

Bond Formation at C8 in the Nucleoside and Nucleotide Purine Scaffold: An Informative Selection

Abstract
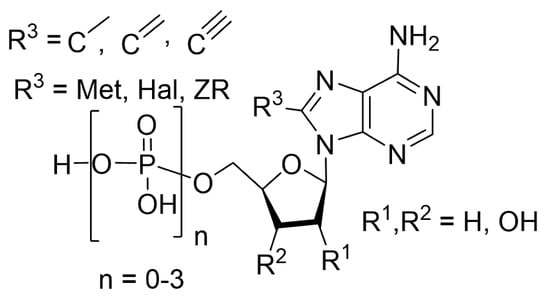
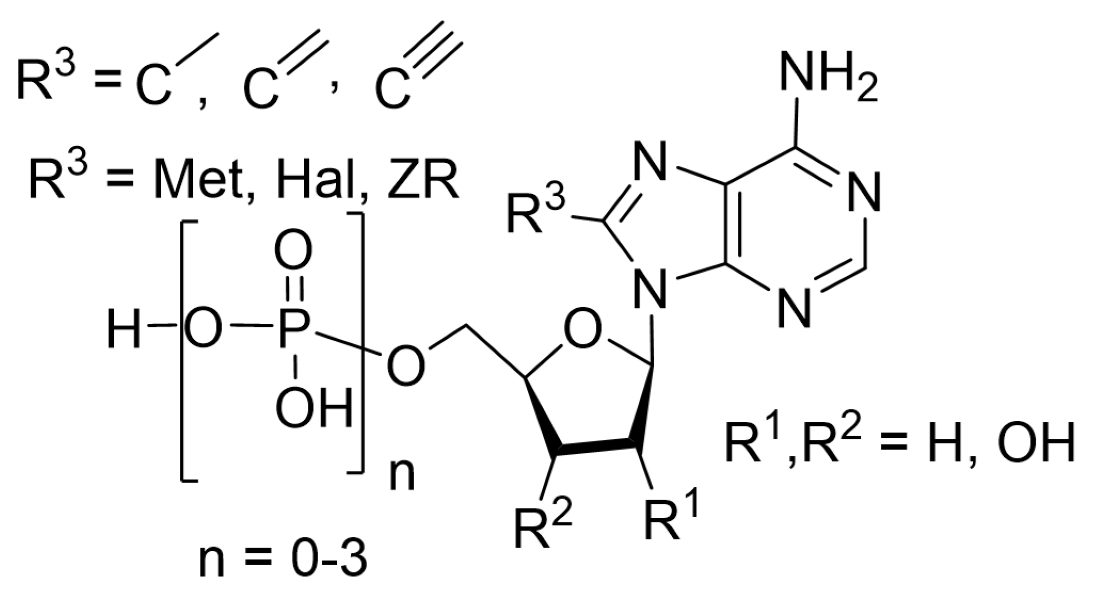{kind=link}
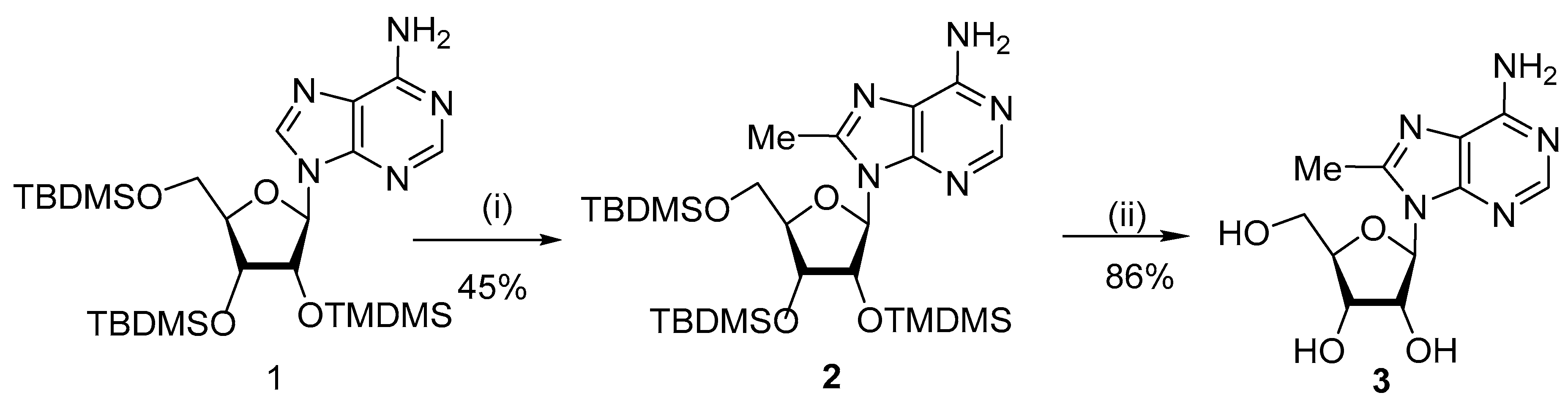{kind=link}
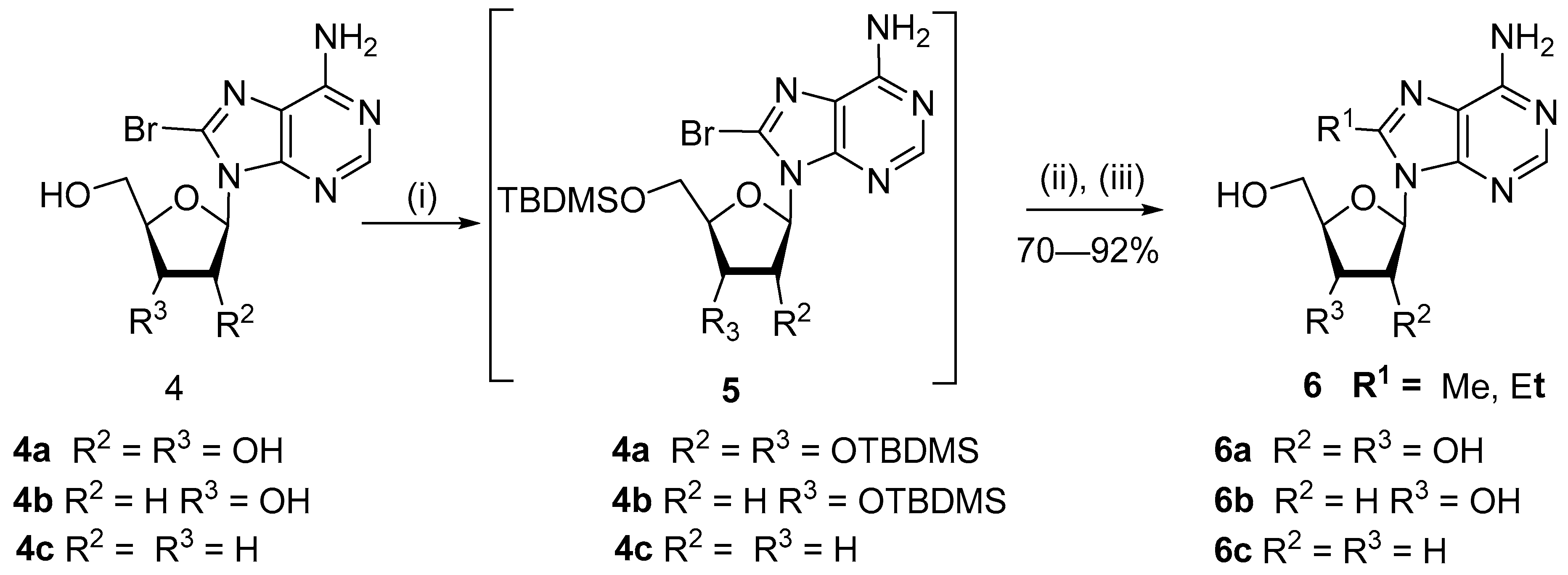{kind=link}
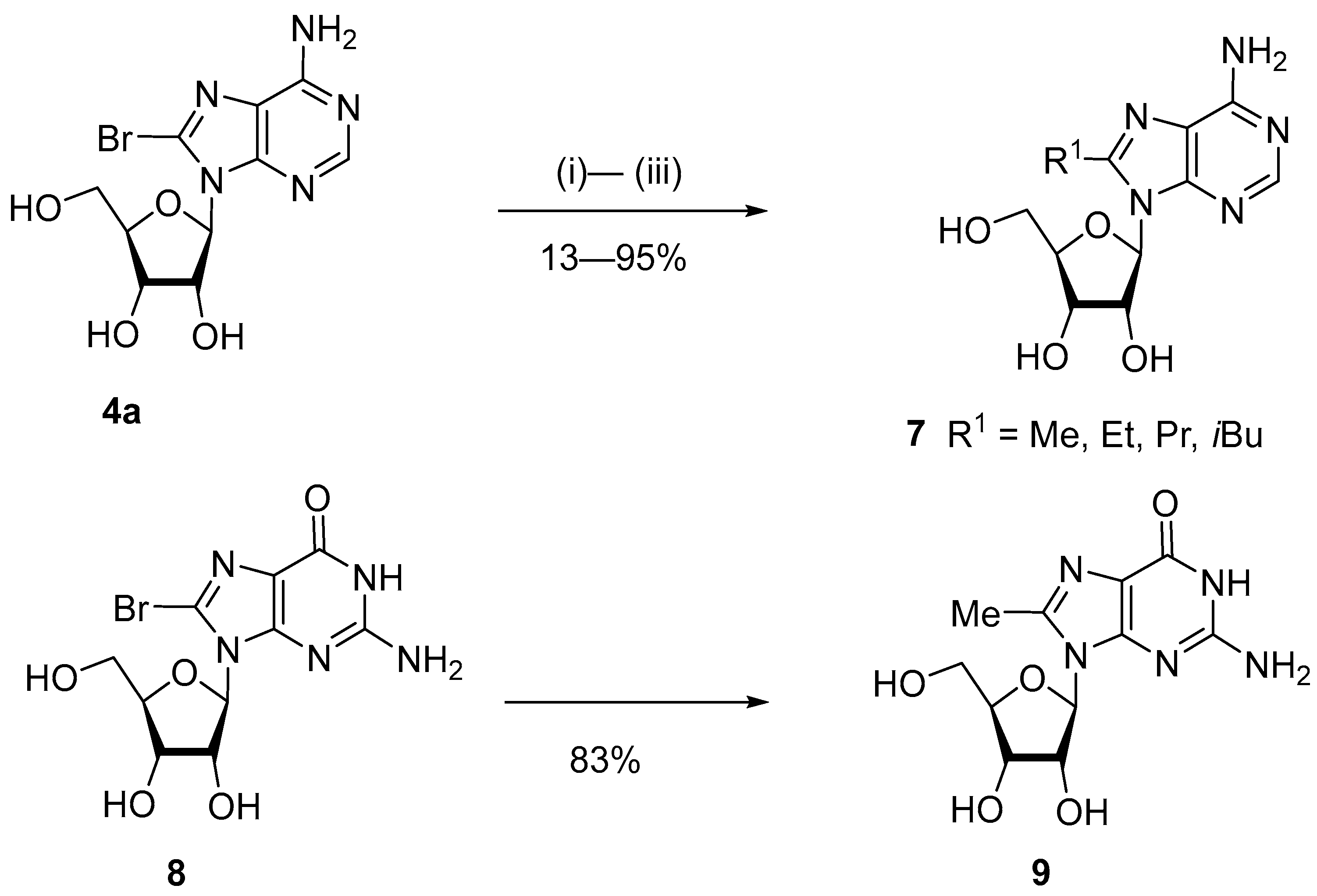{kind=link}
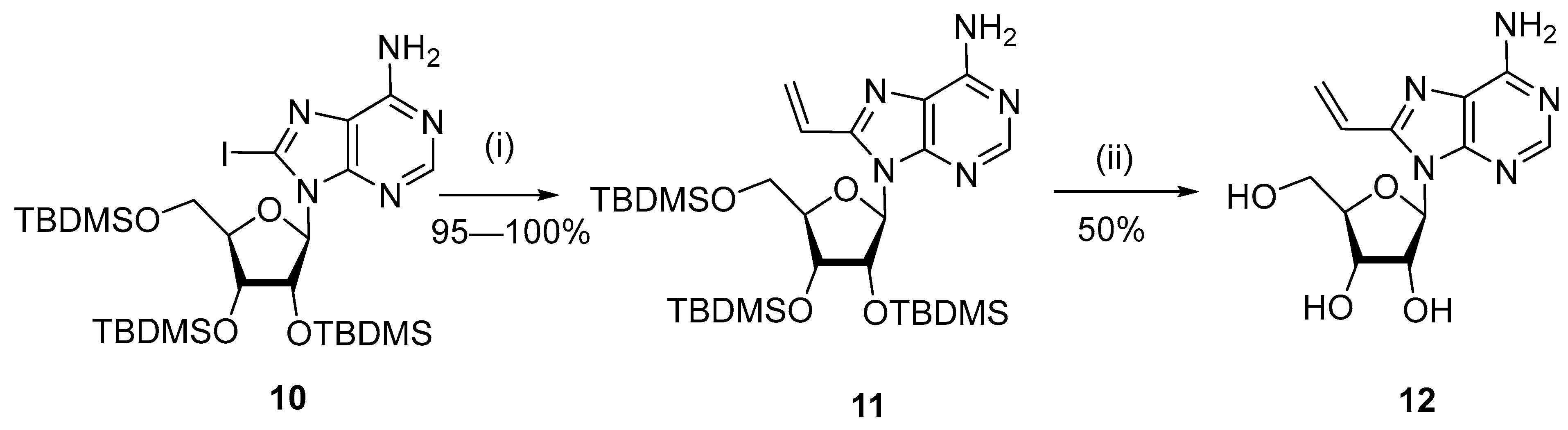{kind=link}
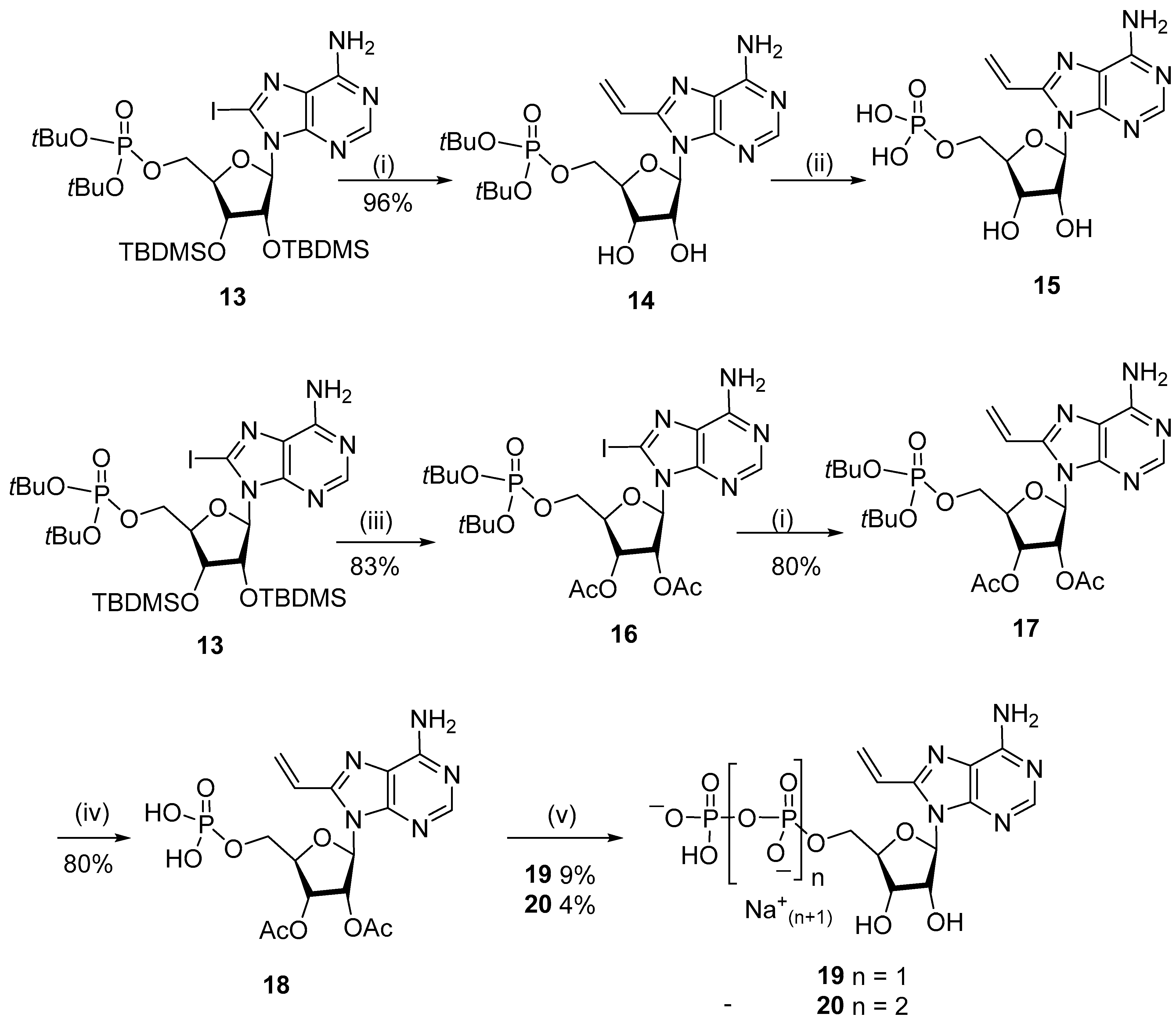{kind=link}
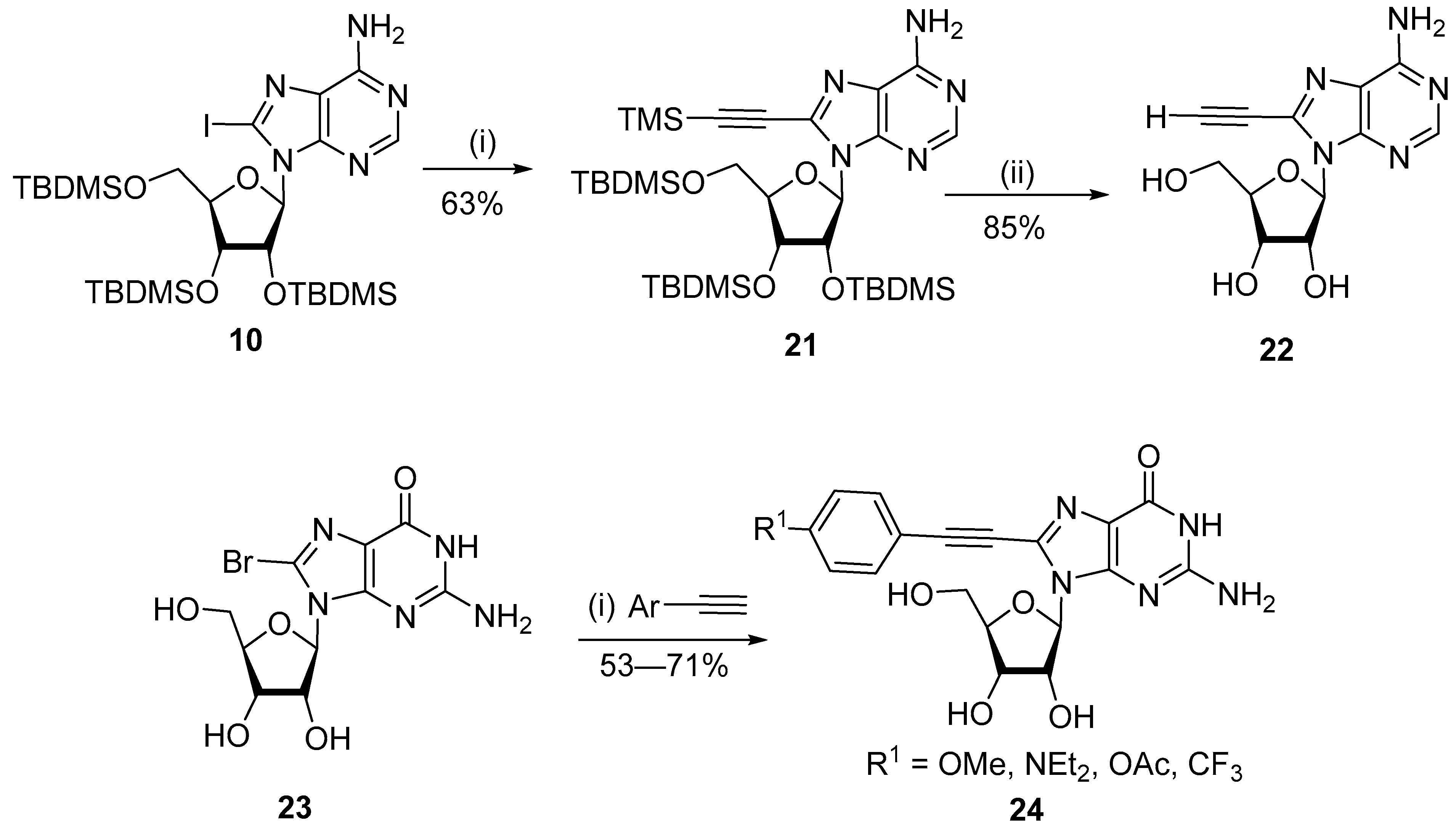{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
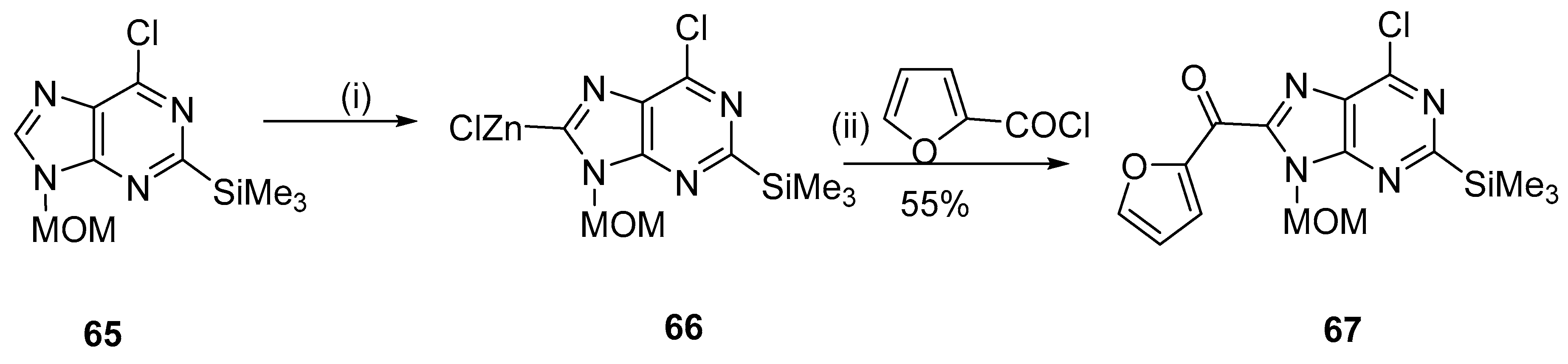{kind=link}
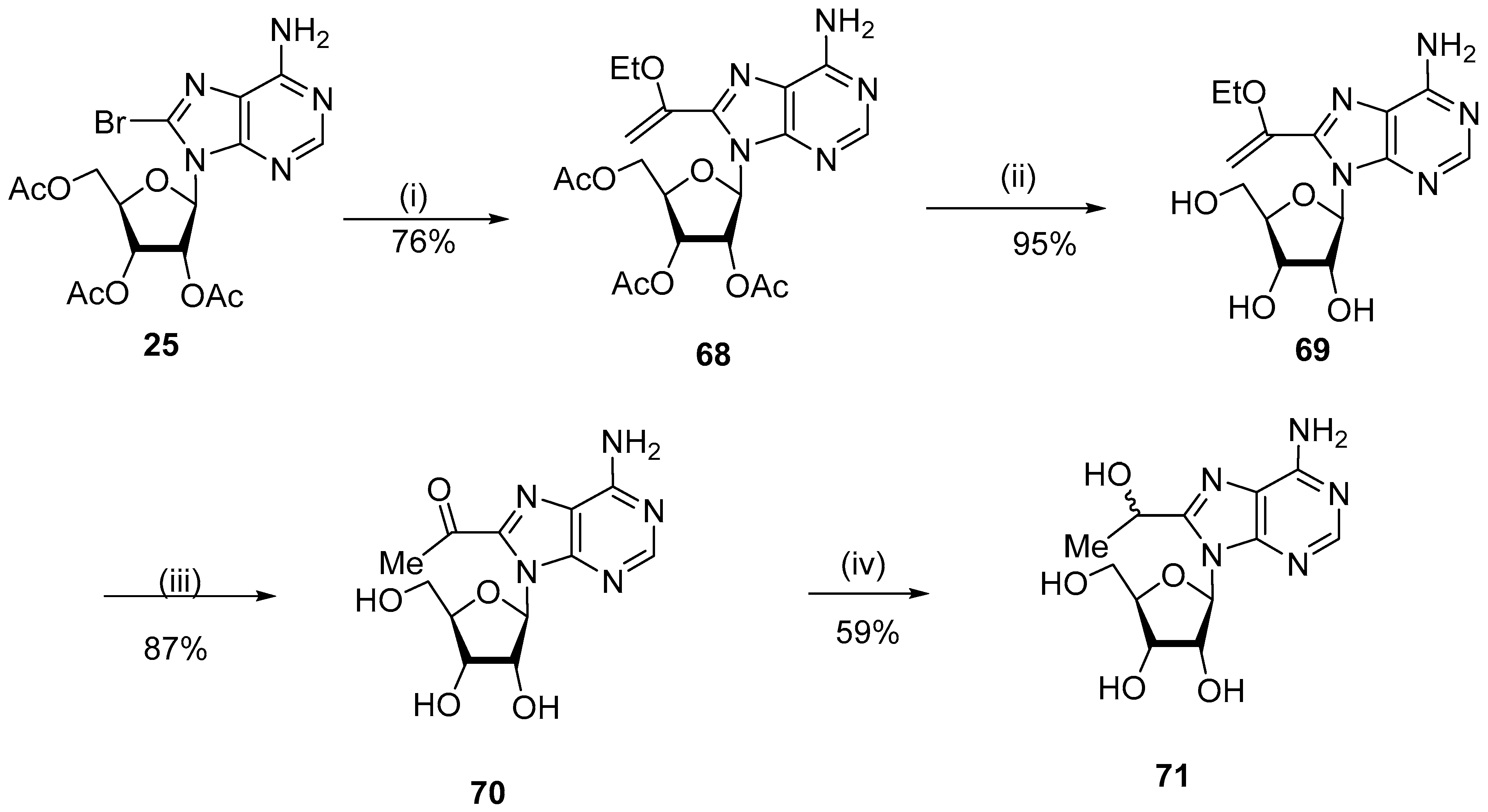{kind=link}
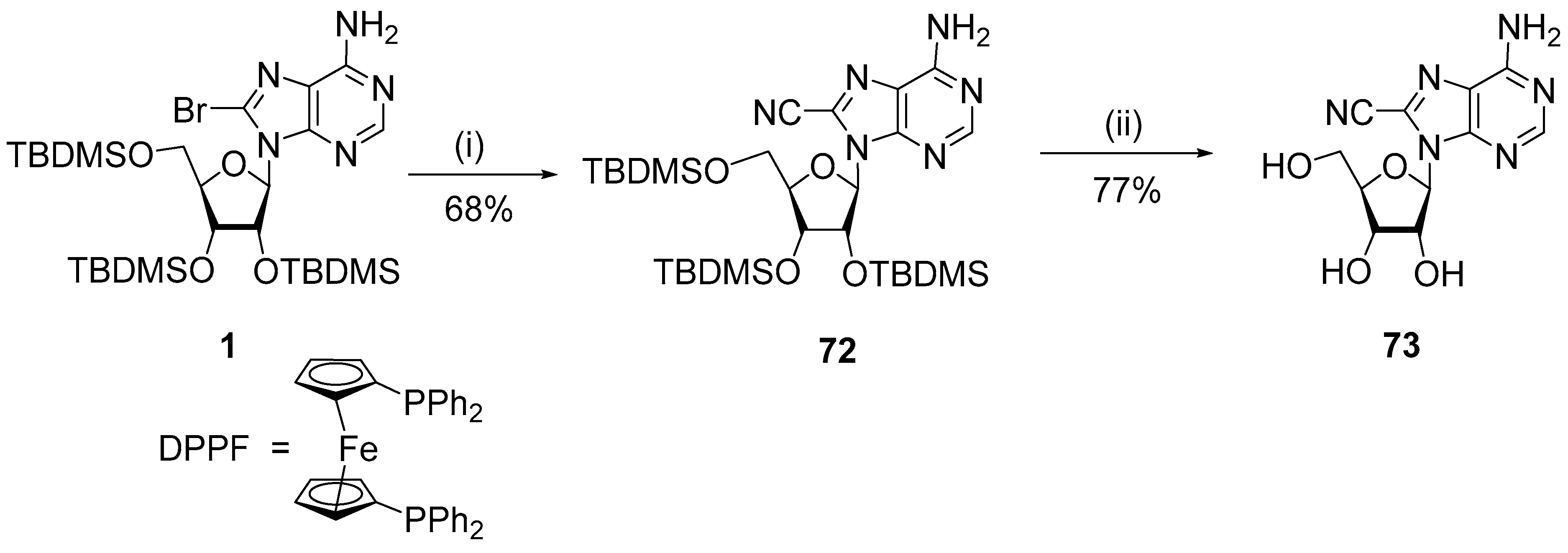{kind=link}
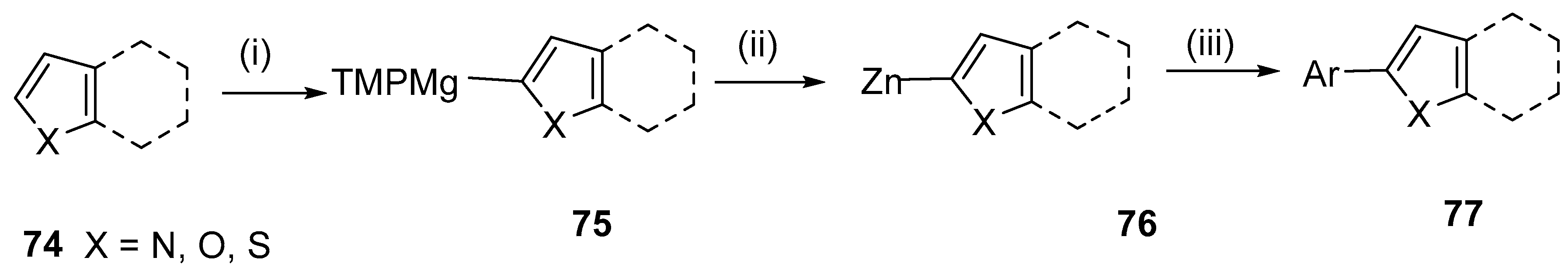{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Carbylations
2.1. Alkylations
2.2. Alkenylations
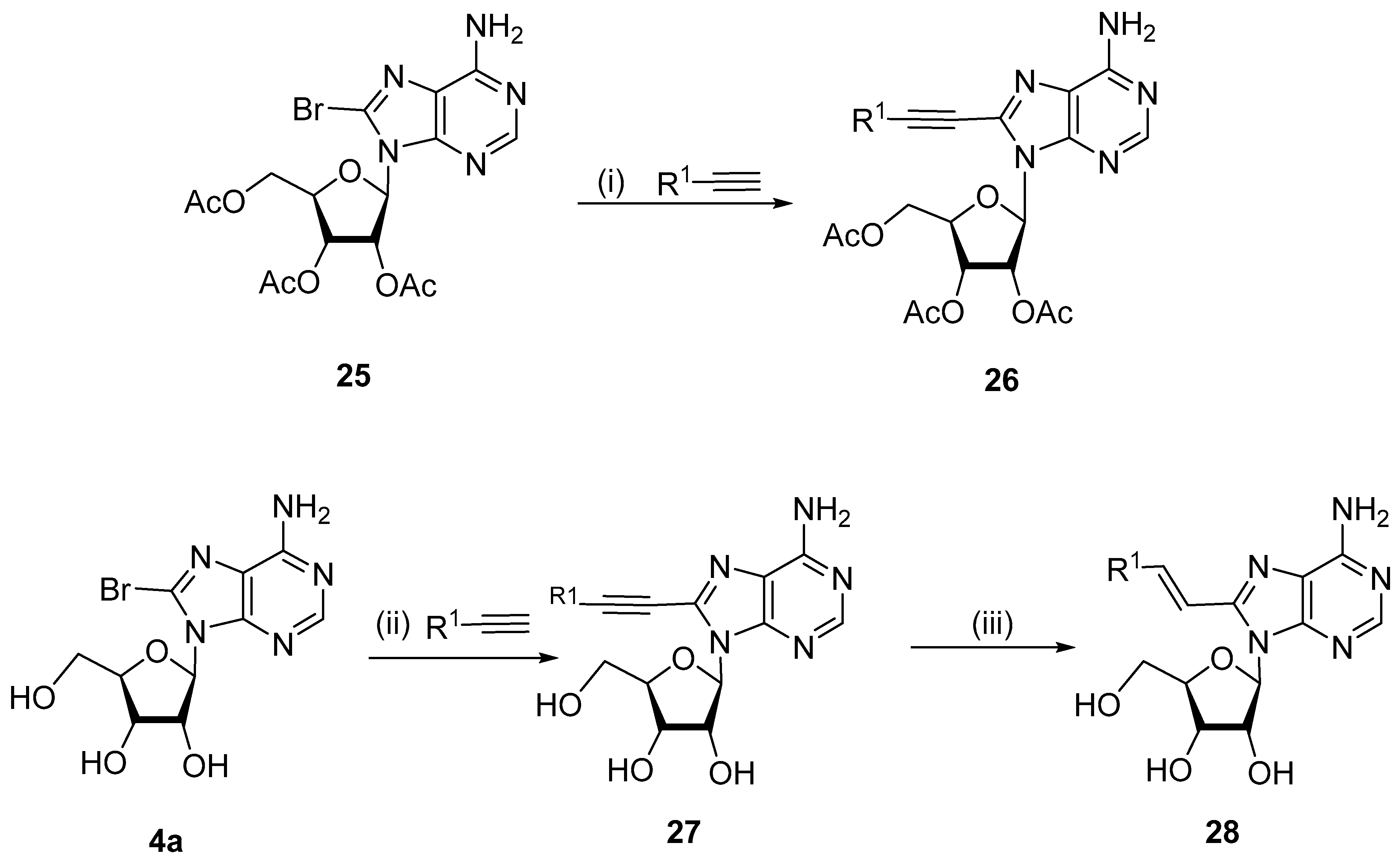
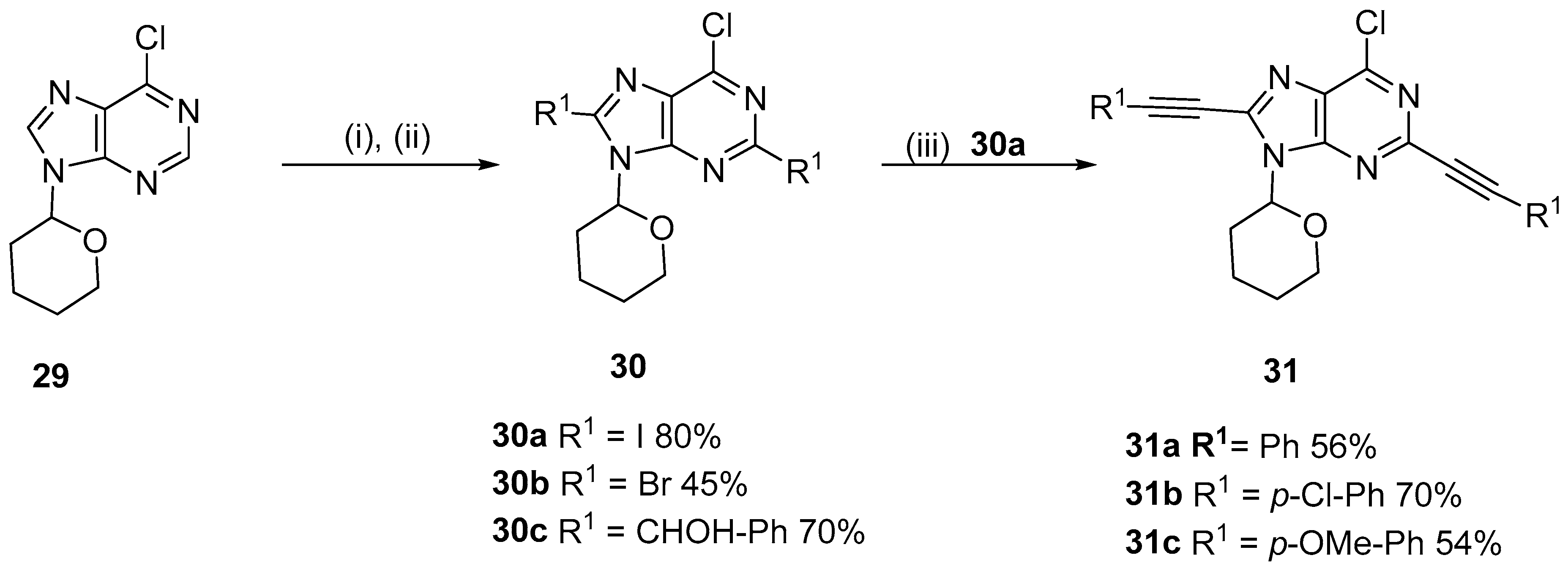
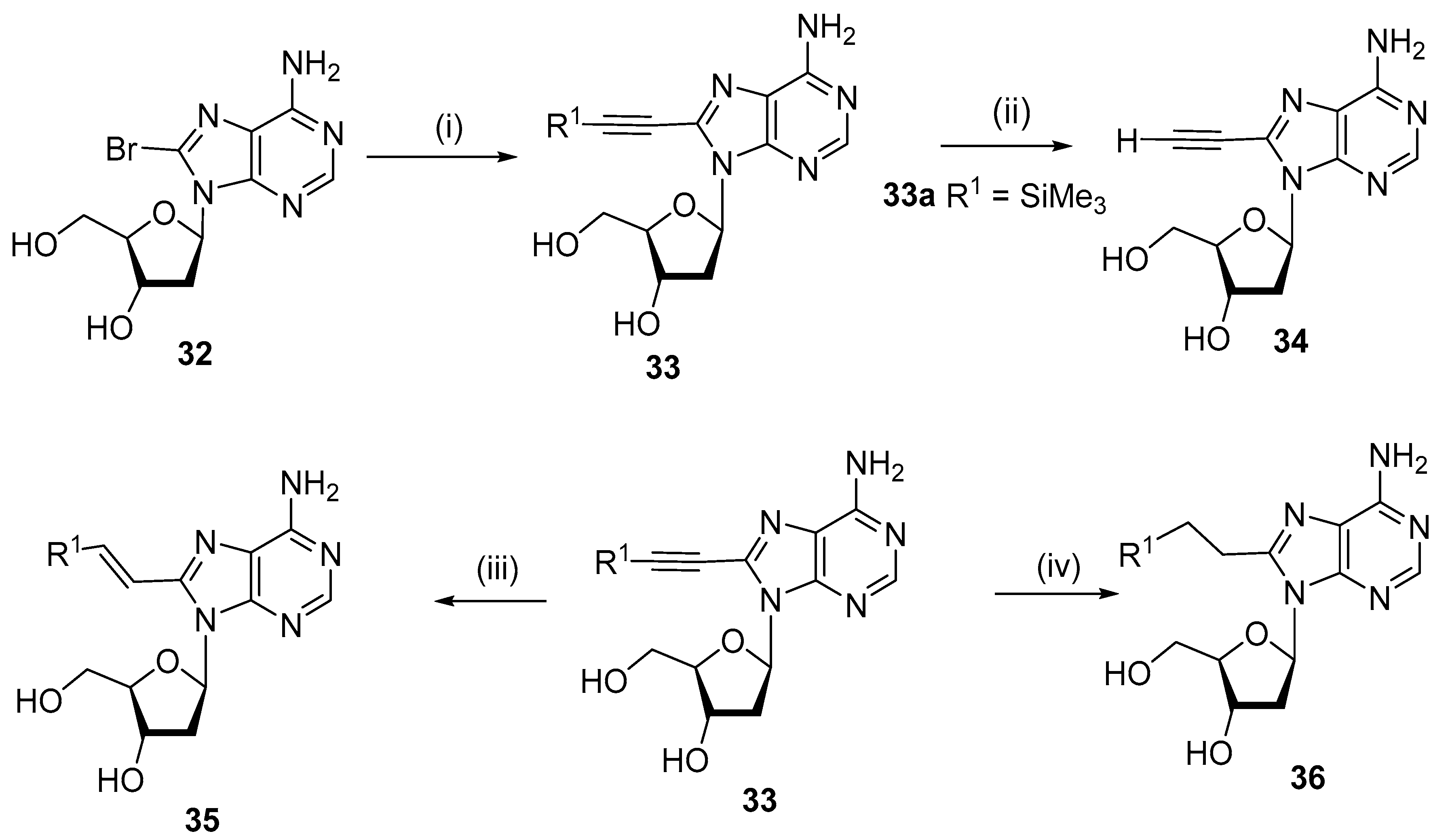
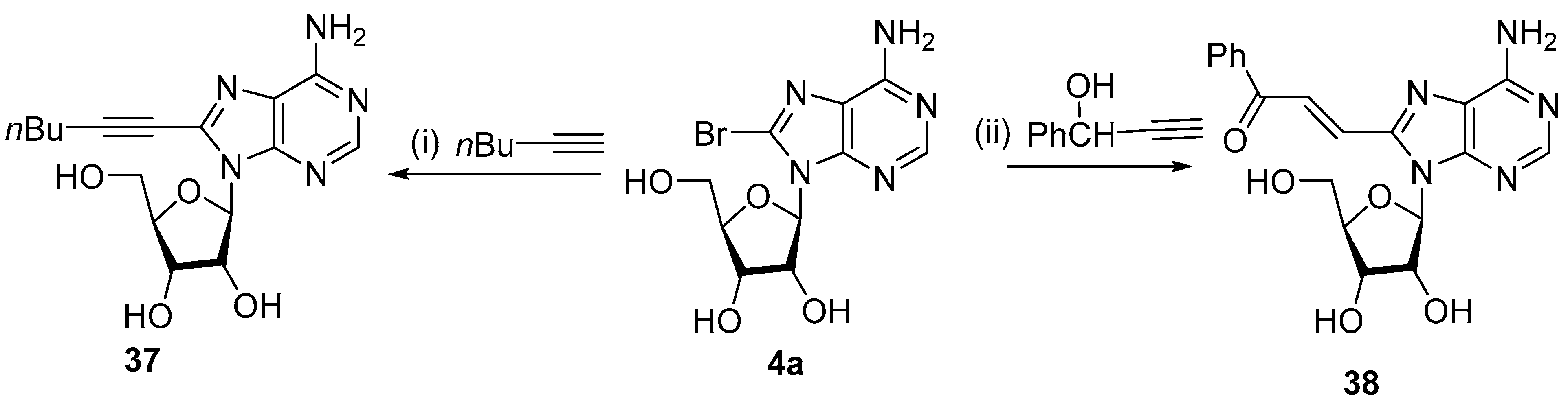
2.3. Alkynylation
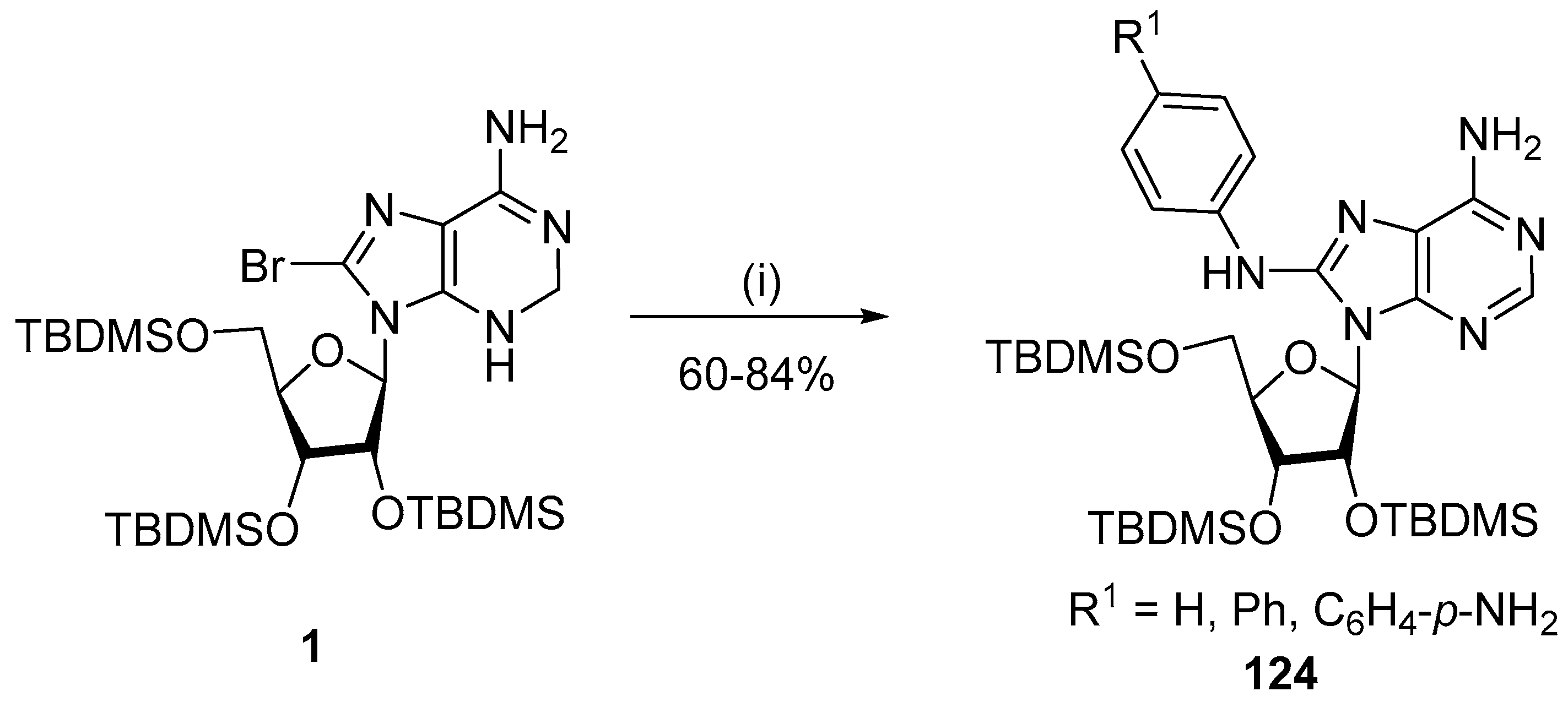
2.4. Arylation and Heteroarylation
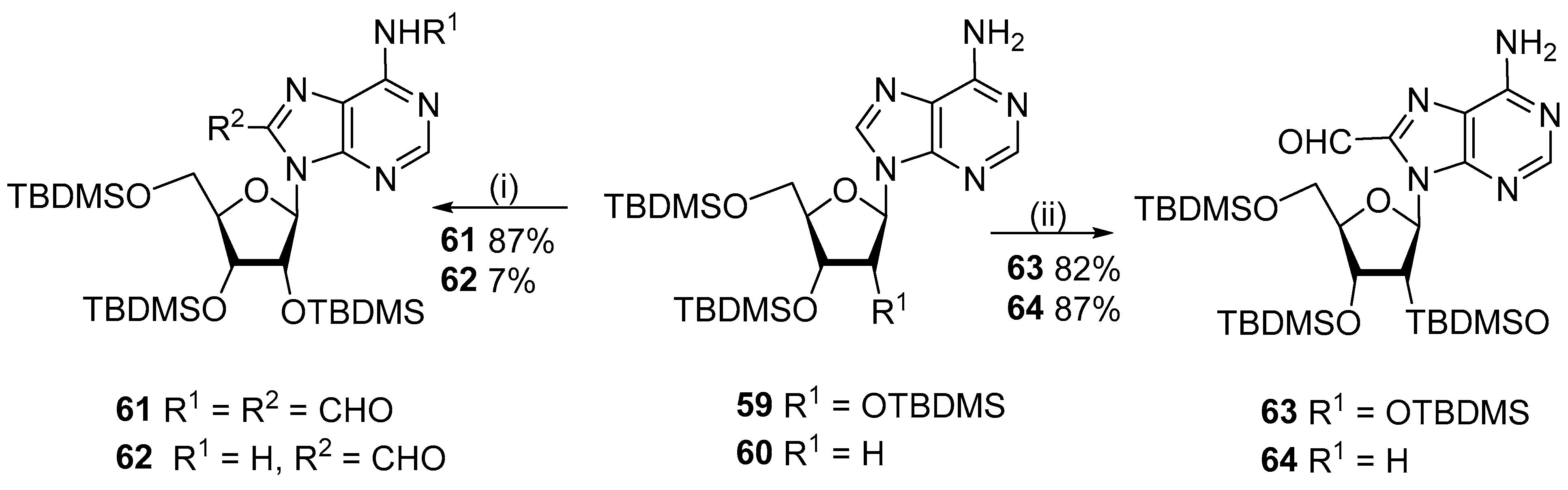
2.5. C8-α-Functionalized C1-Substituents
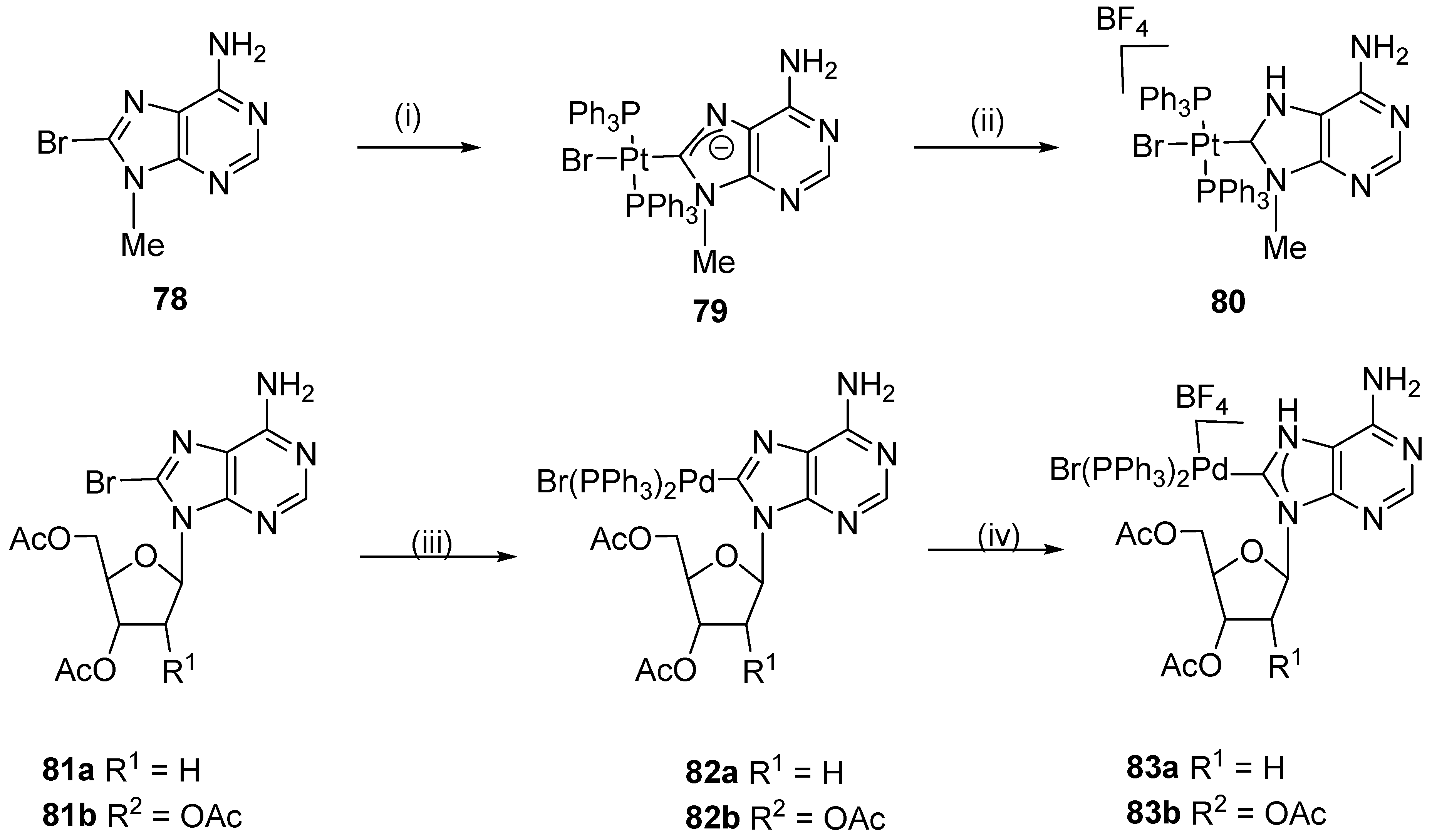
3. Organometalations
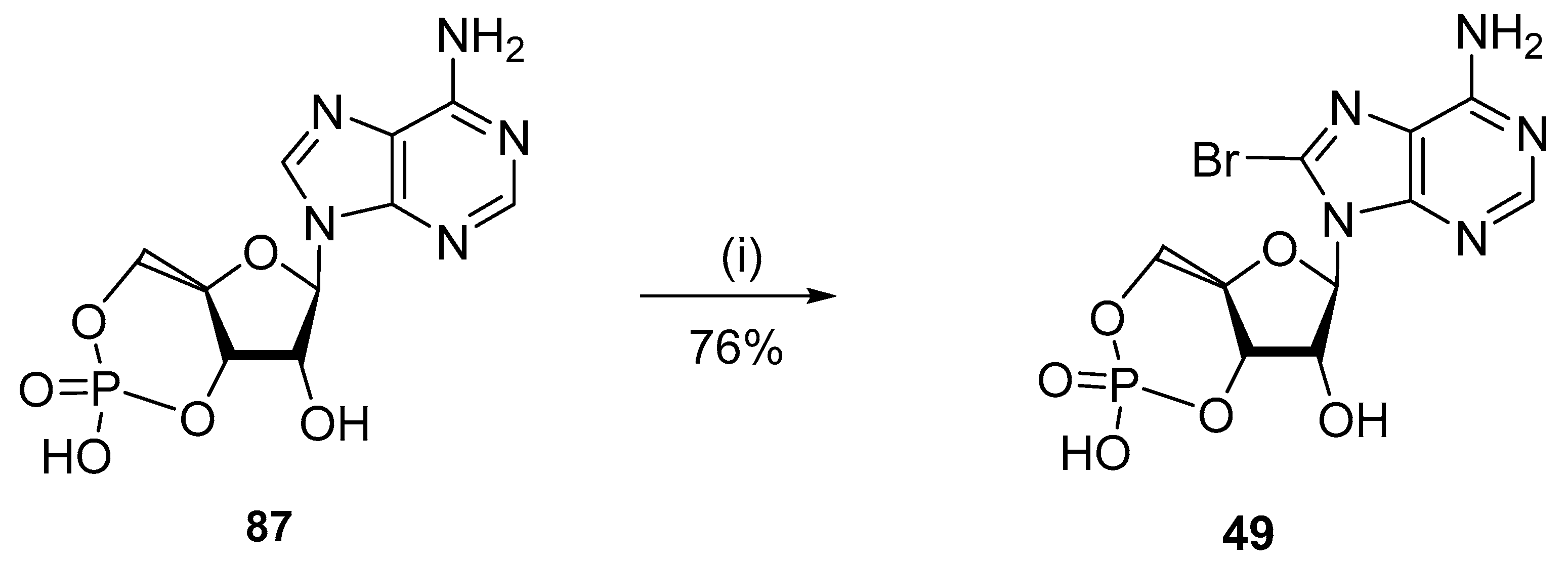
4. Halogenation
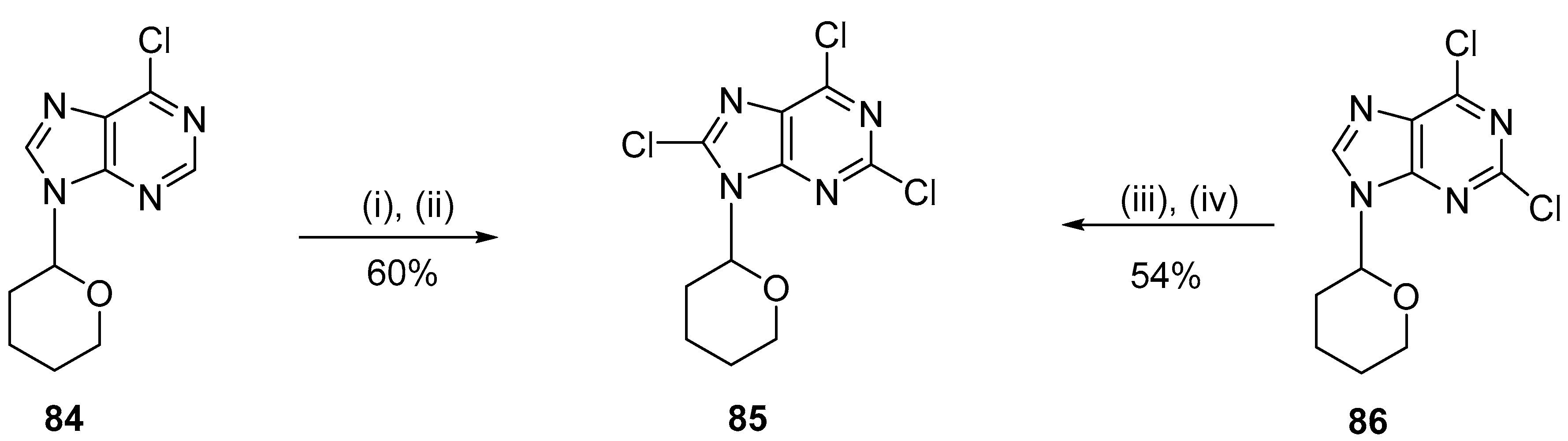
4.1. Chlorination
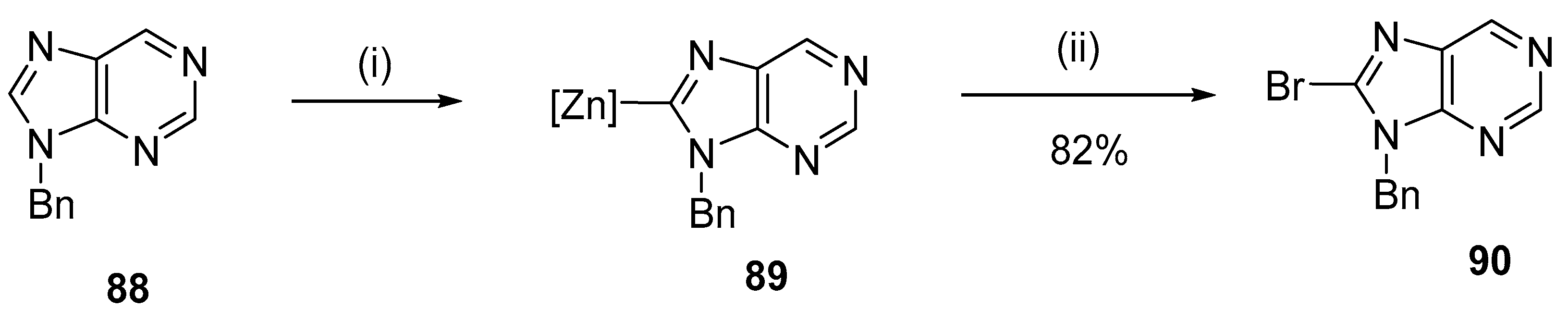
4.2. Bromination
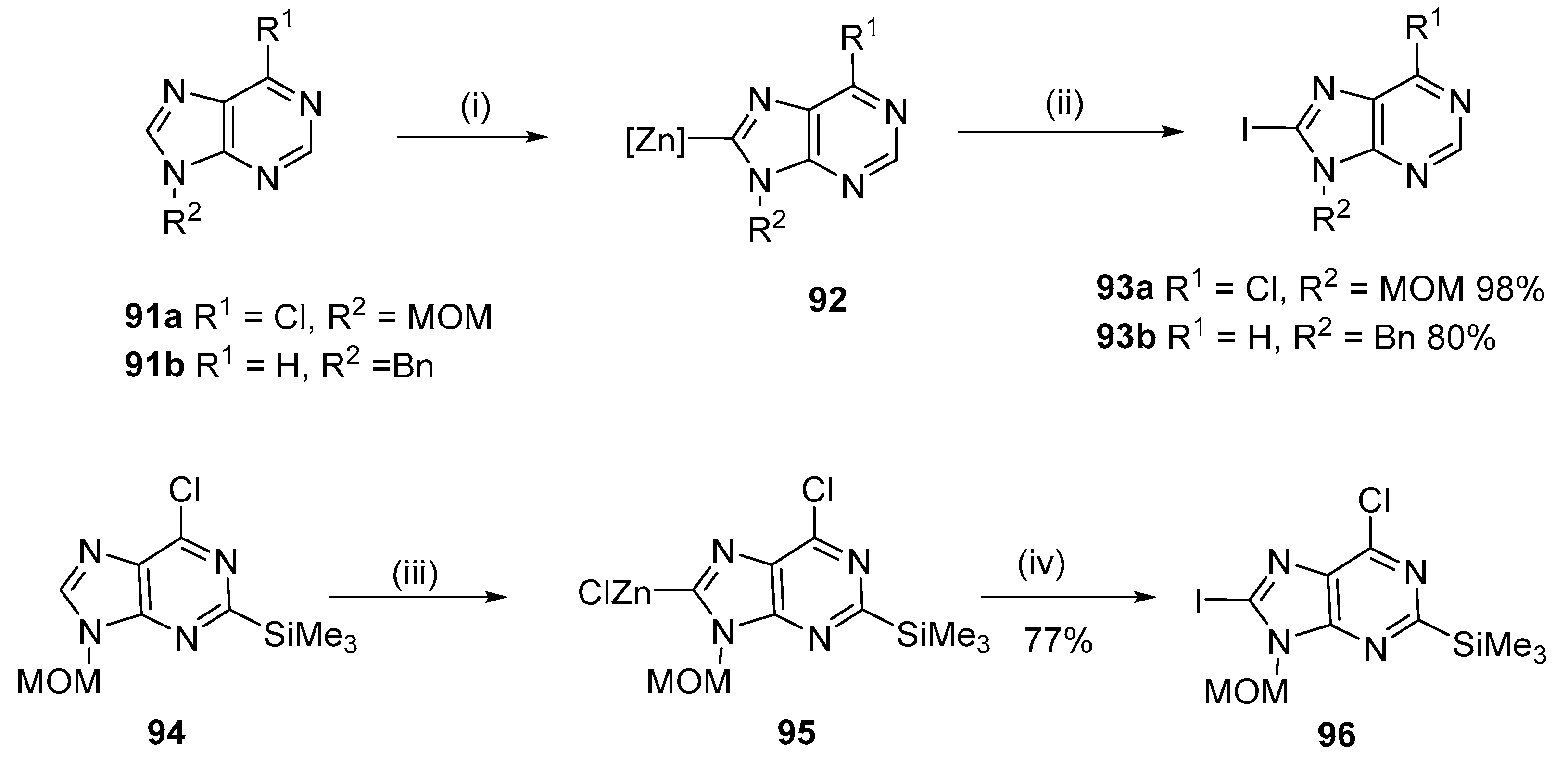
4.3. Iodination
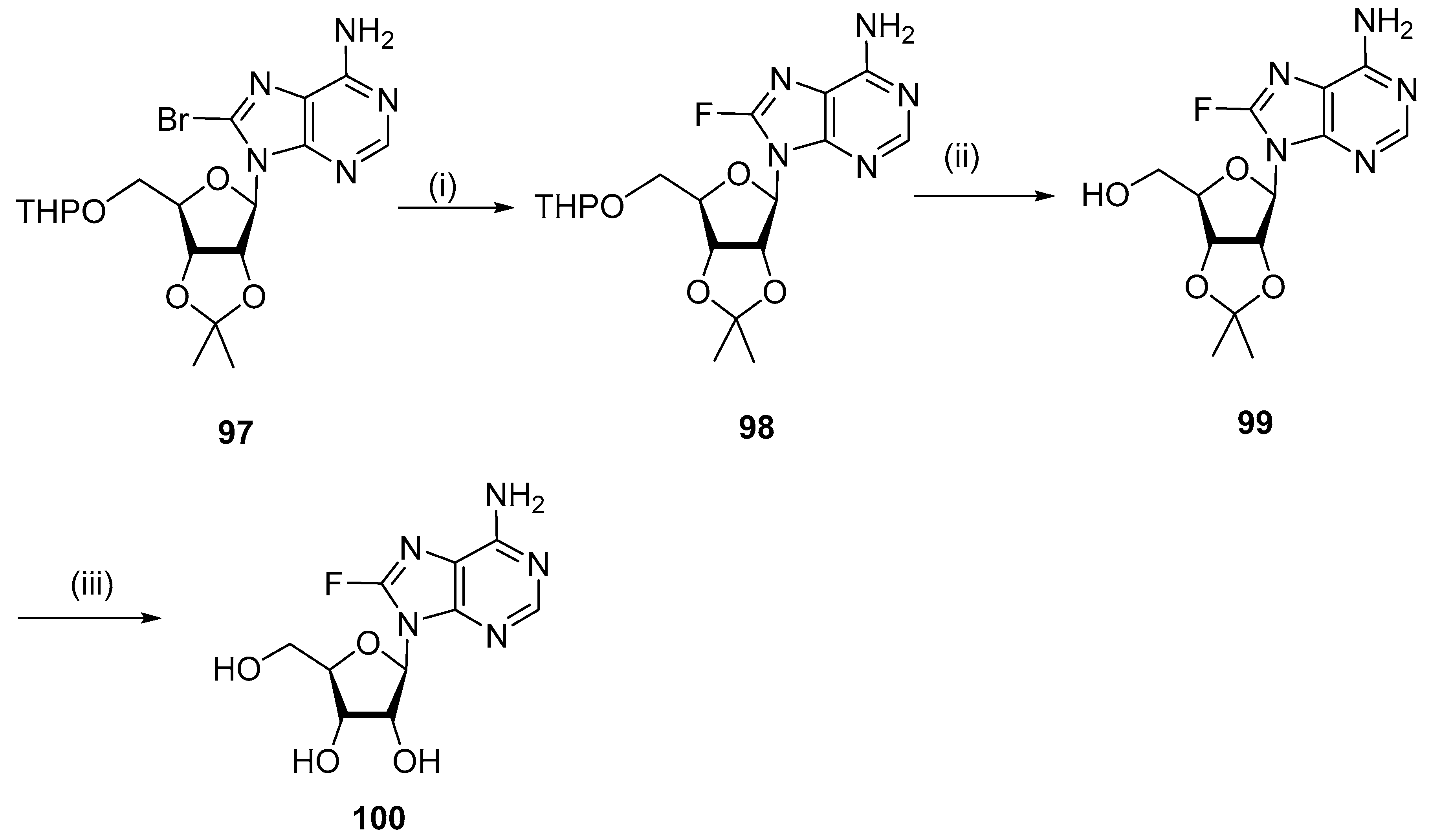
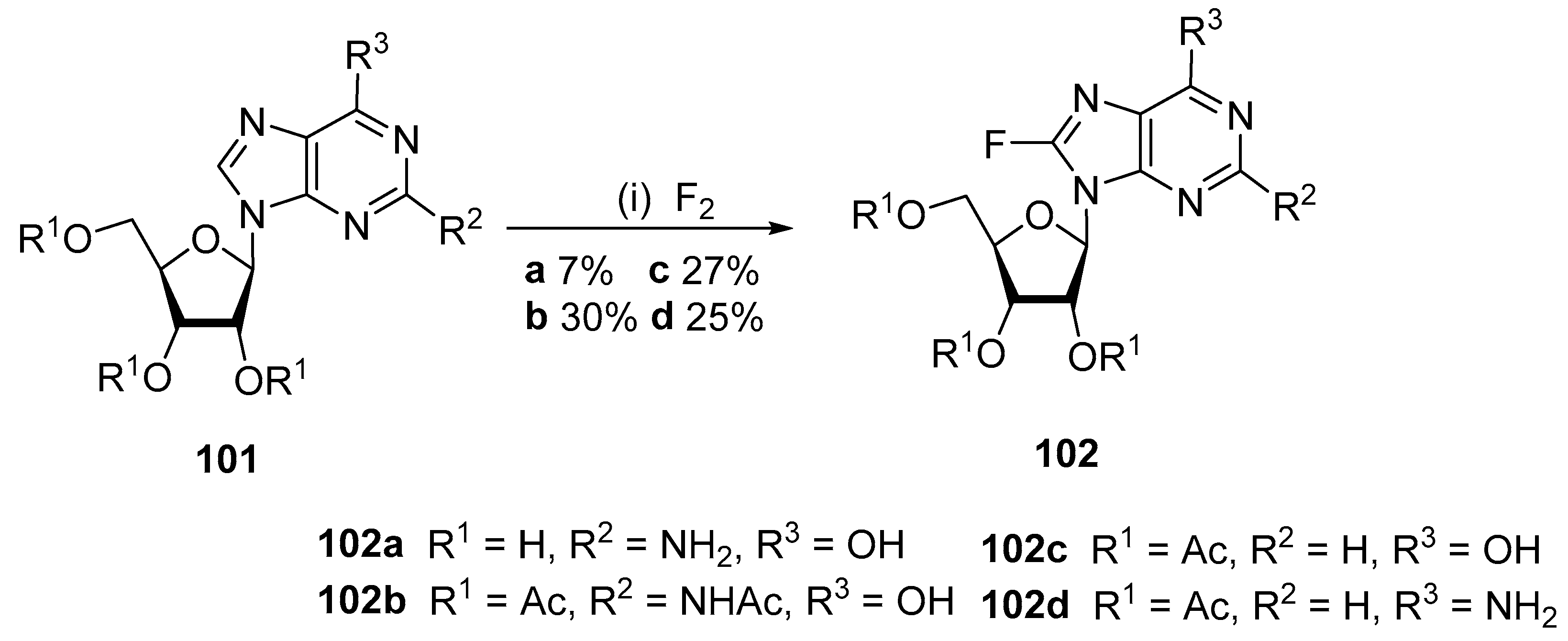
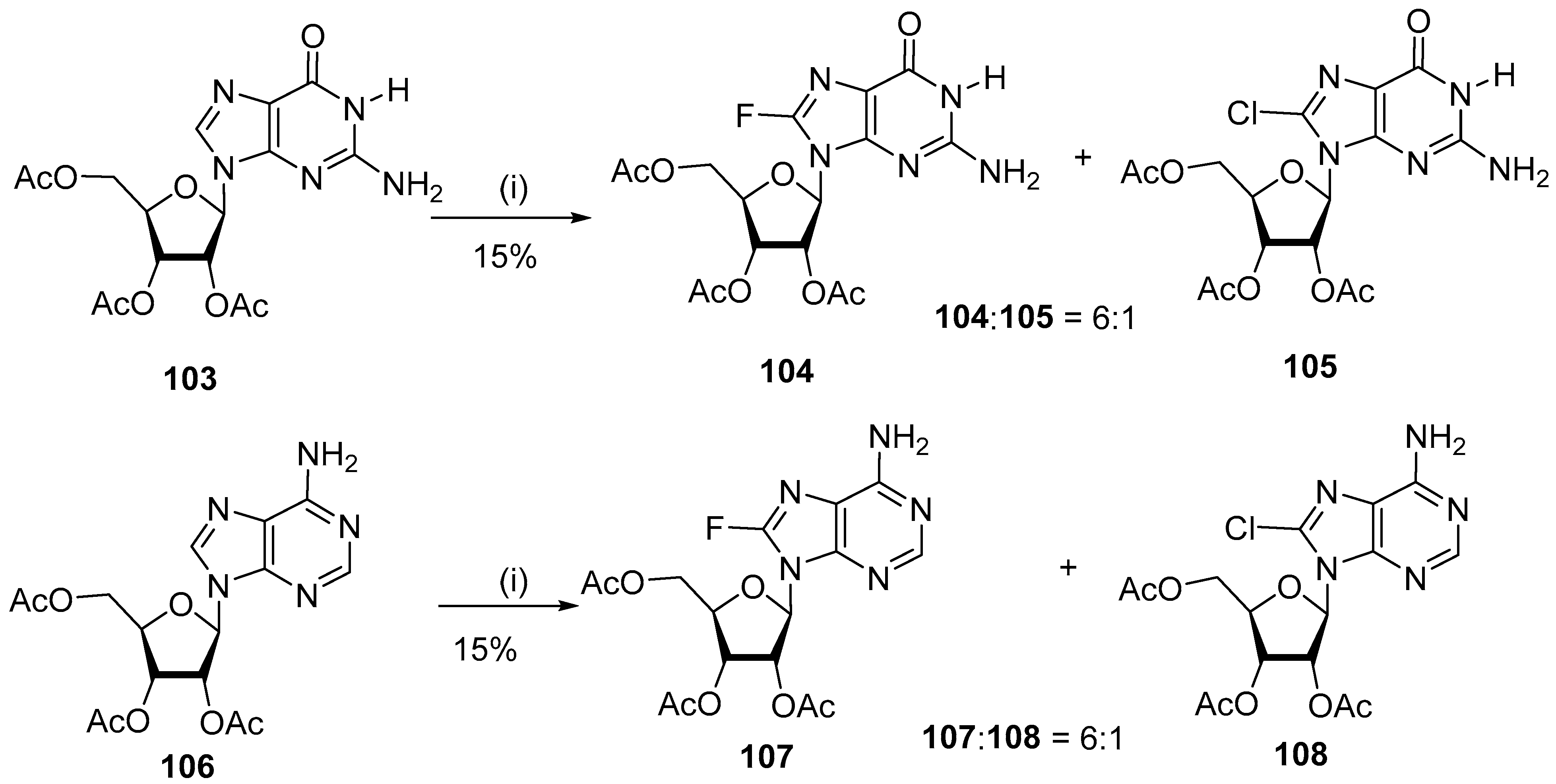
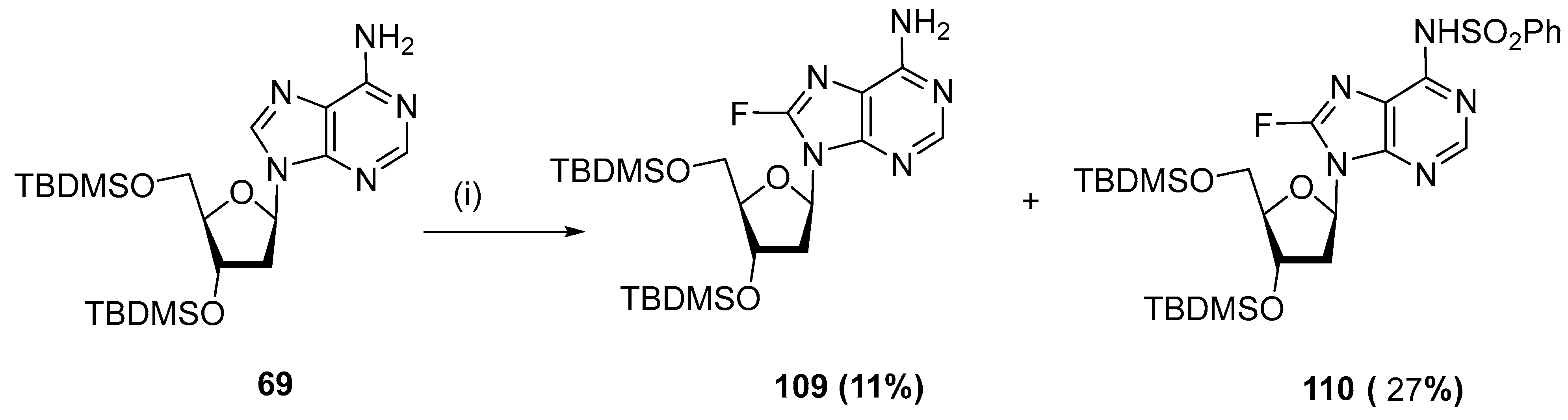
4.4. Fluorination
5. Aza-, Oxa-, and Thia-Carbylations
5.1. Amines and Oxidized Forms
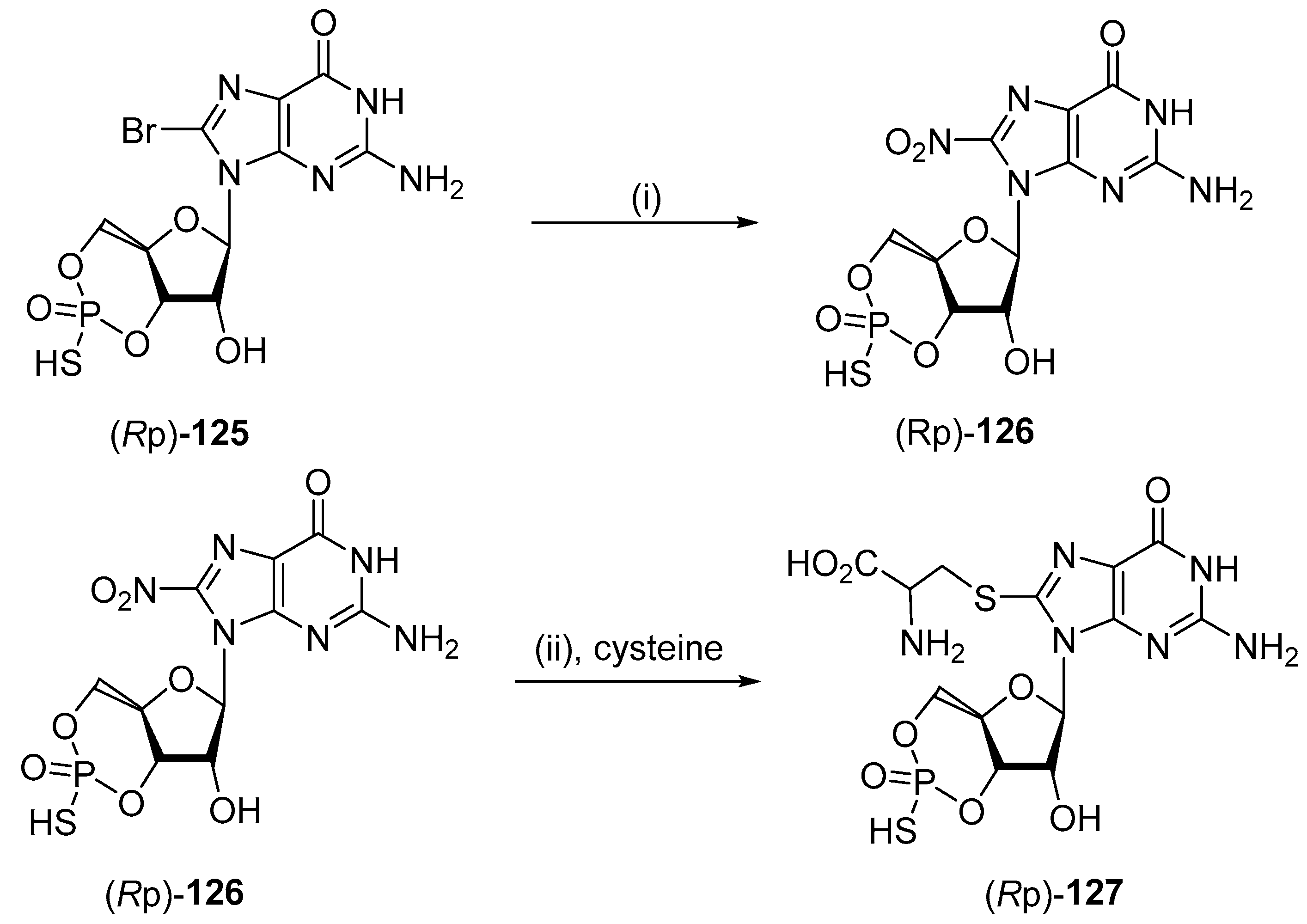
5.2. Nitro Functionalized Derivatives
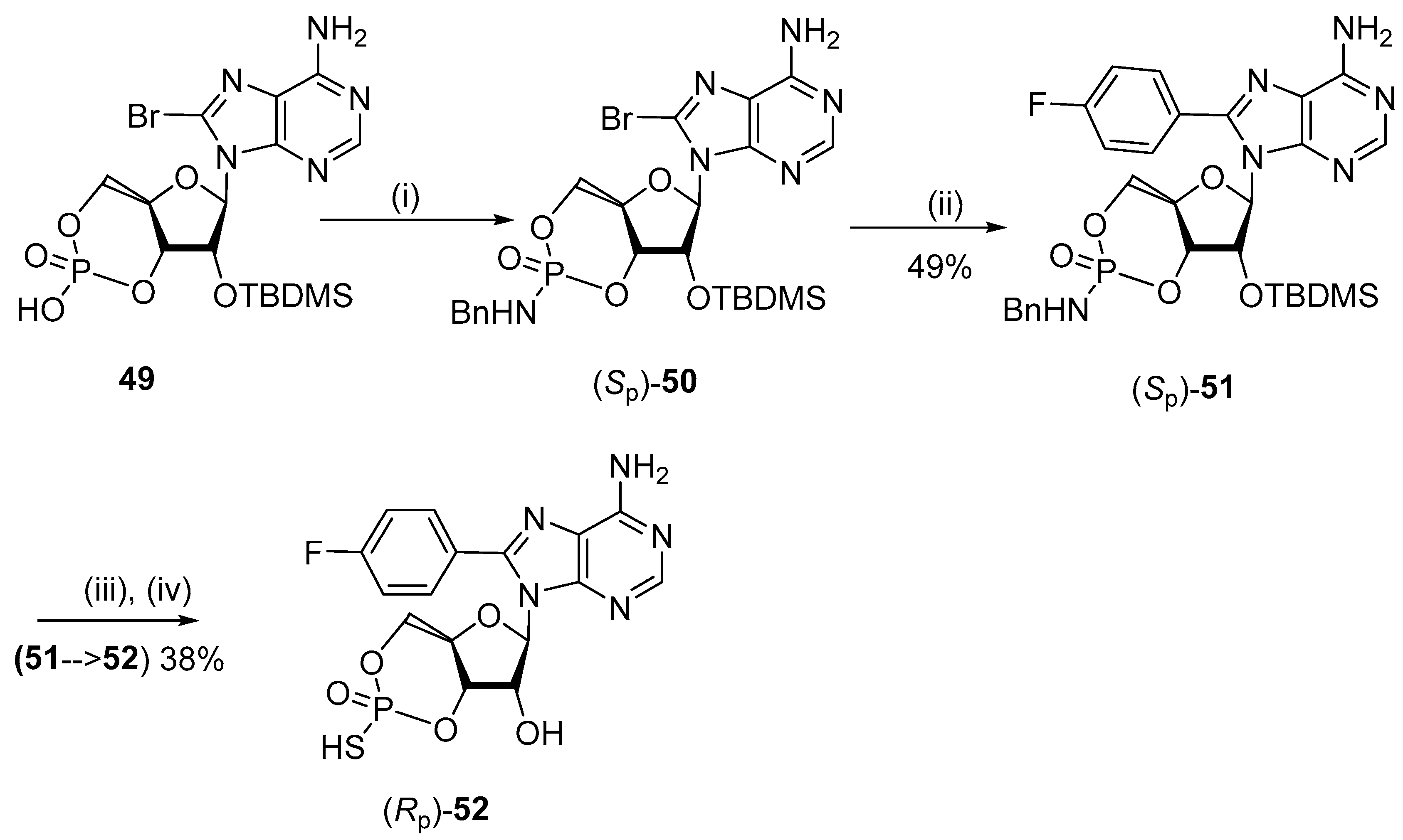
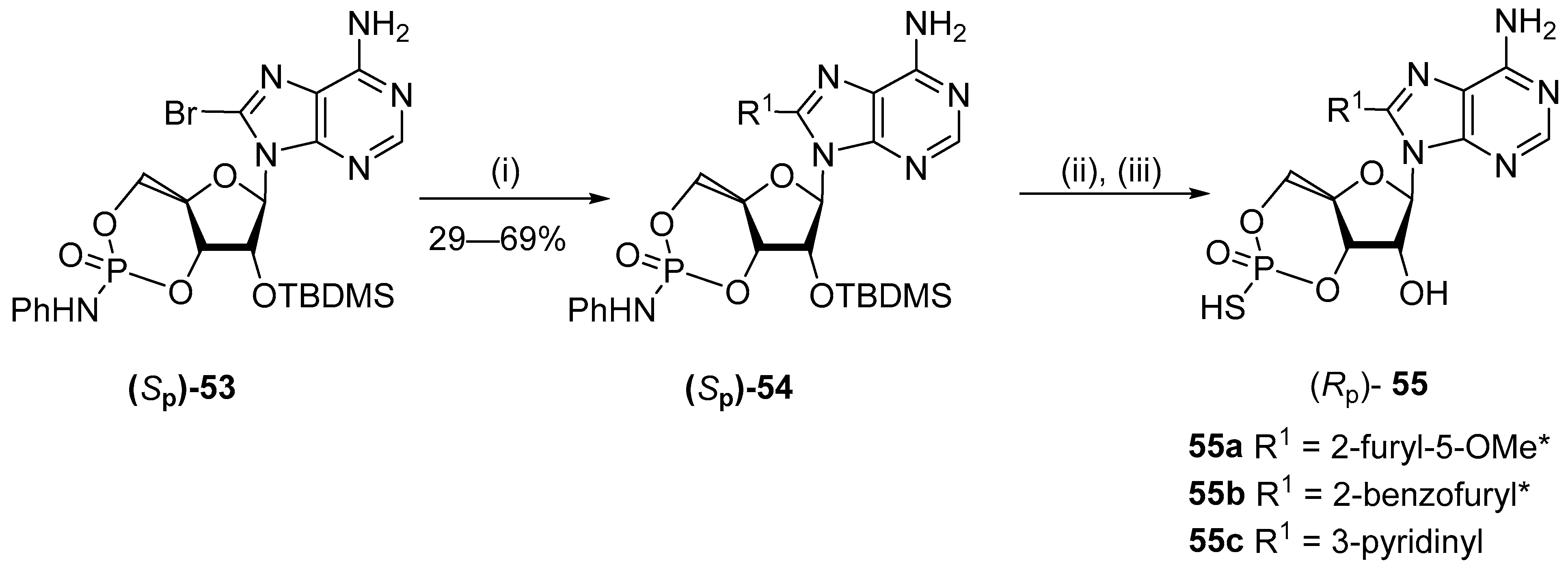
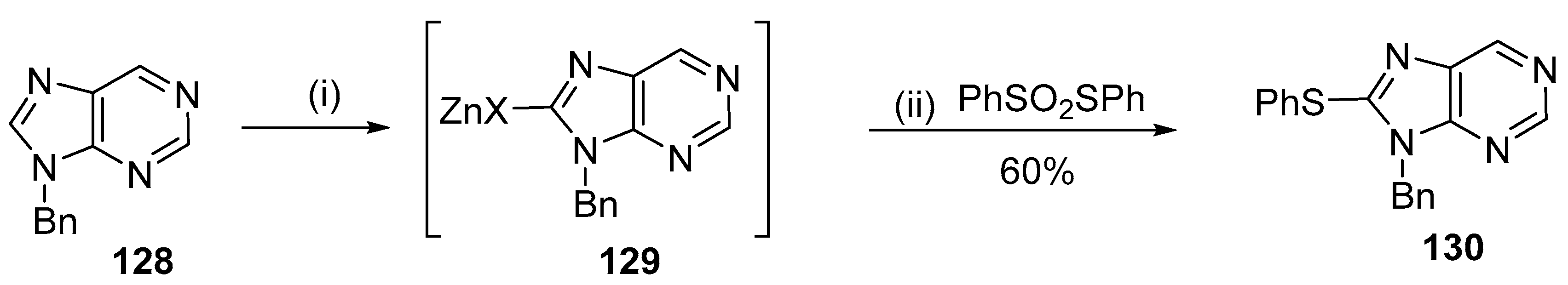
5.3. C8-Sulfenyl Derivatives
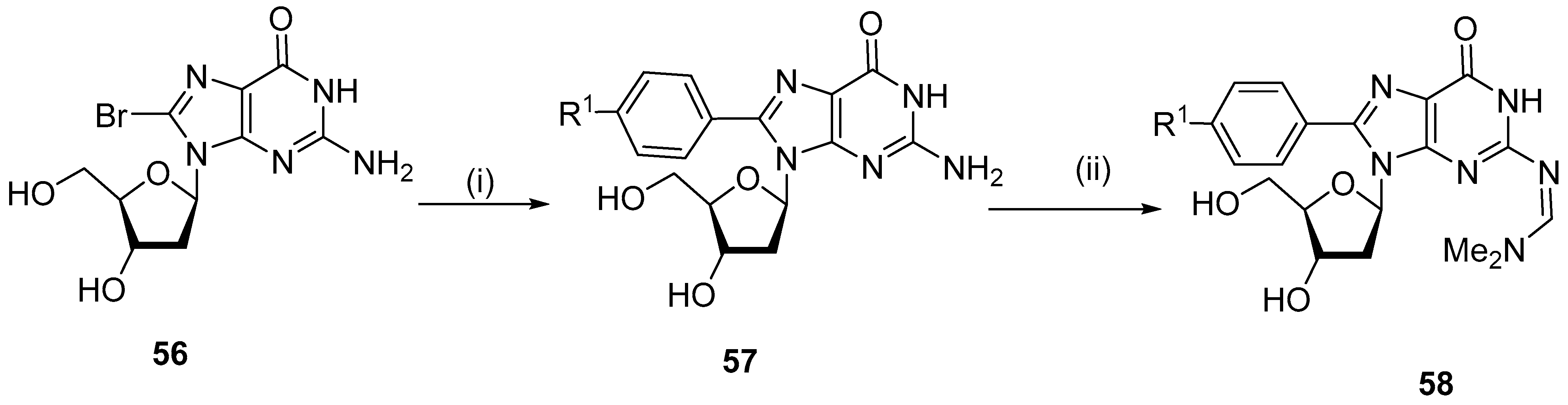
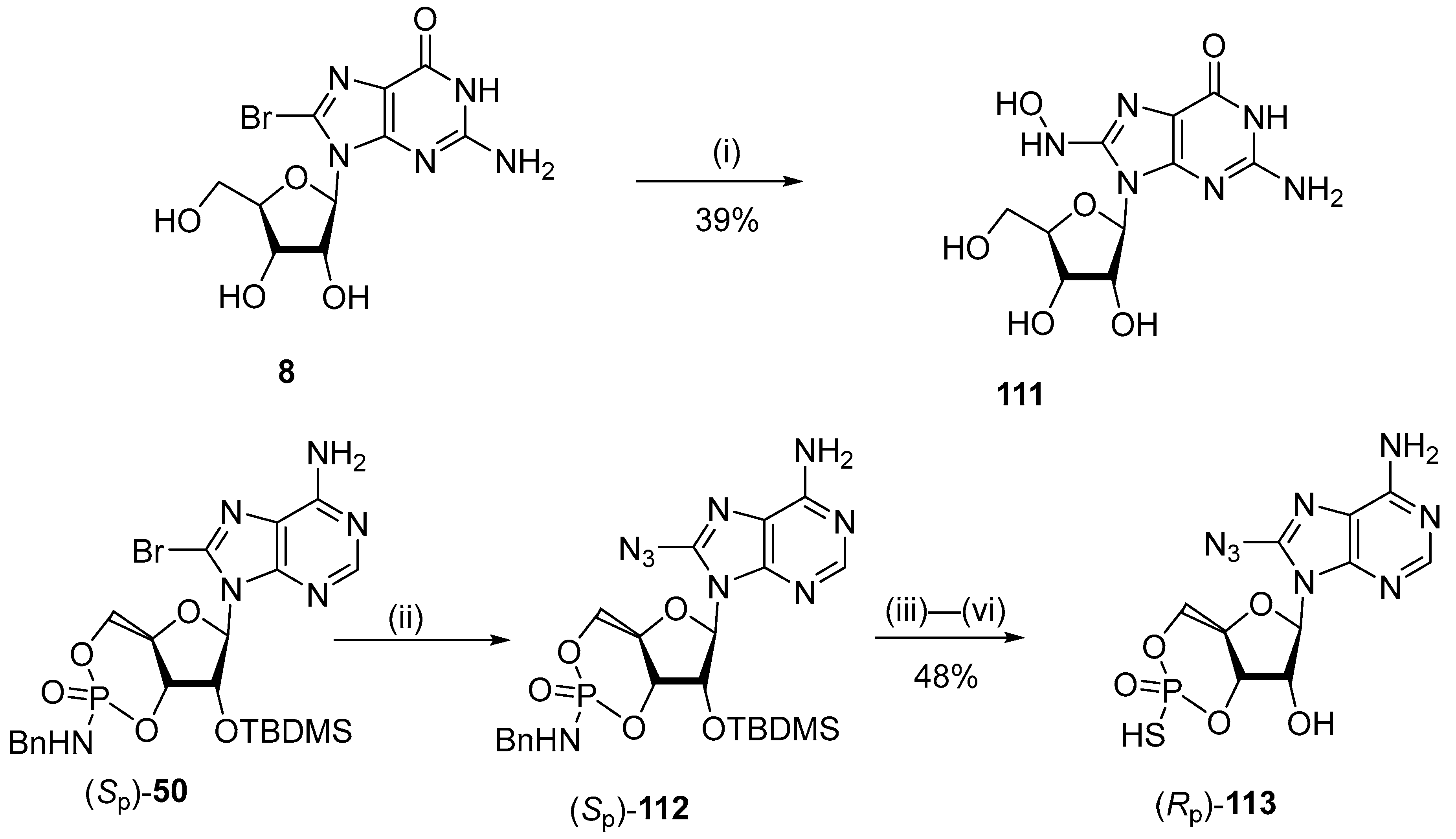
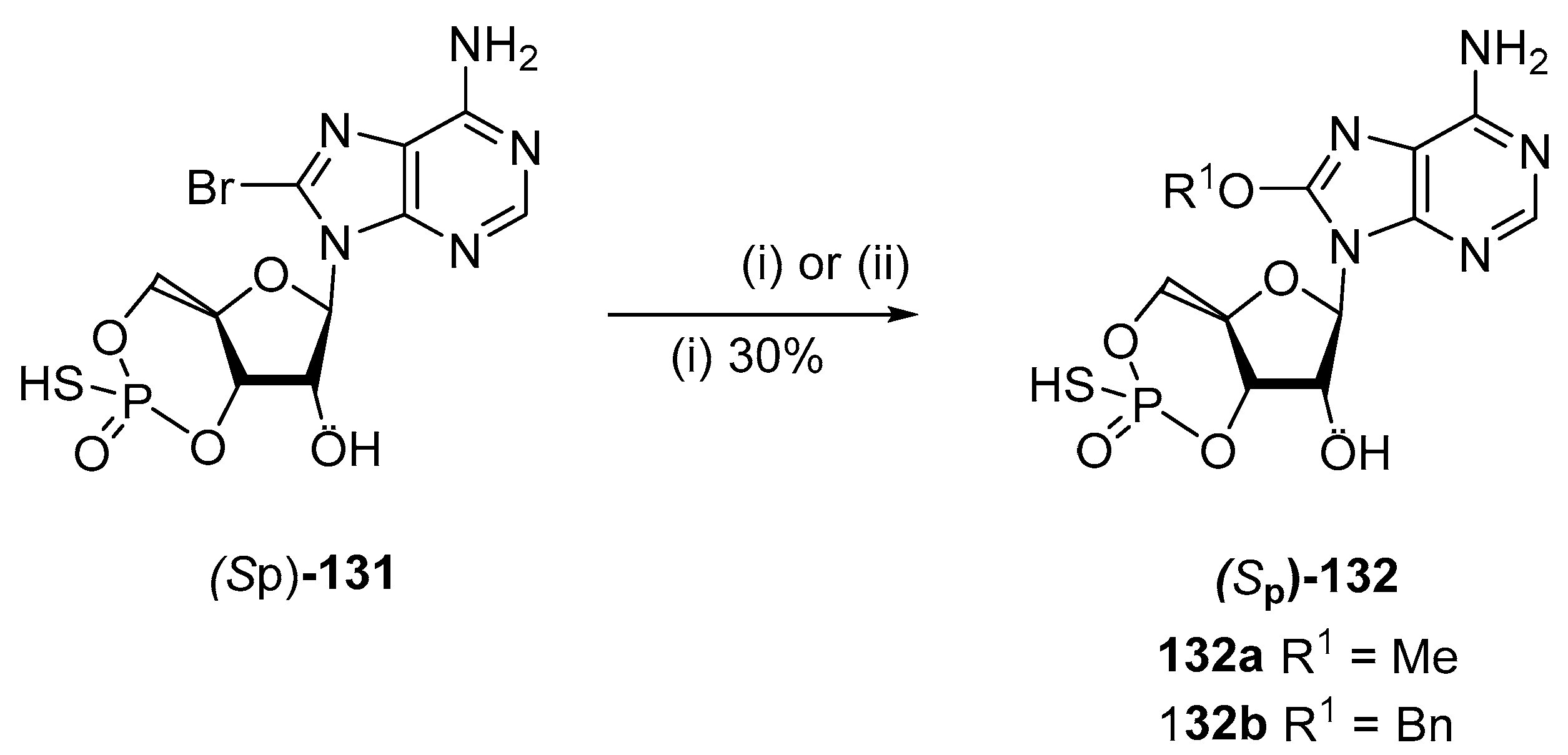
5.4. C8-Hydroxy Derivatives
6. Conclusions
Funding
Conflicts of Interest
References
- Andrei, M.; Bjørnstad, V.; Langli, G.; Rømming, C.; Klaveness, J.; Taskén, K.; Undheim, K. Stereoselective preparation of (Rp)-8-hetaryl-3′,5′-cyclic phosphorothioic acids. Org. Biomol. Chem. 2007, 54, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.K.; Seley-Rathke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Paert II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Haag, B.; Mosrin, M.; Ila, H.; Malakhov, V.; Knochel, P. Regio- and Chemoselective Metalation of Arenes and Heteroarenes Using Hindered Metal Amide Bases. Angew. Chem. Int. Ed. 2011, 50, 9794–9824. [Google Scholar] [CrossRef] [PubMed]
- Undheim, K. Hetarylzinc Cross-Coupling Reactions, Science of Synthesis; Molander, G.A., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany; New York, NY, USA, 2013; pp. 717–761. [Google Scholar]
- Hayakawa, H.; Haraguchi, K.; Tanaka, H.; Miyasaki, T. Direct C-8 Lithiation of Naturally-Occurring Purine Nucleosides. A Simple method for the Synthesis of 8-Carbon-Substituted Purine Nucleosides. Chem. Pharm. Bull. 1987, 35, 723–779. [Google Scholar] [CrossRef][Green Version]
- Van Aerschot, A.A.; Mamos, P.; Weyns, N.J.; Ikeda, S.; De Clercq, E.; Herdewijn, P.A. Antiviral Activity of C-Alkylated Purine Nucleosides Obtained by Cross-Coupling with Tetraalkyltin Reagents. J. Med. Chem. 1993, 36, 2938–2942. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Kitade, Y.; Kanbe, Y.; Maski, Y. Convenient Method for the Synthesis of C-Alkylated Purine Nucleosides: Palladium Catalyzed Cross-Coupling Reactions of Halogenopurine Nucleosides with Trialkylaluminums. J. Org. Chem. 1992, 57, 5268–5270. [Google Scholar] [CrossRef]
- Manfredini, S.; Baraldi, P.G.; Bazzanini, R.; Marangoni, M.; Simoni, D.; Balzarini, J.; De Clercq, E. Synthesis and Cytotoxic Activiyty of 6-Vinyl and 6-Ethynyluridine and 8-Vinyl- and 8-Ethynyladenosine. J. Med. Chem. 1995, 38, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.; Gerez, C.; Tritsch, D.; Fontecave, M.; Biellmann, J.-F.; Burger, A. Synthesis of 8-vinyladenosine 5′-di- and 5′-triphosphate: Evaluation of the diphosphate compound on ribonucleotide reductase. Tetrahedron 2003, 59, 7315–7322. [Google Scholar] [CrossRef]
- Firth, A.C.; Fairlamb, I.J.S.; Darley, K.; Bauman, C.G. Sonogashira alkynylation of unprotected 8-brominated adenosines and guanosines: Fluorescence properties of compact conjugated acetylenes containing a purine ring. Tetrahedron Lett. 2006, 47, 3529–3533. [Google Scholar] [CrossRef]
- Ibrahim, N.; Chevot, F.; Legraverend, M. Regioselective Sonogashira cross-coupling reactions of 6-chloro-2,8-diiodo-9-THP-9H-purine with alkyne derivatives. Tetrahedron Lett. 2011, 52, 305–307. [Google Scholar] [CrossRef]
- Sági, G.; Ötvös, L.; Ikeda, S.; Andrei, G.; Snoeck, R.; De Clerq, E. Synthesis and Antiviral Activities of 8-Alkynyl-, 8-Alkenyl-, and 8- Alkyl-2′-deoxyadenosine Analogues. J. Med. Chem. 1994, 37, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Volpini, R.; Costanci, S.; Lambertucci, C.; Vittori, S.; Klotz, K.-N.; Lorenzen, A.; Cristalli, G. Introduction of Alkynyl Chains on C-8 of Adenosine Led to Very selective Antagonists of the A3 Adenosine Receptor. Bioorg. Med. Chem. Lett. 2001, 11, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Storr, T.E.; Baumann, C.G.; Thatcher, R.J.; De Ornellas, S.; Whitwood, A.C.; Fairlamb, I.J.S. Pd(0)/Cu(I)-Mediated Direct Arylation of 2′-Deoxyadenosines: Mechanistic Role of Cu(I) and Reactivity Comparisons with Related Purine Nucleosides. J. Org. Chem. 2009, 74, 5810–5821. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.; Wagner, G. A facile two-step synthesis of 8-arylated guanosine mono- and triphosphates (8-aryl GXPs). Org. Biomol Chem. 2006, 4, 4526–4532. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.; Wagner, G.R. Suzuki-Miyaura Cross-Coupling of Unprotected Halopurine Nucleosides in Water-Influence of Catalyst and Cosolvent. Synth. Commun. 2006, 36, 3713–3721. [Google Scholar] [CrossRef]
- Western, E.C.; Daft, J.R.; Johnson, E.M.; Gannett, P.M.; Shoughnessay, K.H. Efficient One-Step Suzuki Arylation of Unprotected Halonucleosides, Using Water-Soluble Palladium Calalysts. J. Org. Chem. 2003, 68, 6767–6774. [Google Scholar] [CrossRef] [PubMed]
- Zilberstein, L.; Silberman, A.; Fischer, B. 8-(p-CF3-cinnamyl)-modified purine nucleosides as promising fluorescent probes. Org. Biomol. Chem. 2011, 9, 7763–7773. [Google Scholar] [CrossRef] [PubMed]
- Undheim, K. cAMPS derivatives. A minireview over synthetic medicinal chemistry. Bioorg. Chem. 2019, 91, 103152. [Google Scholar] [CrossRef] [PubMed]
- Vongsutilers, V.; Daft, J.R.; Shaughnessy, K.H.; Gannett, P.M. A General Synthesis of C8-Arylpurine Phosphoramidites. Molecules 2009, 14, 3339–3352. [Google Scholar] [CrossRef] [PubMed]
- Akula, H.K.; Bae, S.; Pradhan, P.; Yang, L.; Zajc, B.; Lakshman, M.K. Diversely C8-functionalized adenine nucleosides via their underexplored carboxaldeshydes. Chem. Commun. 2022, 58, 1744–1747. [Google Scholar] [CrossRef]
- Zimdars, S.; du Jourdin, X.M.; Crestey, F.; Carell, T.; Knochel, P. Trifunctionalization of the Purine Scaffold Using Mg and Zn Organometallic Intermediates. Org. Lett. 2011, 13, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Undheim, K.; Taskén, K.; Klaveness, J.; Langli, G.; Bjørnstad, V. Purine Nucleotide Derivatives. WO 2005/123755 29 December 2005. [Google Scholar]
- Gundersen, L.L. Synthesis of purinecarbonitriles by Pd(0)-catalyzed coupling of halopurines with Zinc cyanide. Acta Chem. Scand. 1996, 50, 58–63. [Google Scholar] [CrossRef]
- Crestey, F.; Zimdars, S.; Knochel, P. Regioselective Functionalization of Purine Derivatives at Position 8 and 6 Using Hindered TMP-Amide Bases of Zn and Mg. Synthesis 2013, 45, 3029–3037. [Google Scholar] [CrossRef]
- Dagousset, G.; Francois, C.; León, T.; Blanc, R.; Sansiaume-Dagousset, E.; Knochel, P. Preparation of Functionalized Lithium, Magnesium, Aluminum, Zinc, Manganese, and Indium Organometallics from Functionalized Organic Halides. Synthesis 2014, 46, 3133–3171. [Google Scholar] [CrossRef]
- Hess, A.; Alandini, N.; Guelen, H.C.; Prohaska, J.P.; Knochel, P. Regioselective magnesiations of functioalized arenes and hetarenes using TMP 2 Mg in hydrocarbons. Chem. Commun. 2022, 58, 8774–8777. [Google Scholar] [CrossRef] [PubMed]
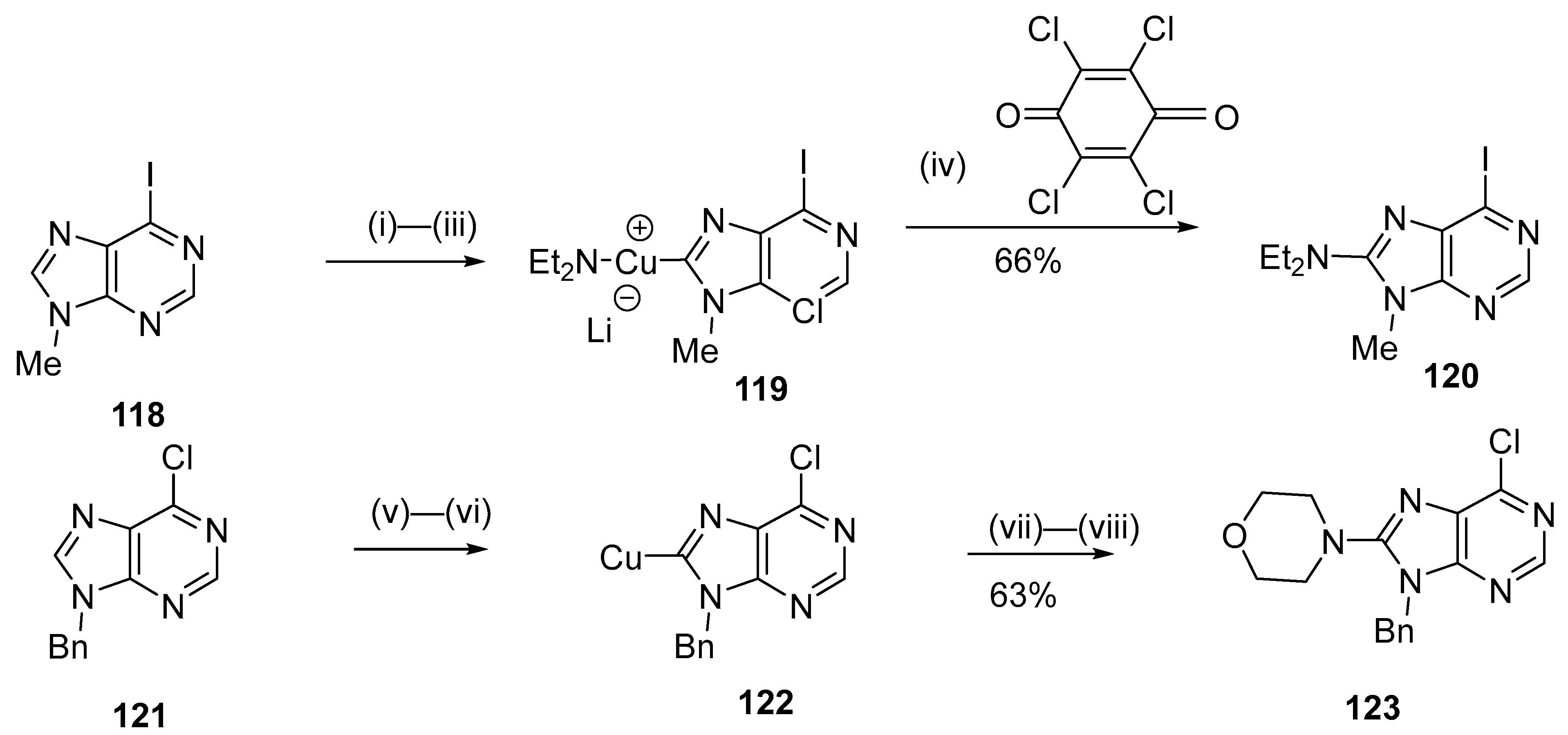
- Brackemeyer, D.; Hervé, A.; Schulte, C.; Brinke, M.; Jahnke, M.C.; Hahn, F.E. A Versatile Methodology for the Regioselective C8-Metalation of Purine Bases. J. Am. Chem. Soc. 2014, 136, 7841–7844. [Google Scholar] [CrossRef] [PubMed]
- Kampert, F.; Brackemeyer, D.; Tan, T.T.; Hahn, F.E. Selective C8-Metalation of Purine Nucleosides via Oxidative Addition. Organometallics 2018, 37, 4181. [Google Scholar] [CrossRef]
- Hocek, M.; Havelkova, M.; Dvorak, D. The Suzuki-Miyaura Cross-Coupling Reactions of 2-, 6- or 8-Halopurines with Boronic Acids Leading to 2-, 6-, or 8-Aryl- and –Alkenylpurine Derivatives. Synthesis 2004, 11, 2869–2876. [Google Scholar] [CrossRef]
- Butora, G.; Schmitt, C.; Levorse, D.A.; Streckfuss, E.; Doss, G.A.; MacCoss, M. The elusive 8-fluoroadenosine: A simple non-enzymatic synthesis and characterization.Regioselective Fluorination. Tetrahedron 2007, 63, 3782–3789. [Google Scholar] [CrossRef]
- Barrio, M.; Namavari, M.; Phelps, M.E.; Saytyamurthy, N. Regioselective Fluorination on treating Guanines with Dilute F2: A Facile Entrance to 8-Fluoroguanine Derivatives. J. Org. Chem. 1996, 61, 6084–86085. [Google Scholar] [CrossRef] [PubMed]
- Barrio, J.R.; Namavari, M.; Phelps, M.E.; Satyamurthy, N. Elemental Fluorine to 8-Fluoropurines in One Step. J. Am. Chem. Soc. 1996, 118, 10408–10411. [Google Scholar] [CrossRef]
- Liu, J.; Barrio, J.R.; Satyamurthy, N. Kinetics and mechanism of the defluorination of 8-fluoropurine nucleosides in basic and acidic media. J. Fluorine Chem. 2006, 127, 1175–1187. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Lagisetty, P.; Zajc, B. Direct Synthesis of 8-Fluoro Purine Nucleosides via Metalation-Fluorination. J. Org. Chem. 2007, 72, 8222–8226. [Google Scholar] [CrossRef] [PubMed]
- Long, R.A.; Robins, R.V.; Townsend, L.B. Purine Nucleosides. XV. The Synthesis of 8-Amino- and 8-Substituted Aminopurine Nucleosides. J. Org. Chem. 1967, 32, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
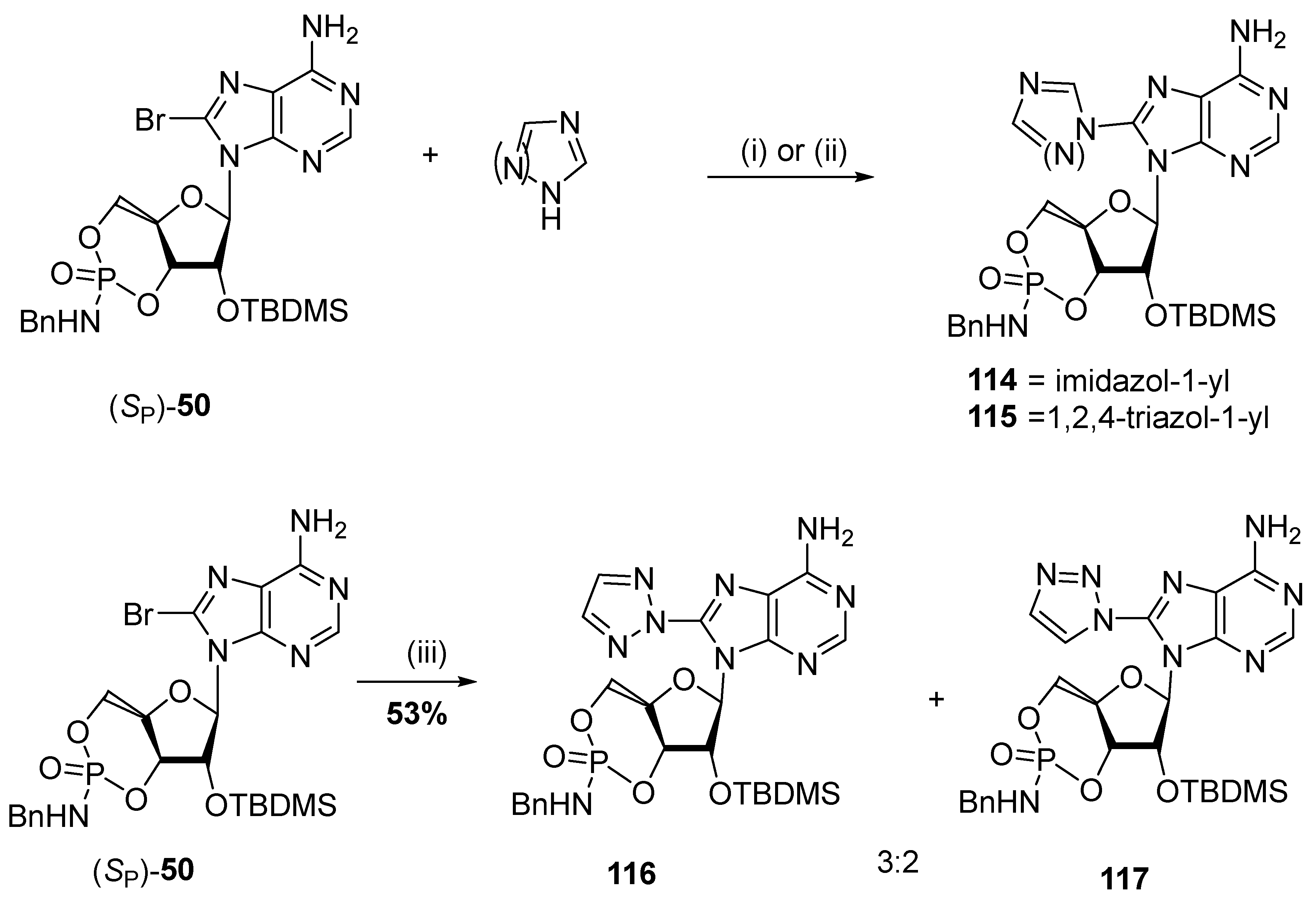
- Andrei, M.; Undheim, K. Azolo substitution into the purine scaffold in nucleoside cyclic 3′,5′-phosphorothioates. Monatshefte Fürchemie Chem. Mon. 2022, 153, 1213–1223. [Google Scholar] [CrossRef]
- Boudet, N.; Dubbaka, S.; Knochel, P. Oxidative Amination of Cuprated Pyrimidine and Purine Derivatives. Org. Lett. 2008, 10, 1715–1718. [Google Scholar] [CrossRef] [PubMed]
- Schoffers, E.; Olsen, P.D.; Means, J.C. Synthesis of C8-Adenosine Adducts of Arylamines Using Palladium Catalysis. Org. Lett. 2001, 3, 4221–4223. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Zhang, T.; Ono, K.; Tsutsuki, H.; Ida, S.; Akashi, S.Z.; Miyata, K.; Oike, Y.; Akaike, T.; Sawa, T. Synthesis and Characterization of 8-Nitroguanosine 3′,5′-Cyclic Monophosphorothioate Rp-Isomer as a Potent Inhibitor of Protein Kinase G1a. Biol. Pharm. Bull. 2017, 40, 365–374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schwede, F.; Bertinetti, D.; Langerijs, C.N.; Hadders, M.A.; Wienk, H.; Ellenbroek, J.H.; de Koning, E.J.P.; Bos, J.L.; Herberg, F.W.; Genieser, H.-G.; et al. Structure-Guided Design of Selected Epac1 and Epac2 Agonists. PLoS Biol. 2015, 13, 100203. [Google Scholar] [CrossRef] [PubMed]






































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Undheim, K. Bond Formation at C8 in the Nucleoside and Nucleotide Purine Scaffold: An Informative Selection. Molecules 2024, 29, 1815. https://doi.org/10.3390/molecules29081815
Undheim K. Bond Formation at C8 in the Nucleoside and Nucleotide Purine Scaffold: An Informative Selection. Molecules. 2024; 29(8):1815. https://doi.org/10.3390/molecules29081815
Chicago/Turabian StyleUndheim, Kjell. 2024. "Bond Formation at C8 in the Nucleoside and Nucleotide Purine Scaffold: An Informative Selection" Molecules 29, no. 8: 1815. https://doi.org/10.3390/molecules29081815
APA StyleUndheim, K. (2024). Bond Formation at C8 in the Nucleoside and Nucleotide Purine Scaffold: An Informative Selection. Molecules, 29(8), 1815. https://doi.org/10.3390/molecules29081815