-6), 4.23 (dd, J5,6′ = 3.5 Hz, J6,6′ = 12.5 Hz, 1H, H-6′), 2.23, 2.19, 2.08, 2.05 (s, 12H, CH3); 13C{1H} NMR (125 MHz, CDCl3) δ 171.79, 170.52, 170.22, 169.63 (C=O), 144.08, 129.72, 121.02 (C-arom), 116.67 (C≡N), 114.87 (C-arom), 68.15, 67.67, 66.95 (C-3, C-4, C-5), 61.50 (C-6), 46.00 (C-2), 21.03, 20.70, 20.67, 20.58 (CH3). Anal. Calcd. for C20H24N2O8: C, 57.14; H, 5.75; N, 6.66. Found: C, 56.97; H, 6.06; N, 6.63. HRMS-(ESI-TOF) m/z [M + H]+ calcd. for C20H25N2O8: 421.1611. Found: 421.1600.
3,4,5,6-Tetra-O-acetyl-2-deoxy-2-(N-phenylacetamido)-d-glucononitrile (43). To a suspension of 3,4,5,6-tetra-O-acetyl-2-deoxy-2-phenylamino-d-glucononitrile (42) (0.2 g, 0.5 mmol) in isopropenyl acetate (5.0 mL), a catalytic amount of p-toluenesulfonic acid was added and the mixture was refluxed for 8 h. Upon cooling, a white solid was separated, which was filtered and dried under a vacuum over silica gel (0.18 g, 81%), and had an m. p. of 195–196 °C. [α]D22 −3.1°; [α]57822 −3.6°; [α]54622 +1.0°; [α]43622 +23.4° (c 0.5, chloroform); IR (KBr) ṽmax/cm−1 2237 (C≡N), 1765, 1744, 1673 (C=O), 1280, 1262, 1224 (C-O-C), 1065, 1036 (C-O); Raman ṽmax/cm−1 2243 (C≡N), 1751, 1671 (C=O); 1H NMR (500 MHz, CDCl3) δ 7.48 (m, 3H, H-arom), 7.33 (d, 2H, H-arom), 5.72 (d, J2,3 = 8.5 Hz, 1H, H-2), 5.67 (dd, J3,4 = 3.0 Hz, J2,3 = 8.5 Hz, 1H, H-4), 5.51 (dd, J3,4 = 3.0 Hz, J4,5 = 8.5 Hz, 1H, H-4), 5.09 (ddd, J5,6 = 3.5 Hz, J5,6′ = 5.5 Hz, J4,5 = 8.5 Hz, 1H, H-5), 4.26 (dd, J5,6 = 3.5 Hz, J6,6′ = 12.5 Hz, 1H, H-6), 4.04 (dd, J5,6′ = 5.5 Hz, J6,6′ = 12.5 Hz, 1H, H-6′), 2.15, 2.05, 1.84 (s, 15H, CH3); 13C{1H} NMR (125 MHz, CDCl3) δ 171.04, 170.53, 169.82, 169.63, 169.23 (C=O), 139.64, 130.20, 129.61, 128.93 (C-arom), 114.40 (C≡N), 68.90, 68.80, 68.48 (C-3, C-4, C-5), 61.55 (C-6), 49.14 (C-2), 22.54, 20.79, 20.67, 20.57, 20.54 (CH3). Anal. Calcd. for C22H27N2O9: C, 57.14; H, 5.62; N, 6.06. Found: C, 57.09; H, 5.76; N, 5.73. HRMS-(ESI-TOF) m/z [M + H]+ calcd. for C22H27N2O9: 463.1717. Found: 463.1709. m/z [M + NH4]+ calcd, for C22H30N3O9: 480.1982. Found: 480.1974.
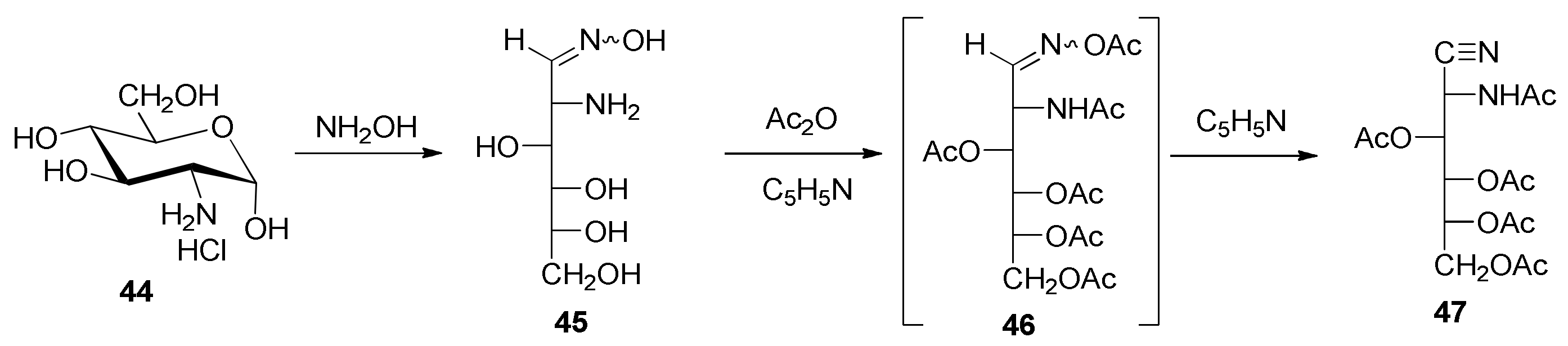
2-Amino-2-deoxy-
d-glucose Oxime Hydrochloride (
45). This compound was synthesized following the procedure described by Restelli and Deulofeu [
61]. IR (KBr) ṽ
max/cm
−1 3500–2400 (OH, NH
+), 1610 (C=N), 1530, 1410, 1100, 1080, 1040, 1010 (C-O), 750;
1H NMR (200 MHz, DMSO-
d6) δ 11.38 (s, 1H, NOH), 8.00 (bs, 2H), 5.31 (bs, 1H), 4.68 (bs, 1H), 4.52 (bs, 1H), (OH, NH), 7.35 (d,
J2,3 = 5.6 Hz, 1H, H-1), 3.86 (m, 2H), 3.47 (m, 5H), 3.25 (d, 1H);
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 144.64 (C=N), 70.78, 70.24, 68.97 (C-3, C-4, C-5), 63.36 (C-6), 52.86 (C-2).
2-Acetamido-3,4,5,6-tetra-
O-acetyl-2-deoxy-
d-glucononitrile (
47). This compound was synthesized by modifying the Restelli and Deulofeu procedure [
61]. A suspension of
45 (1.0 g, 4.2 mmol) in pyridine (6.0 mL) was treated with acetic anhydride (6.0 mL). The mixture was stirred for 24 h at room temperature. After this time, it was poured into ice–water and extracted with dichloromethane (2 × 50 mL), and the organic phase was washed successively with 1
M hydrochloric acid (2 × 50 mL), saturated sodium bicarbonate solution (2 × 50 mL), and distilled water, and dried with anhydrous MgSO
4. The solvent was removed in vacuo to dryness and the resulting residue was crystallized from ethanol. After being stored in the refrigerator for several hours, it was filtered, washed with cold ethanol, and dried under a vacuum over silica gel (0.77 g, 43%). Colorless prisms from ethanol, m. p. 126–129 °C; [α]
D +18.0°; [α]
578 +18.2°; [α]
546 +21.4°; [α]
436 +40.2° (
c 0.5, chloroform) [Lit. [
61] m. p. 126 °C; [α]
D20 +20.5° (
c 4.7, chloroform)]; IR (KBr) ṽ
max/cm
−1 3237 (NH), 2243 (C≡N), 1753, 1730, 1675, 1645 (C=O), 1541 (NH), 1252, 1211 (C-O-C), 1079, 1065, 1051 (C-O); Raman ṽ
max/cm
−1 2243 (C≡N), 1753, 1728, 1643 (C=O);
1H NMR (400 MHz, CDCl
3) δ 6.69 (d,
JNH,2 = 9.8 Hz, 1H, NH), 5.45 (dd,
J2,3 = 6.8 Hz,
JNH,2 = 9.6 Hz, 1H, H-2), 5.36 (dd,
J3,4 = 1.8 Hz,
J4,5 = 9.2 Hz, 1H, H-4), 5.19 (dt,
J5,6 = 4.0 Hz,
J5,6′ = 6.4 Hz,
J4,5 = 9.2 Hz, 1H, H-5), 5.10 (dd,
J3,4 = 1.6 Hz,
J2,3 = 6.8 Hz, 1H, H-3), 4.24 (m, 1H, H-6), 4.23 (m, 1H, H-6′), 2.23, 2.15, 2.08, 2.07, 2.05 (s, 15H, CH
3);
13C{
1H} NMR (100 MHz, CDCl
3) δ 172.31, 170.37, 169.54, 169.39, 169.20 (C=O), 115.77 (C≡N), 67.51, 67.30, 67.00 (C-3, C-4, C-5), 61.42 (C-6), 39.22 (C-2), 22.92, 21.03, 20.68, 20.61, 20.37 (CH
3). HRMS-(ESI-TOF)
m/
z [M + H]
+ calcd. for C
16H
23N
2O
9: 387.1404. Found: 387.1406.
m/
z [M + NH
4]
+ calcd. for C
16H
26N
3O
9: 404.1669. Found: 404.1675.
2-Amino-2-deoxy-
d-
glycero-
l-
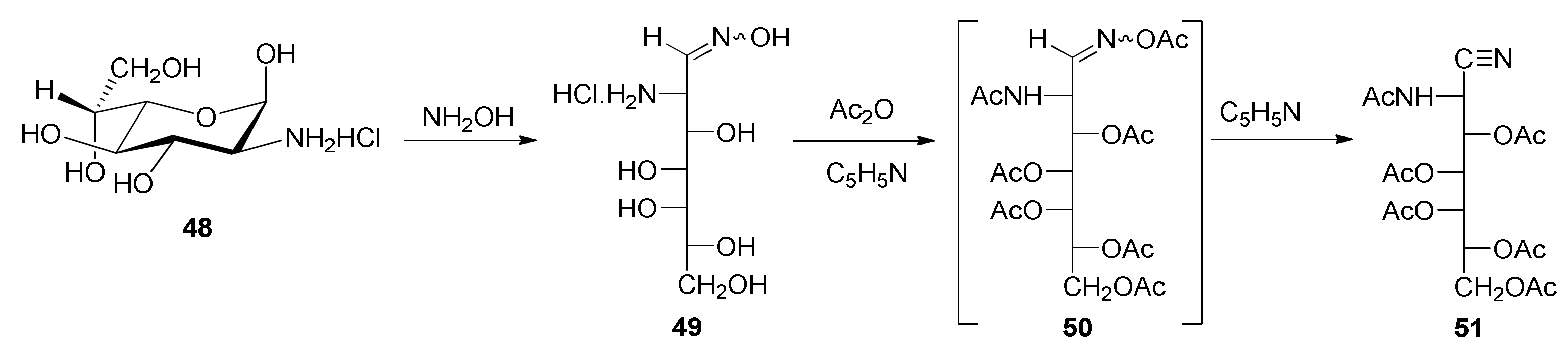
gluco-heptose Oxime Hydrochloride (
49). This compound was obtained in a similar way to that described for the preparation of
45: through the treatment of
48 [
67] with hydroxylamine. Crystallized from aqueous ethanol, it formed colorless prisms, m. p. 157–160 °C (dec.). The product was characterized through its NMR spectra, which were almost coincidental with those of
45.
1H NMR (200 MHz, DMSO-
d6) δ 11.44 (s, 1H, NOH), 8.16 (bs, 2H), 5.27 (bs, 1H), 4.56 (bs, 1H), 4.35 (bs, 1H), 3.72 (m, 1H) (OH, NH), 7.40 (d,
J2,3 = 5.2 Hz, 1H, H-1), 3.92 (m, 2H), 3.50 (m, 6H);
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 144.55 (C=N), 69.74, 69.28, 69.14, 68.97 (C-3, C-4, C-5, C-6), 63.20 (C-7), 52.98 (C-2).
2-Acetamido-3,4,5,6,7-penta-O-acetyl-2-deoxy-d-glycero-l-gluco-heptononitrile (51). 2-Amino-2-deoxy-d-glycero-l-gluco-heptose oxime hydrochloride (49) (0.15 g, 0.6 mmol) in pyridine (1.0 mL) was treated with acetic anhydride (1.1 mL). The mixture was stirred for 24 h at room temperature. After this time, it was poured into ice–water and a white solid was separated by stirring and scraping. It was filtered, washed with cold water, and dried under a vacuum over silica gel (11.6%). The aqueous phase was extracted with dichloromethane (3 × 10 mL) and the organic phase was washed successively with 1M hydrochloric acid (2 × 10 mL), saturated sodium bicarbonate solution (2 × 10 mL), and distilled water, and dried with anhydrous MgSO4. The solvent was removed in vacuo to dryness and the resulting residue was crystallized from ethanol. After being stored in the refrigerator for several hours, it was filtered, washed with cold ethanol, and dried under a vacuum over silica gel (8.1%) (total yield 19.7%). Crystallized from ethanol, it formed colorless prisms, m. p. 154–156 °C; [α]D +22.8°; [α]578 +24.0°; [α]546 +25.5°; [α]436 +40.6° (c 0.5, chloroform); IR (KBr) ṽmax/cm−1 3278 (NH), 2251 (C≡N), 1743, 1690, 1674 (C=O), 1274, 1222 (C-O-C), 1077, 1046, (C-O); Raman ṽmax/cm−1 2245 (C≡N), 1734, 1679 (C=O); 1H NMR (500 MHz, CDCl3) δ 6.67 (d, JNH,2 = 9.5 Hz, 1H, NH), 5.47 (dd, J2,3 = 6.5 Hz, JNH,2 = 10.0 Hz, 1H, H-2), 5.44 (dd, J3,4 = 2.0 Hz, J4,5 = 10.0 Hz, 1H, H-4), 5.39 (ddd, J5,6 = 2.0 Hz, J6,7 = 5.0 Hz, J6,7′ = 7.5 Hz, 1H, H-6), 5.26 (dd, J5,6 = 1.5 Hz, J4,5 = 10.0 Hz, 1H, H-5), 4.91 (dd, J3,4 = 1.5 Hz, J2,3 = 6.5 Hz, 1H, H-3), 4.26 (dd, J6,7 = 5.0 Hz, J7,7′ = 12.0 Hz, 1H, H-7), 3.85 (dd, J6,7′ = 7.5 Hz, J7,7′ = 11.5 Hz, 1H, H-7′), 2.23, 2.17, 2.10, 2.09, 2.07, 2.02 (s, 18H, CH3); 13C{1H} NMR (125 MHz, CDCl3) δ 173.01, 170.31, 170.06, 169.61, 169.49, 169.07 (C=O), 115.75 (C≡N), 67.50, 67.09, 66.71, 66.50 (C-3, C-4, C-5, C-6), 61.90 (C-7), 38.97 (C-2), 22.95, 21.13, 20.62, 20.57, 20.45 (CH3). HRMS-(ESI-TOF) m/z [M + H]+ calcd. for C19H27N2O11: 459.1615. Found: 459.1629.
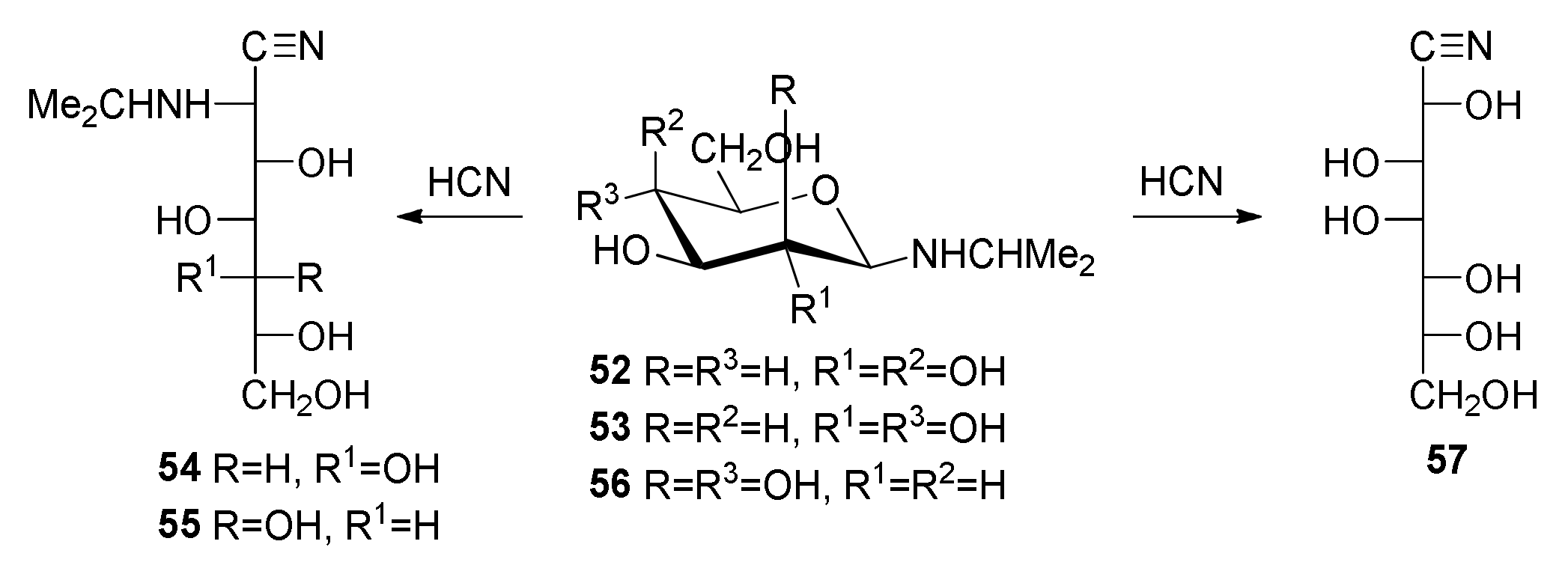
2-Deoxy-2-(
N-isopropylamino)-
d-
glycero-
l-
gluco-heptononitrile (
54) [
50]. White solid crystallized from ethanol, m. p. 130–132 °C (dec.); [α]
D −13.0° (
c 0.5, pyridine); IR (KBr) ṽ
max/cm
−1 3500–3200 (OH, NH), 2210 (C≡N), 1280, 1090, 1040, 1020 (C-O);
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 120.99 (C≡N), 71.69, 70.73 (2C), 69.83 (C-3, C-4, C-5, C-6), 63.95 (C-7), 52.16 (CH, isopropyl), 47.82 (C-2), 23.90, 22.47 (CH
3).
2-Deoxy-2-(
N-isopropylamino)-
d-
glycero-
d-
ido-heptononitrile (
55) and 2-deoxy-2- (
N-isopropylamino)-
d-
glycero-
d-
gulo-heptononitrile (
60) [
50]. White solid crystallized from methanol, m. p. 132–134 °C (dec.). Spectroscopic data of
55:
13C{
1H} NMR (50 MHz, DMSO-
d6) δ 120.29 (C≡N), 72.75, 71.20, 70.04, 69.63 (C-3, C-4, C-5, C-6), 63.67 (C-7), 49.16 (C-2), 46.56 (CH, isopropyl), 23.40, 21.11 (CH
3, isopropyl). Spectroscopic data of
60:
13C{
1H} NMR (50 MHz, DMSO-
d6) 119.98 (C≡N), 71.81, 71.33, 69.63, 69.23 (C-3, C-4, C-5, C-6), 63.45 (C-7), 50.90 (C-2), 46.25 (CH isopr.), 23.57, 21.49 (CH
3, isopropyl).
N-Isopropyl-β-d-mannopyranosylamine (56). To a suspension of d-mannose (5.0 g, 27.7 mmol) in methanol (50 mL), isopropylamine (5.2 mL, 60.0 mmol) was added and the mixture was stirred and heated until dissolution. The solvent was partially evaporated, which often results in crystal formation. When the latter did not occur, the mixture was concentrated and the residue was solidified by triturating it with diethyl ether. The white solid was filtered, washed with ether, and dried in vacuo (5.96 g, 97%). Crystallized from absolute ethanol–diethyl ether, it showed an m. p. of 108–109 °C (dec.); [α]D20 −21° (c 0.5, NH4OH-H2O 1:9); [α]D20 −17° (c 0.5, HCl 2.5 M). IR (KBr) ṽmax/cm−1 3500–3000 (OH), 3280 (NH), 1075, 1045, 1025 (C-O). Anal. Calcd. for C9H19NO5: C, 48.86; H, 8.64; N, 6.33. Found: C, 48.66; H, 8.50; N, 6.11.
d-
glycero-
d-
galacto-Heptononitrile (
57) [
99]. Method (a): To a suspension of
d-mannose (5.4 g, 30.0 mmol) in ethanol (50 mL), isopropylamine (5.2 mL, 60.0 mmol) was added and the mixture was heated until dissolution. After cooling to room temperature, anhydrous hydrogen cyanide (3.0 mL, 80 mmol) was added. A white crystalline solid separated, which was filtered, washed with cold ethanol and diethyl ether, and dried in vacuo (1.1 g, 17%). Crystallized from methanol, it showed an m. p. of 112–113 °C (dec.). Method (b): To a solution of the
N-isopropyl-β-
d-mannopyranosylamine (8.0 g, 36.0 mmol) in ethanol (32 mL), anhydrous hydrogen cyanide (2.8 mL, 74.6 mmol) was added. The reaction mixture was kept overnight in the refrigerator. The resulting white crystalline solid was filtered, washed with cold ethanol and diethyl ether, dried in vacuo (3.4 g, 45%), and recrystallized from methanol. The white solid had an m. p. of 113–115 °C (dec.); [α]
D20 +10.4° (
c. 0.5, pyridine) [Lit. [
99] m. p. 122 °C; [α]
D20 +10.5° (
c. 0.5, pyridine)]. IR (KBr) ṽ
max/cm
−1 3500–3100 (OH), 2220 (C≡N), 1400, 1070, 1000 (C-O);
13C{
1H} NMR (125 MHz, DMSO-
d6) δ 171.72, 170.26, 170.19, 170.04, 169.47 (C=O), 117.87 (C≡N), 68.45, 67.23, 67.18, 65.93 (C-3, C-4, C-5, C-6), 61.79 (C-7), 47.17 (C-2).
3,4,5,6,7-Penta-O-acetyl-2-deoxy-2-(N-isopropylamino)-d-glycero-l-gluco-heptononitrile (58). To a suspension of 2-deoxy-2-(N-isopropylamino)-d-glycero-l-gluco-heptononitrile (54) (0.25 g, 1.0 mmol) in pyridine (1.7 mL), acetic anhydride (1.4 mL) was added and the mixture was kept under stirring until dissolution, and then at 0 °C for 24 h. After this time, the solution was poured onto ice–water and a white solid was separated, which was filtered and washed with cold water (0.38 g, 64%). Crystallized from 96% ethanol, the white solid presented an m. p. of 125–127 °C; [α]D22 +17.2° (c 1.5, pyridine). IR (KBr) ṽmax/cm−1 3330 (NH), 2980, 1740 (C=O), 1365, 1210 (C-O-C), 1040 (C-O); 1H NMR (200 MHz, CDCl3) δ 5.42–5.34 (m, 3H, H-4, H-5, H-6), 4.90 (d, J2,3 = 6.0 Hz, J3,4 = 0 Hz, 1H, H-3), 4.27 (dd, J6,7 = 5.0 Hz, J7,7′ = 11.7 Hz, 1H, H-7), 3.90 (d, J2,3 = 6.0 Hz, 1H, H-2), 3.86 (dd, J6,7′ = 7.2 Hz, J7,7′ = 11.7 Hz, 1H, H-7′), 2.97 (sep, J = 6.2 Hz, 1H, CH isopropyl), 2.18, 2.17, 2.11, 2.09, 2.02 (s, 15H, CH3), 1.13 (d, J = 6.2 Hz, 3H, CH3 isopropyl), 1.01 (d, J = 6.2 Hz, 3H, CH3 isopropyl); 13C{1H} NMR (50 MHz, CDCl3) δ 171.72, 170.26, 170.19, 170.04, 169.47 (C=O), 117.87 (C≡N), 68.45, 67.23, 67.18, 65.93 (C-3, C-4, C-5, C-6), 61.79 (C-7), 47.17 (C-2), 23.24, 21.15, 20.95, 20.53, 20.45, 20.36, 20.28 (CH3). Anal. Calcd. for C20H30N2O10: C, 52.40; H, 6.60; N, 6.11. Found: C, 52.21; H, 6.45; N, 5.90.
3,4,5,6,7-Penta-O-acetyl-2-deoxy-2-(N-isopropylamino)-d-glycero-d-ido-heptononitrile (59) and 3,4,5,6,7-penta-O-acetyl-2-deoxy-2-(N-isopropylamino)-d-glycero-d-gulo- heptononitrile (61). White solid obtained from 55/60, using the procedure described for 58, as a mixture of 59 and 61 in a proportion of ~80:20. IR (KBr) ṽmax/cm−1 3330 (NH), 2980, 1740 (C=O), 1365, 1210 (C-O-C), 1040 (C-O). Spectroscopic data of 59: 1H NMR (200 MHz, CDCl3) δ 5.66 (dd, J3,4 = 6.7 Hz, J4,5 = 3.0 Hz, 1H, H-4), 5.37 (dd, J4,5 = 3.0 Hz, J5,6 = 7.7 Hz, 1H, H-5), 5.23 (dd, J2,3 = 3.8 Hz, J3,4 = 6.7 Hz, 1H, H-3), 4.99 (ddd, J5,6 = 7.7 Hz, J6,7 = 3.2 Hz, J6,7′ = 4.0 Hz, 1H, H-6), 4.23 (dd, J6,7 = 3.2 Hz, J7,7″ = 12.2 Hz, H-7), 4.13 (dd, J6,7′ = 4.0 Hz, J7,7′ = 12.2 Hz, 1H, H-7′), 4.10 (d, J2,3 = 3.8 Hz, 1H, H-2), 3.04 (sep, 1H, CH, isopropyl), 2.17, 2.14, 2.08, 2.05 (s, 15H, CH3, acetate), 1.17 (d, J = 6.2 Hz, 3H, CH3, isopropyl), 1.10 (d, J = 6.2 Hz, 3H, CH3, isopropyl); 13C{1H} NMR (50 MHz, CDCl3) δ 170.00, 169.93, 169.78, 169.24, 168.95 (C=O), 117.26 (C≡N), 70.30, 67.73 (3C) (C-3, C-4, C-5, C-6), 60.66 (C-7), 47.56 (C-2), 46.53 (CH, isopropyl), 23.15, 20.79 (CH3, isopropyl), 20.39, 20.22, 20.13, 20.03, 19.94 (CH3, acetate). Spectroscopic data of 61: 13C{1H} NMR (50 MHz, CDCl3) δ 170.00, 169.93, 169.88, 169.61, 168.87 (C=O), 117.32 (C≡N), 69.21, 69.12, 68.45, 68.34 (C-3, C-4, C-5, C-6), 60.98 (C-7), 49.14 (C-2), 48.73 (CH, isopropyl), 22.97, 20.71 (CH3, isopropyl), 20.79, 20.71, 20.57, 20.22, 20.03 (CH3, acetate).
2,3,4,5,6,7-Hexa-
O-acetyl-
d-
glycero-
d-
galacto-heptononitrile (
63) [
99]. To a stirred solution of
d-
glycero-
d-
galacto-heptononitrile (
57) (1.0 g, 4.1 mmol) in pyridine (5 mL), cooled externally with an ice bath, acetic anhydride (4.0 mL, 27.1 mmol) was added gradually. The reaction mixture was kept overnight in the refrigerator and, subsequently, it was poured onto a water–ice mixture. The resulting solid was filtered, washed with water, and dried under a vacuum over silica gel (1.65 g, 53%). Crystallized from acetone–water, it showed an m. p. of 163–165 °C; [α]
D22 +28.7°; [α]
578 +29.3°; [α]
546 +32.7°; [α]
436 +55.3° (
c 0.3, chloroform) (Lit. [
99] m. p. 124–125 °C).
1H NMR (500 MHz, CDCl
3) δ 5.52 (dd,
J3,4 = 10.0 Hz,
J4,5 = 2.0 Hz, 1H, H-4), 5.50 (dd,
J2,3 = 3.0 Hz,
J3,4 = 10.0 Hz, 1H, H-3), 5.38 (d,
J2,3 = 3.0 Hz, 1H, H-2), 5.38 (dd,
J4,5 = 2.0 Hz,
J5,6 = 9.0 Hz, 1H, H-5), 5.02 (ddd,
J5,6 = 9.0 Hz,
J6,7 = 3.0 Hz,
J6,7′ = 5.0 Hz, 1H, H-6), 4.20 (dd,
J6,7 = 3.0 Hz,
J7,7″ = 12.5 Hz, H-7), 4.00 (dd,
J6,7′ = 5.0 Hz,
J7,7′ = 12.5 Hz, 1H, H-7′), 2.17 (s, 6H, CH
3 acetate), 2.10, 2.07, 2.05, 2.04 (s, 12H, CH
3, acetate);
13C{
1H} NMR (50 MHz, CDCl
3) δ 170.48, 169.80, 169.56, 169.02, 168.76 (C=O), 113.92 (C≡N), 67.81 (C-6), 66.99, 66.89 (C-3,C-5), 66.48 (C-4), 61.75 (C-7), 59.40 (C-2), 20.80, 20.62, 20.48, 20.45, 20.11 (CH
3, acetate). HRMS-(ESI-TOF)
m/
z [M + NH
4]
+ calcd. for C
19H
29N
2O
12: 477.1720. Found: 477.1730.
2,3,4,5,6,7-Hexa-
O-benzoyl-
d-
glycero-
d-
galacto-heptononitrile (
64) [
99]. To a stirred solution of
d-
glycero-
d-
galacto-heptononitrile (1.0 g, 4.1 mmol) in pyridine (5 mL), cooled externally with an ice bath, benzoyl chloride (4.0 mL, 27.1 mmol) was added gradually. The reaction mixture was stored overnight in the refrigerator and, subsequently, it was poured onto a water–ice mixture and extracted with dichloromethane (3 × 20 mL). The organic layer was washed twice with 3
M sulfuric acid; twice with saturated sodium bicarbonate solution; and finally with distilled water. The organic phase was dried with anhydrous sodium sulfate, filtered through activated charcoal, and evaporated to dryness. The residue was evaporated repeatedly with ethanol. The resulting crystalline solid was dried under a vacuum over calcium chloride (1.65 g, 41%). Crystallized from acetone–water, it showed an m. p. of 165 °C; [α]
D22 +30.4° (c 2.0, chloroform) [Lit. [
99] m. p. 161–162 °C; [α]
D22 +30.3° (
c. 2, chloroform)]. IR (KBr) ṽ
max/cm
−1 1715, 1690 (C=O), 1590, 1570, 1435 (phenyl), 1240 (C-O-C), 1070, 1045 (C-O);
1H NMR (200 MHz, CDCl
3) δ 8.04–7.87 (m, 12H, H-arom), 7.56–7.25 (m, 18H, H-arom), 6.31 (dd,
J3,4 = 7.9 Hz,
J4,5 = 2,1 Hz, 1H, H-4), 6.20 (d,
J4,5 = 2.2 Hz,
J5,6 = 7.8 Hz, 2H, H-5), 6.13 (dd,
J2,3 = 4.2 Hz,
J3,4= 7.8 Hz, 1H, H-3), 5.96 (d,
J2,3 = 4.2 Hz, 1H, H-2), 5.79 (m, 1H, H-6), 4.85 (dd,
J6,7 = 3.4 Hz,
J7,7′ = 12.4 Hz, 1H, H-7), 4.45 (dd,
J6,7′ = 5.4 Hz,
J7,7′ = 12.4 Hz, 1H, H-7′);
13C{
1H} NMR (50 MHz, CDCl
3) δ 165.87, 165.21, 164.98, 164.81, 164.57, 164.18 (C=O),134.01, 133.79, 133.25, 133.05, 130.00, 129.91, 129.68, 129.27, 129.09, 128.73, 128.54, 128.49, 128.28, 127.42 (C-arom), 114.05 (C≡N), 69.39, 68.81, 68.22 (C-3,4,5), 62.35 (C-7), 60.67 (C-2).
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}