1. Introduction
Polysaccharides, one of the major classes of natural polymers [
1,
2], have been used for the fabrication of bio-based nanomaterials such as nanoparticles, nanofibers, and nanoscopic hydrogels (nanogels) via the regular assembly of polymeric chains because of their controlled and specific structures [
3,
4,
5,
6]. Polysaccharides are polymers comprising monosaccharide residues linked through glycosidic bonds with regular regio- and stereo-arrangements. Water-soluble polymers such as some polysaccharides and other crosslinkable macromolecules are the most common systems to form hydrogels through the chemical or physical crosslinking of polymeric chains [
7]. Polymeric hydrogels exhibit no solubility in aqueous media owing to their network structures and have high water-holding capacity for developing medical applications [
8,
9]. Particularly, nanogels, which are hydrogels composed of submicron-sized three-dimensional polymeric networks, have increasingly attracted much attention as functional nanomaterials [
10]. Polysaccharide-based nanogels have previously been fabricated via nanoscale network construction from polysaccharide chains [
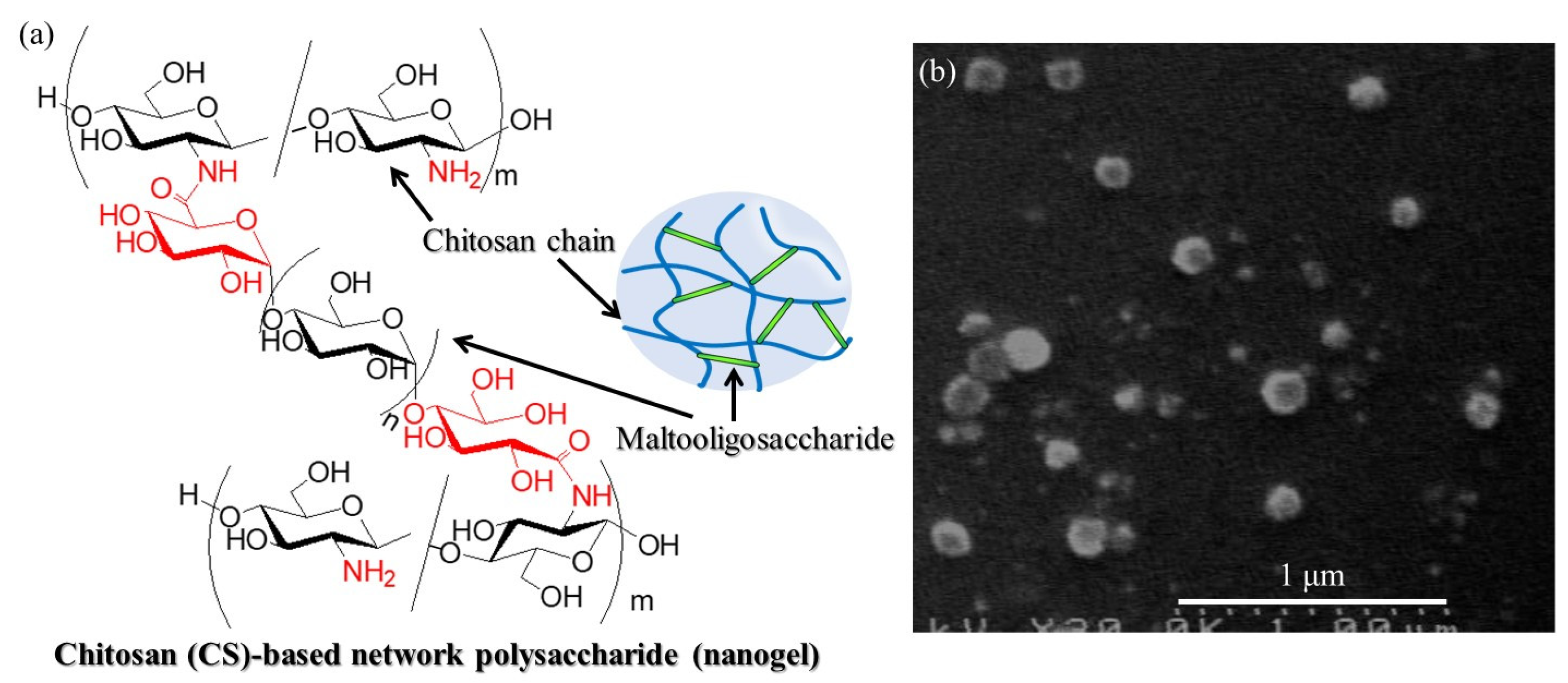
11]. For example, chitosan (CS)-based network polysaccharide nanogels have been prepared, where the chemical structure and nanoscale morphology in the SEM image are shown in
Figure 1a,b, respectively [
12]. CS is a β(1→4)-linked
d-glucosamine (GlcN) polymer and has thus a number of amino groups generated by the deacetylation of
N-acetyl-
d-glucosamine units in an abundant natural polysaccharide, chitin [
13]. Therefore, condensation of water-soluble chitosan (WSCS) with maltooligosaccharides with carboxylate groups at both ends (carboxylate-terminated maltooligosaccharides, GlcA-Glc
n-COONa,
Figure S1) as crosslinkers was conducted in the presence of a condensing agent for chemical crosslinking to construct three-dimensional polymeric networks on the nanoscale. WSCS is a relatively low-molecular-weight CS (approximately 1.0–5.0 × 10
4), which can be prepared by the complete deacetylation of chitin under alkaline conditions and subsequent oxidative depolymerization by treatment with hydrogen peroxide (
Figure S2) [
14,
15].
The preparation procedure for GlcA-Glc
n-COONa has been established in previous studies by means of thermostable glucan phosphorylase (GP, isolated from thermophilic bacteria
Aquifex aeolicus VF5)-catalyzed enzymatic α-glucuronylation using α-
d-glucuronic acid 1-phosphate (GlcA-1-P) and a maltooligosaccharide with a carboxylate group (Glc
6-GlcCOONa) [
16] as glycosyl donor and acceptor, respectively (
Figure S1); the acceptor was obtained by oxidation at the reducing end of maltoheptaose (Glc
7) in the presence of I
2/NaOH [
17]. GP is an enzyme that catalyzes consecutive glycosylations using α-
d-glucose 1-phosphate (Glc-1-P) and maltooligosaccharides as glycosyl donors and acceptors, respectively, according to enzymatic polymerization manner, to produce α(1→4)-glucan, amylose [
18]; the former and latter substrates act as monomers and primers, respectively, in the polymerization. GP shows weak specificity for glycosyl donors, in which non-native sugar 1-phosphate glycosyl donors are recognized as analog substrates of native Glc-1-P, such as GlcA-1-P, in glycosylations with the maltooligosaccharide acceptor to introduce the corresponding sugar residues at the non-reducing end of the acceptor [
19,
20,
21,
22]. Subsequently, GlcA-Glc
n-COONa, prepared by thermostable GP-catalyzed enzymatic α-glucuronylation using GlcA-1-P and Glc
6-GlcCOONa, was used as the crosslinker for the condensation of amino groups in WSCS in the presence of a condensing agent in water to fabricate CS-based network polysaccharides.
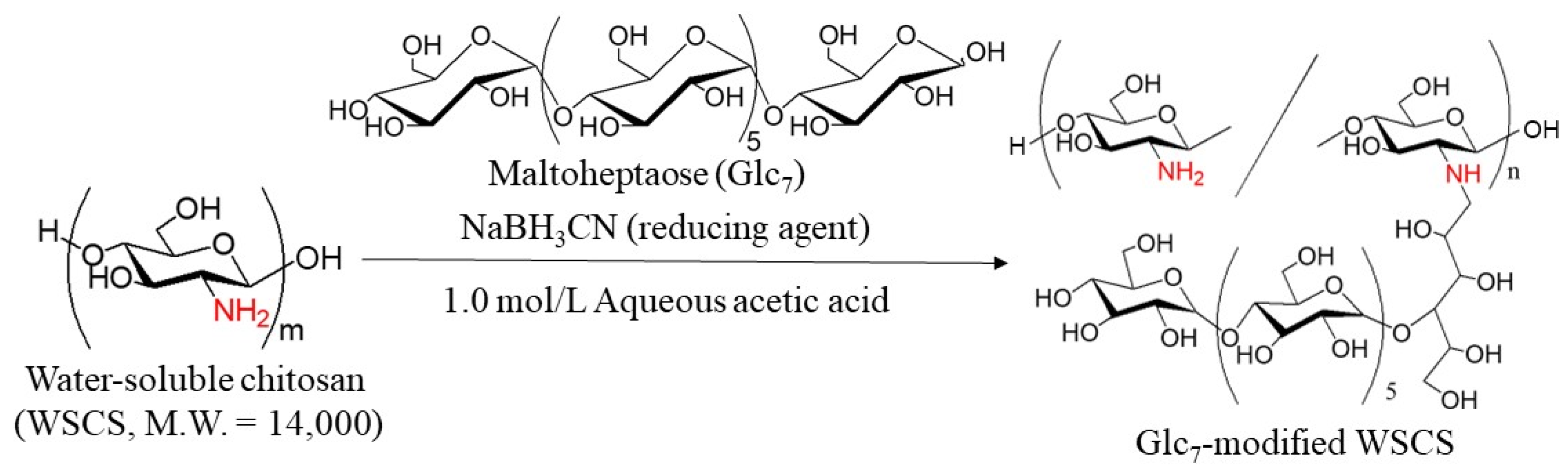
In this study, GP-catalyzed enzymatic assembly of CS-based network polysaccharides was attempted according to the following procedure. Glc
7 primers for GP catalysis were introduced into WSCS by reductive amination using NaBH
3CN as a reducing agent to produce Glc
7-modified WSCS (
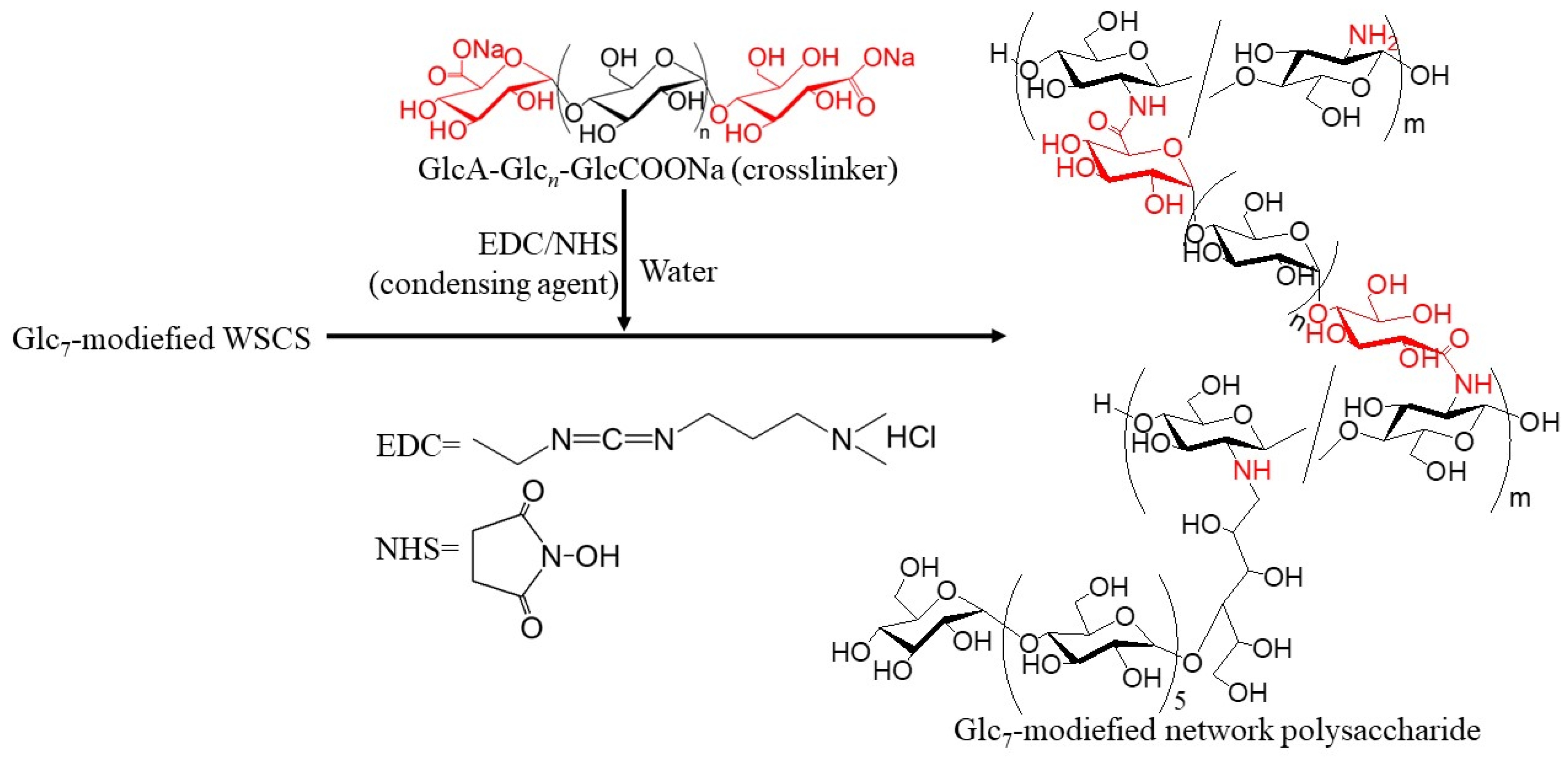
Figure 2). The product was then converted into a network structure via condensation with GlcA-Glc
n-COONa in the presence of a condensing agent (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)/
N-hydroxysuccinimide (NHS)) (
Figure 3). Thermostable GP-catalyzed enzymatic polymerization of Glc-1-P was performed using the Glc
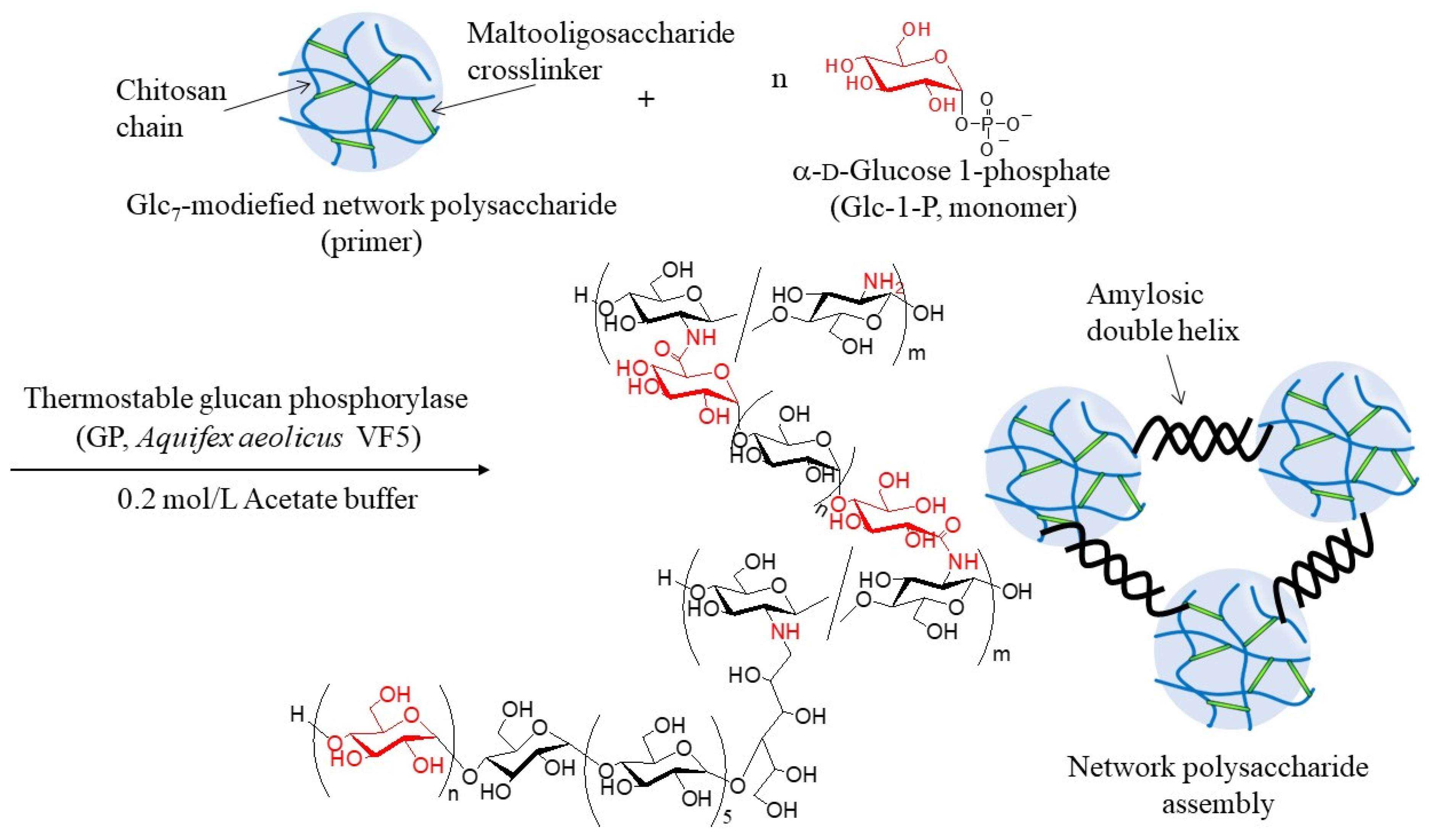
7 primer ends present on WSCS to elongate the amylosic chains (
Figure 4). The elongated chains form well-known amylosic double helices among the network of polysaccharides to produce the assembled products. Depending on the enzymatic polymerization conditions, hydrogels and films formed from the assembled products. Disintegration of the obtained assembly was also conducted by α-amylase-catalyzed enzymatic hydrolysis of the double helical amyloses among the network polysaccharides. Accordingly, the encapsulation and release behaviors of a fluorescent dye, Rhodamine B, using CS-based network polysaccharides were investigated by GP-catalyzed assembly and α-amylase-catalyzed disintegration approaches, respectively.
2. Results and Discussion
The Glc
7 primers were first modified on WSCS, which was prepared by the complete deacetylation of chitin under alkaline conditions and subsequent oxidative depolymerization using hydrogen peroxide (
Figure S2) [
23] by reductive amination in the presence of NaBH
3CN (
Figure 2) [
24]. The reaction was performed in two different feed ratios of amino groups in WSCS, Glc
7, and NaBH
3CN (1:1:5 and 1:2:5, runs 1–3, see
Table 1 below) in 1.0 mol/L aqueous acetic acid for 48 h at 50 °C. Different concentrations of reaction solutions were also employed (12, 18, and 24 mmol/L GlcN units). The products were isolated as insoluble fractions in methanol.
1H NMR analysis was conducted in DCl/D
2O after complete dissolution by acidic hydrolysis of glycosidic linkages in the products because the products were insoluble in common NMR solvents.
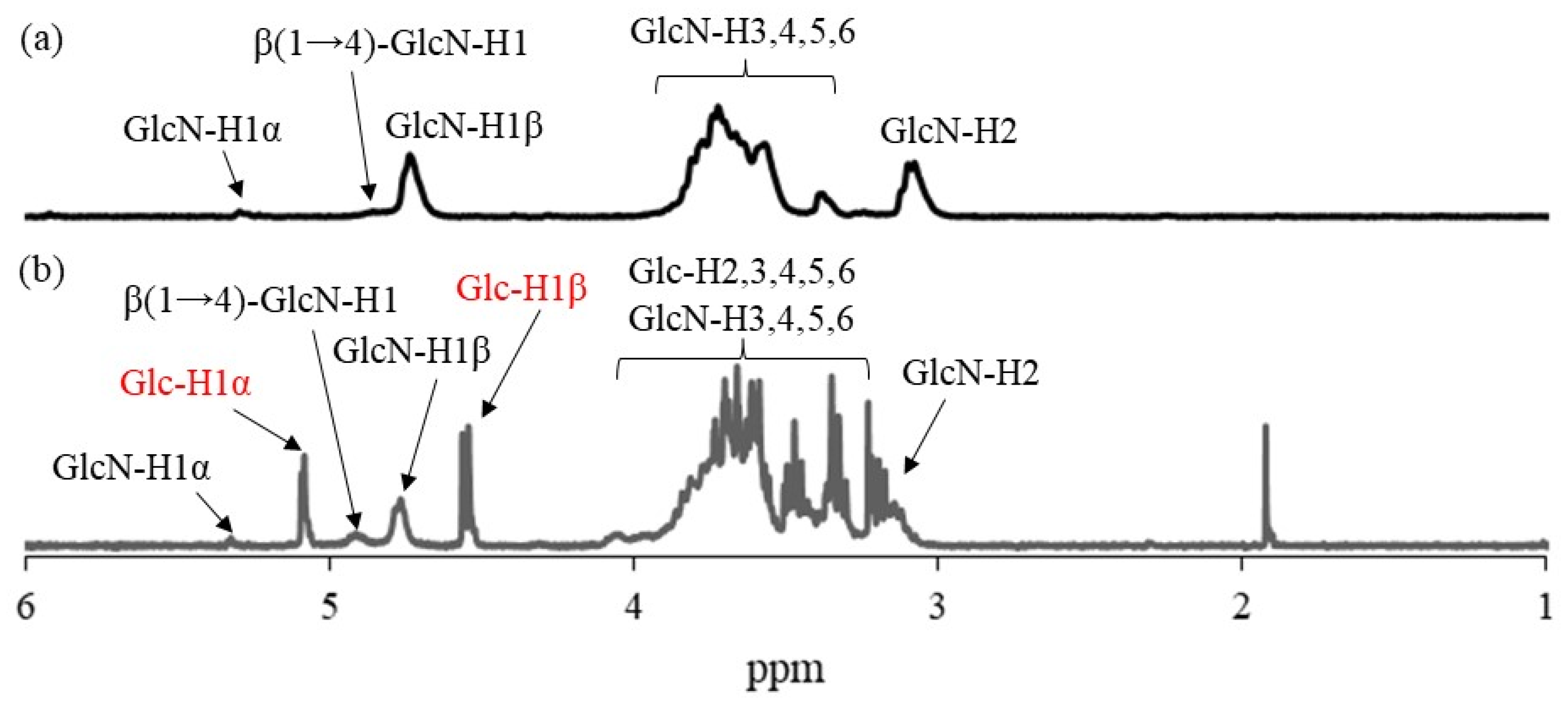
Figure 5 shows the comparison of the
1H NMR spectra of the hydrolysate from run 1 and WSCS in DCl/D
2O. In addition to the signals ascribed to GlcN residues detected in
Figure 5a, the anomeric signals derived from Glc residues (labeled with red characters) produced by acidic hydrolysis of the Glc
7 chains were observed at δ 4.76 and 5.08 in the
1H NMR spectrum of the hydrolysate (
Figure 5b). This NMR result indicates the successful modification of Glc
7 primers on WSCS by reductive amination to obtain Glc
7-modified WSCS. The degree of substitution (DSs) of Glc
7 with the GlcN repeating unit for the product of run 1 was calculated as 1.9% by integrating the ratio of the Glc anomeric signals to the GlcN-H2 signals. The DS values increased according to the GlcN unit concentration (2.7 and 4.2% for the products of runs 2 and 3, respectively).
The residual amino groups in the obtained Glc
7-modified WSCS were subjected to a condensation reaction with the GlcA-Glc
n-COONa crosslinker in water, according to the previously reported procedure, using EDC/NHS as the condensing agent to produce Glc
7-modified network polysaccharides (
Figure 3) [
12]. An aqueous solution of GlcA-Glc
n-COONa, EDC, and NHS at a molar ratio of 0.5:1:1 (carboxylate/condensing agent =1:1) was added to an aqueous solution of Glc
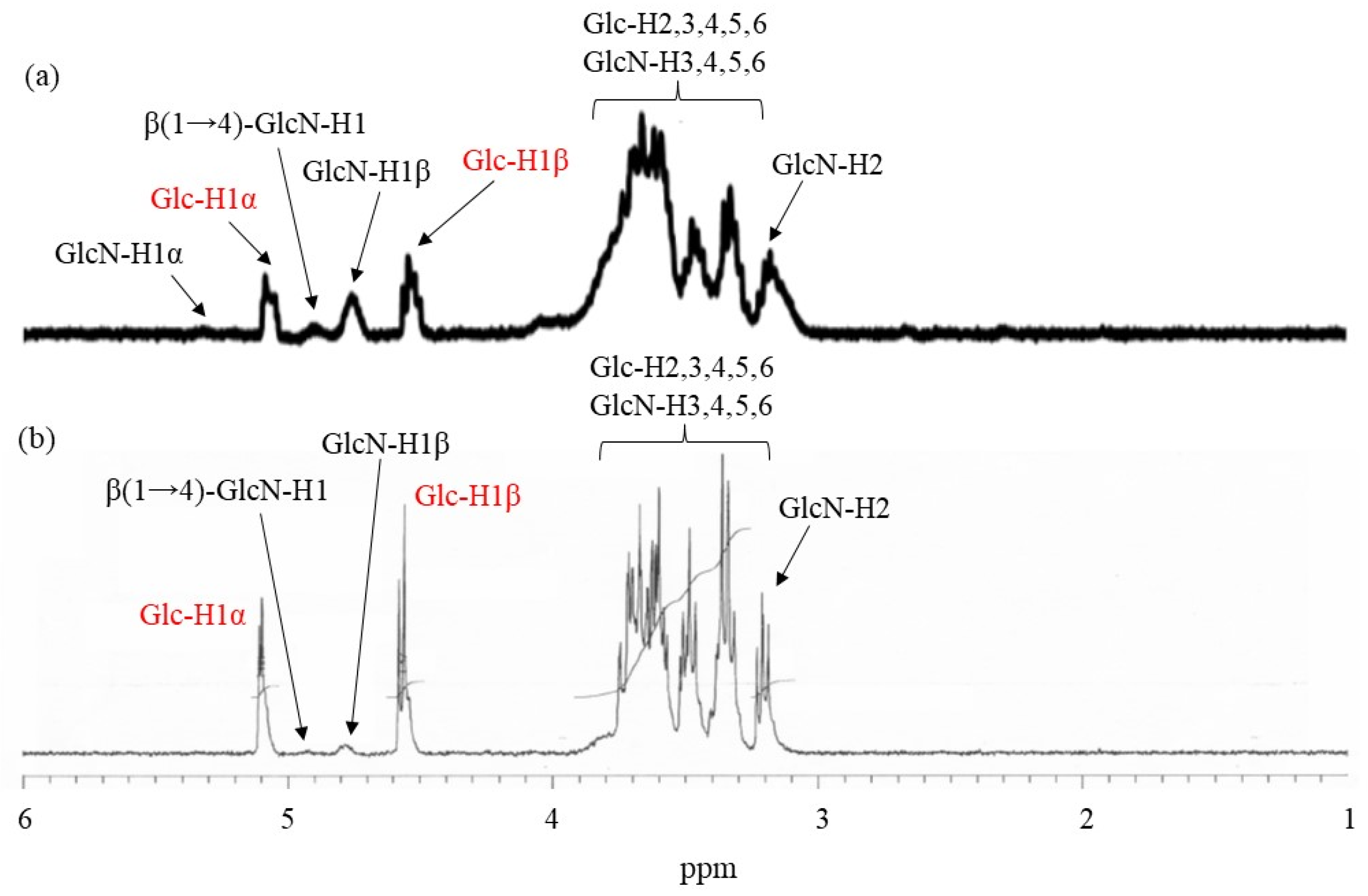
7-modified WSCS (runs 1–3) at a molar ratio of carboxylate to amino groups of 0.05:1, and the mixture was kept for 7 h at room temperature. The products were isolated as insoluble fractions in methanol. The
1H NMR spectrum of the product obtained using Glc
7-modified WSCS from run 1, which was prepared by acid hydrolysis and dissolution in DCl/D
2O (
Figure 6a), indicated a slight increase in the integrated ratio of the Glc anomeric signals (labeled by red characters) to the GlcN anomeric signals compared with that of Glc
7-modified WSCS (
Figure 5b). These data suggest the presence of crosslinker chains composed of Glc residues via a condensation reaction with amino groups in WSCS. For example, the ratio of Glc
7 to the GlcN repeating unit was calculated to be 1.1% using
1H NMR analysis.
The thermostable GP-catalyzed enzymatic polymerization of Glc-1-P was then performed with the Glc
7 primers (DSs = 1.2, 2.7, and 4.2%, runs 1–3) on the obtained network polysaccharides in 0.2 mol/L acetate buffer for 48 h at 50 °C to assemble them via amylosic double helix formation (
Figure 4). Varying Glc-1-P (monomer)/Glc
7 (primer) feed ratios were employed, as listed in
Table 2, because this ratio regulates the molecular weight of the enzymatically produced amylose. As the reaction mixtures at a low feed ratio (i.e., 100) maintained the solution state as polymerization progressed (runs 4, 7, and 8), the products were isolated by dialysis and lyophilization of the reaction mixtures. With an increase in the feed ratio (runs 5, 6, and 9), the reaction mixtures became viscous. Consequently, the reaction mixture turned into a gel at a feed ratio of 500, with a larger DS value of Glc
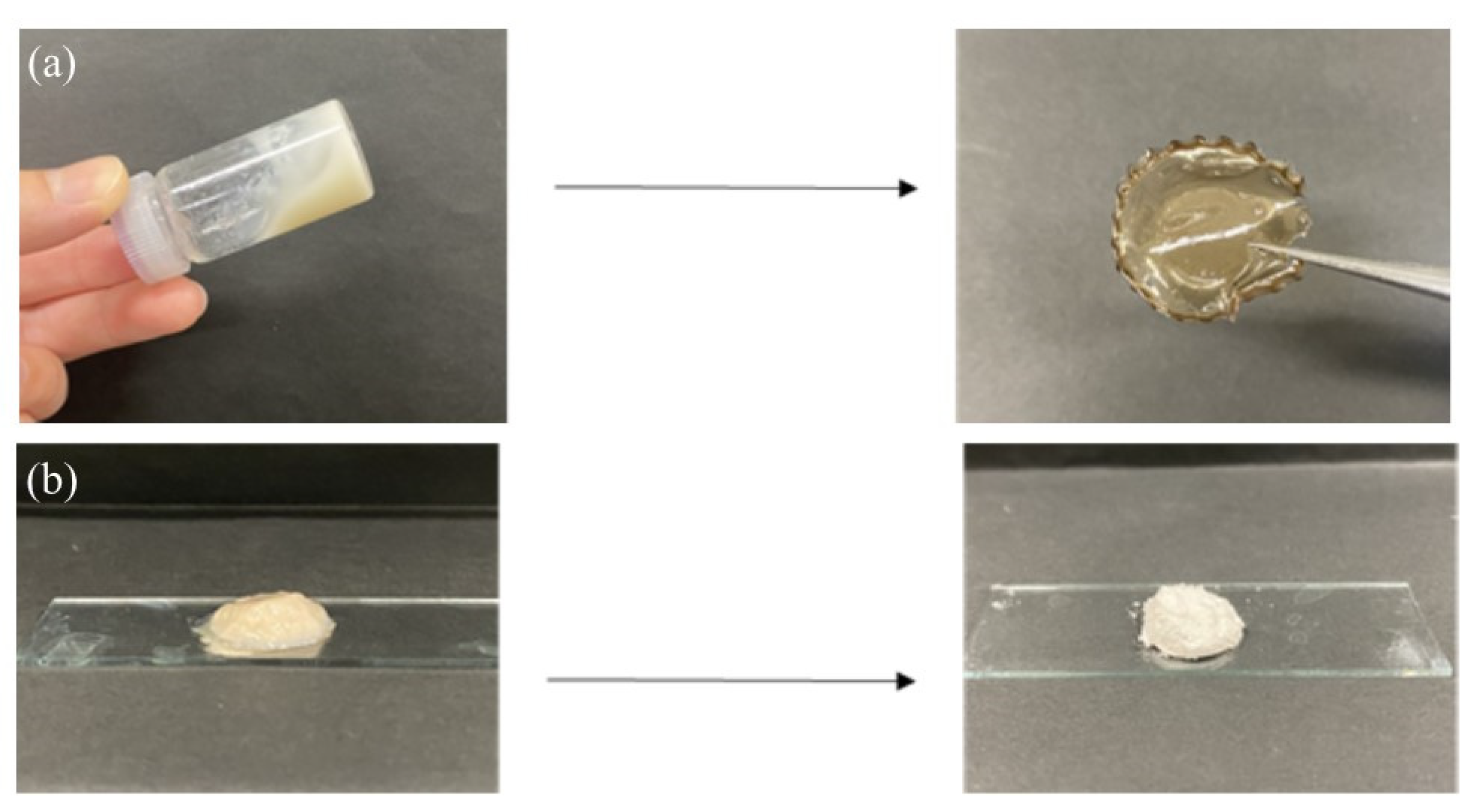
7 (4.2%, run 10). These viscous and gel products were washed with water and lyophilized for purification and isolation. Furthermore, films were formed by casting and drying DMSO solutions of these products, and treatment of the gelling reaction mixture of run 10 with water produced a self-standing hydrogel, as shown in
Figure 7a,b, respectively. Viscous and gel mixtures were produced by GP-catalyzed enzymatic polymerization in the higher monomer (Glc-1-P)/primer (Glc
7) feed ratios and the larger DS values of the Glc
7 primers on WSCS; amyloses with higher molecular weights and larger assembly densities were produced via amylosic double helix formation. The
1H NMR spectroscopic pattern of the samples prepared by acidic hydrolysis and dissolution in DCl/D
2O, representatively shown as the spectrum of the sample from the product of run 4 (
Figure 6b), is similar to that of the Glc
7-modified network WSCS (
Figure 6a); however, the intensities of the Glc anomeric signals (labeled with red characters) to the GlcN anomeric signals in the former spectrum were much higher than those of the latter. The NMR results strongly supported the progress of GP-catalyzed enzymatic polymerization to introduce amylose chains into the network of polysaccharides.
The SEM image of the enzymatic polymerization product (run 4,
Figure 8a) demonstrates the assembling morphology of nanoparticles, suggesting that the elongated amylose chains form double helices among the network polysaccharides; the SEM image of the network polysaccharide nanoparticles is shown in
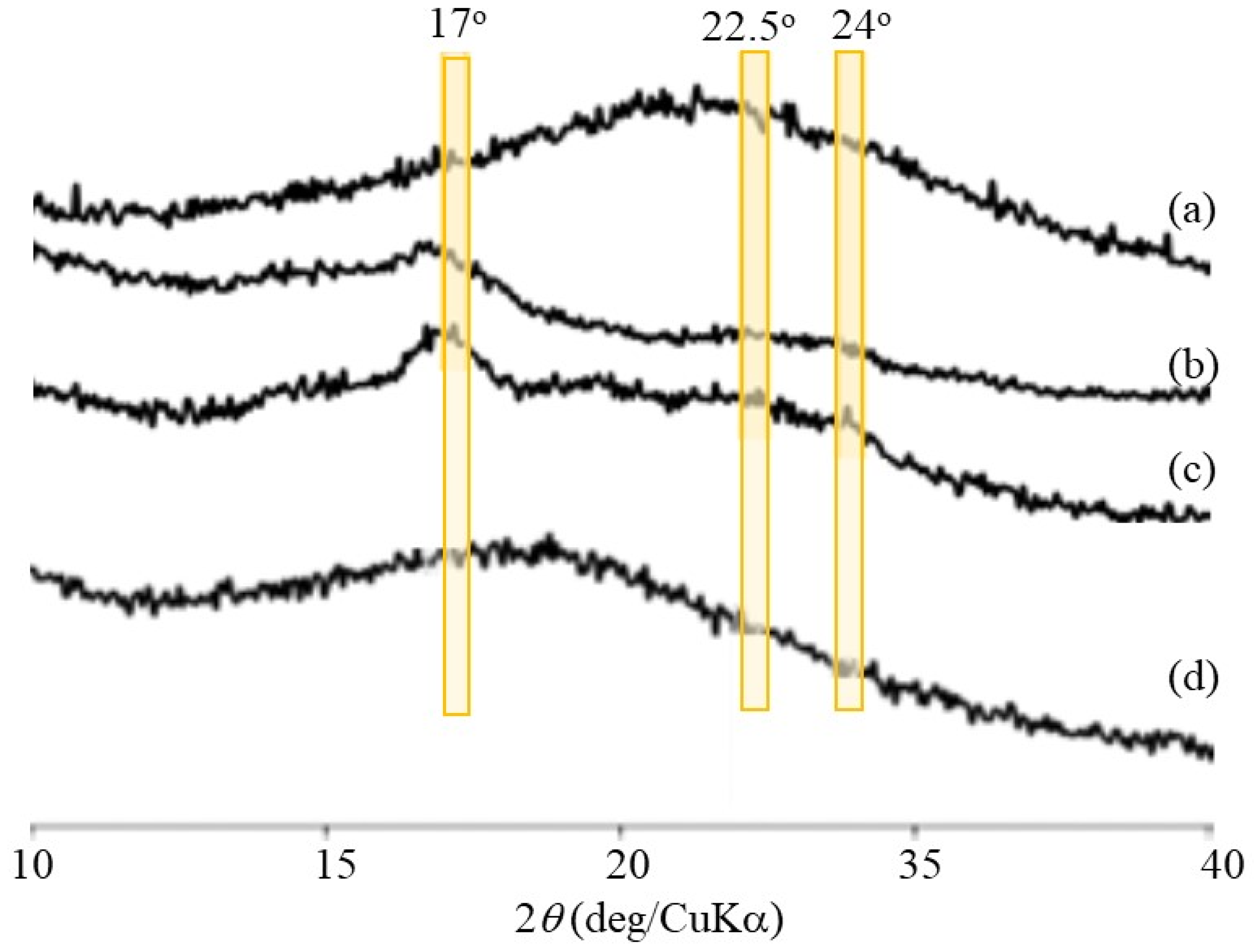
Figure 1b. Powder X-ray diffraction (XRD) measurements were conducted to confirm the presence of a double helix structure in the polysaccharide network assembly. The XRD profile of the Glc
7-modified WSCS did not exhibit any diffraction pattern owing to its amorphous nature (
Figure 9a). In contrast, the XRD profile of the enzymatic polymerization product (run 4) in
Figure 9c clearly shows a typical diffraction pattern (17°, 22.5°, and 24°) assignable to the amylosic double helical crystalline structure, similar to that of a pure amylose sample (
Figure 9b). These XRD results indicate that the CS-based network polysaccharides were efficiently assembled via double helix formation by the amylose chains during GP-catalyzed enzymatic polymerization.
Accordingly, disintegration of the network polysaccharide assembly (run 7) was attempted by α-amylase-catalyzed enzymatic hydrolysis of the double helical amyloses; this is an exo-enzyme that catalyzes random hydrolysis of α-glucan chains, i.e., amylose to produce disaccharides and maltoses. The network polysaccharide assembly (run 7) was treated with α-amylase (from
Bacillus amyloliquefaciens) in 0.2 mol/L acetate buffer at 40 °C, and the reaction mixtures were taken every 2 h. The SEM images of the spin-coated samples clearly show the gradual disintegration of the assembly morphology with prolonged reaction times (
Figure 8b,c). After 6 h of enzymatic treatment, the morphology of the individual nanoparticles was observed using SEM (
Figure 8d). The XRD profile of the sample obtained by dialyzing the reaction mixture after 48 h did not exhibit any amylosic double helical diffraction pattern (
Figure 9d), which was different from that before enzymatic treatment (
Figure 9c). The above results suggest that α-amylase-catalyzed enzymatic hydrolysis of the double helical amyloses efficiently occurred to disintegrate the network polysaccharide assembly.
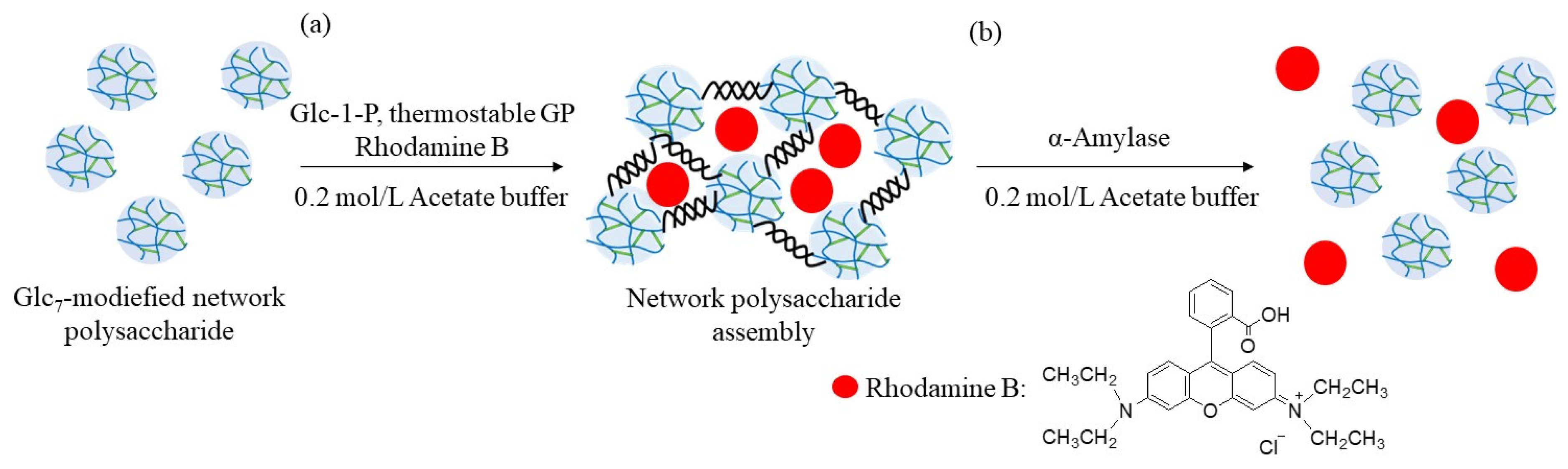
The encapsulation and release behaviors of the fluorescent dye were investigated using the above enzymatic assembly and disintegration processes (
Figure 10). Rhodamine B was selected as the fluorescent dye because it has the ability to form electrostatic interactions with polysaccharides with amino groups (such as WSCS), owing to the presence of an acidic carboxylic acid group, as shown in
Figure 10. Therefore, the abovementioned enzymatic assembly experiment using the Glc
7-modified network polysaccharide of run 7 by thermostable GP-catalyzed enzymatic polymerization was performed in the presence of Rhodamine B (0.2 equiv. with residual amino groups in WSCS) in 0.2 mol/L acetate buffer for 48 h at 50 °C (
Figure 10a). The reaction mixture was dialyzed, and the concentrated dialysate was lyophilized to give the assembling product encapsulating Rhodamine B. The amount of dye present in the outer filtrate was calculated by its fluorescence spectrum when excited at 555 nm. Therefore, the amount of the encapsulated dye in the assembling product was 38% (2.2 mg/6.0 mg), owing to electrostatic interactions between the amino and carboxylic acid groups.
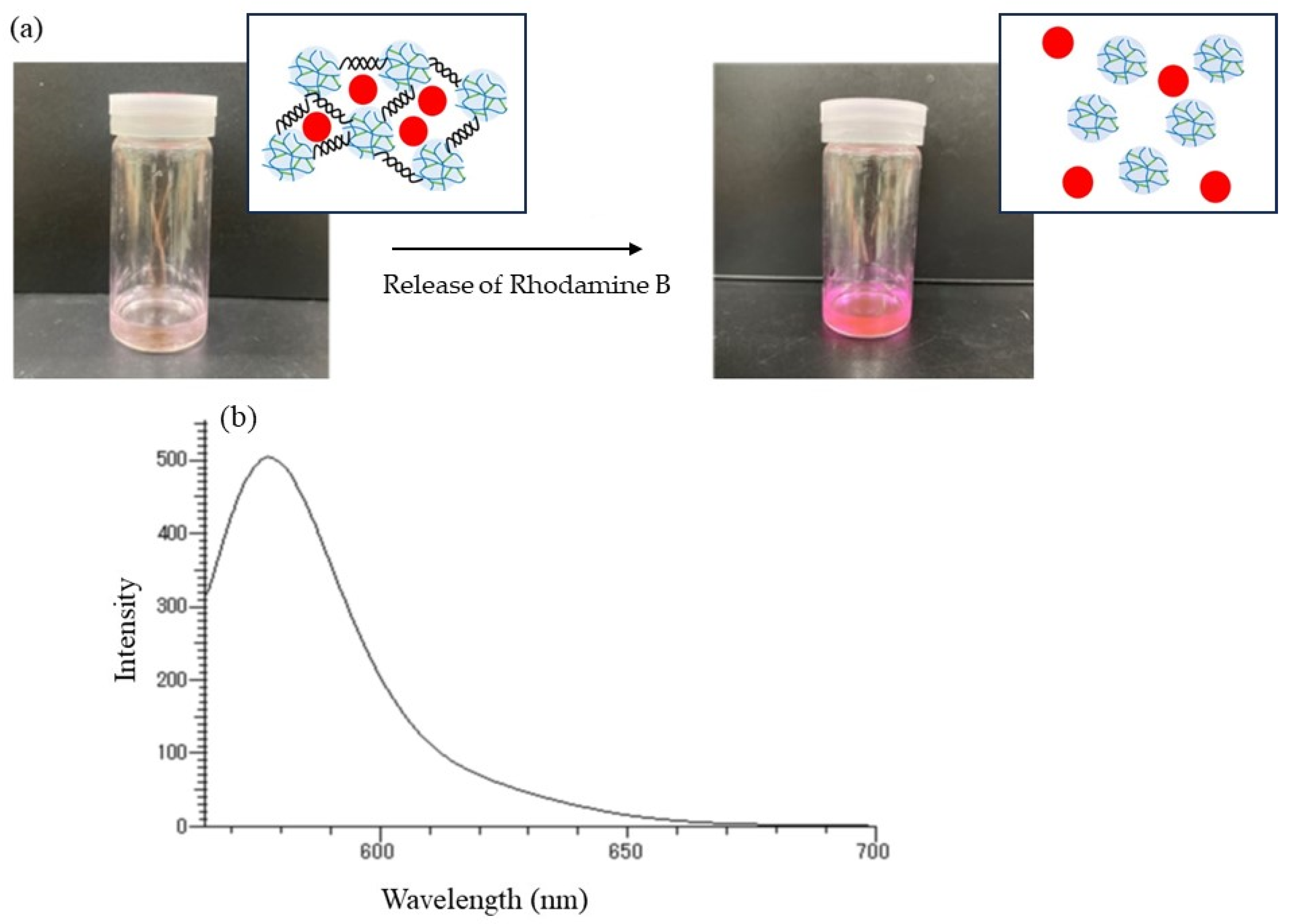
The release behavior of the encapsulated Rhodamine B was then explored by α-amylase-catalyzed enzymatic disintegration of the network polysaccharide assembly. When the mixture of the assembly with 0.2 mol/L acetate buffer was stirred in the presence of α-amylase (from
Bacillus amyloliquefaciens) for 4 h at 40 °C (
Figure 10b); optimal temperatures for hydrolysis of soluble starch by α-amylase from this source were reported to be 40–60 °C [
25], the color gradually changed to red, as shown in
Figure 11a, indicating that Rhodamine B was released into the acetate buffer by enzymatic disintegration. A fluorescence peak at 575 nm, assigned to Rhodamine B, was then detected in the fluorescence spectrum of the reaction mixture excited at 550 nm (
Figure 11b).
3. Materials and Methods
3.1. Materials
Thermostable GP (isolated from
Aquifex aeolicus VF5) was provided by Dr. Takeshi Takaha (Sanwa Starch Co., Ltd., Nara, Japan [
26]). Chitin powder from crab shells was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). GlcA-1-P was prepared by TEMPO-mediated oxidation of Glc-1-P according to a previously reported procedure [
27]. Glc
7 was synthesized by selective hydrolysis of the glycosidic bond of β-cyclodextrin under acidic conditions [
28]. All other reagents and solvents were used as received from commercial sources.
3.2. Synthesis of GlcA-Glcn-GlcCOONa (Figure S1) [16]
A solution of I2 in methanol (3.62 g/24.4 mL) was mixed with an aqueous solution of Glc7 (5.70 g, 4.90 mmol/10 mL) and stirred at 40 °C for 2 h. After the dropwise addition of a solution of NaOH in methanol (2.66 g/75.5 mL), the obtained mixture was stirred for 1 h at 40 °C. The precipitate was isolated via decantation and dried under reduced pressure for 10 h at room temperature. A solution of the product in water (40 mL) was treated with activated carbon for 1 h at room temperature while stirring. After the activated carbon was filtered, the filtrate was concentrated and lyophilized to yield crude Glc6-GlcCOONa (2.38 g).
A mixture of Glc6-GlcCOONa (0.100 g, 0.084 mmol) and GlcA-1-P (0.428 g, 1.26 mmol) in 0.2 mol/L sodium acetate buffer (6.0 mL, pH 6.2) was incubated with thermostable GP (30 U) for 48 h at 50 °C. The reaction solution was concentrated and subjected to an anion exchange column (Amberlite IRA 400 J Cl, Cl− form, eluent: 1.0 mol/L aqueous acetic acid), and the fractions containing the products were collected and neutralized with 2.0 mol/L aqueous Na2CO3. The obtained solution was dialyzed against water (molecular cut off: 1000) and concentrated at approximately 70 °C. After the insoluble deactivated enzyme was removed using cotton plug filtration, the filtrate was lyophilized to give GlcA-Glcn-GlcCOONa (0.0221 g, 0.016 mmol) with a yield of 19.0%. 1H NMR (D2O) δ 3.49 (t, J = 9.6 Hz, GlcA-H4‴), 3.57–4.03 (m, GlcA-H2,3,5, Glc-H2,3,4,5,6, GlcCOONa-H4,5,6), 4.11–4.13 (m, GlcCOONa-H2,3), and 5.17 (m, Glc-H1′), 5.41 (m, Glc-H1″, GlcA-H1‴).
3.3. Preparation of WSCS (Figure S2) [23,29]
A mixture of chitin (10.0 g, 0.049 mol) and 40 wt% aqueous NaOH (500 mL) was stirred for 7 h at 160 °C. The residue was isolated via filtration and washed with water until the filtrate became neutral. After the product was mixed with 10 wt% aqueous acetic acid (500 mL) and left to stand for 12 h at room temperature, the residue was filtered. The filtrate was mixed with a NaOH pellet (85.0 g), and the resulting swollen precipitate was isolated by filtration. A mixture of the product and 40 wt% aqueous NaOH (500 mL) was stirred for 12 h at 160 °C. The residue was isolated via filtration and washed with water until the filtrate became neutral. The product was dried under reduced pressure for 8 h at 70 °C to give fully deacetylated chitosan (5.95 g, 0.0370 mol).
The obtained chitosan (1.0 g, 6.2 mmol) was solubilized in 2.0 wt% aqueous acetic acid (20 mL) by stirring for 6 h at room temperature. After 30 wt% aqueous H2O2 (5.0 mL) was added to the solution, the mixture was stirred for 4 h at 50 °C. The resulting mixture was then neutralized with 10 wt% aqueous NaOH. The resulting residue was removed by centrifugation, and the supernatant was dialyzed against water (molecular cut-off: 1000). The precipitate obtained during dialysis was removed by centrifugation, and the supernatant was lyophilized to obtain WSCS (0.0373 g, 2.3 mmol) with a yield of 37.3%. 1H NMR (D2O) δ 2.89 (br s, H2), 3.63–3.96 (m, H3,4,5,6), and 4.87 (br s, H1). The viscosity average molecular weight (Mv) of the obtained water-soluble chitosan was estimated to be 14,000 using a previous method.
3.4. Synthesis of Glc7-Modified WSCS
The typical experimental procedure was as follows (run 1). After ultrasonication of a mixture of WSCS (40.0 mg, 0.24 mmol for amino groups), Glc7 (553 mg, 0.48 mmol), and NaBH3CN (75.4 mg, 1.2 mmol) with 1.0 mol/L aqueous acetic acid (20 mL), the resulting solution was stirred for 72 h at 50 °C. The reaction mixture was then poured into methanol (300 mL) to precipitate the product. The precipitate was isolated by filtration, washed with methanol, and dried under reduced pressure for 10 h at room temperature to produce Glc7-modified WSCS (140 mg). 1H NMR (DCl/D2O) δ 3.12 (m, GlcN-H2), 3.84–3.23 (m, Glc-H2,3,4,5,6, GlcN-H3,4,5,6), 4.54 (m, GlcH-1β), 4.76 (m, GlcN-H1β), 4.92 (m, β(1→4)-GlcN-H1), 5.08 (m, Glc-H1α), and 5.32 (m, 0.11H, GlcN-H1α).
3.5. Synthesis of Glc7-Modified Network Polysaccharide
A solution of GlcA-Glcn-GlcCOONa (5.9 mg, 4.02 μmol, COONa group; 8.05 μmol), EDC (1.54 mg, 8.00 μmol), and NHS (0.93 mg, 8.00 μmol) in water (0.20 mL), which was prepared by standing the mixture for 1 h at room temperature, was mixed with an aqueous solution of Glc7-modified WSCS (run 1, 30 mg, 0.161 mmol/0.70 mL). The obtained mixture was left to stand for 7 h at room temperature to allow condensation to progress. The reaction mixture was then poured into methanol (30 mL) to produce the precipitate. The precipitated product was isolated by filtration and lyophilized to obtain a network polysaccharide (20.2 mg). 1H NMR (DCl/D2O) δ 3.09 (br s, GlcN-H2), 3.27–3.82 (m, Glc-H2,3,4,5,6, GlcN-H3,4,5,6), 4.54 (m, Glc-H1β), 4.75, (m, GlcN-H1β), 4.87 (m, β(1→4)-GlcN-H1), 5.07 (m, Glc-H1α), and 5.32 (m, GlcN-H1α).
3.6. Thermostable GP-Catalyzed Enzymatic Polymerization Using the Glc7-Modified Network Polysaccharide
The typical experimental procedure was as follows (run 4). A mixture of the Glc
7-modified network polysaccharide obtained by the procedure described in
Section 3.5 (20 mg, 1.9 μm for Glc
7 primer), Glc-1-P (57.8 mg, 0.19 mmol), and thermostable GP (
Aquifex aeolicus VF5, 12 U) in 0.2 mol/L acetate buffer (2.0 mL) was stirred for 48 h at 50 °C. After the reaction mixture was dialyzed (molecular cut-off: 1000) for 12 h, the obtained insoluble material was removed by centrifugation, and the dialysate was lyophilized to give the assembled product (25.9 mg).
1H NMR (DCl/D
2O) δ 3.18 (m, GlcN-H2), 3.75–3.23 (m, Glc-H2,3,4,5,6, GlcN-H3,4,5,6), 4.55 (m, Glc-H1β), 4.78, (m, GlcN-H1β), and 5.10 (m, GlcN-H1α).
When the reaction mixtures became viscous or gelled, fractions insoluble in water, which were obtained by washing them with water, were lyophilized to give the assembled products.
3.7. Amylase-Catalyzed Enzymatic Disintegration of the Assembled Network Polysaccharide
A mixture of the network polysaccharide assembly (run 7, 15 mg) and α-amylase (from Bacillus amyloliquefaciens, 15 U) in 0.2 mol/L acetate buffer (5.0 mL) was stirred at 40 °C. The reaction mixture was collected every 2 h and analyzed using SEM. After 48 h, the reaction mixture was heated for 5 h at 100 °C to deactivate the enzyme. The deactivated enzyme was filtered, and the filtrate was lyophilized. The residue was dialyzed (molecular cut-off: 1000) and the dialysate was lyophilized to obtain the sample for XRD.
3.8. Encapsulation of Rhocamine B
A mixture of Glc7-modified network polysaccharides prepared using Glc7-modified WSCS from run 2 (15 mg), Glc-1-P (544 mg, 0.179 mmol), Rhodamine B (6.0 mg, 0.013 mmol, 0.2 equiv. with residual amino groups in WSCS), and thermostable GP (12 U) was stirred for 48 h at 40 °C. After the reaction mixture was dialyzed (molecular cut-off: 1000) for 5 d, the dialysate was lyophilized to yield the Rhodamine B-encapsulated product (22.2 mg).
3.9. Release of Rhodamine B by α-Amylase Treatment
A mixture of the Rhodamine B-encapsulated network polysaccharide assembly (22.2 mg) and α-amylase (Bacillus amyloliquefaciens, 8.4 U) in 0.2 mol/L acetate buffer (5 mL) was stirred for 4 h at 40 °C. The reaction mixture was then subjected to fluorescence measurements.
3.10. Measurements
1H NMR spectra were recorded using a JEOL ECX400 spectrometer (JEOL, Akishima, Tokyo, Japan). SEM images were obtained using a Hitachi S-4100H electron microscope (Hitachi High-Technologies Corporation, Tokyo, Japan) at an accelerating voltage of 5 kV. Powder XRD measurements were performed using a PANalytical X’Pert Pro MPD instrument (PANalytical B.V., Almelo, The Netherlands) with Ni-filtered Cu-Kα radiation (λ = 0.15418 nm). Fluorescence spectra were recorded on an FP-6300Q3 spectrometer (JASCO Corporation, Hachioji, Tokyo, Japan).
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}